

Abstract
Remodeling of mitochondrial metabolism plays an important role in regulating immune cell fate, proliferation, and activity. Furthermore, given their bacterial ancestry, disruption in mitochondrial fidelity leading to extravasation of their content initiates and amplifies innate immune surveillance with a myriad of physiologic and pathologic consequences. Investigations into the role of mitochondria in the immune system have come to the fore, and appreciation of mitochondrial function and quality control in immune regulation has enhanced our understanding of disease pathogenesis and identified new targets for immune modulation. This mitochondria-centered Review focuses on the role of mitochondrial metabolism and fidelity, as well as the role of the mitochondria as a structural platform, for the control of immune cell polarity, activation, and signaling. Mitochondria-linked disease and mitochondrially targeted therapeutic strategies to manage these conditions are also discussed.
Introduction
Given the preponderance of mitochondria within cells, the late physician-scientist and essayist Lewis Thomas posited that he “could be taken for a very large, motile colony of respiring bacteria” (1). At that time, despite their bacterial origins, there was little appreciation of the integral role of mitochondria in the regulation of immunity. Today, our recognition of the influence of mitochondrial fidelity on immune function extends beyond classical immunology to include mitochondria-linked immune perturbations that contribute to inflammation of degenerative diseases and to autoimmunity (2). In contrast, recognition of the metabolic underpinnings of immune cell function has been evident since the 16th century as manifested by the persistence of the maxim that “starving a fever and feeding a cold” hastens recovery from infectious illnesses (3). Components of this adage have been experimentally validated (4), and intermittent fasting and caloric restriction show evidence of blunting inflammation-linked diseases (5–10).
The field of study exploring the interdependence between metabolism and immune cell fate and function is termed immunometabolism (11–13). Additionally, given the persistence of bacterial signatures within mitochondria, mitochondrial components play diverse roles in triggering immune surveillance programs (2, 14). These aspects of mitochondrial biology and metabolism, in addition to their role in disease pathophysiology and their potential for therapeutic targeting, are the focus of this Review.
Immunometabolism
Metabolic plasticity and immune cell fate.
The bidirectional control between cellular metabolism and innate and adaptive immune cell fate and function has been most extensively explored in the modulation of macrophage polarization and of CD4+ T cell differentiation/activation, respectively (11–13). The metabolic pathways that contribute to immune cell activation and differentiation are schematized in Figure 1A, and examples of how they support immune cell fate are summarized here. On the one hand, inflammatory macrophages (M1) employ aerobic glycolysis for energy production and divert glycolytic intermediates into the pentose phosphate pathway for NADPH synthesis. NADPH in turn is catabolized by NADPH oxidase to generate reactive oxygen species (ROS) to facilitate antimicrobial effects (15). The corresponding preference for glutamine oxidation additionally promotes mitochondrial electron transfer chain (ETC) ROS production (16). On the other hand, alternatively polarized macrophages (M2) rely more on glucose and fat oxidative metabolism to orchestrate reparative functions (17, 18). However, the requirement for fatty acid oxidation in reparative macrophage function has recently been disputed by observations that genetic disruption of macrophage mitochondrial fat uptake did not prevent M2 polarization (19). It should be noted that the binary classification of macrophages is partially an experimental construct, given that macrophage polarity is a spectrum with multiple distinct signatures and functional phenotypes between these “poles” (20).
Figure 1. Overview of metabolic remodeling with immune cell differentiation and activation.

Overview of metabolic remodeling with immune cell differentiation and activation. (A) Major pathways linked to immunometabolism include the generation of ATP from cytosolic glycolysis or from oxidative phosphorylation of pyruvate, fatty acids, and glutamine. Cytosolic NADPH oxidase or mitochondria produce ROS, which can signal, oxidize proteins, or exert antimicrobial effects. The major sites of mitochondrial ROS production linked to immunometabolism are generated at complexes I and III of the ETC. (B) In monocytes, metabolic remodeling is most extensively characterized in the differentiation into the M1 and M2 cell fates. In CD4+ T cells, immunometabolism has been explored at multiple levels of differentiation, proliferation, activation, and migration. B cell immunometabolic control is less well explored; however, metabolic remodeling is important for different B cell fates. FAO, fatty acid oxidation; Glut Ox, glutamine oxidation; Gly, glycolysis; GO, glucose oxidation; PPP, pentose phosphate pathway; RET, reverse electron transport in the electron transfer chain for ROS generation.
The transition from quiescent to activated effector CD4+ T cells is a growth- and energy-demanding event that is accomplished, in part, by a switch from preferential oxidative phosphorylation to an increased reliance on aerobic glycolysis, the pentose phosphate pathway, and glutamine oxidation (21–24). Conversely, regulatory T cells (Tregs) retain reliance on oxidative phosphorylation (25), although they depend on glycolysis for migration (26).
The genetic requirement of these metabolic regulatory effects on immune cell fate is evident where the experimental disruption of glucose uptake impairs effector T cell proliferation and activation (21) and its promotion augments effector T cell function (27) and inflammatory macrophage polarity (28). This reliance on defined metabolic preference is further validated by the promotion of M2 polarization by experimental induction of glucose oxidation (18), and by the facilitation of M1 polarization by genetic disruption of oxidative metabolism and promotion of glycolysis (29).
The characterization of immunometabolism is extending into other immune cells, and metabolic preferences support dendritic cell immunogenic function (30) and B cell quiescence, proliferation, antibody-generating capacity, and survival (31–35). As is discussed in the final section of this Review, therapeutics to modulate substrate preference are being tested in T and B cell–linked disease such as systemic lupus erythematosus. A schematic summarizing metabolic remodeling links to immune cell fate is shown in Figure 1B.
TCA cycle “fragmentation” in immune activation.
The tricarboxylic acid (TCA) cycle generates reducing equivalents for ETC function through oxidation of acetyl-CoA derived from glucose, fat, and amino acids. Immune Toll-like receptor (TLR) and cytokine signaling (36) disrupts coordinated flux of intermediates through the TCA-inciting accumulation of distinct metabolites. Lipopolysaccharide (LPS) signals through TLR4 to upregulate immunoresponsive gene 1 (IRG1), which encodes an enzyme that decarboxylates the TCA intermediate cis-aconitase to produce itaconic acid (37). Itaconic acid exhibits antimicrobial activity (37) and macrophage antiinflammatory effects (38) and functions as an endogenous succinate dehydrogenase (SDH) inhibitor (39). Inhibition of SDH, in turn, promotes succinate accumulation in the TCA cycle in addition to inhibition of complex II of the ETC (38). The accumulation of succinate additionally facilitates transactivation of the proinflammatory cytokine IL-1β (40).
Interestingly, modulation in metabolic intermediate flux through the TCA cycle also controls macrophage polarity (41). Here, levels of specific metabolites confer distinct macrophage functions, as evidenced by succinate and extramitochondrial acetyl-CoA functioning as key intermediates for optimal M1 function. These macrophages exhibit reduced isocitrate dehydrogenase (IDH) transcript levels with diminished IDH enzyme activity (41). This results in reduced citrate conversion to α-ketoglutarate, resulting in citrate accumulation. Excess mitochondrial citrate translocates into the cytosol through the mitochondrial citrate carrier (CIC). Cytosolic citrate conversion to acetyl-CoA promotes production of proinflammatory prostaglandin, nitric oxide, and ROS (42). Interestingly, increased mitochondrial citrate also undergoes decarboxylation to generate itaconic acid, which functions as described above (37–39), and genetic knockdown of CIC impairs LPS-induced M1 activation (42). Alternatively, increased TCA glutamine utilization fuels M2 polarization by upregulating TCA-encoding genes and chemokine production (41). Recent evidence also shows that itaconate directly alkylates cysteine residues on a transcriptional repressor, KEAP1. This in turn enables the transcriptional induction of antioxidant and antiinflammatory encoding genes to limit inflammation and modulate type I IFNs (43). A dimethyl derivative of another TCA intermediate, fumarate, also modulates metabolism via a posttranslational mechanism. Here, dimethyl fumarate covalently modifies catalytic cysteine residues on the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in a process termed “succination.” This blunts glycolysis in myeloid and lymphoid cells, conferring antiinflammatory effects (44). Together these data illustrate the integration of metabolism and immune cell function where inflammatory signaling induces alterations in intermediary metabolism to generate metabolites that serve as signal transducers and posttranslational modifiers to regulate subsequent immune cell function.
ETC perturbations in the control of immune signaling.
A role of LPS signaling in impairing the ETC has been recognized for decades (45), and recently was linked to LPS-induced mitochondrial ROS production (46). A mechanism underpinning the LPS-ROS nexus is TLR4-driven succinate oxidation that orchestrates mitochondrial hyperpolarization with concomitant excess macrophage ROS production. This ROS signaling–induced inflammatory gene programing upregulates IL-1β, and inhibition of succinate oxidation during TLR4 activation promotes antiinflammatory gene expression signatures with induction of the canonical antiinflammatory cytokine IL-10 (16). The observation of blunting of this ROS generation by rotenone-mediated ETC complex I inhibition (16) implicates reverse electron transport (RET) as a mechanism propagating complex I ROS generation (47).
Another identified mechanism is operational following macrophage exposure to live E. coli or bacterial RNA. Here, bacterial components promote disassembly of mitochondrial supercomplexes with disruption of complex I respiration (48). This mechanism depends on phagosomal NADPH oxidase ROS generation to oxidize specific complex I proteins to facilitate complex disassembly (49). Complex I disruption in turn promotes increased respiration through complex II in parallel with IL-1β-induction, and the inhibition of complex II under these conditions reduces IL-1β and increases IL-10 levels (48). Interestingly, activation of TNF-α by E. coli is independent of complex II activity, a finding consistent with prior studies showing that TNF-α production was less sensitive to macrophage metabolic preference (40). Notably, complex I disassembly and its link to complex II activation are independent of LPS and TLR4 signaling (48). Although the TLR4/RET signaling (16) and microbial RNA/NADPH oxidase (48) mechanisms have not been reconciled, they collectively support that numerous mechanisms of ETC disruption regulate IL-1β–driven inflammation. ETC-derived ROS similarly plays a critical role in antigen-specific T cell activation (50).
Mitochondrial cargo in immune activation
The role of mitochondrial constituents in immune activation stems in part from the incorporation of a prokaryotic endosymbiont into more complex (archaeal) bacteria (51). During subsequent evolution into eukaryotic cells, the majority of the engulfed bacterial genes were transferred to the nuclear genome (52). The residual bacterial genome persisted as the mitochondrial genome, and the hypomethylated CpG motifs of mitochondrial DNA (mtDNA) retained immunogenic properties of bacterial CpG DNA motifs (53). Additional mitochondrial features that phenocopy bacteria include protein synthesis, where mtDNA translation is initiated at N-formyl-methionine residues (54), and retention of the phospholipid cardiolipin within the inner mitochondrial membrane (IMM) (55). A consequence of these bacterial remnants is that mitochondrial cargo is viewed as foreign by mammalian intracellular and extracellular immune surveillance programs. These mitochondrial features are in a sense “immune-privileged” and isolated from the cytosol by encasement by the outer mitochondrial membrane (OMM). Loss of integrity of the mitochondria exposes these components to immune pattern recognition receptors (PRRs), functioning as mitochondrial alarmins to trigger immune activation. The canonical immune surveillance programs that recognize signatures/components of mitochondrial stress are discussed below. It should be noted that although these programs are reviewed separately here, the potential for them to be activated contemporaneously is beginning to be recognized (56–58). Additionally, the OMM functions as a structural platform for the assembly of immune regulatory complexes (59–61), and mitochondria–endoplasmic reticulum (ER) contact sites operate as a signaling hub enabling metabolic remodeling to reactivate memory CD8+ T cells (62).
NLRP3 inflammasome.
The inflammasome is an intracellular immune surveillance program that recognizes either pathogen-associated molecular patterns (PAMPs) or host cell–derived damage-associated molecular patterns (DAMPs). The inflammasome is a multiprotein complex that assembles and self-oligomerizes to promote cleavage and activation of canonical cytokines, namely IL-1β and IL-18, to amplify immune responses (63, 64). The nucleotide oligomerization domain–like (NOD-like) receptor family pyrin domain 3 (NLRP3) inflammasome is activated by sterile inflammation associated with crystal-linked diseases like gout, and by metabolic diseases including obesity, diabetes, hyperlipidemia, and cardiovascular disease (65), and its activation further exacerbates these diseases (66–69). For example, in obesity, the inflammasome is initiated through adipose tissue hypertrophy with macrophage infiltration and cytokine secretion; elevated circulating saturated fatty acids, glucose, and lipids; and/or obesity-linked endotoxemia mediated by upregulation of NLRP3 and its canonical cytokines (pro–IL-1β and pro–IL-18) via NF-κB (70–73). Transcriptional induction of NLRP3 inflammasome components is termed “priming,” and mitochondrial ROS signaling contributes to this regulation (74, 75). Priming-induced mitochondrial ROS also facilitates externalization of cardiolipin and promotes the association of NLRP3 and caspase-1 with mitochondria (76).
Following priming, subsequent activation is instigated by assembly and oligomerization of canonical inflammasome constituents (65). NLRP3 complex oligomerization initiates caspase-1 activation and cleavage of pro–IL-1β and pro–IL-18 into bioactive cytokines that function to amplify inflammation (63, 64). Multiple mitochondrial components function as NLRP3 DAMPs following disruption of mitochondrial integrity, including mtDNA (53), cardiolipin (55, 76), and mitochondrial ROS (56, 77). The role of mitochondria in orchestrating this program is further supported by (a) the close association between mitochondria and NLRP3 complex formation, with coordinated perinuclear localization during inflammasome induction (59, 76); (b) the role of the mitochondrial antiviral signaling protein (MAVS) in NLRP3 recruitment to mitochondria following noncrystalline inflammasome activation (60, 78); and (c) evidence that depletion of mtDNA in ρ0 (mtDNA-deficient) cells blunts inflammasome activation (77). Interestingly, mtDNA binds to both the NLRP3 inflammasome and the distinct double-stranded DNA–sensing inflammasome AIM2 (79), whereas oxidized mtDNA interacts exclusively with NLRP3 (79). Finally, mitochondria also function as a structural platform upon which multiple proteins that constitute the NLRP3 inflammasome nucleate and assemble (59, 60, 76, 79, 80). Interestingly, numerous degenerative conditions, including type 2 diabetes, atherosclerosis, chronic kidney disease, and aging, manifest with sterile inflammation and mitochondrial dysfunction (66, 69, 81, 82), although whether these features are mechanistically integrated has not always been experimentally confirmed.
Whether mitochondrial metabolic dysfunction rather than disruption per se activates the NLRP3 inflammasome is less well characterized (75). However, inhibition of mitochondrial respiration, promotion of aerobic glycolysis (75, 83), and/or the selective induction of fatty acid oxidation (84) facilitate inflammasome activation. Additionally, NLRP3 activators diminish mitochondrial membrane potential and reduce intracellular NAD+ levels, thereby blunting SIRT2 deacetylase activity. The consequent acetylation of the SIRT2 target α-tubulin facilitates trafficking of NLRP3 inflammasome components to spatially align and bind to mitochondria and ER to facilitate inflammasome activation (80).
Investigation into whether the mitochondrial permeability transition pore (mPTP) regulates extrusion of mitochondrial content into the cytoplasm has also been explored. Although the structure of the mPTP remains contentious (85, 86), its ability to facilitate release of high–molecular weight molecules (approximately 1.5 kDa) through the IMM is established, as is the role of increased intracellular calcium (Ca2+) in opening the pore (87), and the inhibition of opening by cyclosporin A (CsA) (85, 86). The contribution of the mPTP to NLRP3 activation is supported by inflammasome activation as a result of increasing intracellular Ca2+ (88, 89) and by CsA blunting of mtDNA extrusion (77). The investigation of the mechanisms underpinning mtDNA extrusion through the mPTP has not been comprehensively defined. Nevertheless, given its proximity to ROS and its lack of protective histones, mtDNA is susceptible to oxidative stress with formation of 8-hydroxydeoxyguanosine modifications. These base modifications preferentially reside on mtDNA fragments rather than on intact circular mtDNA (90), and mPTP activation is linked to mtDNA fragment extrusion in a CsA-dependent manner (91). Additionally, the extent of this leak appears to be dependent on mtDNA fragment size (92). CsA similarly blunts NLRP3 activation in response to numerous ROS-dependent and ROS-independent inflammasome triggers (55). Finally, the interdependence of different immune activation triggers is further illustrated by the demonstration that E. coli–induced complex II–driven respiration is dependent on NLRP3 (48), although the mechanisms underpinning this are unclear.
mtDNA TLR9 signaling.
Intracellular TLR9 is a distinct immune surveillance PRR that recognizes unmethylated DNA derived from bacteria, viruses, and extruded mtDNA (93, 94) to initiate a MyD88 signaling– and NF-κB transactivation–dependent inflammatory cascade (95). Interestingly, impaired mitochondrial turnover (94), traumatic injury (54), and nonalcoholic steatohepatitis (96) promote mtDNA extravasation, triggering TLR9 signaling that results in organ-specific (94, 96) or systemic inflammation (54). Although the mechanisms underpinning recognition of nucleotides by TLR9 and their subsequent activation are better characterized for bacterial and viral nucleotides, the mtDNA-induced programs probably mirror these mechanisms. One potential mechanism whereby mtDNA engages TLR9 is via trafficking within mitochondrial-derived vesicles to the endosomes (97). This mechanism, or other mechanisms that traffic mtDNA, are postulated to facilitate antigen presentation given that TLR9, which resides on the ER, is recruited to the endosome-lysosome compartments (98) to undergo proteolytic cleavage and receptor activation (99). In general, the CpG motifs that function as ligands for TLR9 activation sequester in the endosome-lysosome compartment before triggering this signaling pathway. In contrast, extracellular free or microparticle-encased mtDNA activates this pathway via canonical immune cell surface PRRs or via the immunoglobulin superfamily member receptor for advanced glycation end products (RAGE), which undergoes endocytosis to initiate TLR9-dependent signaling (100, 101). Interestingly, this RAGE-mediated inflammatory pathway regulates mitochondrial bioenergetics (102), although whether this potential mitochondrial feedback loop is triggered by mtDNA release does not appear to have been investigated.
cGAS-STING signaling.
The cytosolic DNA-sensing cGAS/STING (cyclic GMP–AMP synthase linked to stimulator of interferon genes) pathway is activated in response to exogenous viral infections (103) and in response to endogenous DNA leakage associated with cancer and aging (104). The detection of cytoplasmic DNA by cGAS generates cyclic dinucleotide cGAMP as a second messenger to activate STING. The downstream phosphorylation and activation of IFN regulatory factor 3 (IRF3) transactivate expression of IFN-β and canonical IFN-stimulated genes. In parallel with NLRP3 activation and TLR9 signaling, mtDNA activates cGAS/STING and type I IFN signaling (2). An intriguing route for the escape of mtDNA during cGAS/STING activation has been identified through the OMM in the context of apoptosis, in which Bak and Bax apoptotic channels release mtDNA (105, 106). Normally this leakage is inert because mtDNA is lysed by concurrent activation of apoptotic caspases (105, 106). However, when apoptotic caspases are inhibited or genetically disrupted, type I IFN signaling is initiated and manifests as enhanced resistance to viral infections (106). The enigma remains of how the IMM is breached given that this membrane is not necessarily disrupted during apoptosis (107). A recent finding shows that the OMM proteins Bak and Bax generate macropores that facilitate IMM herniation with the efflux of mtDNA (108). The possibility that this mechanism is operational in initiating cGAS/STING signaling is intriguing, but as yet unproven.
Nevertheless, the molecular machinery orchestrating mtDNA-initiated cGAS/STING activation has been further characterized following genetic disruption of transcription factor A, mitochondrial (TFAM), a regulatory protein that controls mitochondria transcription and translational and mitochondrial nucleoid integrity (109). Haploinsufficient TFAM cells exhibit disrupted mtDNA repair capacity and distorted mtDNA distribution in parallel with cGAS/STING activation (110). The role of mtDNA in TFAM deficiency-induced inflammation was validated by evidence of blunted antiviral signaling following depletion of mtDNA through dideoxycytosine-mediated inhibition of mtDNA replication. In contrast, disruption of mitochondrial quality control programs (expanded upon below) in TFAM-heterozygous cells exacerbated type I IFN signaling (110). Interestingly, and possibly owing to the role of TFAM in controlling nucleoid packaging, distinct mtDNA fragments from the D-loop regulatory region were implicated in cGAS/STING activation (111). Whether the D-loop mtDNA region confers specificity for IFN signaling requires exploration. In parallel with caspase-defective apoptotic signaling, reduction in TFAM levels also primes antiviral signaling in response to pathogen invasion and viral challenge.
Mitochondrial antiviral signaling.
An additional PRR family linked to mitochondria are retinoic acid–inducible gene I–like (RIG-I–like) receptors (RLRs) (14). An OMM-anchored adaptor molecule, termed the mitochondrial antiviral signaling protein (MAVS), is an essential platform for antiviral RLR signal transduction (61). The MAVS C-terminal transmembrane domain resembles other OMM protein transmembrane domains, and its depletion negated OMM MAVS localization (61). This transmembrane domain was shown to facilitate MAVS dimerization, providing an interface for direct binding and activation of downstream immune-effector molecules (112). Although mechanisms orchestrating recruitment of RLR signaling molecules to mitochondria remain uncertain, other OMM proteins function as MAVS cofactors. The TOM70 protein, which functions as an OMM pre-protein receptor for mitochondrial protein import, binds to MAVS, and the subsequent interaction between TOM70 and the chaperone HSP90 orchestrates binding of RLR effector molecules to coordinate antiviral signaling (113). Interestingly, this requirement for mitochondrial localization to activate the MAVS signalosome is exploited by viruses, as epitomized by hepatitis C virus, which encodes a serine protease that cleaves MAVS from the OMM to prevent antiviral signaling (114).
An additional mtDNA immune triggering pathway has recently been described. Here, exposure of T cells, B cells, NK cells, monocytes, and neutrophils to distinct oligonucleotides promulgates ejection of mtDNA out of the cell, which creates filamentous web-like structures that activate type I IFN signaling in adjacent cells (115). This process is insensitive to inhibition by TLR9, cGAS/STING, AIM2 inflammasome, ROS, or the permeability transition pore inhibitors (115). Although this process requires further characterization, it adds a previously unrecognized trigger for rapid immune activation. The contribution of mitochondrial components to immune regulation is shown in Figure 2.
Figure 2. Mitochondria-linked activation of immune pathways.
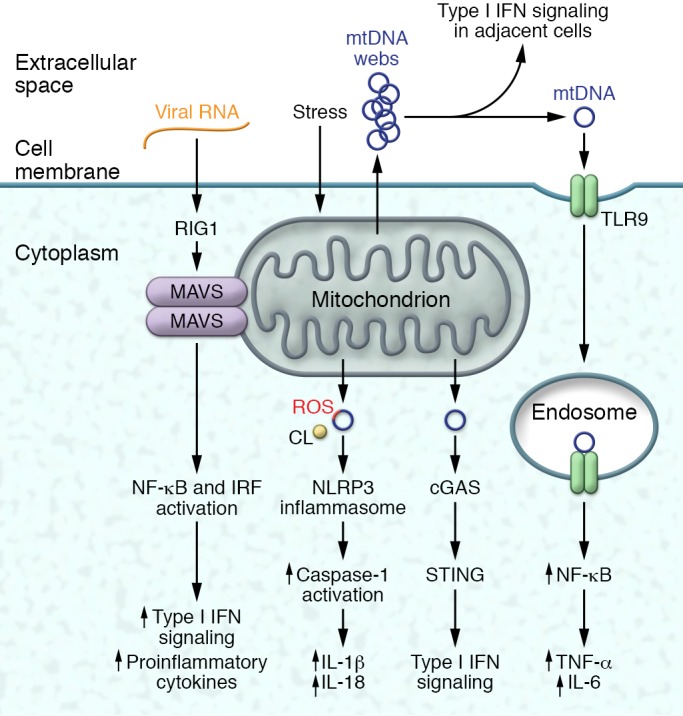
Mitochondrial content and structure can play integral roles in the activation of inflammatory signaling in response to stress effects or viral infections. Extracellular mtDNA can activate NF-κB–driven inflammation via the intracellular TLR9 receptor. Alternatively, released mtDNA can initiate type I IFN signaling in adjacent immune cells. In response to mitochondrial stressors, ROS-damaged mtDNA, mitochondrial ROS, and the release of cardiolipin (CL) from the IMM can activate the NLRP3 inflammasome to promote IL-1β and IL-18 signaling. The loss of mitochondrial integrity with the extrusion of mtDNA can also activate the cGAS/STING pathway and type I IFN signaling. Finally, the mitochondrion functions as a platform for the dimerization of MAVS to activate a combination of NF-κB and IFN regulator signaling in response to viral infections.
Taken together, there is evidence of considerable overlap between mitochondrial cargo–triggered immune surveillance programs, and it is conceivable that they may function independently and/or may be coordinately activated (56, 57). Additional studies are required to assess whether shared or distinct mechanisms of mtDNA extrusion and mitochondrial ROS signaling (65, 116), as well as different mtDNA species, dictate engagement of these various intracellular immune surveillance sensors.
Mitochondrial quality control programs
It stands to reason that if disruption of mitochondrial integrity propagates innate immune sensing and activation, enhancing mitochondrial quality control should confer immune resistance. Although an expansive discussion of these mitochondrial quality control programs is beyond the scope of this Review, emerging evidence supporting this proposition is briefly reviewed. The quality of mitochondria is controlled by numerous regulatory programs, including control of mitochondrial biogenesis (117) and dynamics (118), autophagy (119), and proteostasis (120, 121), and the role of “nutrient-sensing” programs to sustain mitochondrial fidelity (122).
Mitochondrial dynamics.
The control of mitochondrial fission and fusion plays an important role in controlling mitochondrial integrity, metabolism, and levels of mtDNA (118). Furthermore, mitochondrial dynamics facilitate turnover of damaged mitochondrial components in coordination with other housekeeping programs including autophagy and proteostasis. The molecular machinery controlling mitochondrial dynamics is well characterized (123), and their genetic disruption is linked to immune modulation. This is epitomized by observations that knockdown of the OMM fusion protein mitofusin 2 (Mfn2) abrogated viral infection–mediated NLRP3 inflammasome activation (124) and that modulation of Mfn2 levels controlled CD4+ T cell activation (125). A non-canonical function of Mfn2 similarly inhibits antiviral signaling through interaction with MAVS (126), and genetic induction of mitochondrial fission to remove damaged mtDNA abrogates type I IFN signaling in TFAM-depleted cells (110). The regulation of mitochondrial dynamics is also tightly coupled to metabolic remodeling in T cells in that genetically driving mitochondrial fusion in glycolytic effector T cells forces reprogramming toward oxidative metabolism and memory T cell function (127). Interestingly, the disruption of mitochondrial fusion in nonimmune cells can also evoke inflammation through TLR9 signaling in response to mtDNA leakage (128).
Mitochondrial autophagy.
The recycling of complete or damaged components of mitochondria occurs in isolation (mitophagy) or as a component of broader recycling of cellular content (macroautophagy/autophagy) to maintain mitochondrial quality control. The molecular machinery governing both mitophagy and autophagy is well defined, and the molecular or pharmacologic disruption of these programs demonstrates their link to innate immunity (129). The genetic depletion of autophagy mediators disrupts autophagy and mitophagy with subsequent extrusion of mtDNA into the cytosol. The consequences of reduced mitochondrial clearance activate NLRP3 inflammasome and RLR type I IFN production (59, 77, 116). Conversely, activation of a bacterially induced inflammasome family member, NLRC4, by Pseudomonas aeruginosa that exhibited mitochondrial damage is abrogated by autophagy induction (130). The control of mitochondrial content by autophagy is also operational in sustaining the regenerative potential of hematopoietic stem cells (HSCs). Here, aging-associated reductions in autophagy increase mitochondrial content and metabolic activity, resulting in accelerated myeloid differentiation and reduced HSC renewal (131). Mitophagy is also regulated, in part, by the E3 ligase parkin (132), and mutations in the parkin-encoding gene PARK2 lead to early-onset Parkinson disease (PD) (133). Neuroinflammation is increasingly recognized in PD (134), and parkin regulates inflammatory pathways (135–137). However, whether the parkin-linked neuroinflammation is linked to mitophagy does not appear to have been investigated.
Sirtuins.
The sirtuin family of deacylase enzymes, including three major NAD+-dependent deacetylases, play important roles in mitochondrial homeostasis (122). Briefly, the mitochondrial regulatory functions of these deacetylases can be exemplified by (a) SIRT1 contribution to nuclear regulatory control of mitochondrial biogenesis and to nutrient-sensing signal transduction (122); (b) SIRT2-mediated modulation of mitochondrial subcellular localization and control of nutrient-sensing signal transduction (138, 139); and (c) SIRT3 deacetylation and regulation of mitochondrial metabolic and homeostatic proteins (140). Studies linking these sirtuin effects to immune modulation are limited. The plurality of SIRT1 immunoregulatory effects is not directly linked to mitochondrial function per se, but rather is linked via deacetylation and inactivation of the RelA/p65 subunit of the canonical inflammatory NF-κB transactivator and via activation of the nutrient-sensing kinase AMPK, which modulates bioenergetics and activates autophagy (141). Nevertheless, SIRT1 activation skews macrophages to their reparative polarity via increased fatty acid oxidation (142). SIRT2 deacetylates and destabilizes microtubules, resulting in disrupted intracellular organelle trafficking. As previously described, inflammasome-mediated SIRT2 inhibition optimized microtubule-directed spatial alignment of mitochondria to amplify the NLRP3 inflammasome (80). Activation of SIRT3 has been shown to deacetylate and activate mitochondrial superoxide dismutase (SOD2), which inhibits the NLRP3 inflammasome by blunting mitochondrial ROS generation and by reducing mtDNA leakage (6, 56). The absence of SIRT3 similarly augments fatty acid–induced renal tubular inflammation via impaired mitochondrial oxidative metabolism and increased mitochondrial ROS (143).
Additional mitochondrial regulatory control programs linked to inflammation include regulation of mitochondrial biogenesis and modulation of uncoupling protein 2 (UCP2). Interestingly, IFN-γ drives a transcriptional regulatory program governing mitochondrial biogenesis (144). Induction of biogenesis augments mitochondrial respiratory function and ROS to control bacterial infections, and genetic disruption of mitochondrial biogenesis reduces ROS production with diminished murine bacterial-clearance capacity (144). At a distinct regulatory level, the inner mitochondrial membrane UCP2 modulates myeloid cell mitochondrial ROS. Here, genetic ablation of UCP2 heightens TLR4-initiated ROS-sensitive inflammatory signaling (145). Moreover, UCP2-KO mice display increased myeloid mitochondrial ROS and increased resistance against intracellular microbial infections (146).
Human disease, mitochondria, and immune modulation
In addition to the readily apparent disruption of mitochondrial integrity in response to intracellular viral and bacterial infections as well as traumatic cellular injury (54), the pathophysiology of autoimmune and inflammatory diseases is similarly linked to immunometabolism and to mitochondrial immunogenicity.
Systemic lupus erythematosus.
The autoimmune disease systemic lupus erythematosus (SLE) is characterized by multiorgan damage from dysregulated innate and adaptive immunity. Immune cell metabolic remodeling, mitochondrial ROS, and mtDNA perturbations contribute to this pathophysiology (147). Immunometabolism signatures include evidence that SLE-linked cytokine B cell–activating factor (148) promotes B cell metabolic remodeling with enhanced glycolysis and a greater antibody generation capacity (33), and that CD4+ T cells from humans with lupus and mouse models exhibit increased oxidative phosphorylation and glycolysis compared with controls (149).
Mitochondrial ROS and mtDNA concurrently contribute to neutrophil extracellular trap (NET) formation, in which ROS-induced oxidized mtDNA incorporates into, and amplifies, NETosis. Oxidized mtDNA also activates STING-dependent type I IFN signaling to exacerbate lupus (150). The roles of immunometabolism and mitochondrial-linked immune activation in SLE is schematized in Figure 3.
Figure 3. Metabolic remodeling and mitochondrial mediators of SLE.
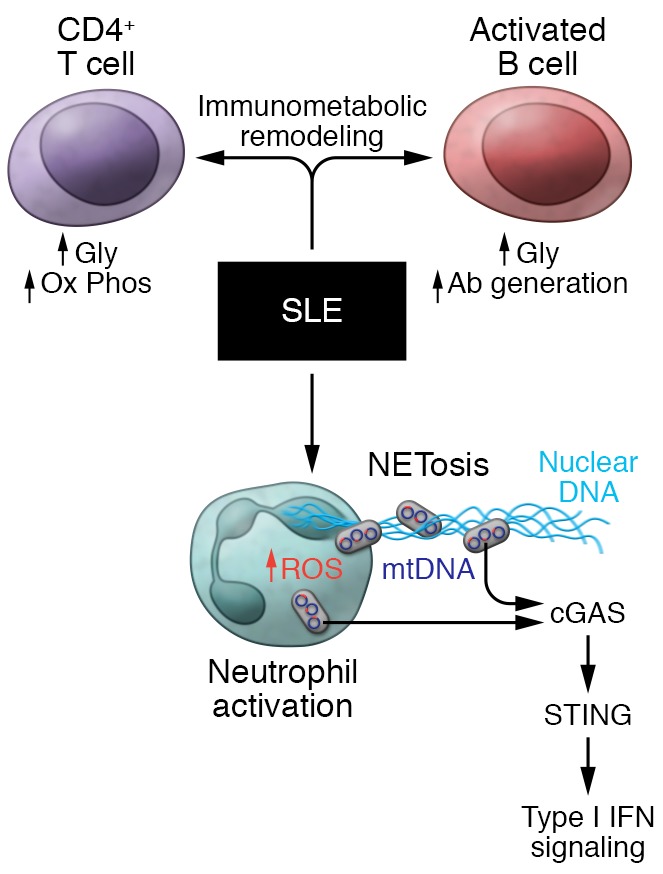
SLE pathology encompasses a broad array of immunometabolism- and mitochondria-initiated events. Immunometabolic remodeling is evident in both effector T cells, driving cytokine production, and in B cells, promoting antibody generation. Activation of neutrophils by immune complexes increases mitochondrial ROS, which promotes the oxidation of mtDNA. Subsequently, the concurrent extrusion of ROS-damaged mtDNA and nuclear/genomic DNA from NETs triggers mtDNA-dependent, STING-mediated type I IFN signaling. In parallel, the ROS-damaged mtDNA can also trigger cGAS/STING signaling in immune cells in a mechanism distinct from NETosis.
Rheumatoid arthritis and other autoimmune diseases.
Rheumatoid arthritis (RA) is predominantly characterized by inflammation of the joint synovium resulting in a polyarthritis. The immunogenic role of mitochondria in this pathophysiology is supported by markedly increased mitochondrial ROS production in circulating monocytes (151) and evidence of excessive mtDNA levels in the synovial fluid of RA patients (152, 153). Furthermore, the intra-articular injection of mtDNA but not nuclear DNA evokes arthritis in animal models (153), and in humans, levels of joint mtDNA correlate with inflammatory biomarkers of disease severity (151, 152). It has yet to be determined whether these mitochondrial triggers initiate or amplify RA.
Additional immune diseases linked to mitochondria include fibromyalgia, a disease in which cytochrome B gene mutations correlate with NLRP3 inflammasome activity (154). Additionally, antibodies directed against nuclear-encoded mitochondrial enzymes, termed anti-mitochondrial antibodies, are pathognomonic biomarkers of primary biliary cirrhosis (155).
Mitochondrial haplotypes have also been linked to inflammatory diseases such as age-related macular degeneration (156), and mtDNA-dependent inflammation is associated with cardiovascular and liver disease and linked to immunosenescence and other aging-associated degenerative conditions (2).
Targeting mitochondria to control inflammation
Given the importance of mitochondrial metabolic remodeling in immune cell polarity and activation and the impact of mitochondrial integrity on inflammation, the modulation of these mitochondrial effects is an intriguing target to regulate immune activity.
Metabolic modulators of immune function.
Numerous compounds that remodel metabolic pathways could be potential targets to control immunometabolism. The use of these compounds to temper human diseases or for evaluation in murine disease models is beginning to be explored, and examples are briefly reviewed below.
Similar to findings that succinate oxidation promotes M1 polarity, dimethyl malonate (DMM) and 3-nitroproprionic acid blunted SDH activity and ameliorated E. coli infection in mice, and DMM similarly blunted LPS-induced murine sepsis (16, 48). As described in the TCA cycle section, the TCA intermediates dimethyl fumarate and itaconate directly modulate metabolic and immune regulatory proteins via posttranslational modifications (43, 44). In an extension of these findings, heptelidic acid, which covalently modifies the same catalytic cysteine residues on GAPDH to inhibit its activity, blunts autoimmune encephalopathy in mice (44), and the cell-permeable itaconate derivative 4-octyl itaconate counteracts LPS-induced inflammation in macrophages and in mice (43).
Furthermore, given the parallel induction of mitochondrial oxidative phosphorylation and glycolysis in SLE CD4+ T cells, a combination of metformin to block mitochondrial respiration and 2-deoxyglucose (2-DG) to reduce glucose uptake and glycolysis was tested in primary human cells and in murine lupus models (149, 157). Given separately, metformin or 2-DG prevented murine T cell activation, and metformin alone blunted human lupus CD4+ T cell IFN-γ secretion (149, 157), whereas the combined agents were required to reverse murine lupus (157).
Interventions to enhance mitochondrial integrity.
Given that NAD+ functions as a cofactor in sirtuin activation, numerous NAD+ metabolic intermediate precursors have been employed to test their effects on improving mitochondrial integrity in disease (158–160). Nicotinamide riboside (NR), a precursor in the NAD+ salvage pathway, activated SIRT3 targets and blunted the NLRP3 inflammasome in primary human peripheral blood mononuclear cells (6) and in a murine model of hepatic steatosis (161). Additionally, administration of the NAD+ precursor nicotinamide mononucleotide similarly blunted pancreatic islet IL-1β production in diabetogenic mice (162). Finally, the administration of a neutralizing antibody to disrupt the CD38 NADase increased T cell NAD+ levels, conferred metabolic remodeling, and augmented antitumor functioning of these effector cells (163).
TLR9 antagonists.
TLR9 antagonists are being developed for clinical use, and their efficacy following mtDNA extrusion–mediated TLR9 activation was evident in disease models including protection against murine mitophagy-defective myocarditis and nonalcoholic steatohepatitis (94, 96). TLR9 antagonists similarly blunt mtDNA-induced NETosis in primary human neutrophils (164) and reduce autoimmune symptoms in a chemically induced murine lupus (165).
Mitochondrial ROS inhibitors.
The role of mitochondrial ROS in immune modulation has been described throughout this Review, and the effect of blunting mitochondrial ROS on immune modulation has been explored. Examples of this include administering the mitochondrial-targeted SOD mimetic mitoTEMPO to blunt the inflammasome in human macrophages (6) and to ameliorate type I IFN signaling in murine lupus (150).
Caloric-restricted diets.
Although intermittent fasting has been found to blunt inflammatory diseases and markers of innate immune activation, the mechanisms underpinning this regulation are incompletely characterized (5, 7–10). To explore the mitochondrial contribution to this biology, prolonged fasting was interrogated in human subjects and in a mouse model and showed that the NLRP3 inflammasome was blunted, in part, via SIRT3 activation with improvement in mitochondrial respiration and enhanced mitochondrial fidelity (6, 56).
In light of the recognition of the role of mitochondria in immune modulation, numerous ongoing studies will exploit this link to explore the role of mitochondrial modulation to control immune responses.
Conclusions
The field of immunometabolism is in its infancy, and our understanding of how diverse immune surveillance programs integrate with mitochondrial fidelity is identifying novel targets for the modulation of innate immune activity. Further characterization of these pathways should cement the centrality of the mitochondrion in directing health and disease. An important caveat in this field is the significant differences between murine and human immune systems (11, 57, 63, 166, 167). This caveat reinforces our need to sustain the exploration of human subjects to understand the role of mitochondria in orchestrating immune function and pathology (168). Conversely, the relative accessibility of human immune cells affords the opportunity for rapid advancement in the exploration and interrogation of mitochondria to understand their role in immune function. Finally, given our expanding understanding of the role of mitochondria in immune activation, the commentary by Lewis Thomas that “mitochondria are stable and responsible lodgers, and I choose to trust them” (169) should include the proviso, “if they retain their fidelity and appropriate location within cells.”
Acknowledgments
This Review is mitochondria-centric, focusing on the central role of mitochondria the regulation of immune cell polarity, activation, and immune signaling. Immune pathway signaling has not been extensively reviewed. Additionally, space constraints have limited inclusion of all original articles supporting the concepts explored. MNS is supported by the NIH, National Heart, Lung, and Blood Institute Division of Intramural Research (ZIA-HL006047-07 and ZIA-HL005102-12)
Version 1. 07/30/2018
Electronic publication
Version 2. 08/31/2018
Print issue publication
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(9):3651–3661.https://doi.org/10.1172/JCI120845.
References
- 1. Thomas L. Organelles as organisms. In: The lives of a cell — notes of a biology watcher. London, United Kingdom: Penguin Books; 1978:67–71. [Google Scholar]
- 2.West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. 2017;17(6):363–375. doi: 10.1038/nri.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fischetti M. Fact or fiction?: feed a cold, starve a fever. New York, New York, USA: Scientific American; 2016. https://www.scientificamerican.com/article/fact-or-fiction-feed-a-cold/ Accessed June 19, 2018.
- 4.Wang A, et al. Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell. 2016;166(6):1512–1525.e12. doi: 10.1016/j.cell.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi IY, Lee C, Longo VD. Nutrition and fasting mimicking diets in the prevention and treatment of autoimmune diseases and immunosenescence. Mol Cell Endocrinol. 2017;455:4–12. doi: 10.1016/j.mce.2017.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Traba J, et al. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J Clin Invest. 2015;125(12):4592–4600. doi: 10.1172/JCI83260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi IY, et al. A diet mimicking fasting promotes regeneration and reduces autoimmunity and multiple sclerosis symptoms. Cell Rep. 2016;15(10):2136–2146. doi: 10.1016/j.celrep.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei M, et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci Transl Med. 2017;9(377):eaai8700. doi: 10.1126/scitranslmed.aai8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fraser DA, Thoen J, Djoseland O, Forre O, Kjeldsen-Kragh J. Serum levels of interleukin-6 and dehydroepiandrosterone sulphate in response to either fasting or a ketogenic diet in rheumatoid arthritis patients. Clin Exp Rheumatol. 2000;18(3):357–362. [PubMed] [Google Scholar]
- 10.Johnson JB, et al. Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma. Free Radic Biol Med. 2007;42(5):665–674. doi: 10.1016/j.freeradbiomed.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van den Bossche J, O’Neill LA, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol. 2017;38(6):395–406. doi: 10.1016/j.it.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 12.Olenchock BA, Rathmell JC, Vander Heiden MG. Biochemical underpinnings of immune cell metabolic phenotypes. Immunity. 2017;46(5):703–713. doi: 10.1016/j.immuni.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell. 2017;169(4):570–586. doi: 10.1016/j.cell.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11(6):389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haschemi A, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012;15(6):813–826. doi: 10.1016/j.cmet.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mills EL, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167(2):457–470.e13. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vats D, et al. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006;4(1):13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan Z, et al. Pyruvate dehydrogenase kinase 1 participates in macrophage polarization via regulating glucose metabolism. J Immunol. 2015;194(12):6082–6089. doi: 10.4049/jimmunol.1402469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nomura M, Liu J, II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, Finkel T. Fatty acid oxidation in macrophage polarization. Nat Immunol. 2016;17(3):216–217. doi: 10.1038/ni.3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murray PJ, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Macintyre AN, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20(1):61–72. doi: 10.1016/j.cmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerriets VA, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. 2015;125(1):194–207. doi: 10.1172/JCI76012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang R, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35(6):871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caro-Maldonado A, Gerriets VA, Rathmell JC. Matched and mismatched metabolic fuels in lymphocyte function. Semin Immunol. 2012;24(6):405–413. doi: 10.1016/j.smim.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Angelin A, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 2017;25(6):1282–1293.e7. doi: 10.1016/j.cmet.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kishore M, et al. Regulatory T cell migration is dependent on glucokinase-mediated glycolysis. Immunity. 2017;47(5):875–889 e810. doi: 10.1016/j.immuni.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacobs SR, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180(7):4476–4486. doi: 10.4049/jimmunol.180.7.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freemerman AJ, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem. 2014;289(11):7884–7896. doi: 10.1074/jbc.M113.522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ko CW, Counihan D, Wu J, Hatzoglou M, Puchowicz MA, Croniger CM. Macrophages with a deletion of the phosphoenolpyruvate carboxykinase 1 (Pck1) gene have a more proinflammatory phenotype. J Biol Chem. 2018;293(9):3399–3409. doi: 10.1074/jbc.M117.819136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pearce EJ, Everts B. Dendritic cell metabolism. Nat Rev Immunol. 2015;15(1):18–29. doi: 10.1038/nri3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jellusova J, et al. Gsk3 is a metabolic checkpoint regulator in B cells. Nat Immunol. 2017;18(3):303–312. doi: 10.1038/ni.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mambetsariev N, Lin WW, Wallis AM, Stunz LL, Bishop GA. TRAF3 deficiency promotes metabolic reprogramming in B cells. Sci Rep. 2016;6:35349. doi: 10.1038/srep35349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caro-Maldonado A, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol. 2014;192(8):3626–3636. doi: 10.4049/jimmunol.1302062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H, et al. ATP-degrading ENPP1 is required for survival (or persistence) of long-lived plasma cells. Sci Rep. 2017;7(1):17867. doi: 10.1038/s41598-017-18028-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lam WY, et al. Mitochondrial pyruvate import promotes long-term survival of antibody-secreting plasma cells. Immunity. 2016;45(1):60–73. doi: 10.1016/j.immuni.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Infantino V, Iacobazzi V, Menga A, Avantaggiati ML, Palmieri F. A key role of the mitochondrial citrate carrier (SLC25A1) in TNFα- and IFNγ-triggered inflammation. Biochim Biophys Acta. 2014;1839(11):1217–1225. doi: 10.1016/j.bbagrm.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michelucci A, et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A. 2013;110(19):7820–7825. doi: 10.1073/pnas.1218599110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lampropoulou V, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 2016;24(1):158–166. doi: 10.1016/j.cmet.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cordes T, et al. Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J Biol Chem. 2016;291(27):14274–14284. doi: 10.1074/jbc.M115.685792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tannahill GM, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jha AK, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42(3):419–430. doi: 10.1016/j.immuni.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 42.Infantino V, et al. The mitochondrial citrate carrier: a new player in inflammation. Biochem J. 2011;438(3):433–436. doi: 10.1042/BJ20111275. [DOI] [PubMed] [Google Scholar]
- 43.Mills EL, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018;556(7699):113–117. doi: 10.1038/nature25986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kornberg MD, et al. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science. 2018;360(6387):449–453. doi: 10.1126/science.aan4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGivney A, Bradley SG. Action of bacterial endotoxin and lipid A on mitochondrial enzyme activities of cells in culture and subcellular fractions. Infect Immun. 1979;25(2):664–671. doi: 10.1128/iai.25.2.664-671.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.West AP, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472(7344):476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong HS, Dighe PA, Mezera V, Monternier PA, Brand MD. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J Biol Chem. 2017;292(41):16804–16809. doi: 10.1074/jbc.R117.789271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garaude J, et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat Immunol. 2016;17(9):1037–1045. doi: 10.1038/ni.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guarás A, et al. The CoQH2/CoQ ratio serves as a sensor of respiratory chain efficiency. Cell Rep. 2016;15(1):197–209. doi: 10.1016/j.celrep.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 50.Sena LA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38(2):225–236. doi: 10.1016/j.immuni.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andersson SG, et al. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396(6707):133–140. doi: 10.1038/24094. [DOI] [PubMed] [Google Scholar]
- 52.Archibald JM. Endosymbiosis and eukaryotic cell evolution. Curr Biol. 2015;25(19):R911–R921. doi: 10.1016/j.cub.2015.07.055. [DOI] [PubMed] [Google Scholar]
- 53.West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll-like receptors. Annu Rev Cell Dev Biol. 2006;22:409–437. doi: 10.1146/annurev.cellbio.21.122303.115827. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Q, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iyer SS, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39(2):311–323. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Traba J, et al. Prolonged fasting suppresses mitochondrial NLRP3 inflammasome assembly and activation via SIRT3-mediated activation of superoxide dismutase 2. J Biol Chem. 2017;292(29):12153–12164. doi: 10.1074/jbc.M117.791715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gaidt MM, et al. The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell. 2017;171(5):1110–1124.e18. doi: 10.1016/j.cell.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kerur N, et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat Med. 2018;24(1):50–61. doi: 10.1038/nm.4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 60.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153(2):348–361. doi: 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122(5):669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 62.Bantug GR, et al. Mitochondria-endoplasmic reticulum contact sites function as immunometabolic hubs that orchestrate the rapid recall response of memory CD8+ T cells. Immunity. 2018;48(3):542–555.e6. doi: 10.1016/j.immuni.2018.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481(7381):278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 64.Henao-Mejia J, Elinav E, Strowig T, Flavell RA. Inflammasomes: far beyond inflammation. Nat Immunol. 2012;13(4):321–324. doi: 10.1038/ni.2257. [DOI] [PubMed] [Google Scholar]
- 65.Traba J, Sack MN. The role of caloric load and mitochondrial homeostasis in the regulation of the NLRP3 inflammasome. Cell Mol Life Sci. 2017;74(10):1777–1791. doi: 10.1007/s00018-016-2431-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62(1):194–204. doi: 10.2337/db12-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim HY, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med. 2014;20(1):54–61. doi: 10.1038/nm.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Youm YH, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013;18(4):519–532. doi: 10.1016/j.cmet.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. 2012;18(3):363–374. doi: 10.1038/nm.2627. [DOI] [PubMed] [Google Scholar]
- 71.Kheirandish-Gozal L, et al. Lipopolysaccharide-binding protein plasma levels in children: effects of obstructive sleep apnea and obesity. J Clin Endocrinol Metab. 2014;99(2):656–663. doi: 10.1210/jc.2013-3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goossens GH, et al. Expression of NLRP3 inflammasome and T cell population markers in adipose tissue are associated with insulin resistance and impaired glucose metabolism in humans. Mol Immunol. 2012;50(3):142–149. doi: 10.1016/j.molimm.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 73.Stienstra R, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A. 2011;108(37):15324–15329. doi: 10.1073/pnas.1100255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bauernfeind F, Bartok E, Rieger A, Franchi L, Núñez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol. 2011;187(2):613–617. doi: 10.4049/jimmunol.1100613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Won JH, Park S, Hong S, Son S, Yu JW. Rotenone-induced impairment of mitochondrial electron transport chain confers a selective priming signal for NLRP3 inflammasome activation. J Biol Chem. 2015;290(45):27425–27437. doi: 10.1074/jbc.M115.667063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Elliott EI, et al. Cutting edge: mitochondrial assembly of the NLRP3 inflammasome complex is initiated at priming. J Immunol. 2018;200(9):3047–3052. doi: 10.4049/jimmunol.1701723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Park S, et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol. 2013;191(8):4358–4366. doi: 10.4049/jimmunol.1301170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shimada K, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Misawa T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14(5):454–460. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- 81.Yu EP, Bennett MR. Mitochondrial DNA damage and atherosclerosis. Trends Endocrinol Metab. 2014;25(9):481–487. doi: 10.1016/j.tem.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 82.Granata S, et al. NLRP3 inflammasome activation in dialyzed chronic kidney disease patients. PLoS One. 2015;10(3):e0122272. doi: 10.1371/journal.pone.0122272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xie M, et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun. 2016;7:13280. doi: 10.1038/ncomms13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moon JS, et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat Med. 2016;22(9):1002–1012. doi: 10.1038/nm.4153. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 85.Bernardi P, Rasola A, Forte M, Lippe G. The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol Rev. 2015;95(4):1111–1155. doi: 10.1152/physrev.00001.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gutiérrez-Aguilar M, Baines CP. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochim Biophys Acta. 2015;1850(10):2041–2047. doi: 10.1016/j.bbagen.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rasola A, Bernardi P. Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium. 2011;50(3):222–233. doi: 10.1016/j.ceca.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 88.Murakami T, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A. 2012;109(28):11282–11287. doi: 10.1073/pnas.1117765109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rada B, Park JJ, Sil P, Geiszt M, Leto TL. NLRP3 inflammasome activation and interleukin-1β release in macrophages require calcium but are independent of calcium-activated NADPH oxidases. Inflamm Res. 2014;63(10):821–830. doi: 10.1007/s00011-014-0756-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Suter M, Richter C. Fragmented mitochondrial DNA is the predominant carrier of oxidized DNA bases. Biochemistry. 1999;38(1):459–464. doi: 10.1021/bi9811922. [DOI] [PubMed] [Google Scholar]
- 91.Patrushev M, Kasymov V, Patrusheva V, Ushakova T, Gogvadze V, Gaziev A. Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell Mol Life Sci. 2004;61(24):3100–3103. doi: 10.1007/s00018-004-4424-1. [DOI] [PubMed] [Google Scholar]
- 92.García N, Chávez E. Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci. 2007;81(14):1160–1166. doi: 10.1016/j.lfs.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 93.Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol. 2006;7(1):49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 94.Oka T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485(7397):251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.De Nardo D. Toll-like receptors: activation, signalling and transcriptional modulation. Cytokine. 2015;74(2):181–189. doi: 10.1016/j.cyto.2015.02.025. [DOI] [PubMed] [Google Scholar]
- 96.Garcia-Martinez I, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest. 2016;126(3):859–864. doi: 10.1172/JCI83885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Matheoud D, et al. Parkinson’s disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell. 2016;166(2):314–327. doi: 10.1016/j.cell.2016.05.039. [DOI] [PubMed] [Google Scholar]
- 98.Latz E, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5(2):190–198. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- 99.Avalos AM, et al. Cell-specific TLR9 trafficking in primary APCs of transgenic TLR9-GFP mice. J Immunol. 2013;190(2):695–702. doi: 10.4049/jimmunol.1202342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tian J, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8(5):487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 101.Sirois CM, et al. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J Exp Med. 2013;210(11):2447–2463. doi: 10.1084/jem.20120201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kang R, et al. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene. 2014;33(5):567–577. doi: 10.1038/onc.2012.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17(10):1142–1149. doi: 10.1038/ni.3558. [DOI] [PubMed] [Google Scholar]
- 104.Dou Z, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550(7676):402–406. doi: 10.1038/nature24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.White MJ, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159(7):1549–1562. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rongvaux A, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159(7):1563–1577. doi: 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kanneganti TD, Kundu M, Green DR. Innate immune recognition of mtDNA — an undercover signal? Cell Metab. 2015;21(6):793–794. doi: 10.1016/j.cmet.2015.05.019. [DOI] [PubMed] [Google Scholar]
- 108.McArthur K, et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. 2018;359(6378):eaao6047. doi: 10.1126/science.aao6047. [DOI] [PubMed] [Google Scholar]
- 109.Bonawitz ND, Clayton DA, Shadel GS. Initiation and beyond: multiple functions of the human mitochondrial transcription machinery. Mol Cell. 2006;24(6):813–825. doi: 10.1016/j.molcel.2006.11.024. [DOI] [PubMed] [Google Scholar]
- 110.West AP, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520(7548):553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang Y, et al. MicroRNA-9 inhibits NLRP3 inflammasome activation in human atherosclerosis inflammation cell models through the JAK1/STAT signaling pathway. Cell Physiol Biochem. 2017;41(4):1555–1571. doi: 10.1159/000470822. [DOI] [PubMed] [Google Scholar]
- 112.Tang ED, Wang CY. MAVS self-association mediates antiviral innate immune signaling. J Virol. 2009;83(8):3420–3428. doi: 10.1128/JVI.02623-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu XY, Wei B, Shi HX, Shan YF, Wang C. Tom70 mediates activation of interferon regulatory factor 3 on mitochondria. Cell Res. 2010;20(9):994–1011. doi: 10.1038/cr.2010.103. [DOI] [PubMed] [Google Scholar]
- 114.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A. 2005;102(49):17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ingelsson B, et al. Lymphocytes eject interferogenic mitochondrial DNA webs in response to CpG and non-CpG oligodeoxynucleotides of class C. Proc Natl Acad Sci U S A. 2018;115(3):E478–E487. doi: 10.1073/pnas.1711950115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A. 2009;106(8):2770–2775. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cherry AD, Piantadosi CA. Regulation of mitochondrial biogenesis and its intersection with inflammatory responses. Antioxid Redox Signal. 2015;22(12):965–976. doi: 10.1089/ars.2014.6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. 2016;212(4):379–387. doi: 10.1083/jcb.201511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Okamoto K, Kondo-Okamoto N. Mitochondria and autophagy: critical interplay between the two homeostats. Biochim Biophys Acta. 2012;1820(5):595–600. doi: 10.1016/j.bbagen.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 120.Hallberg BM, Larsson NG. Making proteins in the powerhouse. Cell Metab. 2014;20(2):226–240. doi: 10.1016/j.cmet.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 121.Jensen MB, Jasper H. Mitochondrial proteostasis in the control of aging and longevity. Cell Metab. 2014;20(2):214–225. doi: 10.1016/j.cmet.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sack MN, Finkel T. Mitochondrial metabolism, sirtuins, and aging. Cold Spring Harb Perspect Biol. 2012;4(12):a013102. doi: 10.1101/cshperspect.a013102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17(4):491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc Natl Acad Sci U S A. 2013;110(44):17963–17968. doi: 10.1073/pnas.1312571110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhao GJ, et al. Up-regulation of mitofusin-2 protects CD4+ T cells from HMGB1-mediated immune dysfunction partly through Ca(2+)-NFAT signaling pathway. Cytokine. 2012;59(1):79–85. doi: 10.1016/j.cyto.2012.03.026. [DOI] [PubMed] [Google Scholar]
- 126.Yasukawa K, et al. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci Signal. 2009;2(84):ra47. doi: 10.1126/scisignal.2000287. [DOI] [PubMed] [Google Scholar]
- 127.Buck MD, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. 2016;166(1):63–76. doi: 10.1016/j.cell.2016.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rodríguez-Nuevo A, et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 2018;37(10):e96553. doi: 10.15252/embj.201796553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Harris J, Deen N, Zamani S, Hasnat MA. Mitophagy and the release of inflammatory cytokines. Mitochondrion. 2018;41:2–8. doi: 10.1016/j.mito.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 130.Jabir MS, et al. Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy. 2015;11(1):166–182. doi: 10.4161/15548627.2014.981915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ho TT, et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature. 2017;543(7644):205–210. doi: 10.1038/nature21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Narendra D, Walker JE, Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb Perspect Biol. 2012;4(11):a011338. doi: 10.1101/cshperspect.a011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85(2):257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tansey MG, Goldberg MS. Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37(3):510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Müller-Rischart AK, et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell. 2013;49(5):908–921. doi: 10.1016/j.molcel.2013.01.036. [DOI] [PubMed] [Google Scholar]
- 136.Frank-Cannon TC, et al. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci. 2008;28(43):10825–10834. doi: 10.1523/JNEUROSCI.3001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chung JY, et al. Elevated TRAF2/6 expression in Parkinson’s disease is caused by the loss of Parkin E3 ligase activity. Lab Invest. 2013;93(6):663–676. doi: 10.1038/labinvest.2013.60. [DOI] [PubMed] [Google Scholar]
- 138.Tang X, et al. SIRT2 acts as a cardioprotective deacetylase in pathological cardiac hypertrophy. Circulation. 2017;136(21):2051–2067. doi: 10.1161/CIRCULATIONAHA.117.028728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lu Z, Chen Y, Aponte AM, Battaglia V, Gucek M, Sack MN. Prolonged fasting identifies heat shock protein 10 as a Sirtuin 3 substrate: elucidating a new mechanism linking mitochondrial protein acetylation to fatty acid oxidation enzyme folding and function. J Biol Chem. 2015;290(4):2466–2476. doi: 10.1074/jbc.M114.606228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Webster BR, Lu Z, Sack MN, Scott I. The role of sirtuins in modulating redox stressors. Free Radic Biol Med. 2012;52(2):281–290. doi: 10.1016/j.freeradbiomed.2011.10.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013;25(10):1939–1948. doi: 10.1016/j.cellsig.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 142.Liu TF, Vachharajani VT, Yoza BK, McCall CE. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J Biol Chem. 2012;287(31):25758–25769. doi: 10.1074/jbc.M112.362343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Koyama T, et al. SIRT3 attenuates palmitate-induced ROS production and inflammation in proximal tubular cells. Free Radic Biol Med. 2011;51(6):1258–1267. doi: 10.1016/j.freeradbiomed.2011.05.028. [DOI] [PubMed] [Google Scholar]
- 144.Sonoda J, et al. Nuclear receptor ERRα and coactivator PGC-1β are effectors of IFN-γ-induced host defense. Genes Dev. 2007;21(15):1909–1920. doi: 10.1101/gad.1553007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Emre Y, Hurtaud C, Nübel T, Criscuolo F, Ricquier D, Cassard-Doulcier AM. Mitochondria contribute to LPS-induced MAPK activation via uncoupling protein UCP2 in macrophages. Biochem J. 2007;402(2):271–278. doi: 10.1042/BJ20061430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Arsenijevic D, et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet. 2000;26(4):435–439. doi: 10.1038/82565. [DOI] [PubMed] [Google Scholar]
- 147.Lightfoot YL, Blanco LP, Kaplan MJ. Metabolic abnormalities and oxidative stress in lupus. Curr Opin Rheumatol. 2017;29(5):442–449. doi: 10.1097/BOR.0000000000000413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Carter LM, Isenberg DA, Ehrenstein MR. Elevated serum BAFF levels are associated with rising anti-double-stranded DNA antibody levels and disease flare following B cell depletion therapy in systemic lupus erythematosus. Arthritis Rheum. 2013;65(10):2672–2679. doi: 10.1002/art.38074. [DOI] [PubMed] [Google Scholar]
- 149.Yin Y, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7(274):274ra18. doi: 10.1126/scitranslmed.aaa0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lood C, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22(2):146–153. doi: 10.1038/nm.4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Miesel R, Murphy MP, Kröger H. Enhanced mitochondrial radical production in patients which rheumatoid arthritis correlates with elevated levels of tumor necrosis factor α in plasma. Free Radic Res. 1996;25(2):161–169. doi: 10.3109/10715769609149921. [DOI] [PubMed] [Google Scholar]
- 152.Hajizadeh S, DeGroot J, TeKoppele JM, Tarkowski A, Collins LV. Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res Ther. 2003;5(5):R234–R240. doi: 10.1186/ar787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol. 2004;75(6):995–1000. doi: 10.1189/jlb.0703328. [DOI] [PubMed] [Google Scholar]
- 154.Cordero MD, et al. Mutation in cytochrome b gene of mitochondrial DNA in a family with fibromyalgia is associated with NLRP3-inflammasome activation. J Med Genet. 2016;53(2):113–122. doi: 10.1136/jmedgenet-2015-103392. [DOI] [PubMed] [Google Scholar]
- 155.Gershwin ME, et al. Primary biliary cirrhosis: an orchestrated immune response against epithelial cells. Immunol Rev. 2000;174:210–225. doi: 10.1034/j.1600-0528.2002.017402.x. [DOI] [PubMed] [Google Scholar]
- 156.Kenney MC, et al. Inherited mitochondrial DNA variants can affect complement, inflammation and apoptosis pathways: insights into mitochondrial-nuclear interactions. Hum Mol Genet. 2014;23(13):3537–3551. doi: 10.1093/hmg/ddu065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yin Y, et al. Glucose oxidation is critical for CD4+ T cell activation in a mouse model of systemic lupus erythematosus. J Immunol. 2016;196(1):80–90. doi: 10.4049/jimmunol.1501537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Frederick DW, et al. Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab. 2016;24(2):269–282. doi: 10.1016/j.cmet.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yoshino J, Baur JA, Imai SI. NAD+ intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 2018;27(3):513–528. doi: 10.1016/j.cmet.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rajman L, Chwalek K, Sinclair DA. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metab. 2018;27(3):529–547. doi: 10.1016/j.cmet.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lee HJ, Hong YS, Jun W, Yang SJ. Nicotinamide riboside ameliorates hepatic metaflammation by modulating NLRP3 inflammasome in a rodent model of type 2 diabetes. J Med Food. 2015;18(11):1207–1213. doi: 10.1089/jmf.2015.3439. [DOI] [PubMed] [Google Scholar]
- 162.Caton PW, Kieswich J, Yaqoob MM, Holness MJ, Sugden MC. Nicotinamide mononucleotide protects against pro-inflammatory cytokine-mediated impairment of mouse islet function. Diabetologia. 2011;54(12):3083–3092. doi: 10.1007/s00125-011-2288-0. [DOI] [PubMed] [Google Scholar]
- 163.Chatterjee S, et al. CD38-NAD+ axis regulates immunotherapeutic anti-tumor T cell response. Cell Metab. 2018;27(1):85–100.e8. doi: 10.1016/j.cmet.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Itagaki K, et al. Mitochondrial DNA released by trauma induces neutrophil extracellular traps. PLoS One. 2015;10(3):e0120549. doi: 10.1371/journal.pone.0120549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Perego J, et al. Guanabenz inhibits TLR9 signaling through a pathway that is independent of eIF2α dephosphorylation by the GADD34/PP1c complex. Sci Signal. 2018;11(514):eaam8104. doi: 10.1126/scisignal.aam8104. [DOI] [PubMed] [Google Scholar]
- 166.Seok J, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110(9):3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gaidt MM, et al. Human monocytes engage an alternative inflammasome pathway. Immunity. 2016;44(4):833–846. doi: 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 168.Young NS. Mouse medicine and human biology. Semin Hematol. 2013;50(2):88–91. doi: 10.1053/j.seminhematol.2013.03.024. [DOI] [PubMed] [Google Scholar]
- 169.Thomas L. Notes of a biology-watcher. N Engl J Med. 1971;284(19):1082–1083. doi: 10.1056/NEJM197105132841908. [DOI] [PubMed] [Google Scholar]