

Abstract
The biological basis of human aging remains one of the greatest unanswered scientific questions. Increasing evidence, however, points to a role for alterations in mitochondrial function as a potential central regulator of the aging process. Here, we focus primarily on three aspects of mitochondrial biology that link this ancient organelle to how and why we age. In particular, we discuss the role of mitochondria in regulating the innate immune system, the mechanisms linking mitochondrial quality control to age-dependent pathology, and the possibility that mitochondrial-to-nuclear signaling might regulate the rate of aging.
Introduction
While prokaryotes can be viewed as immortal, their seemingly more advanced cousins, eukaryotes, all are cloaked in a more complex but mortal shell. What regulates this mortality and controls why humans age is undoubtedly multifactorial. Here, however, we focus on one appealing cause of aging that can be traced back to an unwitnessed event that occurred over 1.5 billion years ago. The event in question involved the entry of an immortal prokaryote into a eukaryote cell (1). This beneficial union, or symbiogenesis, would eventually lead the invading prokaryote to evolve into what we now more commonly call the mitochondria. The notion that the seed of our mortality can somehow be linked to this unlikely billion-year-plus marriage has been appreciated for quite some time. In the 1920s, Raymond Pearl noted that metabolic rates appeared to inversely correlate with lifespan (2). Building on this, Denham Harman, in the 1950s, proposed the free radical theory of aging. Harman’s conjecture was that the generation of reactive oxygen species (ROS), which he viewed as likely coming from the mitochondria, gave rise to the subsequent accumulation of damaged proteins, lipids, and DNA, thereby fueling aging and age-related diseases in an inevitable but stochastic process (3). While the notion that increased mitochondrial ROS directly causes aging has fallen into disfavor (4), in its wake, other aspects of mitochondrial biology have grown increasingly more appealing. Some links between aspects of mitochondrial biology and aging have been the subject of many excellent recent reviews (5–8). Here, we focus on those aspects of mitochondrial physiology that have perhaps the strongest connection to human aging. These processes include the emerging role that mitochondria play in inflammation; how dysregulation of mitochondrial quality control and age-related mitochondrial dysfunction contributes to aging, age-related mitochondrial dysfunction, and the notion of retrograde signaling; and why a little mitochondrial stress might ultimately be a good thing.
Mitochondria and the inflammation of aging
Aging is associated with evidence of an activation of the innate immune system that leads to a condition known as “inflammaging” (9). This is particularly true of elderly individuals who age less successfully than their peers, as the both the frail and the pre-frail exhibit higher circulating levels of inflammatory mediators such as C-reactive protein, IL-6, and fibrinogen (10). These relationships exist even in the absence of associated age-related diseases (e.g., cancer, cardiovascular disease, etc.), suggesting a potential causative relationship between activation of the immune system and accelerated aging (11). Perhaps because of its stability and ease of measurement, the data are most compelling with respect to circulating IL-6, as elevated IL-6 serum levels are predictive of incident disability, frailty, walking speed, and overall mortality (10, 12, 13). Analysis of T cells from elderly individuals suggests that although basal levels of cytokine production are elevated, invoked responses to a cytokine challenge are reduced (14). This appears to be due to a chronic activation of JAK-STAT signaling in the circulating immune cells of elderly patients who demonstrate markers of chronic inflammation (14).
Besides evidence for augmented basal cytokine signaling, chronic activation of the inflammasome with elevated IL-1β levels appears to identify a subset of elderly individuals with higher rates of essential hypertension, arterial stiffness, and all-cause mortality (15). Consistent with this, evidence for activation of the NLRP3 inflammasome is seen in a wide range of age-related conditions, including atherosclerosis (16), Alzheimer’s disease (17), and metabolic syndrome (18). Interestingly, a number of preclinical observations suggest that the activation of the inflammasome may be maladaptive. For instance, in mice, genetic deletion of NLRP3 appears to reduce the inflammation associated with aging and slow the age-dependent incidence of insulin resistance, cognitive decline, and frailty (19, 20). In that regard, the success of the recent CANTOS trial using canakinumab, a humanized antibody that neutralizes IL-1β, might be instructive. In this trial, over 10,000 patients who had suffered a previous myocardial infarction were randomized to placebo or one of three doses of canakinumab. Interestingly, this antiinflammatory strategy appeared to reduce subsequent cardiovascular events (21). Secondary analysis moreover suggested that the development of fatal lung cancers might also be reduced (22). This supports the notion that activation of the innate immune system in the elderly might have broad deleterious effects that are amenable to targeted therapies.
There is increasing evidence that mitochondria might play an important role in the inflammaging phenotype (23). The immune system is capable of sensing and responding to tissue damage and views the release of intracellular molecules in an analogous fashion to dangerous pathogens. This “danger theory” argues that certain molecules released from senescent or dying cells might constitute signals that trigger an immune response (24), often termed damage-associated molecular patterns (DAMPs). In that context, the mitochondria, derived several billions of years ago from the endosymbiosis of α-proteobacteria into a eukaryotic cell, represent a smorgasbord of potential immune-stimulating DAMPs. Putative mitochondrial DAMPs include the release of mitochondrial DNA (mtDNA), N-formyl peptides generated by translation of mitochondrial-encoded protein, and unique lipid species such as cardiolipin, a unique phospholipid enriched on the inner mitochondrial membrane (25). In the case of circulating mtDNA, immune activation occurs, in part, by activation of Toll-like receptor 9 (TLR9) signaling, while for mitochondrial proteins, activation occurs through binding to formyl peptide receptor-1. This was first demonstrated in the case of trauma, where the release of mtDNA and formyl peptides activated circulating neutrophils to mediate tissue injury (26). These early experiments were also able to show that, without any tissue injury, direct infusion of mitochondrial DAMPs could recapitulate much of the neutrophil-mediated organ injury seen following a traumatic event, including a rise in IL-6 levels without any tissue injury (26). Interestingly, circulating mtDNA appears to increase gradually with age after the fifth decade of life (27). This level of circulating mtDNA appears sufficient to elicit augmented cytokine production from cultured monocytes (27). Moreover, in patients, levels of circulating mtDNA correlate with serum inflammatory markers and may have a genetic underpinning (27).
DAMPs and the related pathogen-associated molecular patterns (PAMPs) can also trigger the assembly of the inflammasome, a high–molecular weight protein complex that functions to activate caspase-1 (28). As previously mentioned, inflammasome activation appears to identify a subset of at-risk elderly individuals (15), and genetic inhibition of this pathway in mice appears to protect animals from certain age-related deficits (19). While a number of distinct forms of inflammasome complexes exist, the majority of work has centered on NLRP3, since it appears to respond to the broadest range of stimuli. Stimulation of the NLRP3 inflammasome results in the activation of caspase-1, which in turn results in the proteolytic activation of potent cytokines such as IL-1β and IL-18 (28). Interestingly, caspase-1 activation can also induce mitochondrial damage (29). This relationship is just one of many between the inflammasome and the mitochondria. For instance, MAVS, the mitochondria-associated adaptor molecule that is found on the outer mitochondrial membrane, can serve as a platform for inflammasome activation (30). There is also evidence that the mitochondrial phospholipid cardiolipin can directly bind and activate NLRP3 (31). A number of other mitochondrial-derived signals have been implicated in inflammasome activation. A wealth of evidence suggests that ROS generated in the mitochondria participate in NLRP3 activation (32), although the absolute requirement for oxidants is unclear, as is the precise mechanism or target that is dependent on redox modification (33). Release of mtDNA into the cytosol can also activate the inflammasome (34). This, however, differs from previously discussed circulating mtDNA, which exerts its effects by binding and activating TLR9 signaling (Figure 1).
Figure 1. Immune activation by mtDNA release through both intracellular and extracellular pathways.
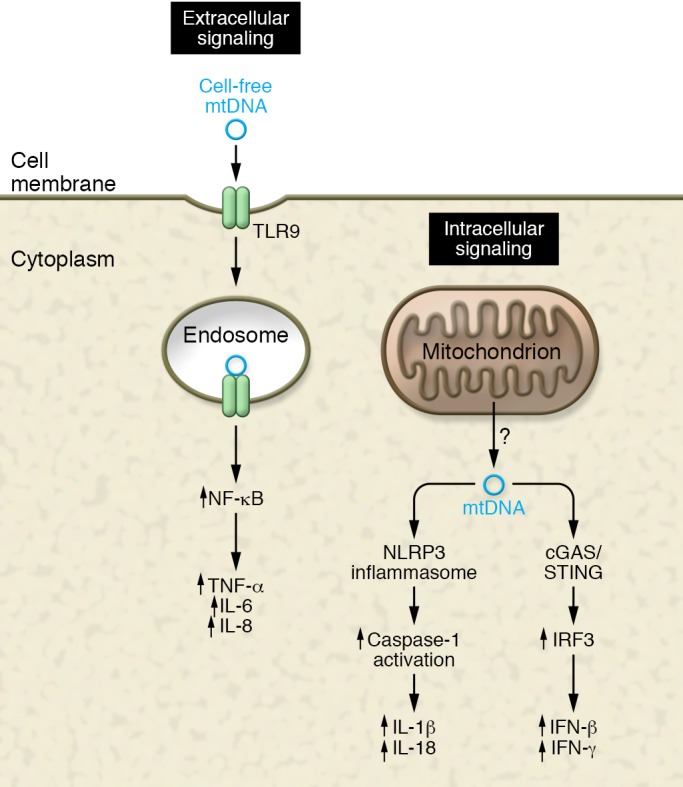
The innate immune system has at least three sensors for detecting mtDNA: TLR9 for sensing circulating (extracellular) mtDNA, and the NLRP3 inflammasome and the cGAS/STING pathway for sensing cytoplasmic (intracellular) mtDNA release. A host of downstream signaling pathways and cytokines are induced following activation of each of these distinct pathways. The precise mechanism through which mtDNA is released into the cytosol or the circulation remains uncertain.
How mtDNA finds its way into either the cytosol or the circulation remains a bit mysterious. Certainly this is understandable in the case mentioned previously involving massive trauma, or other forms of cell death (26). Yet circulating mtDNA can be readily detected in human serum under a wide range of conditions, including normal aging (35). One possibility is that the release of mtDNA is kept in check by quality control mechanisms, particularly through autophagic/mitophagic removal of dysfunctional mitochondria. Consistent with this notion, disrupting autophagy increased free mtDNA in the cytosol and increased IL-1β secretion (36). Similarly, recent evidence suggests that in macrophages, mitophagy acts to restrain inflammasome activation by again removing damaged mitochondria that could act as NLRP3 activators via the release of ROS or mtDNA (37). Moreover, a very recent report has demonstrated that the cytokine IL-10 can promote mitophagy in macrophages by inhibiting the mTOR pathway and inducing metabolic reprogramming in this cell type (38). The absence of IL-10 was shown to lead to dysregulated NLRP3 activation and increased IL-1β production (38). This is consistent with the altered mitochondrial morphology seen in IL-10–deficient mice (39), perhaps the most widely used and established mouse model for frailty (40). Together, these observations suggest an important intersection between mitophagy, inflammasome activation, and the prevention of age-associated impairment.
While we have already discussed how mtDNA can activate TLR9 signaling and inflammasome activation, there appears to be yet another means through which mtDNA can activate the immune system. Given its evolutionary origins, it is not surprising that, like foreign bacterial and viral DNA, mtDNA can also be recognized by cyclic GMP-AMP synthase (cGAS), a cytosolic sensor of double-stranded DNA (dsDNA). Once bound to dsDNA, cGAS induces signaling through the adaptor protein STING, leading to activation of the transcription factor IRF3. In turn, IRF3 orchestrates the induction of type I IFNs and IFN-stimulated genes, a process that is critical in the response to viral infection. Interestingly, mouse embryonic fibroblasts that are haploinsufficient for the mitochondrial transcription factor TFAM show a reduction in mtDNA content and evidence of a constitutive activation of the cGAS/STING/IRF3 pathway (41). These observations are broadly consistent with previous reports that had linked TFAM to inflammation in microglia (42). Remarkably, infection with certain herpesviruses also caused depletion of mtDNA in cells (41). Similar to what was observed in TFAM+/– cells, viral infection also triggered mitochondrial-dependent IRF3 activation. Obviously, while this strategy of releasing mtDNA to augment antiviral defenses has short-term appeal, the implications for triggering long-term deleterious consequences are also evident. Since aging involves involution of the thymus, decreased repertoire of T cells, and evidence for chronic and persistent viral infections (43), this mitochondrial-initiated, virally triggered pathway might play a significant role in the associated inflammation seen in the elderly.
Finally, there are other important ways in which mitochondria could participate in the observed age-dependent sterile inflammation. Increasingly, the tricarboxylic acid (TCA) metabolites generated by mitochondrial metabolism appear to be able to shape the immune response. The activation of macrophages by the Gram-negative bacterial product lipopolysaccharide (LPS) switches their metabolism from oxidative phosphorylation to glycolysis. This metabolic shift has been shown to result in the buildup of the TCA metabolite succinate (44). Interestingly, this increase in succinate can stabilize HIF-1α by inhibiting prolyl hydroxylase domain (PHD) enzyme activity that normally regulates HIF-1α stability. This succinate-dependent HIF-1α induction can in turn regulate IL-1β production (44). Mitochondrial-produced succinate can also work as a chemokine, as specific G protein–coupled succinate receptors exist on both immune and nonimmune cells (45). Besides its role in HIF-1α stabilization, succinate accumulation in the setting of LPS stimulation can also be metabolized by a process known as reverse electron transport (46). This reverse flow of electrons from mitochondrial complex II to complex I is known to produce ROS. Reverse electron transport appears to increase as animals age and may actually be beneficial and extend lifespan in certain settings (47). However, in the context of LPS stimulation of macrophages, the combination of a high mitochondrial membrane potential along with succinate oxidation drives mitochondrial ROS production, producing a proinflammatory response (46). Other examples have also demonstrated that mitochondrial membrane potential may actually be an important determinant of immune responsiveness (48). Moreover, succinate appears to be just one of many mitochondrial-derived metabolites that might modulate the immune response and thereby contribute to the age-dependent alteration in immunity. Other examples include a regulatory role for mitochondrial fatty acid oxidation, as well as for other individual metabolites including citrate, acetate, acetyl-CoA, and itaconate (49). Evidence suggests that these metabolic pathways and individual metabolites can each shape the immune response and potentially link alterations in mitochondrial function with age-dependent alteration in immune function. Interestingly, in model organisms, such as yeast and worms, levels of TCA metabolites appear to regulate overall lifespan (50, 51).
Mitochondrial DNA and aging
Levels of mitochondrial mutations certainly increase with age (52). Yet the abundance of mitochondrial genomes, ranging from hundreds to thousands of copies per cell, makes the mere presence of a detected mutation more difficult to functionally interpret. Indeed, next-generation sequencing methods have determined that a low level of mutant mitochondrial genomes (termed heteroplasmy), which are inherited from mother to child, are readily apparent in almost everyone (53). Yet the functional implications of these sequencing results are less clear. While mtDNA encodes critical proteins required for electron transport, it is generally believed that any pathogenic mtDNA mutation would need to reach a threshold of more than 60% and perhaps closer to 90% in a given cell or tissue to have a measurable bioenergetic effect (6). Most likely, the increase in mtDNA mutational load is a correlate of aging rather than being primarily responsible for the aging phenotype. Nonetheless, experimental evidence suggests that the germline load of mtDNA mutations can shape the subsequent rate of aging (54). Moreover, there are engineered mouse models, such as the proofreading-deficient, mtDNA polymerase (POLγ) mouse, that do accumulate extremely high levels of mitochondrial mutations. Notably, these animals also exhibit a shortened lifespan, along with certain visible age-related phenotypes including gray hair and kyphosis (55–57). Thus, mtDNA mutations can, at high levels, drive mammalian aging. This is also evident in human subjects who carry mutations in the mitochondrial replication machinery. Indeed, over 300 pathological mutations exist in human POLγ, and these mutations are linked to the development of a wide spectrum of conditions, including Parkinson’s disease and other age-related pathologies (58). Another potentially more common example is the growing realization that patients with chronic HIV infection exhibit aspects of accelerated aging, including increased frailty, augmented cardiovascular disease, and significantly higher rates of bone fractures (59). While these conditions might be secondary to the chronic low-level inflammation associated with viral infection, it is of interest to note that certain HIV therapeutics have known off-target effects, including an ability to inhibit POLγ activity (60). This raises the possibility that this growing clinical syndrome might phenocopy aspects of the proofreading-deficient POLγ mouse. Indeed, HIV-positive patients treated with nucleoside analog drugs have increased levels of mitochondrial mutations that correlate with the duration of antiretroviral therapy (61, 62).
Mitophagy and aging
Analysis of mitochondrial function in tissues, such as the skeletal muscle of older subjects, reveals a decline in mitochondrial respiratory capacity to roughly 50% of what is seen in younger subjects and a reduction in ATP (63, 64). These mitochondrial deficits track closely with functional decline in muscle strength and are believed by most to be a causal factor for age-related sarcopenia (65). While, as mentioned above, in certain rare cases, accumulation of mtDNA mutations might contribute to this functional deficit, it is likely that other mechanisms are at play. An increase in the steady-state levels of dysfunction could be due to an increase in age-dependent damage to mitochondria, an age-dependent decline in the removal of dysfunctional mitochondria, or some combination of both mechanisms. The removal of damaged but intact mitochondria occurs through the process of mitophagy. We have already briefly discussed this process in the setting of inflammasome activation, where mitophagy, by keeping the level of damaged mitochondria low, prevents immune activation (36–38). The molecular regulation of mitophagy is incompletely understood. One known stimulus is the accumulation of ubiquitinated proteins on the outer mitochondrial membrane. This process can be catalyzed by the E3-ubiquitin ligase parkin, which in turn is activated by PTEN-inducible putative kinase 1 (PINK1) (66). In cultured cells, mitochondrial dysfunction has been shown to stabilize PINK1 levels, thereby activating this cascade (67). The fact that both parkin and PINK1 are found as causes of inherited forms of Parkinson’s disease has strengthened the link between mitophagy and age-related pathologies (68). Nonetheless, important caveats remain, suggesting our knowledge is far from complete. First, at least in mice, germline deletion of Parkin has relatively subtle effects. In particular, these animals do not develop Parkinson’s disease, although recent observations have demonstrated that when crossed to the proofreading-deficient POLγ mouse, Parkin–/– mice do exhibit neuronal cell death in the substantia nigra, the specific dopaminergic region of the brain affected in Parkinson’s disease (69). This suggests that there may be separate programs for basal mitophagy and stress-induced mitophagy. This conjecture is also supported by recent technical improvements that have led to the ability to more accurately monitor in vivo mitophagy, including the development of the fluorescence-based mt-Keima (70) and the mito-QC reporter mouse (71). Adaptation of these techniques has demonstrated that in flies, the absence of Pink1 or parkin did not alter basal mitophagy (72). Similarly, in mice expressing the mito-QC reporter, there was no effect of Pink1 deletion on basal mitophagic flux (73). Thus, the precise in vivo role of PINK1 and parkin remains to be fully discerned.
While definitive roles for PINK1 and parkin have not been established, the biochemical underpinnings of mitophagy are clearly expanding. For instance, during development, certain tissues such as the oocyte, reticulocyte, and the ocular lens target all or part of their normal-functioning mitochondria for removal. In reticulocytes, the inner mitochondrial BCL2-related protein NIX (also known as BNIP3L) appears to play an essential role in mitochondrial clearance (74, 75). In reticulocytes, as well as in the clearance of paternal mitochondria in the Caenorhabditis elegans oocyte, this process is seemingly parkin-independent (76). This suggests that different tissues might have different mechanisms for mitophagic removal. This is also consistent with the wide variation seen in mitophagic flux between tissues (70).
The recognition of ubiquitinated and presumably damaged mitochondria occurs through adaptor proteins that link the mitochondria to the isolation membrane/phagophore of the expanding autophagosome. In the reticulocyte, there is evidence that NIX might play this role (77). Recently, another inner mitochondrial protein, prohibitin 2, was also identified as a mitophagy adaptor protein (78). In this case, parkin-dependent ubiquitination trigged proteosomal-mediated outer mitochondrial membrane rupture, exposing prohibitin 2 to stimulate mitophagy (78). Interestingly, the prohibitin family had been previously linked to a wide array of age-related maladies, in part owing to its ability to regulate mitochondrial ROS levels (79). Other classic autophagy adaptors such as p62, NBR1, NDP52, Tax1BP1, and optineurin also can play a role, with the strongest evidence to date implicating NDP52 and optineurin as important mitophagy regulators (80).
There is evidence that the level of mitophagy markedly declines in mammalian tissues during normal aging (70, 81). It is tempting to speculate that this decline underlies the known accumulation of damaged mitochondria and provides a mechanism for the observed functional deficits and increased oxidative stress commonly observed in aging tissues and organs. In animal models in which essential autophagy genes are deleted (e.g., Atg7), dysfunctional mitochondria accumulate (81, 82). Interestingly, in these various models, disruption of autophagy produces a marked rise in ROS levels, and the functional deficits observed in these tissues can be significantly mitigated by antioxidant therapies (81, 82). This suggests that a decline in autophagy, and more specifically mitophagy, might fuel the vicious circle of oxidative stress–induced, age-related tissue damage (ref. 83 and Figure 2). Model organisms have also strengthened the connection between mitophagy and aging. For instance, various genetic perturbations that extend the lifespan of C. elegans all appear to increase mitophagy (84). Moreover, this increase in mitophagy is required for the observed increased longevity (84). Similarly, in flies, overexpression of parkin can extend lifespan (85). In mammals, strategies that extend survival, such as calorie restriction in rodents, appear to preserve mitochondrial morphology (86). In addition, certain naturally occurring compounds such as the polyamine spermidine appear to increase autophagy and mitophagy, resulting in an increase in lifespan and a protection from age-related diseases (87). Similarly, urolithin A, a metabolite of ellagitannins, compounds enriched in pomegranate juice, also stimulate mitophagy and appear to extend the lifespan of worms and improve muscle function in old mice (88). These studies imply that more targeted, and presumably more effective, approaches to stimulate mitophagy might have widespread beneficial effects in combating age-related functional decline.
Figure 2. Aging impairs mitophagic removal of stressed mitochondria.
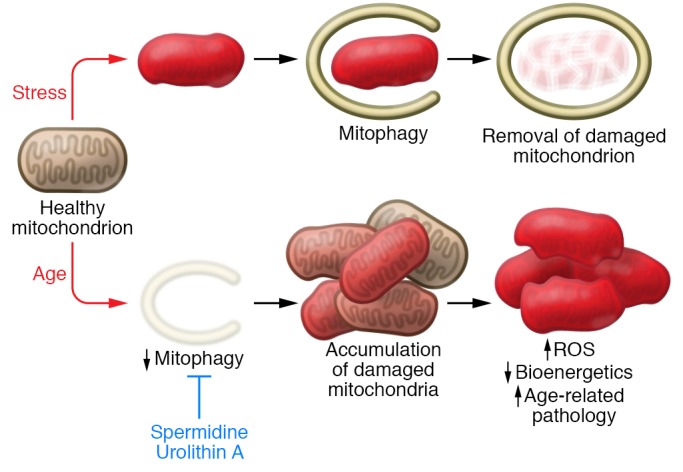
The age-dependent decline in mitophagy might provide the mechanism to explain the known interrelationship between increased ROS, decreased bioenergetic capacity, and age-dependent functional decline. Agents such as spermidine and urolithin A appear to stimulate mitophagy and thereby prevent age-related pathologies, which are described in more detail in the main text.
Retrograde signaling: from mitochondria to nucleus
It is important to stress that the removal of mitochondria through mitophagy represents only one aspect of mitochondrial quality control. Lysosomal destruction of an entire mitochondrion comes at a high thermodynamic cost, and therefore efforts short of complete destruction are likely to predominate when damage is less severe (Figure 3). One approach appears to be that, rather than complete removal, only a small region of the mitochondrion, presumably the most heavily damaged, is targeted for removal. This can occur through the creation of mitochondrial-derived vesicles (MDVs), a parkin-dependent process (89). Even more efficient are ways that damaged mitochondria can signal back to the nucleus to orchestrate a nuclear transcriptional response that reduces mitochondrial stress and thereby prevent subsequent damage. These retrograde signaling pathways are increasingly being seen as critical for maintaining functional capacity in aging tissues. In this context, mitochondrial quality control can be viewed as a broad spectrum of responses where the initial response (retrograde signaling, MDVs, or mitophagy) depends on the nature and magnitude of the underlying stress. The fact that parkin appears to play a role in both MDV formation and mitophagy argues that, at least at a molecular level, these processes are not wholly distinct. At present, the relative importance of these pathways to mediate aging or age-related pathology is largely unknown.
Figure 3. A range of quality control mechanisms exist to deal with stress in the mitochondria.
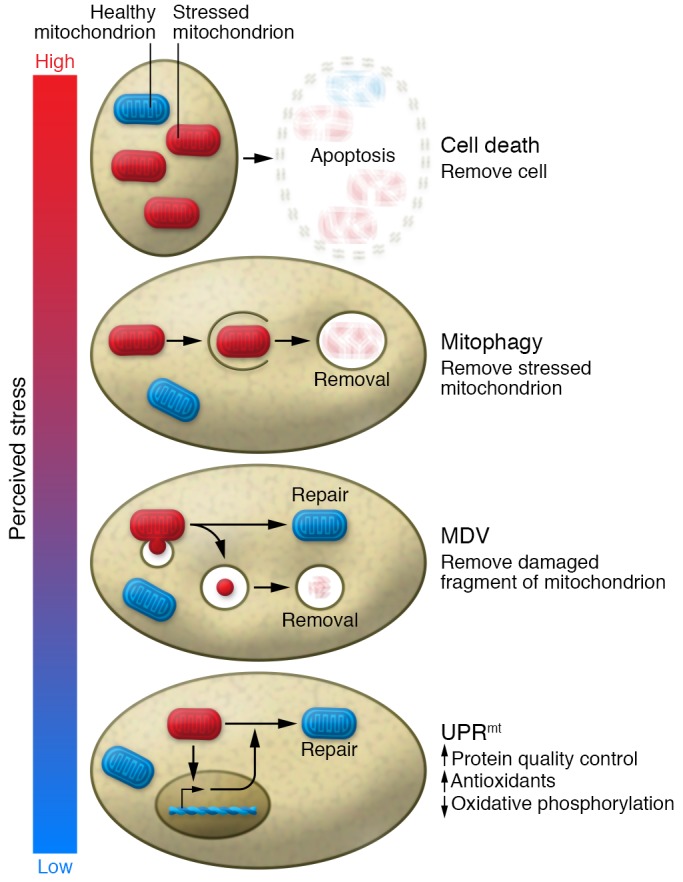
The magnitude of these mechanisms ranges in accordance with the severity of the perceived stress. From lowest to highest magnitude of response, they include activation of the UPRmt to initiate a transcriptional program to potentially relieve the stress; removal of part of the mitochondria into a mitochondrial-derived vesicle (MDV) in hopes of preserving the undamaged part; activation of mitophagy to remove the entire damaged mitochondria; and induction of cell death through apoptosis or necrosis to remove the entire damaged cell. As such, the magnitude of response can be titrated to the level of perceived stress.
In the context of aging, the impetus to understand retrograde signaling originated from a series of counterintuitive observations in model organisms where genome-wide or targeted RNAi screens revealed that impairing mitochondrial electron transport paradoxically resulted in increased lifespan (90, 91). Even more counterintuitive were studies in which the release of mitochondrial ROS, long thought to be the cause of aging, actually appeared to extend lifespan (92, 93). These observations are also supported by observations in mammalian models. While severe mitochondrial dysfunction is either incompatible with life or a cause of significant morbidity, some mouse models with a modest increase in mitochondrial ROS levels or defects in oxidative phosphorylation capacity, like their worm counterparts, appear to also have an increase in lifespan (94, 95).
These sets of observations have concentrated efforts to understand how the cell knows its mitochondria are encountering stress, and to define the nuclear response to an SOS sent from this critical organelle. What has emerged from these studies is a still-incomplete picture, but a number of tantalizing clues suggest this retrograde intracellular communication may be critically important. One such pathway that fits within this rubric is the mitochondrial unfolded protein response (UPRmt), a coordinated response classically triggered by mtDNA depletion or by protein misfolding in the mitochondrial matrix (96). The initial genetic characterization of this pathway has been most extensively explored in worms, in which mitochondrial dysfunction results in the transcription factor ATFS-1 being directed away from the mitochondria and into the nucleus (97). While initial studies of the transcriptional output of the UPRmt mostly centered on mitochondrial chaperones, which allow the stabilization of misfolded matrix proteins, it now appears this response is more comprehensive, with the induction of hundreds of genes including proteases, antioxidant enzymes, and genes involved in mitochondrial protein import, mitochondrial dynamics, and cellular metabolism (96). To date, the bulk of this analysis has been done in model organisms, particularly the worm C. elegans. There are reasons to suspect that significant differences exist in the corresponding mammalian UPRmt. For instance, there is likely not a precise single mammalian equivalent of ATFS-1, although the bZIP transcription factors ATF4 and ATF5 appear to be important in coordinating a similar response in mammals (98, 99). Similarly, factors known to play a role in C. elegans do not appear to play similar roles in mammals. As one example, in the worm, the mitochondrial ClpP protease plays a central role in the UPRmt response (100), while the mammalian ortholog does not appear to significantly regulate this response (101).
The connection between the UPRmt response and lifespan came from analysis of long-lived worm mutants with defects in their electron transport. Remarkably, in these models, activation of the UPRmt response was essential for the observed lifespan increase but this longevity response appeared to be induced in a non–cell-autonomous fashion (102). In particular, mitochondrial dysfunction in neurons caused a UPRmt response in the intestines, implicating a soluble factor that the authors deemed a mitokine (102). Although the precise nature of the mitokine(s) remains obscure, in the worm, neurotransmitters like serotonin as well as neuropeptides appear to be involved (103, 104). There is also an epigenetic memory associated with these effects, as mitochondrial dysfunction only produces lifespan extension in the worm when it occurs during a specific developmental window (102). Later studies have implicated histone methyltransferases (105) and the Jumonji family of histone lysine demethylase as important epigenetic regulators of the UPRmt (106). Moreover, the expression of the mouse Jumonji orthologs appears to correlate with activation of the mammalian UPRmt response and, more importantly, with longevity in the BXD mouse genetic reference panel, a collection of over 150 distinct mouse lines (106). In addition, there is also evidence that the mTOR kinase, linked to lifespan regulation in a wide range of organisms, can also regulate the UPRmt response (107). That said, the absolute requirement for the UPRmt response in regulating lifespan has not been established (108).
Besides longevity, the UPRmt response is increasingly implicated in age-related diseases. For instance, in model organisms, toxic polyglutamine repeat protein aggregates seen in Huntington’s disease can bind directly to mitochondria and induce the UPRmt response (103). A similar role for the UPRmt response has been observed in Alzheimer’s disease, where the UPRmt activation is seen in individuals with cognitive impairment (109). In model organisms, boosting the UPRmt response appears to lessen the severity of amyloid-β proteotoxicity (109). In addition, a number of pharmacological interventions linked to increasing lifespan, including metformin, resveratrol, rapamycin, and NAD+ supplementation, all appear to activate retrograde signaling from the mitochondria (110–112).
Conclusion
The mitochondria have traditionally been viewed as simple, autonomous energetic factories whose waste product, ROS, fueled the aging process. As our knowledge of this organelle has expanded, so too have the connections between mitochondrial function and aging biology. Besides what has been discussed here, we should note that there is also an important role for mitochondria dysfunction in cellular senescence, important links between altered mitochondrial dynamics and biogenesis in age-related conditions, and an emerging role for mitochondrial dysfunction to regulate age-related stem and progenitor cell decline (6, 8, 113). These areas, when added to what was presented here, firmly link mitochondrial biology to aging biology. Nonetheless, important gaps in our knowledge remain. For instance, while circulating mtDNA might trigger an innate immune response, it is unclear how much this phenomenon contributes to the inflammaging phenotype. Similarly, what role mitochondrial-dependent inflammasome activation plays in aging remains largely unknown. In addition, while a general decline in mitophagy occurs with age (70), and while this could drive the age-dependent accumulation of dysfunctional mitochondria, firm genetic proof that a decline in mitophagy contributes to age-dependent mitochondrial dysfunction, much less age-related functional decline, remains elusive. The absence of obvious effects of PINK1 or parkin deletion on in vivo mitophagy (72, 73) suggests that our understanding of this process is limited. For instance, largely missing from the current analysis is an understanding of how mitochondrial deubiquitinases might contribute to this process (114). In that regard, USP30, a mitochondrial deubiquitinase that antagonizes parkin’s function and appears to rescue Pink1-deficient flies, may warrant more attention (115).
Finally, the mammalian UPRmt response remains poorly understood and its implications largely unexplored. The powerful genetics of C. elegans demonstrates that this pathway is clearly important in this model organism. Yet, could this be a case where we are being misled by the idiosyncrasies of a purely postmitotic organism (e.g., C. elegans) and/or a case where evolution has found better or different answers? Indeed, an important caveat to all the analysis discussed here is that the link between mitochondria, metabolism, and aging may be species-dependent. For instance, arguments have been advanced that the profound lifespan effects of caloric restriction observed in rodents may not be as evident in long-lived species (e.g., humans) in which energy depot storage, reproductive span, and reproductive costs are markedly different (116). Similar arguments could also be advanced for the relative role of mitochondria in short-lived organisms with a high metabolic rate compared with long-lived, lower-metabolic-rate species.
Whatever the magnitude of effects in humans, further dissection of mitochondrial signaling pathways seems essential to understand the best points for therapeutic manipulation. The concept of a little mitochondrial stress being beneficial is attractive, but it may be difficult to clearly define the boundary between therapeutic mitochondrial hormesis (mitohormesis) and bioenergetic collapse. This is perhaps best illustrated by the historical experience with dinitrophenol, a mitochondrial uncoupler and an inducer of mitochondrial stress. In the 1930s, nearly 100,000 Americans were taking or had taken this drug, which produced an increase in metabolic rate and a subsequent capacity for profound weight loss (117). Unfortunately, due to its narrow therapeutic index, it also produced concomitant muscle, liver, and cardiovascular toxicity, including cardiac arrest (117, 118). These historical efforts serve as a cautionary tale and suggest that “drugging” the mitochondria remains a formidable challenge. That said, a new cadre of biologically active molecules targeting mitochondrial quality control or retrograde signaling are being described and carry the promise of clinical applicability to a wide swath of age-related conditions. These mitochondria-centric small molecules, some found in high abundance in naturally occurring substances such as blue cheese (spermidine) or pomegranate juice (urolithin A), provide a literal new diet of opportunities to potentially tinker with human aging. That’s exciting and welcome news to hopefully spice up a relationship that hasn’t seen much change over the last few billion years.
Acknowledgments
This work was supported by funds from the Leducq Foundation and the Progeria Research Foundation.
Version 1. 07/30/2018
Electronic publication
Version 2. 07/31/2018
Author name correction
Version 3. 08/31/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(9):3662–3670.https://doi.org/10.1172/JCI120842.
Contributor Information
Arnon Blum, Email: ABlum@poria.health.gov.il.
Jie Liu, Email: JIELIU@pitt.edu.
Toren Finkel, Email: finkelt@upmc.edu.
References
- 1.Sagan L. On the origin of mitosing cells. J Theor Biol. 1967;14(3):255–274. doi: 10.1016/0022-5193(67)90079-3. [DOI] [PubMed] [Google Scholar]
- 2.Speakman JR. Body size, energy metabolism and lifespan. J Exp Biol. 2005;208(pt 9):1717–1730. doi: 10.1242/jeb.01556. [DOI] [PubMed] [Google Scholar]
- 3.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 4.Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radic Biol Med. 2011;51(2):327–336. doi: 10.1016/j.freeradbiomed.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 5.Sebastián D, Palacín M, Zorzano A. Mitochondrial dynamics: coupling mitochondrial fitness with healthy aging. Trends Mol Med. 2017;23(3):201–215. doi: 10.1016/j.molmed.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Kauppila TES, Kauppila JHK, Larsson NG. Mammalian mitochondria and aging: an update. Cell Metab. 2017;25(1):57–71. doi: 10.1016/j.cmet.2016.09.017. [DOI] [PubMed] [Google Scholar]
- 7.van de Ven RAH, Santos D, Haigis MC. Mitochondrial sirtuins and molecular mechanisms of aging. Trends Mol Med. 2017;23(4):320–331. doi: 10.1016/j.molmed.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korolchuk VI, Miwa S, Carroll B, von Zglinicki T. Mitochondria in cell senescence: is mitophagy the weakest link? EBioMedicine. 2017;21:7–13. doi: 10.1016/j.ebiom.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franceschi C, et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128(1):92–105. doi: 10.1016/j.mad.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 10.Soysal P, et al. Inflammation and frailty in the elderly: a systematic review and meta-analysis. Ageing Res Rev. 2016;31:1–8. doi: 10.1016/j.arr.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 11.Newman AB, et al. Associations of subclinical cardiovascular disease with frailty. J Gerontol A Biol Sci Med Sci. 2001;56(3):M158–M166. doi: 10.1093/gerona/56.3.M158. [DOI] [PubMed] [Google Scholar]
- 12.Ferrucci L, et al. Change in muscle strength explains accelerated decline of physical function in older women with high interleukin-6 serum levels. J Am Geriatr Soc. 2002;50(12):1947–1954. doi: 10.1046/j.1532-5415.2002.50605.x. [DOI] [PubMed] [Google Scholar]
- 13.Adriaensen W, Matheï C, Vaes B, van Pottelbergh G, Wallemacq P, Degryse JM. Interleukin-6 as a first-rated serum inflammatory marker to predict mortality and hospitalization in the oldest old: a regression and CART approach in the BELFRAIL study. Exp Gerontol. 2015;69:53–61. doi: 10.1016/j.exger.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 14.Shen-Orr SS, et al. Defective signaling in the JAK-STAT pathway tracks with chronic inflammation and cardiovascular risk in aging humans. Cell Syst. 2016;3(4):374–384.e4. doi: 10.1016/j.cels.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furman D, et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat Med. 2017;23(2):174–184. doi: 10.1038/nm.4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saresella M, et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener. 2016;11:23. doi: 10.1186/s13024-016-0088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32(8):373–379. doi: 10.1016/j.it.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Youm YH, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013;18(4):519–532. doi: 10.1016/j.cmet.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldberg EL, Dixit VD. Drivers of age-related inflammation and strategies for healthspan extension. Immunol Rev. 2015;265(1):63–74. doi: 10.1111/imr.12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ridker PM, et al. Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 22.Ridker PM, et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10105):1833–1842. doi: 10.1016/S0140-6736(17)32247-X. [DOI] [PubMed] [Google Scholar]
- 23.Krysko DV, et al. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011;32(4):157–164. doi: 10.1016/j.it.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 24.Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4(6):469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- 25.Dela Cruz CS, Kang MJ. Mitochondrial dysfunction damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion. 2018;41:37–44. doi: 10.1016/j.mito.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Q, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pinti M, et al. Circulating mitochondrial DNA increases with age and is a familiar trait: implications for “inflamm-aging”. Eur J Immunol. 2014;44(5):1552–1562. doi: 10.1002/eji.201343921. [DOI] [PubMed] [Google Scholar]
- 28.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 29.Yu J, et al. Inflammasome activation leads to caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci U S A. 2014;111(43):15514–15519. doi: 10.1073/pnas.1414859111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153(2):348–361. doi: 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iyer SS, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39(2):311–323. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 33.van der Burgh R, Boes M. Mitochondria in autoinflammation: cause, mediator or bystander? Trends Endocrinol Metab. 2015;26(5):263–271. doi: 10.1016/j.tem.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 34.Shimada K, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Picca A, et al. Circulating mitochondrial DNA at the crossroads of mitochondrial dysfunction inflammation during aging muscle wasting disorders. Rejuvenation Res. doi: 10.1089/rej.2017.1989. doi: 10.1089/rej.2017.1989. [published online ahead of print January 2, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong Z, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164(5):896–910. doi: 10.1016/j.cell.2015.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356(6337):513–519. doi: 10.1126/science.aal3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ko F, et al. Impaired mitochondrial degradation by autophagy in the skeletal muscle of the aged female interleukin 10 null mouse. Exp Gerontol. 2016;73:23–27. doi: 10.1016/j.exger.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kane AE, Hilmer SN, Mach J, Mitchell SJ, de Cabo R, Howlett SE. Animal models of frailty: current applications in clinical research. Clin Interv Aging. 2016;11:1519–1529. doi: 10.2147/CIA.S105714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.West AP, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520(7548):553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Little JP, et al. Mitochondrial transcription factor A (Tfam) is a pro-inflammatory extracellular signaling molecule recognized by brain microglia. Mol Cell Neurosci. 2014;60:88–96. doi: 10.1016/j.mcn.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 43.Brunner S, Herndler-Brandstetter D, Weinberger B, Grubeck-Loebenstein B. Persistent viral infections and immune aging. Ageing Res Rev. 2011;10(3):362–369. doi: 10.1016/j.arr.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 44.Tannahill GM, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rubic T, et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol. 2008;9(11):1261–1269. doi: 10.1038/ni.1657. [DOI] [PubMed] [Google Scholar]
- 46.Mills EL, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167(2):457–470.e13. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scialò F, et al. Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 2016;23(4):725–734. doi: 10.1016/j.cmet.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sukumar M, et al. Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab. 2016;23(1):63–76. doi: 10.1016/j.cmet.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol. 2017;18(5):488–498. doi: 10.1038/ni.3704. [DOI] [PubMed] [Google Scholar]
- 50.Edwards CB, Copes N, Brito AG, Canfield J, Bradshaw PC. Malate and fumarate extend lifespan in Caenorhabditis elegans. PLoS One. 2013;8(3):e58345. doi: 10.1371/journal.pone.0058345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCormick MA, et al. A comprehensive analysis of replicative lifespan in 4,698 single-gene deletion strains uncovers conserved mechanisms of aging. Cell Metab. 2015;22(5):895–906. doi: 10.1016/j.cmet.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cortopassi GA, Arnheim N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990;18(23):6927–6933. doi: 10.1093/nar/18.23.6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Payne BA, et al. Universal heteroplasmy of human mitochondrial DNA. Hum Mol Genet. 2013;22(2):384–390. doi: 10.1093/hmg/dds435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ross JM, et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature. 2013;501(7467):412–415. doi: 10.1038/nature12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kujoth GC, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309(5733):481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 56.Trifunovic A, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429(6990):417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 57.Mito T, et al. Mitochondrial DNA mutations in mutator mice confer respiration defects and B-cell lymphoma development. PLoS One. 2013;8(2):e55789. doi: 10.1371/journal.pone.0055789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DeBalsi KL, Hoff KE, Copeland WC. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res Rev. 2017;33:89–104. doi: 10.1016/j.arr.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van Epps P, Kalayjian RC. Human immunodeficiency virus and aging in the era of effective antiretroviral therapy. Infect Dis Clin North Am. 2017;31(4):791–810. doi: 10.1016/j.idc.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 60.Sohl CD, et al. Probing the structural and molecular basis of nucleotide selectivity by human mitochondrial DNA polymerase gamma. Proc Natl Acad Sci U S A. 2015;112(28):8596–8601. doi: 10.1073/pnas.1421733112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Payne BA, et al. Mitochondrial aging is accelerated by anti-retroviral therapy through the clonal expansion of mtDNA mutations. Nat Genet. 2011;43(8):806–810. doi: 10.1038/ng.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gardner K, Hall PA, Chinnery PF, Payne BA. HIV treatment and associated mitochondrial pathology: review of 25 years of in vitro, animal, and human studies. Toxicol Pathol. 2014;42(5):811–822. doi: 10.1177/0192623313503519. [DOI] [PubMed] [Google Scholar]
- 63.Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. J Physiol (Lond) 2000;526(pt 1):203–210. doi: 10.1111/j.1469-7793.2000.t01-1-00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Santanasto AJ, et al. Skeletal muscle mitochondrial function and fatigability in older adults. J Gerontol A Biol Sci Med Sci. 2015;70(11):1379–1385. doi: 10.1093/gerona/glu134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Picca A, et al. Update on mitochondria and muscle aging: all wrong roads lead to sarcopenia. Biol Chem. 2018;399(5):421–436. doi: 10.1515/hsz-2017-0331. [DOI] [PubMed] [Google Scholar]
- 66.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sekine S, Youle RJ. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018;16(1):2. doi: 10.1186/s12915-017-0470-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85(2):257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pickrell AM, et al. Endogenous parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron. 2015;87(2):371–381. doi: 10.1016/j.neuron.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun N, et al. Measuring in vivo mitophagy. Mol Cell. 2015;60(4):685–696. doi: 10.1016/j.molcel.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McWilliams TG, et al. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol. 2016;214(3):333–345. doi: 10.1083/jcb.201603039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee JJ, et al. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol. 2018;217(5):1613–1622. doi: 10.1083/jcb.201801044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McWilliams TG, et al. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 2018;27(2):439–449.e5. doi: 10.1016/j.cmet.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schweers RL, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104(49):19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sandoval H, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454(7201):232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157(1):65–75. doi: 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Novak I, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11(1):45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wei Y, Chiang WC, Sumpter R, Jr , Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168(1–2):224–238.e10. doi: 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Theiss AL, Sitaraman SV. The role and therapeutic potential of prohibitin in disease. Biochim Biophys Acta. 2011;1813(6):1137–1143. doi: 10.1016/j.bbamcr.2011.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lazarou M, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.García-Prat L, et al. Autophagy maintains stemness by preventing senescence. Nature. 2016;529(7584):37–42. doi: 10.1038/nature16187. [DOI] [PubMed] [Google Scholar]
- 82.Wu JJ, et al. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging (Albany NY) 2009;1(4):425–437. doi: 10.18632/aging.100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yan Y, Finkel T. Autophagy as a regulator of cardiovascular redox homeostasis. Free Radic Biol Med. 2017;109:108–113. doi: 10.1016/j.freeradbiomed.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. 2015;521(7553):525–528. doi: 10.1038/nature14300. [DOI] [PubMed] [Google Scholar]
- 85.Rana A, Rera M, Walker DW. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc Natl Acad Sci U S A. 2013;110(21):8638–8643. doi: 10.1073/pnas.1216197110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Khraiwesh H, et al. Mitochondrial ultrastructure and markers of dynamics in hepatocytes from aged, calorie restricted mice fed with different dietary fats. Exp Gerontol. 2014;56:77–88. doi: 10.1016/j.exger.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eisenberg T, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016;22(12):1428–1438. doi: 10.1038/nm.4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ryu D, et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med. 2016;22(8):879–888. doi: 10.1038/nm.4132. [DOI] [PubMed] [Google Scholar]
- 89.Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 2014;33(19):2142–2156. doi: 10.15252/embj.201488104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003;33(1):40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- 91.Dillin A, et al. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298(5602):2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- 92.Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8(12):e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zarse K, et al. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012;15(4):451–465. doi: 10.1016/j.cmet.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dell’agnello C, et al. Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Hum Mol Genet. 2007;16(4):431–444. doi: 10.1093/hmg/ddl477. [DOI] [PubMed] [Google Scholar]
- 95.Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, Hekimi S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005;19(20):2424–2434. doi: 10.1101/gad.1352905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Melber A, Haynes CM. UPRmt regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018;28(3):281–295. doi: 10.1038/cr.2018.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337(6094):587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Quirós PM, et al. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol. 2017;216(7):2027–2045. doi: 10.1083/jcb.201702058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr Biol. 2016;26(15):2037–2043. doi: 10.1016/j.cub.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13(4):467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 101.Seiferling D, et al. Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Rep. 2016;17(7):953–964. doi: 10.15252/embr.201642077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144(1):79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Berendzen KM, et al. Neuroendocrine coordination of mitochondrial stress signaling and proteostasis. Cell. 2016;166(6):1553–1563.e10. doi: 10.1016/j.cell.2016.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shao LW, Niu R, Liu Y. Neuropeptide signals cell non-autonomous mitochondrial unfolded protein response. Cell Res. 2016;26(11):1182–1196. doi: 10.1038/cr.2016.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tian Y, et al. Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt) Cell. 2016;165(5):1197–1208. doi: 10.1016/j.cell.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Merkwirth C, et al. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell. 2016;165(5):1209–1223. doi: 10.1016/j.cell.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Khan NA, et al. mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab. 2017;26(2):419–428.e5. doi: 10.1016/j.cmet.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 108.Bennett CF, et al. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat Commun. 2014;5:3483. doi: 10.1038/ncomms4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sorrentino V, et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature. 2017;552(7684):187–193. doi: 10.1038/nature25143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.De Haes W, et al. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc Natl Acad Sci U S A. 2014;111(24):E2501–E2509. doi: 10.1073/pnas.1321776111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang H, et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352(6292):1436–1443. doi: 10.1126/science.aaf2693. [DOI] [PubMed] [Google Scholar]
- 112.Houtkooper RH, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497(7450):451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen H, Chan DC. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. 2017;26(1):39–48. doi: 10.1016/j.cmet.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bingol B, Sheng M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic Biol Med. 2016;100:210–222. doi: 10.1016/j.freeradbiomed.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 115.Bingol B, et al. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510(7505):370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- 116.Shanley DP, Kirkwood TB. Caloric restriction does not enhance longevity in all species and is unlikely to do so in humans. Biogerontology. 2006;7(3):165–168. doi: 10.1007/s10522-006-9006-1. [DOI] [PubMed] [Google Scholar]
- 117.Colman E. Dinitrophenol and obesity: an early twentieth-century regulatory dilemma. Regul Toxicol Pharmacol. 2007;48(2):115–117. doi: 10.1016/j.yrtph.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 118.Yen M, Ewald MB. Toxicity of weight loss agents. J Med Toxicol. 2012;8(2):145–152. doi: 10.1007/s13181-012-0213-7. [DOI] [PMC free article] [PubMed] [Google Scholar]