

Abstract
Over 40 years ago, Loeb and colleagues proposed that errors in DNA replication produce a mutator phenotype that is involved in generating the multiple mutations required for tumor development. In this issue of the JCI, Li, Castrillon, and colleagues describe a mouse model containing a single base change in the gene encoding replicative DNA polymerase ε (POLE) that mimics the “ultramutator” phenotype recently reported in many human tumors. Their seminal accomplishment validates Loeb’s hypothesis and the use of mutational signatures to understand the origins and potentially the treatment of human tumors, and it offers an exciting opportunity to further explore the mechanisms responsible for normal DNA replication fidelity and their perturbations.
Single base substitution in the mouse model of DNA polymerase
In humans, the POLEP286R mutation is associated with several tumor types whose genomes have an “ultramutator” phenotype (1, 2), wherein the genomic base substitution frequency is much higher than anticipated by loss of the proofreading activity of DNA polymerase ε (POL ε) alone. Moreover, the POLEP286R mutation is present in only 1 copy of the POLE gene and is dominant over the other, WT allele. Li et al. (3) have constructed a mouse model containing a single base substitution mutation that changes a proline to an arginine in the N-terminal domain of Pole. This domain encodes the 3′ exonuclease that proofreads mismatches made during nuclear DNA replication. The authors report that fibroblasts from heterozygous PoleP286R mice senesce earlier than do those from WT animals, but that markers of DNA damage are not elevated. Impressively, the mice harbor the heterozygous PoleP286R mutation from highly malignant tumors of diverse lineages, and these appear with short latency. Importantly, these tumors have the highest base substitution frequencies of any monoallelic mouse model of human cancer observed to date, up to 100 substitutions per megabase. In addition, only two of a predicted 41 mice lacking the WT allele of Pole were live-born, and they harbored independent malignancies with even larger numbers of mutations. Overall, the data imply that the PoleP286R mutation does not block DNA replication but results in much higher levels of replication-dependent point mutations that are initially biologically silent but quickly result in widespread tumorigenesis. These conclusions strongly support Loeb’s mutator hypothesis for cancer (4); they are consistent with epidemiological data published last year implying that DNA replication errors may account for two-thirds of the mutations found in human cancers (5), and they validate the use of human tumor mutational signatures seen in numerous recent studies aimed at understanding the origins and, potentially, the treatment of tumors.
As nicely discussed by Li et al. for their new mouse model and also considered recently for humans (2, 6), the ultramutator phenotype potentially provides new opportunities for developing more effective treatments for tumors exhibiting this phenotype. As an initial thought, it might be interesting to determine whether any of the many mutations that accumulate in tumors from the heterozygous mice, or perhaps even those in the rare hemizygous mice, are also seen among extragenic “escape from error extinction” (eex) mutants found that suppress the mutator phenotype exhibited by proofreading-deficient yeast Pol ε (7). If so, those mutations could be suppressors of the extremely high mutability observed in PoleP286R cells that allow the ultramutator mice to live long enough to develop tumors, potentially offering insights into how those tumors might be treated.
Fidelity of DNA replication
The new mouse model also presents opportunities to obtain fundamental mechanistic insights into DNA replication fidelity. Replication fidelity depends on three major processes: the ability of DNA replicases to select the correct nucleotide, exonucleolytic proofreading at a growing replication fork when an incorrect nucleotide is inadvertently incorporated, and DNA mismatch repair to correct mismatches that escape proofreading (8).
An effect on nucleotide selectivity?
Among the many mammalian DNA polymerases identified, Pol ε is one of three polymerases tasked with the bulk of nuclear DNA replication (9). The crystal structure of the catalytic domain of Pol ε (ref. 10 and Figure 1) reveals that proline 286 is located in the amino-terminal domain harboring the proofreading exonuclease active site. This proline is adjacent to and interacts with the thumb domain of the polymerase that contacts the duplex primer stem of the growing DNA. Given this location and the key role for prolines in determining the structures of proteins (ref. 11 and references therein), it is possible that substitution of arginine for proline at this location alters the polymerase structure and reduces nucleotide selectivity at the polymerase active site located some distance away, thereby contributing to the ultramutagenesis observed in the PoleP286R-mutant mouse.
Figure 1. X-ray crystal structure of DNA polymerase ε.
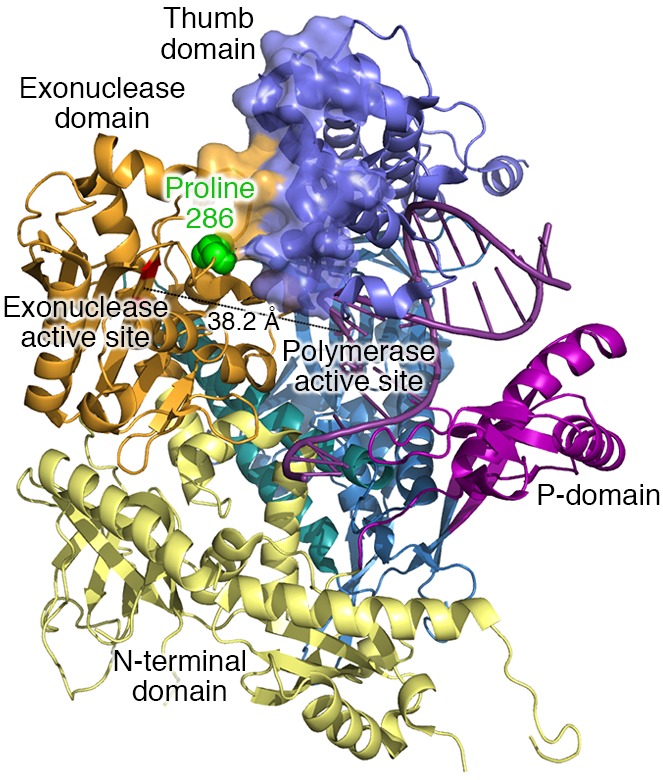
X-ray crystal structure of the 142-kDa N-terminal region of the catalytic subunit of yeast DNA polymerase ε (10). Yeast proline 301 is homologous to human proline 286, which is highlighted in green. The catalytic residues for the 3′ exonuclease activity are shown in red. They are located 38.2 Å away from the polymerase active site. See text for further descriptions.
Effects on proofreading and processivity?
The POLEP286R mutation in humans also clearly reduces the catalytic rate of the exonuclease reaction in POLE (12). Curiously, however, the homologous PoleP301R mutation in yeast results in a mutator effect that is much greater than that seen in an exonuclease-deficient Pol ε mutant that is catalytically dead (13). This suggests that something more than a simple defect in proofreading by P286R Pol ε alone reduces replication fidelity. One possibility based on studies in yeast (refs. 14 and 15 and discussed more recently in ref. 16) suggests that when a replicase generates mismatches, some of those errors are removed by the proofreading activity associated with another polymerase. If so, then in addition to suppressing proofreading by the mutant Pol ε itself, additional reduction of such “extrinsic proofreading” could contribute to the ultramutator phenotype observed in the heterozygous Pol2P286R mutant. One way for this to happen would be to change the processivity of replication, i.e., the number of consecutive dNTPs incorporated by a replicase during a single cycle of association with DNA – polymerization – dissociation from DNA. Processivity depends on polymerase residues that interact with the DNA substrate, among which may be certain residues in the N-terminal domain of Pol ε clearly associated with ultramutagenesis (17). Is it possible that the P286R Pol ε mutant enzyme has increased processivity? If so, this might explain the paucity of insertion/deletion mutations observed in the Pol2P286R mice (3), because processivity correlates with replication fidelity for insertion/deletion errors (18). More important, enhanced processivity could promote base-base mismatch extension, while excluding proofreading, by the heterozygous P286R Pol ε mutant polymerase itself and/or proofreading by the remaining WT Pol ε or by Pol δ.
Effects due to altered dNTP pools?
An additional and nonexclusive hypothesis is that the presence of the Pol2P286R may lead to changes in the absolute and/or relative concentrations of dNTPs. When dNTP pools become unbalanced (e.g., excess dGTP over dATP), nucleotide selectivity can be affected, such that replicases generate more mismatches (e.g., via misinsertion of dGTP rather than dATP opposite template T) to increase the mutation rate. In addition to unbalanced dNTP pools, absolute dNTP concentrations are important. For example, the noncatalytic C-terminal domain of Pol ε is essential for checkpoint control, a process that halts replication and increases all four dNTP concentrations to ultimately allow replication and promote yeast survival at the expense of a mutator phenotype (19). In a recent review article, Barbari and Shcherbakova (6) mention the unpublished observation that hypermutability in yeast is not caused by expansion of dNTP pools, so this latter explanation may be less likely. Nonetheless, it would be interesting to examine dNTP concentrations in Pol2P286R mouse cells, because even small and/or transient changes in dNTP concentrations can strongly affect replication fidelity.
Effects on mismatch repair?
It is also possible that the Pol2P286R mutation may affect a third determinant of replication fidelity, DNA mismatch repair. Although the major eukaryotic DNA polymerase used to correctly resynthesize DNA after a mismatch has been excised is thought to be Pol δ (reviewed in ref. 20), a role for Pol ε in mismatch repair has also been suggested (e.g., see ref. 21). Perhaps more important, because mismatch repair in E. coli can be readily saturated by excess replication errors due to a proofreading defect (22), it seems possible that mismatch repair in mammals could also be saturated in the Pol2P286R mutant, thereby contributing to the ultramutator phenotype. Consistent with compromised mismatch repair, Hodel et al. (23) recently reported that suppression of mismatch repair was sufficient to drive explosive mutation accumulation in human cells lacking a single Pole proofreading allele, and Haradhvala et al. (24) recently identified two previously unexplained Cosmic database mutational signatures in tumors arising from loss of polymerase proofreading followed by subsequent loss of mismatch repair.
Wide-ranging implications and applications of this approach
As discussed by Li et al., the approach they have taken in mice is potentially applicable to study any of several other ultramutators described in the human database, including other POLE ultramutators suggested to bind to DNA and whose homologs in yeast reduce replication fidelity (17). The ultramutator phenotype of the Pol2P286R mutant might also partially reflect altered interactions of the POLE catalytic subunit with any of several other proteins at the replication fork, including the three noncatalytic subunits of the POLE holoenzyme, or any of dozens of other proteins involved in replication (25), several of which are known to affect genome stability. It is also possible that expression levels of mutant and WT Pole may vary in a transient manner, thereby contributing to the ultramutator phenotype observed in tumors. Finally, while the current evidence strongly suggests that the Pol2P286R-mutant mouse has highly error-prone replication that promotes cancer, Pol ε is also reported to function in several DNA repair reactions that can affect genome stability, including base excision repair, nucleotide excision repair, and repair of dsDNA breaks (reviewed in ref. 1). Regardless of whether the final explanation(s) are related to the above considerations or something yet to be appreciated, Li et al. have now provided an outstanding opportunity to further explore the contribution of ultramutagenesis to carcinogenesis and its treatment and to better understand eukaryotic DNA replication fidelity.
Acknowledgments
Research in the Kunkel laboratory is funded by Projects Z01 ES065070 and Z01 ES065089 to TAK from the Division of Intramural Research of the National Institutes of Health, National Institute of Environmental Health Sciences.
I sincerely thank Joseph Dahl for assistance in preparing Figure 1 and Joseph Dahl (NIEHS) and Katarzyna Bebenek (NIEHS) for their advice on the content of this article.
Version 1. 08/20/2018
Electronic publication
Version 2. 08/31/2018
Print issue publication
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(9):3754–3756. https://doi.org/10.1172/JCI123021.
See the related article at Polymerase-mediated ultramutagenesis in mice produces diverse cancers with high mutational load.
References
- 1.Rayner E, et al. A panoply of errors: polymerase proofreading domain mutations in cancer. Nat Rev Cancer. 2016;16(2):71–81. doi: 10.1038/nrc.2015.12. [DOI] [PubMed] [Google Scholar]
- 2.Campbell BB, et al. Comprehensive analysis of hypermutation in human cancer. Cell. 2017;171(5):1042–1056.e10. doi: 10.1016/j.cell.2017.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li HD, et al. Polymerase-mediated ultramutagenesis in mice produces diverse cancers with high mutational load. J Clin Invest. 2018;128(9):4179–4191. doi: 10.1172/JCI122095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loeb LA, Springgate CF, Battula N. Errors in DNA replication as a basis of malignant changes. Cancer Res. 1974;34(9):2311–2321. [PubMed] [Google Scholar]
- 5.Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017;355(6331):1330–1334. doi: 10.1126/science.aaf9011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barbari SR, Shcherbakova PV. Replicative DNA polymerase defects in human cancers: Consequences, mechanisms, and implications for therapy. DNA Repair (Amst) 2017;56:16–25. doi: 10.1016/j.dnarep.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams LN, Herr AJ, Preston BD. Emergence of DNA polymerase ε antimutators that escape error-induced extinction in yeast. Genetics. 2013;193(3):751–770. doi: 10.1534/genetics.112.146910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kunkel TA, Erie DA. Eukaryotic mismatch repair in relation to DNA replication. Annu Rev Genet. 2015;49:291–313. doi: 10.1146/annurev-genet-112414-054722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stillman B. Reconsidering DNA Polymerases at the replication fork in eukaryotes. Mol Cell. 2015;59(2):139–141. doi: 10.1016/j.molcel.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hogg M, et al. Structural basis for processive DNA synthesis by yeast DNA polymerase ε. Nat Struct Mol Biol. 2014;21(1):49–55. doi: 10.1038/nsmb.2712. [DOI] [PubMed] [Google Scholar]
- 11.Morgan AA, Rubenstein E. Proline: the distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS One. 2013;8(1):e53785. doi: 10.1371/journal.pone.0053785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shinbrot E, et al. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014;24(11):1740–1750. doi: 10.1101/gr.174789.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kane DP, Shcherbakova PV. A common cancer-associated DNA polymerase ε mutation causes an exceptionally strong mutator phenotype, indicating fidelity defects distinct from loss of proofreading. Cancer Res. 2014;74(7):1895–1901. doi: 10.1158/0008-5472.CAN-13-2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pavlov YI, Frahm C, Nick McElhinny SA, Niimi A, Suzuki M, Kunkel TA. Evidence that errors made by DNA polymerase α are corrected by DNA polymerase delta. Curr Biol. 2006;16(2):202–207. doi: 10.1016/j.cub.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Flood CL, Rodriguez GP, Bao G, Shockley AH, Kow YW, Crouse GF. Replicative DNA polymerase δ but not ε proofreads errors in Cis and in Trans. PLoS Genet. 2015;11(3):e1005049. doi: 10.1371/journal.pgen.1005049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.St Charles JA, Liberti SE, Williams JS, Lujan SA, Kunkel TA. Quantifying the contributions of base selectivity, proofreading and mismatch repair to nuclear DNA replication in Saccharomyces cerevisiae. DNA Repair (Amst) 2015;31:41–51. doi: 10.1016/j.dnarep.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barbari SR, Kane DP, Moore EA, Shcherbakova PV. Functional analysis of cancer-associated DNA polymerase ε variants in Saccharomyces cerevisiae. G3 (Bethesda) 2018;8(3):1019–1029. doi: 10.1534/g3.118.200042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bebenek K, Kunkel TA. Streisinger revisited: DNA synthesis errors mediated by substrate misalignments. Cold Spring Harb Symp Quant Biol. 2000;65:81–91. doi: 10.1101/sqb.2000.65.81. [DOI] [PubMed] [Google Scholar]
- 19.Williams LN, et al. dNTP pool levels modulate mutator phenotypes of error-prone DNA polymerase ε variants. Proc Natl Acad Sci U S A. 2015;112(19):E2457–E2466. doi: 10.1073/pnas.1422948112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106(2):302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 21.Tran HT, Gordenin DA, Resnick MA. The 3′→5′ exonucleases of DNA polymerases delta and epsilon and the 5′→3′ exonuclease Exo1 have major roles in postreplication mutation avoidance in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19(3):2000–2007. doi: 10.1128/MCB.19.3.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fijalkowska IJ, Schaaper RM. Mutants in the Exo I motif of Escherichia coli dnaQ: defective proofreading and inviability due to error catastrophe. Proc Natl Acad Sci U S A. 1996;93(7):2856–2861. doi: 10.1073/pnas.93.7.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hodel KP, et al. Explosive mutation accumulation triggered by heterozygous human Pol ε proofreading-deficiency is driven by suppression of mismatch repair. Elife. 2018;(7):e32692. doi: 10.7554/eLife.32692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haradhvala NJ, et al. Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat Commun. 2018;9(1):1746. doi: 10.1038/s41467-018-04002-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeeles JTP, Janska A, Early A, Diffley JFX. How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol Cell. 2017;65(1):105–116. doi: 10.1016/j.molcel.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]