

Abstract
Mitochondrial dysfunction has been implicated in the development of heart failure. Oxidative metabolism in mitochondria is the main energy source of the heart, and the inability to generate and transfer energy has long been considered the primary mechanism linking mitochondrial dysfunction and contractile failure. However, the role of mitochondria in heart failure is now increasingly recognized to be beyond that of a failed power plant. In this Review, we summarize recent evidence demonstrating vicious cycles of pathophysiological mechanisms during the pathological remodeling of the heart that drive mitochondrial contributions from being compensatory to being a suicide mission. These mechanisms include bottlenecks of metabolic flux, redox imbalance, protein modification, ROS-induced ROS generation, impaired mitochondrial Ca2+ homeostasis, and inflammation. The interpretation of these findings will lead us to novel avenues for disease mechanisms and therapy.
The heart is an organ with high energy demands. The majority of the ATP consumed by the heart (~95%) is derived from oxidative metabolism in the mitochondria, organelles that occupy approximately one-third of the volume of adult cardiomyocytes and are colloquially known as the “powerhouses” of the cell. Mitochondrial dysfunction has been widely observed in the failing heart irrespective of etiology. Although this dysfunction is recognized as a maladaptive response, specific mechanisms connecting mitochondrial dysfunction and the development or progression of heart failure are complex and are not fully understood. Initially, decreased energy supply is considered the main consequence of mitochondrial dysfunction. However, recent advances have identified a number of mechanisms that contribute to the pathogenesis of heart failure beyond the notion of “power plant” failure. Here we will review some of the development in this area with an emphasis on the crosstalk between impaired bioenergetics and the maladaptive signaling circuit triggered by mitochondrial dysfunction in the pathophysiology of heart failure.
Overview of mitochondrial function
Mitochondria are double-membraned organelles found in nearly all eukaryotic cells. A primary function of the mitochondrion is to generate ATP through oxidative phosphorylation. Oxidative phosphorylation takes place in the inner mitochondrial membrane, during which reducing equivalents, e.g., NADH and FADH2, are transferred from the carrier molecules to the electron transport chain (ETC) while protons are pumped to the intermembrane space. Because the inner mitochondrial membrane is impermeable to most ions and small molecules, proton pumping generates a membrane potential that is used to convert ADP to ATP by the ATP synthase. Thus, the inner membrane impermeability and the mitochondrial membrane potential are critical properties of functioning mitochondria.
To maintain oxidative phosphorylation, a variety of carbon substrates are metabolized via specific pathways that eventually converge on the tricarboxylic acid (TCA) cycle to produce reduced equivalents, e.g., NADH and FADH2. Oxidative phosphorylation also generates reactive oxygen species (ROS), which were first considered by-products but were later found to possess many (patho)physiological functions (Figure 1). Moreover, oxidative metabolism in the mitochondria is not limited to ATP generation. In certain cell types, such as brown adipocytes, uncoupling proteins use the mitochondrial membrane potential for thermogenesis (1). Intermediary metabolism in the mitochondria provides metabolites for multiple biological processes, such as biosynthesis and protein modifications (refs. 2, 3, and Figure 1). Furthermore, mitochondria regulate cell signaling by modulating redox state, supplying cofactors for biochemical reactions and generating ligands for signaling transduction (4–6).
Figure 1. An overview of mitochondrial function in health and disease.
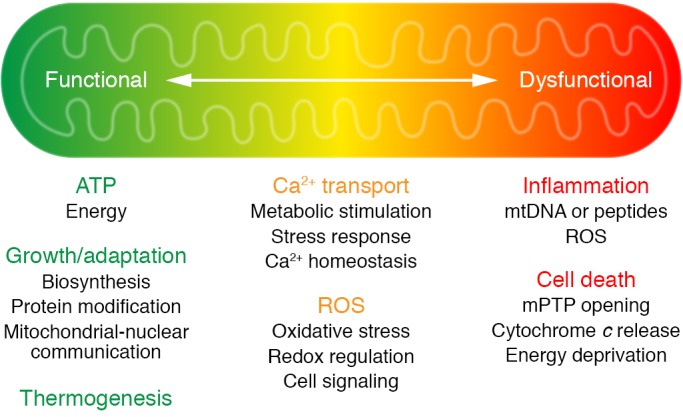
Mitochondria are known as the powerhouse of the cell. Under normal conditions, oxidative metabolism in mitochondria produces ATP; it also produces heat in certain specialized cell types, such as brown adipocytes. In addition to generating ATP, intermediate metabolism in the mitochondria produces metabolites for biosynthesis, protein modification, and signal transduction. Oxidative phosphorylation is coupled with generation of reactive oxygen species (ROS), which can either serve as molecular signals or cause cell damage and cell death. Mitochondrial metabolism is stimulated by calcium, but under pathological conditions, calcium overload can trigger the opening of the mitochondrial permeability transition pore (mPTP). The release of mitochondrial content, such as cytochrome c, induces apoptosis, or the loss of membrane potential (a consequence of prolonged mPTP opening) causes ATP deprivation and necrosis. Leak of damage-associated molecular patterns (DAMPs), such as mitochondrial DNA and peptides, or excessive ROS generation also causes inflammation that results in further tissue damage. The transition of mitochondria from a powerhouse to a death engine is key to the pathogenesis of many diseases, including heart failure (also see Figure 3).
Mitochondria possess active calcium transport systems, and multiple enzymes in the oxidative metabolism pathways are activated by calcium. Thus, calcium is an important second messenger connecting contractile function and mitochondrial metabolism (7–11). Moreover, mitochondrial uptake of calcium has been proposed as a mechanism that maintains intracellular calcium homeostasis (12). The identification of mitochondrial calcium uniporter (MCU) in 2011 (13, 14) stimulated an array of investigations on the role of mitochondrial calcium in health and diseases.
The roles of mitochondria are not limited to supporting life; rather, they are also actively involved in initiating cell death. Stress conditions that lead to calcium or ROS overload trigger the opening of the mitochondrial permeability transition pore (mPTP), leading to loss of mitochondrial membrane potential (15–18). This results in failure to produce ATP and release of mitochondrial proteins such as cytochrome c, which trigger cell death through necrotic and apoptotic pathways, respectively (refs. 16, 17, 19, and Figure 1). It has been shown that mitochondria-initiated cell death is an important mechanism in heart failure (20), and the links between mitochondrial calcium content and cardiac dysfunction during chronic stress have been a focus of investigation over the past decade. Mitochondrial DNA and/or ROS have been shown to be triggers of inflammatory response (21–24). Regulation of innate immunity by mitochondrial function has been increasingly recognized in both cardiac and noncardiac diseases (21–25). As sterile inflammation is common in heart failure, the role of mitochondrial function has also emerged as an important pathogenic mechanism, which will be discussed in detail below. Collectively, these observations suggest that molecular mechanisms promoting the transition of mitochondria from energy-producing to death-initiating functions (Figure 1) are key to identifying disease mechanisms and therapeutic targets.
Mitochondrial dysfunction and energy starvation in heart failure
If ATP synthesis were ceased in a healthy human heart, stored ATP would sustain heartbeat for only a few seconds (26). It is therefore essential that the rate of ATP consumption is matched with the rate of ATP synthesis on a beat-to-beat basis. This is accomplished by oxidative metabolism in mitochondria using fatty acids as the primary fuel (27, 28). During pathological heart remodeling, cardiac metabolism is reprogrammed toward increased reliance on glucose with a significant increase of glycolysis, whereas fatty acid oxidation is downregulated (29). Notably, ATP generated from glycolysis alone contributes less than 5% of the total ATP consumed in a normal adult heart (29). Thus, upregulating glycolysis is not an effective method of increasing energy supply.
On the energy demand side, pathological remodeling increases energy expenditure through unfavorable cardiac geometry, increased neurohormonal stimulation, and impaired calcium handling (30, 31). Collectively, these changes disturb the homeostasis between energy supply and demand, resulting in a stress on myocardial energetics (Figure 2). The hypothesis that a failing heart is energy starved is consistent with such a scenario and has inspired decades of research to date (refs. 28, 32–34). Clinical evidence supporting the energy starvation hypothesis in heart failure comes from the observation that therapeutic measures to reduce energy consumption, such as vasodilators and beta blockers, improve survival in heart failure, while treatments that increase energy demand of the heart, such as inotropes, worsen the outcome (refs. 35–37 and Figure 2). Few strategies of increasing ATP supply have been tried clinically thus far (38, 39).
Figure 2. Mismatch of energy demand and generation drives the development of heart failure.

In a healthy heart, energy production meets energy demand on a beat-by-beat basis. Pathological remodeling of the heart results in inefficiencies that increase energy demand but concomitantly reduce the capacity for energy supply. The subsequent metabolic remodeling in an attempt to regain energy homeostasis temporarily sustains the ATP level in the heart but likely drives the heart to failure via maladaptive circuits that produce mitochondrial stress. Current heart failure therapy aims at reducing the energy demand to alleviate the mismatch. Strategies that antagonize metabolic remodeling and/or mitochondrial stress signaling cascade could offer novel therapies. FAO, fatty acid oxidation.
The high-energy phosphate content of the failing heart is reduced. This is first manifested as a decrease of the energy reserve compound phosphocreatine (PCr) resulting in a lower PCr/ATP ratio (40–43). It is puzzling, however, that the ATP content in a failing heart is largely maintained until the end stage (40, 42–45) despite the early signs of a mismatch between energy supply and demand. One interpretation of this observation is that myocardial energy status is sustained by “adaptive” mechanisms mobilized during the progression of heart failure (46, 47). The nature of these mechanisms is poorly understood. Whether they are ultimately adaptive warrants reevaluation. As the stressed heart goes on a downward spiral to failure despite a relatively stable ATP level, it raises a possibility that mechanisms used during cardiac remodeling to restore energy homeostasis, which are either originating from or targeted to mitochondria, contribute to the vicious cycles that drive cardiac remodeling to heart failure (Figure 2). This leads to the question of whether the demise of the stressed heart is caused by energy starvation or the effort to fight starvation. In sections below, recent studies examining the metabolic and/or mitochondrial responses in the chronically stressed heart are reviewed to determine whether efforts to sustain energy homeostasis can be potentially costly to the failing heart and thus accelerate heart failure. These areas are also potential therapeutic opportunities (Figure 2).
Bottleneck of metabolic flux in heart failure
Cardiomyocytes possess a high capacity for oxidative metabolism and are capable of using a variety of carbon substrates for ATP synthesis. In a normal heart, the flux through intermediate metabolism and oxidative phosphorylation can increase substantially to meet energy demand during high-performance activities such as exercise (48, 49). The heart is also metabolically flexible, able to adapt to fuel availability by switching its substrate preference (27). If this is the case, what are the bottlenecks that render failing hearts incapable of using these mechanisms to maintain energy homeostasis during chronic stress?
A hallmark of metabolic remodeling in pathological hypertrophy is downregulation of fatty acid oxidation and increased utilization of glucose. Prior studies showed that a greater reliance on glucose in the failing heart led to increased glycolysis that had been uncoupled from oxidation, causing increases in lactate production and anaplerosis (29, 50). It also drives a greater flux of glucose into accessory pathways, all of which reduce the efficiency of ATP synthesis and exacerbate pathological remodeling (27, 29, 50). Promoting glucose oxidation, either directly or by inhibiting fatty acid oxidation, has been proposed as a therapeutic strategy (28, 51). Partly replacing fatty acids by glucose for energy provision was considered beneficial because ATP generation from glucose could moderately increase oxygen efficiency (52, 53). However, the capacity for glucose-derived ATP production is rather limited in adult hearts (27). Increased glucose uptake and metabolism also enables growth signaling that promotes the pathological remodeling of the heart. A recent study showed that high intracellular glucose inhibits branched-chain amino acid (BCAA) catabolism, resulting in BCAA accumulation during the development of cardiac hypertrophy (54). Such a metabolic response is required for mTOR activation and cardiomyocyte hypertrophy. These findings imply that shifts in substrate preference to glucose support growth at the cost of contractile performance. In support of this notion, enhancing glucose uptake capacity to a superphysiological level was shown to improve myocardial energetics as well as stimulate pathological hypertrophy (55, 56).
Fatty acid is the predominant fuel for the adult heart. Its long carbon chain makes fatty acid the most effective substrate for energy provision. Downregulation of fatty acid oxidation pathways and accumulation of incompletely oxidized fatty acids were observed in an early stage of heart failure, suggesting a mismatch between fatty acid supply and oxidation (57). Promoting fatty acid utilization has been shown to be beneficial in a number of models of heart failure (58–60). However, the benefit of sustaining fatty acid oxidation is likely beyond energy supply. Recent evidence suggests that sustaining fatty acid oxidation during pathological hypertrophy prevents increased reliance on glucose and thus suppresses the effects of glucose on pathological growth (58, 61). Inability to oxidize fatty acids under conditions of increased lipid availability, such as obesity and diabetes, could lead to accumulation of lipotoxic metabolites (62). Moreover, incomplete fatty acid oxidation has been implicated in the development of insulin resistance in skeletal muscle, although similar studies in the heart are lacking (63, 64).
More recently, it was observed that ketone body oxidation was increased in the failing hearts of both human and animal models (65, 66). This increase is associated with elevated circulating ketone levels in patients with heart failure (65, 67), suggesting a change in systemic metabolism. While emerging work suggests that increased ketone body oxidation is cardioprotective (68), its underlying mechanisms remain to be elucidated. Besides providing energy to compensate for reduced fatty acid oxidation, other potential mechanisms have been proposed as benefits of increased ketone body utilization, including modulation of oxidative stress and posttranslational modifications (68–70). Furthermore, oxidation of fatty acids or ketone body reduces glucose use by the heart (58, 71, 72); thus, the benefit of increasing ketone body utilization in the failing heart could also be attributed to reduced reliance on glucose.
Mitochondrial oxidation reduction and protein modification
Mitochondrial proteins can be modified through acylation of lysine residues by thioester-CoAs, such as acetyl-CoA, succinyl-CoA, malonyl-CoA, etc., produced by intermediary metabolism (73). The most studied modification is acetylation, although modifications by other acyl-CoAs also occur in the mitochondria (Figure 3). Increased mitochondrial protein acetylation has been found in the failing hearts of animal models and patients (74–78). A large number of proteins, including those involved in substrate oxidation, e.g., pyruvate dehydrogenase and fatty acid oxidation enzymes, TCA cycle enzymes, and ETC proteins, have been shown to be hyperacetylated (75, 76). Protein hyperacetylation has been shown to decrease activities of succinate dehydrogenase, pyruvate dehydrogenase, ATP synthase, and malate-aspartate shuttle enzymes; additionally, hyperacetylation of oligomycin sensitivity–conferring protein (OSCP) increased the sensitivity to mPTP opening (75, 76, 78–82). Moreover, inhibition of malate-aspartate shuttle by acetylation impairs the transport of cytosolic NADH into mitochondria, thereby disrupting cytosolic redox state and glycolytic ATP production during the transition from cardiac hypertrophy to failure (76, 83).
Figure 3. Maladaptive mechanisms connecting mitochondrial dysfunction and progression of heart failure.

Inadequate stimulation of mitochondrial metabolism increases ATP generation at the expense of triggering maladaptive responses such as imbalance among substrate supply, catabolism, and oxidative phosphorylation (OXPHOS), as well as increased protein modifications by acylation such as acetylation (LysAc). Increased availability of acyl-CoA is a driver for protein modification, while the mismatch between NADH production and oxidation decreases the NAD+/NADH ratio, compromising the sirtuin deacetylase function. These effects collectively increase protein acetylation in the failing heart. Increased protein acylation, especially acetylation, impairs energy metabolism through negative feedback to substrate metabolism and OXPHOS. Further stimulation of mitochondrial metabolism under these conditions increases the risk of calcium overload, leads to greater ROS generation, and induces mPTP opening. Increased protein acetylation also weakens antioxidant defense and sensitizes the mPTP to calcium or ROS. In the face of increased oxidative stress, effort to maintain the mitochondrial antioxidant system (e.g., the Gpx or Prx pathway) may divert energy metabolism away from ATP generation through nicotinamide nucleotide transhydrogenase (Nnt). Oxidative damage causes ROS-induced ROS release, leading to further injury of mitochondria. Failure to remove the damaged mitochondria results in the leak of DAMPs, such as mitochondrial DNA or peptides, that trigger an inflammatory response. NAD+/NADH redox imbalance also promotes NLRP3 inflammasome activation. The vicious cycle of these mechanisms ultimately drives mitochondria from being energy-producing to death-initiating organelles.
The cause of mitochondrial protein hyperacetylation in the failing heart is less understood. One possibility is the presence of excessive acyl-CoAs. It has been shown that protein acylation can either be mediated by an acyltransferase or occur nonenzymatically (73, 84), and in the latter case, the concentration of the acyl-CoAs is an important determinant of the modification. Reduced fatty acid oxidation in failing hearts is associated with an increased level of short-chain acyl-CoAs in the myocardium (57, 66); thus, a mismatch in the supply and oxidation of acetyl-CoA may lead to increased protein acetylation (Figure 3).
Another potential mechanism of protein hyperacetylation is reduced protein deacetylation, which is catalyzed by the sirtuin family of NAD+-dependent deacetylases (Figure 3). Three sirtuin proteins are localized to the mitochondria: SIRT3, SIRT4, and SIRT5, among which SIRT3 is the predominant deacetylase (73). SIRT4 and SIRT5 have weak deacetylase function but possess demalonylase and desuccinylase activities or ADP-ribosylation function (73). Downregulation of SIRT3 has been shown in the failing heart (78, 85). Furthermore, sirtuin activity is dependent on NAD+ availability. Diminished NAD+ level and reduced NAD+/NADH ratio have been observed in hearts with mitochondrial dysfunction and/or pathological hypertrophy (75, 86, 87). A lower NAD+/NADH ratio decreases enzymatic activities for substrate catabolism, and decreased NAD+ level suppresses NAD+-dependent protein deacetylation, resulting in mitochondrial protein hyperacetylation and impaired function (88). These changes orchestrate a negative regulatory circuit that ultimately limits the overall metabolic flux and increases the mitochondrial susceptibility to stress (refs. 76, 87, and Figure 3). Increasing intracellular NAD+ level by pharmacological or genetic approaches normalizes the NAD+/NADH ratio, restores protein acetylation, and improves cardiac function in mouse models of heart failure (76, 87, 89).
There is, however, still a large gap in our knowledge of how acylation of each individual lysine residue of a specific protein contributes to its ultimate functional change. Most studies have not quantified the occupancy of the modification. Evaluation of the impact of acetylation on enzymatic activity has only been done with overexpression of acetyl-mimetic or acetyl-resistant mutations that replace the target lysine residue. Furthermore, regulation of metabolism by protein acylation likely operates through a network of proteins. The ultimate impact on metabolism may not be predicted from the functional change of an individual protein. For example, increased protein acetylation has been associated with opposite changes in fatty acid oxidation in different organs (90–92) or in different diseases (90, 93). Notably, a number of proteins also show decreased acetylation in heart failure (75), the significance of which is not understood. Furthermore, the observation of decreased NAD+/NADH ratio in the failing heart is initially counterintuitive to the concept of an energy-starved heart. It has been reported that myocytes isolated from failing hearts present a higher NAD+/NADH ratio during rapid electrical pacing, suggesting an inability to maintain NADH production and energy exhaustion (94). Similarly, the end-stage failing heart shows lower levels of fatty acyl-CoA levels, suggesting that mitochondria are depleted of fuel supply (66). These findings seem to present a picture of transition from a desynchronized metabolic network during the development of heart failure to an overall impairment of energy metabolism at late stages. Mechanisms that initiate and promote this transition are likely multiple and are only beginning to be understood.
Mitochondrial Ca2+ and the development of heart failure
Dysregulation of Ca2+ homeostasis is a hallmark of heart failure. Impaired Ca2+ reuptake by the sarcoplasmic reticulum (SR) and increased Ca2+ leak through ryanodine receptors in failing hearts result in reduced cytosolic Ca2+ transients during excitation but increased cytosolic Ca2+ at baseline. Since the SR and mitochondria are in close proximity, it is speculated that mitochondria act as a Ca2+ sink under pathological conditions and the resultant Ca2+ overload contributes to mitochondrial dysfunction (95–97). A number of metabolic enzymes in the mitochondria are activated by Ca2+, such as pyruvate dehydrogenase, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase (7, 9, 10). Ca2+ also regulates other proteins involved in oxidative phosphorylation, ROS scavenging, or mPTP opening (8, 9, 11). These roles make Ca2+ an important regulator of mitochondrial function as well as a potential player in the development of heart failure (Figure 3).
However, the exact amount of Ca2+ taken up by mitochondria on a beat-by-beat basis in normal and failing hearts is unclear. Whether mitochondria serve as a major Ca2+ sink is also controversial (12, 98). Few studies have directly examined mitochondrial Ca2+ dynamics in heart failure. The majority of data demonstrating Ca2+ toxicity in mitochondria are derived from experiments manipulating intracellular Ca2+ homeostasis using genetic approaches. Indeed, cytosolic Ca2+ overload in a number of animal models results in mitochondrial phenotypes similar to that seen in heart failure, e.g., the opening of mPTP, increased mitochondrial oxidative stress, collapse of mitochondrial membrane potential, impaired ATP production, and necrosis of cardiomyocytes (16, 18, 20, 95). Whether a comparable level of mitochondrial Ca2+ overload occurs in heart failure, and if so, through what mechanisms, remain to be determined.
One major player in the control of mitochondrial Ca2+ uptake is the recently identified mitochondrial Ca2+ uniporter (MCU) complex (13, 14). Deletion of the MCU eliminates mitochondrial Ca2+ uptake in vitro (99). MCU-null mice on C57BL/6 background die around E11.5–E13.5 but are viable and grossly normal on mixed backgrounds (100). Cardiac-specific deletion of the MCU in adult mice also produces a rather mild phenotype, thus calling into question the physiological relevance of mitochondrial Ca2+ uptake via the MCU (101, 102). The MCU has low affinity but high capacity to transport Ca2+, which results in a sigmoidal relationship between mitochondrial Ca2+ uptake and Ca2+ concentrations outside mitochondria (103). Such characteristics allow the MCU to rapidly uptake Ca2+ during the peak Ca2+ transient but prevent uptake under basal conditions. The basal Ca2+ level is elevated in the failing heart, while the amplitude of Ca2+ transients is reduced. Since the basal Ca2+ level in cardiomyocytes is in the nanomolar range while the Ca2+ transient reaches the micromolar level, how these changes alter mitochondrial Ca2+ load is difficult to predict. Direct measurement of mitochondrial Ca2+ load in failing hearts is sparse. In a mouse model with defective oxidative phosphorylation induced by cardiac-specific deletion of mitochondrial transcription factor A (TFAM), increased mitochondrial Ca2+ was observed (104). Both increased activity of the MCU and diminished efflux via Na+/Ca2+ were found to be responsible for increased mitochondrial Ca2+ in these hearts. Increased mitochondrial Ca2+ in this model improves energy deficits in an in vitro assay, suggesting that increasing Ca2+ content in the mitochondria is a compensatory measure to stimulate energy production (104). Similar findings are also reported in cardiomyocytes from pressure overload–induced heart failure (94). However, the TFAM mutant mice develop cardiomyopathy later in life, suggesting that recruitment of Ca2+ into the mitochondria, which is initially a compensatory response to energy deficit, is detrimental over the long term (Figure 3).
The mitochondrial Na+/Ca2+ exchanger (NCLX) is proposed to be responsible for the efflux of Ca2+ into the cytosol (105). Deletion of NCLX in the mouse heart results in mitochondrial Ca2+ overload, necrotic cell death, and sudden death of the animal. These observations confirm that NCLX is critical in controlling mitochondrial Ca2+ level and reinforce the notion that mitochondrial Ca2+ overload is detrimental (106). On the other hand, overexpression of NCLX, although associated with significantly increased Ca2+ efflux, does not change mitochondrial Ca2+ content, suggesting that additional mechanisms are in play to maintain mitochondrial Ca2+ homeostasis.
In support of the mitochondrial Ca2+ overload hypothesis, overexpression of NCLX in mouse hearts is beneficial in cardiac remodeling after myocardial infarction (106). In contrast, another study suggests that increased activity of mitochondrial Na+/Ca2+ exchange in a hamster heart failure model results in decreased mitochondrial matrix Ca2+, impaired energy supply, and increased myocyte death (107). In models of diabetic cardiomyopathy, measures that increase mitochondrial Ca2+ level appear to be beneficial, which argues against the Ca2+ overload hypothesis (108, 109). Mitochondrial Ca2+ uptake is also reduced in human myocardium from explanted end-stage failing hearts (110). However, this decrease could reflect the overall impairment of mitochondrial function at the end stage of the disease, rather than its pathogenesis.
Mitochondrial ROS in heart failure
ROS were originally viewed as a by-product of mitochondrial respiration. Electron leakage from multiple sites of complex I and complex III in the ETC results in partial reduction of oxygen to superoxide (111). It has been shown that superoxide generated in complex I is released into mitochondrial matrix, whereas superoxide generated by complex III can be deposited into either mitochondrial matrix or intermembrane space (111). At either location, superoxide is quickly dismutated to hydrogen peroxide by superoxide dismutase (SOD), with SOD2 being the primary mitochondrial isoform. Mitochondrial hydrogen peroxide is removed by the antioxidant systems of peroxiredoxin (Prx) and glutathione peroxidase (Gpx) in the mitochondria (112). Depending on the amount of mitochondrial ROS (mtROS) and the conditions under which it is generated, mtROS can play a physiological or pathological role (113–117). Damage caused by mtROS has been shown as a major pathogenic mechanism in heart failure. Furthermore, the signaling role of mtROS is being increasingly recognized in cellular response to mitochondrial stress (Figure 3).
In the failing heart, damage caused by excessive mtROS is evident in human patients and animal models (118–120). Mitochondria-targeted ROS scavenging has demonstrated benefit in animal models of heart failure (118, 121, 122). Age-related cardiac pathology was delayed in mitochondrial catalase–transgenic mice, in which ectopic overexpression of catalase reduced mitochondrial hydrogen peroxide (123). Reduction of mitochondrial hydrogen peroxide by overexpression of the mitochondrial Prx or Gpx system is beneficial for hearts under pathological stress, whereas the deficiency of antioxidant systems shows adverse consequences (122, 124). Operation of mitochondrial Prx and Gpx systems requires continuous reduction of thioredoxin 2 (Trx2) or conversion of oxidized glutathione to glutathione using NADPH. In mammals, the supply of mitochondrial NADPH is dependent on isocitrate dehydrogenase 2 (IDH2) and nicotinamide nucleotide transhydrogenase (Nnt). Impairments of these enzyme activities could contribute to oxidative stress and the development of heart failure (125–127). However, these enzyme reactions produce NADPH by diverting substrate metabolism away from generating NADH for ATP production. A recent study suggests that increased oxidative stress in the failing mouse heart leads to excessive activation of Nnt, which supplies NADPH for antioxidant (126). This compensatory response turns out to be detrimental, as the Nnt reaction directs NADH away from ATP production, resulting in impaired myocardial energetics (Figure 3).
During the development of heart failure, increased oxidative stress in mitochondria could be the cause and/or the consequence of mitochondrial dysfunction. Stimulation of mitochondrial respiration for ATP production is associated with increased ROS generation due to increased ETC activity. Running counter to the notion that increased energy demand in the chronically stressed heart should stimulate mitochondrial ATP synthesis, recent studies suggest that ATP synthase activity is inhibited in the failing heart because of increased protein acetylation (78). Inhibition of ATP synthase activity not only impedes ATP production but also impairs electron flow and enhances ROS emission (128, 129). As discussed above, mitochondrial protein hyperacetylation has been observed in the failing human heart and animal models of heart failure. Whether this is a common mechanism for increased ROS production remains to be investigated. Other mechanisms, including mitochondrial Ca2+ overload or failure to remove damaged mitochondria due to defective mitochondrial protein quality control, are also implicated in the increase of mtROS during pathological remodeling (95, 130, 131). Mitochondrial damage incurred by initial increase of oxidative stress will cause further increases of ROS generation and more severe damage, resulting in so-called ROS-induced ROS release in mitochondria (18). Animal models of heart failure demonstrate downregulation of the ROS scavenging system, such as decreased protein expression and reduced activity of SOD2, which occurs due to protein modification (75, 132). Thus, increased mtROS can be attributed to both increased ROS generation and decreased scavenging. Changes of SOD activity in the failing human heart are, however, mixed, showing either decreases or no change of SOD activities (133–136).
Mitochondrial ROS have been shown to affect a broad range of cellular functions in the context of heart failure (Figure 3). Excessive ROS triggers mPTP opening and causes cell death (18). Increased oxidative stress is associated with mtDNA damage and impaired mitochondrial biogenesis (120, 137). Oxidative modification of proteins and lipids resulted in decreased enzyme activities that compromise the metabolic capacity of the mitochondria (138). mtDNA leakage, a consequence of ROS-induced damage, is a potential trigger of inflammation (discussed in greater detail below). Furthermore, superoxide reactions with NO produce highly toxic peroxynitrate and at the same time reduce the bioavailability of NO (112). In recent years, mtROS was also shown as a molecular mediator of hypoxia signaling, MAP kinase pathway, inflammation, and retrograde communication between mitochondria and nucleus (115, 116). The specific role of these mechanisms during the development of heart failure needs to be fully elucidated.
Mitochondrial dysfunction and inflammation in heart failure
Heart failure is associated with heightened inflammatory response, but clinical trials targeting the proinflammatory cytokines have yielded disappointing outcomes (139, 140). Thus, a better understanding of the mechanisms underlying the inflammatory response is required to develop effective therapy. Inflammation associated with most cases of heart failure is “sterile,” i.e., with no infection by microorganisms (140), suggesting a state of activated innate immunity. In the absence of pathogen invasion, the release of endogenous molecules containing damage-associated molecular patterns (DAMPs) triggers an inflammatory response (21, 141, 142). Mitochondrial DAMPs retain at least two unique molecular features that are evolutionary endosymbionts derived from bacteria: mtDNA rich in unmethylated CpG motifs and N-formyl peptides. Emerging evidence suggests that mtDNA and N-formyl peptides act on TLR9 and formyl peptide receptors, respectively, to induce inflammatory responses through the assembly of the NLRP3 inflammasome (143–146).
It is increasingly recognized that NLRP3 inflammasome activation is an important mechanism linking mitochondria function and integrity to innate immunity (147). During the development of heart failure, increased mitochondrial damage associated with higher oxidative stress and impaired quality control through autophagy/mitophagy contributes to cardiac inflammation (21, 147, 148). Release of mtDNA into circulation is found in mice after transverse aortic constriction surgery, and elevated levels of circulating mtDNA are also observed in patients after myocardial infarction (149); both can trigger activation of circulating immune cells. It is also observed that mtDNA-induced inflammatory responses do not require the release of mtDNA into circulation, suggesting that cells that reside within the heart, including macrophages and myocytes, contribute to pathogenesis (21). In addition to the release of mtDNA, recent studies suggest that mitochondrial redox state and NAD+ availability modulate NLRP3 activity (150, 151). Given the emerging role of NAD+/NADH redox imbalance in the development of heart failure, this mechanism likely presents another link between mitochondrial dysfunction and inflammation of failing heart.
Therapeutic implications
Mitochondrial function has been considered a therapeutic target for a variety of diseases, including heart failure. Prior efforts, primarily focused on reducing oxidative stress and improving bioenergetics, have yielded limited clinical success despite positive results from preclinical studies (38, 39). A new generation of ROS-scavenging compounds that demonstrated improved tissue permeability and subcellular targeting are currently being tested in humans (e.g., NCT03506633, NCT02966665, NCT01925937, ClinicalTrials.gov). The lack of effective compounds to manipulate cardiac substrate metabolism in vivo is also a barrier to translating the concept of metabolic modulation into clinical practice. Recent studies have tested the efficacy of perhexiline on surrogate end points in heart failure patients. Perhexiline is thought to act by inhibiting mitochondrial carnitine palmitoyltransferase-1, thus increasing glucose oxidation through inhibition of fatty acid oxidation. Short-term treatment with perhexiline has improved symptoms and bioenergetics in heart failure patients (51, 152, 153). However, the clinical benefits of perhexiline have recently been attributed to pleiotropic mechanisms, as perhexiline did not alter cardiac substrate utilization in heart failure patients (153, 154).
Along the lines of metabolic modulation, unexpected cardiovascular benefits of SGLT2 inhibition were recently observed in a diabetes patient population (155, 156) and suggest an intriguing opportunity to modulate glucose and ketone body metabolism in the heart. SGLT2 is a sodium/glucose cotransporter responsible for glucose reabsorption in the kidney. Inhibition of the transporter reduces blood glucose level in type 2 diabetes patients. Interestingly, administration of SGLT2 inhibitors demonstrated a significant reduction of cardiovascular death and hospitalized heart failure in these patients that had not been observed with other glycemic control measures. SGLT2 inhibition removes glucose from the body and triggers ketosis, whereas traditional treatment controls blood glucose level by promoting tissue glucose uptake. It is thus speculated that switching cardiac metabolism from glucose to ketone bodies protects against heart failure. However, patients with diabetes present different metabolic signatures during the development of heart failure than do nondiabetic patients. Ongoing trials (e.g., NCT02653482, NCT03485222, ClinicalTrials.gov) will evaluate the effects of SGLT2 inhibition in nondiabetic heart failure patients. Moreover, SGLT2 inhibition affects multiple mechanisms in cardiovascular regulation; the role of metabolic modulation versus other effects requires further evaluation (157).
As discussed above, a novel target for mitochondrial dysfunction is the balance of NAD+/NADH redox, which regulates substrate oxidation, NAD+-dependent protein deacetylation, and mitochondria-initiated inflammatory response. Thus, restoration of NAD+/NADH redox is a promising breakpoint for the vicious cycle of mitochondrial metabolic imbalance in heart failure progression. A number of studies have shown benefits of increasing the NAD+ level and normalizing the NAD+/NADH ratio in failing hearts. Using pharmacological or genetic approaches to target NAD+ salvage pathway, these studies found that increasing intracellular NAD+ level enhanced mitochondrial stress tolerance, improved myocardial energetics, reduced the contractile dysfunction caused by pressure overload or adrenergic stimulation, and improved cardiac function in mouse models of mitochondrial cardiomyopathy (76, 87, 89, 158). Several NAD+ precursors are available for human use. A recent clinical study described the pharmacokinetics of nicotinamide riboside (NR), one of the NAD+ precursors, in healthy volunteers, and provided dosing information for NR to raise blood NAD+ level (159). Prior studies also showed that NR was safe and well tolerated in normal individuals (159, 160). Additional information on the safety and tolerability of NAD+ precursors in heart failure patients is required before its efficacy on heart failure progression can be determined.
Conclusion
Heart failure is a complex clinical syndrome that represents the final outcome of failed compensation for cardiac injury caused by a variety of etiologies. Mitochondrial dysfunction develops as the result of unsuccessful adaptation to energy stress in the heart, which, in turn, perpetuates a maladaptive spiral to further cardiac damage. As summarized here, a number of mitochondria-mediated mechanisms have been identified that drive the demise of the heart prior to the ultimate ATP depletion. These observations will not only provide important links between cardiac stress and energy starvation during the development of heart failure but will also inspire therapeutic approaches for mitochondrial dysfunction.
Acknowledgments
This work was supported in part by NIH grants HL118989, HL129510, HL126209, HL110349, and HL142628 (to RT) and American Heart Association Postdoctoral Fellowship 18POST33990352 (to BZ).
Version 1. 08/20/2018
Electronic publication
Version 2. 08/31/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(9):3716–3726.https://doi.org/10.1172/JCI120849.
Contributor Information
Bo Zhou, Email: zb817@u.washington.edu.
Rong Tian, Email: rongtian@u.washington.edu.
References
- 1.Cunningham SA, Wiesinger H, Nicholls DG. Quantification of fatty acid activation of the uncoupling protein in brown adipocytes and mitochondria from the guinea-pig. Eur J Biochem. 1986;157(2):415–420. doi: 10.1111/j.1432-1033.1986.tb09683.x. [DOI] [PubMed] [Google Scholar]
- 2.Ahn CS, Metallo CM. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015;3(1):1. doi: 10.1186/s40170-015-0128-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 2015;21(6):805–821. doi: 10.1016/j.cmet.2015.05.014. [DOI] [PubMed] [Google Scholar]
- 4.Aguiar CJ, et al. Succinate causes pathological cardiomyocyte hypertrophy through GPR91 activation. Cell Commun Signal. 2014;12:78. doi: 10.1186/s12964-014-0078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu M, Liu H, Dudley SC. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res. 2010;107(8):967–974. doi: 10.1161/CIRCRESAHA.110.220673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raimundo N. Mitochondrial pathology: stress signals from the energy factory. Trends Mol Med. 2014;20(5):282–292. doi: 10.1016/j.molmed.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Denton RM, Richards DA, Chin JG. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem J. 1978;176(3):899–906. doi: 10.1042/bj1760899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glancy B, Willis WT, Chess DJ, Balaban RS. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry. 2013;52(16):2793–2809. doi: 10.1021/bi3015983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hopper RK, et al. Mitochondrial matrix phosphoproteome: effect of extra mitochondrial calcium. Biochemistry. 2006;45(8):2524–2536. doi: 10.1021/bi052475e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCormack JG, Denton RM. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem J. 1979;180(3):533–544. doi: 10.1042/bj1800533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Territo PR, Mootha VK, French SA, Balaban RS. Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase. Am J Physiol Cell Physiol. 2000;278(2):C423–C435. doi: 10.1152/ajpcell.2000.278.2.C423. [DOI] [PubMed] [Google Scholar]
- 12.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13(9):566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 13.De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476(7360):336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baughman JM, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476(7360):341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baumgartner HK, et al. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J Biol Chem. 2009;284(31):20796–20803. doi: 10.1074/jbc.M109.025353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernardi P, Di Lisa F. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol. 2015;78:100–106. doi: 10.1016/j.yjmcc.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192(7):1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94(3):909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kinnally KW, Peixoto PM, Ryu SY, Dejean LM. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim Biophys Acta. 2011;1813(4):616–622. doi: 10.1016/j.bbamcr.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakayama H, et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117(9):2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oka T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485(7397):251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 25.Jin Z, Wei W, Yang M, Du Y, Wan Y. Mitochondrial complex I activity suppresses inflammation and enhances bone resorption by shifting macrophage-osteoclast polarization. Cell Metab. 2014;20(3):483–498. doi: 10.1016/j.cmet.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin Z, Wei W, Yang M, Du Y, Wan Y. Mitochondrial complex I activity suppresses inflammation and enhances bone resorption by shifting macrophage-osteoclast polarization. Cell Metab. 2014;20(3):483–498. doi: 10.1016/j.cmet.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kolwicz SC, Jr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. 2013;113(5):603–616. doi: 10.1161/CIRCRESAHA.113.302095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90(1):207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 29.Allard MF, Schönekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. 1994;267(2 pt 2):H742–H750. doi: 10.1152/ajpheart.1994.267.2.H742. [DOI] [PubMed] [Google Scholar]
- 30.von Lueder TG, Kotecha D, Atar D, Hopper I. Neurohormonal blockade in heart failure. Card Fail Rev. 2017;3(1):19–24. doi: 10.15420/cfr.2016:22:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorski PA, Ceholski DK, Hajjar RJ. Altered myocardial calcium cycling and energetics in heart failure — a rational approach for disease treatment. Cell Metab. 2015;21(2):183–194. doi: 10.1016/j.cmet.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neubauer S. The failing heart — an engine out of fuel. N Engl J Med. 2007;356(11):1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 33.Rosca MG, et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. 2008;80(1):30–39. doi: 10.1093/cvr/cvn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taegtmeyer H, Lubrano G. Rethinking cardiac metabolism: metabolic cycles to refuel and rebuild the failing heart. F1000Prime Rep. 2014;6:90. doi: 10.12703/P6-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.SOLVD Investigators, Yusuf S, Pitt B, Davis CE, Hood WB, Cohn JN. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991;325(5):293–302. doi: 10.1056/NEJM199108013250501. [DOI] [PubMed] [Google Scholar]
- 36.Doughty RN, MacMahon S, Sharpe N. Beta-blockers in heart failure: promising or proved? J Am Coll Cardiol. 1994;23(3):814–821. doi: 10.1016/0735-1097(94)90773-0. [DOI] [PubMed] [Google Scholar]
- 37.Tariq S, Aronow WS. Use of inotropic agents in treatment of systolic heart failure. Int J Mol Sci. 2015;16(12):29060–29068. doi: 10.3390/ijms161226147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown DA, et al. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. 2017;14(4):238–250. doi: 10.1038/nrcardio.2016.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang W, Karamanlidis G, Tian R. Novel targets for mitochondrial medicine. Sci Transl Med. 2016;8(326):326rv3. doi: 10.1126/scitranslmed.aac7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao R, Nascimben L, Friedrich J, Gwathmey JK, Ingwall JS. Decreased energy reserve in an animal model of dilated cardiomyopathy. Relationship to contractile performance. Circ Res. 1996;78(5):893–902. doi: 10.1161/01.RES.78.5.893. [DOI] [PubMed] [Google Scholar]
- 41.Neubauer S, et al. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. 1997;96(7):2190–2196. doi: 10.1161/01.CIR.96.7.2190. [DOI] [PubMed] [Google Scholar]
- 42.Tian R, Nascimben L, Ingwall JS, Lorell BH. Failure to maintain a low ADP concentration impairs diastolic function in hypertrophied rat hearts. Circulation. 1997;96(4):1313–1319. doi: 10.1161/01.CIR.96.4.1313. [DOI] [PubMed] [Google Scholar]
- 43.Weiss RG, Gerstenblith G, Bottomley PA. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc Natl Acad Sci U S A. 2005;102(3):808–813. doi: 10.1073/pnas.0408962102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith CS, Bottomley PA, Schulman SP, Gerstenblith G, Weiss RG. Altered creatine kinase adenosine triphosphate kinetics in failing hypertrophied human myocardium. Circulation. 2006;114(11):1151–1158. doi: 10.1161/CIRCULATIONAHA.106.613646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Starling RC, Hammer DF, Altschuld RA. Human myocardial ATP content and in vivo contractile function. Mol Cell Biochem. 1998;180(1–2):171–177. [PubMed] [Google Scholar]
- 46.Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95(2):135–145. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- 47.Tian R, Ingwall JS. Energetic basis for reduced contractile reserve in isolated rat hearts. Am J Physiol. 1996;270(4 pt 2):H1207–H1216. doi: 10.1152/ajpheart.1996.270.4.H1207. [DOI] [PubMed] [Google Scholar]
- 48.Goodwin GW, Taegtmeyer H. Improved energy homeostasis of the heart in the metabolic state of exercise. Am J Physiol Heart Circ Physiol. 2000;279(4):H1490–H1501. doi: 10.1152/ajpheart.2000.279.4.H1490. [DOI] [PubMed] [Google Scholar]
- 49.Kaijser L, Berglund B. Myocardial lactate extraction and release at rest and during heavy exercise in healthy men. Acta Physiol Scand. 1992;144(1):39–45. doi: 10.1111/j.1748-1716.1992.tb09265.x. [DOI] [PubMed] [Google Scholar]
- 50.Pound KM, et al. Substrate-enzyme competition attenuates upregulated anaplerotic flux through malic enzyme in hypertrophied rat heart and restores triacylglyceride content: attenuating upregulated anaplerosis in hypertrophy. Circ Res. 2009;104(6):805–812. doi: 10.1161/CIRCRESAHA.108.189951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dyck JR, et al. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ Res. 2004;94(9):e78–e84. doi: 10.1161/01.RES.0000129255.19569.8f. [DOI] [PubMed] [Google Scholar]
- 52.Burkhoff D, Weiss RG, Schulman SP, Kalil-Filho R, Wannenburg T, Gerstenblith G. Influence of metabolic substrate on rat heart function and metabolism at different coronary flows. Am J Physiol. 1991;261(3 pt 2):H741–H750. doi: 10.1152/ajpheart.1991.261.3.H741. [DOI] [PubMed] [Google Scholar]
- 53.Korvald C, Elvenes OP, Myrmel T. Myocardial substrate metabolism influences left ventricular energetics in vivo. Am J Physiol Heart Circ Physiol. 2000;278(4):H1345–H1351. doi: 10.1152/ajpheart.2000.278.4.H1345. [DOI] [PubMed] [Google Scholar]
- 54.Shao D, et al. Glucose promotes cell growth by suppressing branched-chain amino acid degradation. Nat Commun. 2018;9(1):2935. doi: 10.1038/s41467-018-05362-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luptak I, Yan J, Cui L, Jain M, Liao R, Tian R. Long-term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation. 2007;116(8):901–909. doi: 10.1161/CIRCULATIONAHA.107.691253. [DOI] [PubMed] [Google Scholar]
- 56.Pereira RO, et al. Inducible overexpression of GLUT1 prevents mitochondrial dysfunction and attenuates structural remodeling in pressure overload but does not prevent left ventricular dysfunction. J Am Heart Assoc. 2013;2(5):e000301. doi: 10.1161/JAHA.113.000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lai L, et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail. 2014;7(6):1022–1031. doi: 10.1161/CIRCHEARTFAILURE.114.001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kolwicz SC, Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res. 2012;111(6):728–738. doi: 10.1161/CIRCRESAHA.112.268128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chess DJ, Lei B, Hoit BD, Azimzadeh AM, Stanley WC. Effects of a high saturated fat diet on cardiac hypertrophy and dysfunction in response to pressure overload. J Card Fail. 2008;14(1):82–88. doi: 10.1016/j.cardfail.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Okere IC, et al. High-fat diet prevents cardiac hypertrophy and improves contractile function in the hypertensive dahl salt-sensitive rat. Clin Exp Pharmacol Physiol. 2005;32(10):825–831. doi: 10.1111/j.1440-1681.2005.04272.x. [DOI] [PubMed] [Google Scholar]
- 61.Choi YS, et al. Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion. J Mol Cell Cardiol. 2016;100:64–71. doi: 10.1016/j.yjmcc.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab. 2012;15(6):805–812. doi: 10.1016/j.cmet.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koves TR, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7(1):45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 64.Ussher JR, et al. Insulin-stimulated cardiac glucose oxidation is increased in high-fat diet-induced obese mice lacking malonyl CoA decarboxylase. Diabetes. 2009;58(8):1766–1775. doi: 10.2337/db09-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aubert G, et al. The failing heart relies on ketone bodies as a fuel. Circulation. 2016;133(8):698–705. doi: 10.1161/CIRCULATIONAHA.115.017355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bedi KC, et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation. 2016;133(8):706–716. doi: 10.1161/CIRCULATIONAHA.115.017545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lommi J, et al. Blood ketone bodies in congestive heart failure. J Am Coll Cardiol. 1996;28(3):665–672. doi: 10.1016/0735-1097(96)00214-8. [DOI] [PubMed] [Google Scholar]
- 68.Uchihashi M, et al. Cardiac-specific Bdh1 overexpression ameliorates oxidative stress and cardiac remodeling in pressure overload-induced heart failure. Circ Heart Fail. 2017;10(12):e004417. doi: 10.1161/CIRCHEARTFAILURE.117.004417. [DOI] [PubMed] [Google Scholar]
- 69.Murray AJ, et al. Novel ketone diet enhances physical and cognitive performance. FASEB J. 2016;30(12):4021–4032. doi: 10.1096/fj.201600773R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shimazu T, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339(6116):211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gormsen LC, et al. Ketone body infusion with 3-hydroxybutyrate reduces myocardial glucose uptake and increases blood flow in humans: a positron emission tomography study. J Am Heart Assoc. 2017;6(3):e005066. doi: 10.1161/JAHA.116.005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation. 2009;119(21):2818–2828. doi: 10.1161/CIRCULATIONAHA.108.832915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carrico C, Meyer JG, He W, Gibson BW, Verdin E. The mitochondrial acylome emerges: proteomics, regulation by sirtuins, and metabolic and disease implications. Cell Metab. 2018;27(3):497–512. doi: 10.1016/j.cmet.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grillon JM, Johnson KR, Kotlo K, Danziger RS. Non-histone lysine acetylated proteins in heart failure. Biochim Biophys Acta. 2012;1822(4):607–614. doi: 10.1016/j.bbadis.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Horton JL, et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight. 2016;2(1):e84897. doi: 10.1172/jci.insight.84897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee CF, et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation. 2016;134(12):883–894. doi: 10.1161/CIRCULATIONAHA.116.022495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vadvalkar SS, Baily CN, Matsuzaki S, West M, Tesiram YA, Humphries KM. Metabolic inflexibility and protein lysine acetylation in heart mitochondria of a chronic model of type 1 diabetes. Biochem J. 2013;449(1):253–261. doi: 10.1042/BJ20121038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang X, et al. MicroRNA-195 regulates metabolism in failing myocardium via alterations in sirtuin 3 expression and mitochondrial protein acetylation. Circulation. 2018;137(19):2052–2067. doi: 10.1161/CIRCULATIONAHA.117.030486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Finley LW, et al. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS One. 2011;6(8):e23295. doi: 10.1371/journal.pone.0023295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hafner AV, et al. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010;2(12):914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jing E, et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes. 2013;62(10):3404–3417. doi: 10.2337/db12-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang H, et al. SIRT3-dependent GOT2 acetylation status affects the malate-aspartate NADH shuttle activity and pancreatic tumor growth. EMBO J. 2015;34(8):1110–1125. doi: 10.15252/embj.201591041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lewandowski ED, O’Donnell JM, Scholz TD, Sorokina N, Buttrick PM. Recruitment of NADH shuttling in pressure-overloaded and hypertrophic rat hearts. Am J Physiol Cell Physiol. 2007;292(5):C1880–C1886. doi: 10.1152/ajpcell.00576.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scott I, Webster BR, Li JH, Sack MN. Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem J. 2012;443(3):655–661. doi: 10.1042/BJ20120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen T, et al. Mouse SIRT3 attenuates hypertrophy-related lipid accumulation in the heart through the deacetylation of LCAD. PLoS One. 2015;10(3):e0118909. doi: 10.1371/journal.pone.0118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Diguet N, et al. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation. 2018;137(21):2256–2273. doi: 10.1161/CIRCULATIONAHA.116.026099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Karamanlidis G, et al. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013;18(2):239–250. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Walker MA, Tian R. NAD(H) in mitochondrial energy transduction: implications for health and disease. Curr Opin Physiol. 2018;3:101–109. doi: 10.1016/j.cophys.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Martin AS, et al. Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich’s ataxia cardiomyopathy model. JCI Insight. 2017;2(14):e93885. doi: 10.1172/jci.insight.93885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Alrob OA, et al. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc Res. 2014;103(4):485–497. doi: 10.1093/cvr/cvu156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hirschey MD, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464(7285):121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Thapa D, et al. Acetylation of mitochondrial proteins by GCN5L1 promotes enhanced fatty acid oxidation in the heart. Am J Physiol Heart Circ Physiol. 2017;313(2):H265–H274. doi: 10.1152/ajpheart.00752.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fukushima A, et al. Acetylation contributes to hypertrophy-caused maturational delay of cardiac energy metabolism. JCI Insight. 2018;3(10):e99239. doi: 10.1172/jci.insight.99239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu T, O’Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103(3):279–288. doi: 10.1161/CIRCRESAHA.108.175919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Santulli G, Xie W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A. 2015;112(36):11389–11394. doi: 10.1073/pnas.1513047112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Odagiri K, et al. Local control of mitochondrial membrane potential, permeability transition pore and reactive oxygen species by calcium and calmodulin in rat ventricular myocytes. J Mol Cell Cardiol. 2009;46(6):989–997. doi: 10.1016/j.yjmcc.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 97.O-Uchi J, et al. Adrenergic signaling regulates mitochondrial Ca2+ uptake through Pyk2-dependent tyrosine phosphorylation of the mitochondrial Ca2+ uniporter. Antioxid Redox Signal. 2014;21(6):863–879. doi: 10.1089/ars.2013.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Williams GS, Boyman L, Lederer WJ. Mitochondrial calcium and the regulation of metabolism in the heart. J Mol Cell Cardiol. 2015;78:35–45. doi: 10.1016/j.yjmcc.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Holmström KM, et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J Mol Cell Cardiol. 2015;85:178–182. doi: 10.1016/j.yjmcc.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pan X, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15(12):1464–1472. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kwong JQ, et al. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 2015;12(1):15–22. doi: 10.1016/j.celrep.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Luongo TS, et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 2015;12(1):23–34. doi: 10.1016/j.celrep.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Patron M, et al. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell. 2014;53(5):726–737. doi: 10.1016/j.molcel.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sommakia S, et al. Mitochondrial cardiomyopathies feature increased uptake and diminished efflux of mitochondrial calcium. J Mol Cell Cardiol. 2017;113:22–32. doi: 10.1016/j.yjmcc.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Boyman L, Williams GS, Khananshvili D, Sekler I, Lederer WJ. NCLX: the mitochondrial sodium calcium exchanger. J Mol Cell Cardiol. 2013;59:205–213. doi: 10.1016/j.yjmcc.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Luongo TS, et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature. 2017;545(7652):93–97. doi: 10.1038/nature22082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kuo TH, Zhu L, Golden K, Marsh JD, Bhattacharya SK, Liu BF. Altered Ca2+ homeostasis and impaired mitochondrial function in cardiomyopathy. Mol Cell Biochem. 2002;238(1-2):119–127. doi: 10.1023/a:1019967323419. [DOI] [PubMed] [Google Scholar]
- 108.Diaz-Juarez J, et al. Expression of the mitochondrial calcium uniporter in cardiac myocytes improves impaired mitochondrial calcium handling and metabolism in simulated hyperglycemia. Am J Physiol Cell Physiol. 2016;311(6):C1005–C1013. doi: 10.1152/ajpcell.00236.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ji L, et al. MICU1 alleviates diabetic cardiomyopathy through mitochondrial Ca2+-dependent antioxidant response. Diabetes. 2017;66(6):1586–1600. doi: 10.2337/db16-1237. [DOI] [PubMed] [Google Scholar]
- 110.Michels G, et al. Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels. Circulation. 2009;119(18):2435–2443. doi: 10.1161/CIRCULATIONAHA.108.835389. [DOI] [PubMed] [Google Scholar]
- 111.Wong HS, Dighe PA, Mezera V, Monternier PA, Brand MD. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J Biol Chem. 2017;292(41):16804–16809. doi: 10.1074/jbc.R117.789271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ribas V, García-Ruiz C, Fernández-Checa JC. Glutathione and mitochondria. Front Pharmacol. 2014;5:151. doi: 10.3389/fphar.2014.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Alleman RJ, Katunga LA, Nelson MA, Brown DA, Anderson EJ. The “Goldilocks Zone” from a redox perspective—adaptive vs. deleterious responses to oxidative stress in striated muscle. Front Physiol. 2014;5:358. doi: 10.3389/fphys.2014.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Frasier CR, Moore RL, Brown DA. Exercise-induced cardiac preconditioning: how exercise protects your achy-breaky heart. J Appl Physiol. 2011;111(3):905–915. doi: 10.1152/japplphysiol.00004.2011. [DOI] [PubMed] [Google Scholar]
- 115.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35(9):505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Javadov S, Jang S, Agostini B. Crosstalk between mitogen-activated protein kinases and mitochondria in cardiac diseases: therapeutic perspectives. Pharmacol Ther. 2014;144(2):202–225. doi: 10.1016/j.pharmthera.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Song M, Chen Y, Gong G, Murphy E, Rabinovitch PS, Dorn GW. Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ Res. 2014;115(3):348–353. doi: 10.1161/CIRCRESAHA.115.304384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dai DF, et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res. 2011;108(7):837–846. doi: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Goh KY, et al. Impaired mitochondrial network excitability in failing guinea-pig cardiomyocytes. Cardiovasc Res. 2016;109(1):79–89. doi: 10.1093/cvr/cvv230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. 2010;106(9):1541–1548. doi: 10.1161/CIRCRESAHA.109.212753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Graham D, et al. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54(2):322–328. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- 122.Huang Q, et al. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation. 2015;131(12):1082–1097. doi: 10.1161/CIRCULATIONAHA.114.012725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schriner SE, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308(5730):1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 124.Ardanaz N, et al. Lack of glutathione peroxidase 1 accelerates cardiac-specific hypertrophy and dysfunction in angiotensin II hypertension. Hypertension. 2010;55(1):116–123. doi: 10.1161/HYPERTENSIONAHA.109.135715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ku HJ, Ahn Y, Lee JH, Park KM, Park JW. IDH2 deficiency promotes mitochondrial dysfunction and cardiac hypertrophy in mice. Free Radic Biol Med. 2015;80:84–92. doi: 10.1016/j.freeradbiomed.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 126.Nickel AG, et al. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 2015;22(3):472–484. doi: 10.1016/j.cmet.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 127.Wang F, et al. A small molecule inhibitor of mutant IDH2 rescues cardiomyopathy in a D-2-hydroxyglutaric aciduria type II mouse model. J Inherit Metab Dis. 2016;39(6):807–820. doi: 10.1007/s10545-016-9960-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Boudina S, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56(10):2457–2466. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 129.Chouchani ET, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515(7527):431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hoshino A, et al. Oxidative post-translational modifications develop LONP1 dysfunction in pressure overload heart failure. Circ Heart Fail. 2014;7(3):500–509. doi: 10.1161/CIRCHEARTFAILURE.113.001062. [DOI] [PubMed] [Google Scholar]
- 131.Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci U S A. 2011;108(23):9572–9577. doi: 10.1073/pnas.1106291108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zou X, et al. Manganese superoxide dismutase (SOD2): is there a center in the universe of mitochondrial redox signaling? J Bioenerg Biomembr. 2017;49(4):325–333. doi: 10.1007/s10863-017-9718-8. [DOI] [PubMed] [Google Scholar]
- 133.Bäumer AT, Flesch M, Wang X, Shen Q, Feuerstein GZ, Böhm M. Antioxidative enzymes in human hearts with idiopathic dilated cardiomyopathy. J Mol Cell Cardiol. 2000;32(1):121–130. doi: 10.1006/jmcc.1999.1061. [DOI] [PubMed] [Google Scholar]
- 134.Borchi E, et al. Enhanced ROS production by NADPH oxidase is correlated to changes in antioxidant enzyme activity in human heart failure. Biochim Biophys Acta. 2010;1802(3):331–338. doi: 10.1016/j.bbadis.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 135.Dieterich S, Bieligk U, Beulich K, Hasenfuss G, Prestle J. Gene expression of antioxidative enzymes in the human heart: increased expression of catalase in the end-stage failing heart. Circulation. 2000;101(1):33–39. doi: 10.1161/01.CIR.101.1.33. [DOI] [PubMed] [Google Scholar]
- 136.Sam F, et al. Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J Card Fail. 2005;11(6):473–480. doi: 10.1016/j.cardfail.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 137.Ide T, et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88(5):529–535. doi: 10.1161/01.RES.88.5.529. [DOI] [PubMed] [Google Scholar]
- 138.Roussel J, et al. Palmitoyl-carnitine increases RyR2 oxidation and sarcoplasmic reticulum Ca2+ leak in cardiomyocytes: role of adenine nucleotide translocase. Biochim Biophys Acta. 2015;1852(5):749–758. doi: 10.1016/j.bbadis.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 139.Gullestad L, Ueland T, Vinge LE, Finsen A, Yndestad A, Aukrust P. Inflammatory cytokines in heart failure: mediators and markers. Cardiology. 2012;122(1):23–35. doi: 10.1159/000338166. [DOI] [PubMed] [Google Scholar]
- 140.Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res. 2015;116(7):1254–1268. doi: 10.1161/CIRCRESAHA.116.302317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Iyer SS, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39(2):311–323. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rubic T, et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol. 2008;9(11):1261–1269. doi: 10.1038/ni.1657. [DOI] [PubMed] [Google Scholar]
- 143.Wenceslau CF, McCarthy CG, Szasz T, Goulopoulou S, Webb RC. Mitochondrial N-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am J Physiol Heart Circ Physiol. 2015;308(7):H768–H777. doi: 10.1152/ajpheart.00779.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang Q, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dela Cruz CS, Kang MJ. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion. 2018;41:37–44. doi: 10.1016/j.mito.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Horng T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014;35(6):253–261. doi: 10.1016/j.it.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shimada K, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zang QS, et al. Specific inhibition of mitochondrial oxidative stress suppresses inflammation and improves cardiac function in a rat pneumonia-related sepsis model. Am J Physiol Heart Circ Physiol. 2012;302(9):H1847–H1859. doi: 10.1152/ajpheart.00203.2011. [DOI] [PubMed] [Google Scholar]
- 149.Bliksøen M, et al. Increased circulating mitochondrial DNA after myocardial infarction. Int J Cardiol. 2012;158(1):132–134. doi: 10.1016/j.ijcard.2012.04.047. [DOI] [PubMed] [Google Scholar]
- 150.Traba J, et al. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J Clin Invest. 2015;125(12):4592–4600. doi: 10.1172/JCI83260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Misawa T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14(5):454–460. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- 152.Abozguia K, et al. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation. 2010;122(16):1562–1569. doi: 10.1161/CIRCULATIONAHA.109.934059. [DOI] [PubMed] [Google Scholar]
- 153.Beadle RM, et al. Improvement in cardiac energetics by perhexiline in heart failure due to dilated cardiomyopathy. JACC Heart Fail. 2015;3(3):202–211. doi: 10.1016/j.jchf.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 154.George CH, Mitchell AN, Preece R, Bannister ML, Yousef Z. Pleiotropic mechanisms of action of perhexiline in heart failure. Expert Opin Ther Pat. 2016;26(9):1049–1059. doi: 10.1080/13543776.2016.1211111. [DOI] [PubMed] [Google Scholar]
- 155.d’Emden M, Amerena J, Deed G, Pollock C, Cooper ME. SGLT2 inhibitors with cardiovascular benefits: transforming clinical care in Type 2 diabetes mellitus. Diabetes Res Clin Pract. 2018;136:23–31. doi: 10.1016/j.diabres.2017.11.023. [DOI] [PubMed] [Google Scholar]
- 156.Vettor R, Inzucchi SE, Fioretto P. The cardiovascular benefits of empagliflozin: SGLT2-dependent and -independent effects. Diabetologia. 2017;60(3):395–398. doi: 10.1007/s00125-016-4194-y. [DOI] [PubMed] [Google Scholar]
- 157.Martens P, Mathieu C, Verbrugge FH. Promise of SGLT2 inhibitors in heart failure: diabetes and beyond. Curr Treat Options Cardiovasc Med. 2017;19(3):23. doi: 10.1007/s11936-017-0522-x. [DOI] [PubMed] [Google Scholar]
- 158.Yamamoto T, Byun J, Zhai P, Ikeda Y, Oka S, Sadoshima J. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS One. 2014;9(6):e98972. doi: 10.1371/journal.pone.0098972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Airhart SE, et al. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS One. 2017;12(12):e0186459. doi: 10.1371/journal.pone.0186459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Trammell SA, et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016;7:12948. doi: 10.1038/ncomms12948. [DOI] [PMC free article] [PubMed] [Google Scholar]