

Abstract
Quantitative abnormalities of the von Willebrand factor–factor VIII (VWF-FVIII) complex associate with inherited bleeding or thrombotic disorders. Receptor-mediated interactions between plasma VWF-FVIII and phagocytic or immune cells can influence their hemostatic and immunogenic activities. Genetic association studies have demonstrated that variants in the STAB2 gene, which encodes the scavenger receptor stabilin-2, associate with plasma levels of VWF-FVIII. However, the mechanistic basis and pathophysiological consequences of this association are unknown. We have demonstrated that stabilin-2–expressing cells bind and internalize human VWF and FVIII in a VWF-dependent manner, and stabilin-2–deficient mice displayed prolonged human VWF-FVIII half-life compared with controls. The stabilin-2 variant p.E2377K significantly decreased stabilin-2 expression and impaired VWF endocytosis in a heterologous expression system, and common STAB2 variants associated with plasma VWF levels in type 1 von Willebrand disease patients. STAB2-deficient mice displayed a decreased immunogenic response to human VWF-FVIII complex, while coinfusion of human VWF-FVIII with the stabilin-2 ligand hyaluronic acid attenuated the immune response to exogenous FVIII. Collectively, these data suggest that stabilin-2 functions as both a clearance and an immunoregulatory receptor for VWF-FVIII, making stabilin-2 a novel molecular target for modification of the half-life of VWF-FVIII and the immune response to VWF-FVIII concentrates.
Keywords: Genetics, Hematology
Keywords: Coagulation, endothelial cells
Introduction
The coagulation factors von Willebrand factor (VWF) and factor VIII (FVIII) circulate in the plasma as a noncovalent complex, with VWF serving as a carrier to protect FVIII from accelerated proteolysis and clearance (1). VWF regulates primary hemostasis by facilitating platelet-platelet and platelet–endothelial cell interactions, while FVIII is an essential coagulation cofactor in the extrinsic tenase complex, facilitating secondary hemostasis. Quantitative abnormalities in plasma levels of VWF and FVIII strongly influence the function of the hemostatic system. Elevated levels of VWF-FVIII are associated with an increased risk for venous (2, 3) and arterial thrombosis (4, 5), while the inherited bleeding disorders type 1 and type 3 von Willebrand disease and hemophilia A are characterized by quantitative deficiencies in VWF and FVIII, respectively.
Plasma levels of VWF-FVIII are regulated by mechanisms that influence the rate at which they are synthesized and secreted, and by pathways that facilitate the clearance of VWF-FVIII from the plasma. VWF is synthesized by the vascular endothelium and megakaryocytes, and is released either constitutively or on demand from endothelial cell Weibel-Palade bodies or platelet α-granules. The majority of FVIII is synthesized and secreted by sinusoidal and lymphatic endothelial cell beds (6–9). In contrast, pathways that contribute to VWF-FVIII clearance have been only partially characterized. Animal studies using radiolabeled ligands have identified the liver and the spleen as the major sites of VWF-FVIII clearance (10, 11). However, given its relative size and proportionate blood flow, it is presumed that the liver is the predominant site for this function. Previously, in vivo uptake of VWF and/or FVIII has been largely restricted to studies of receptors expressed by hepatocytes and macrophages (10, 12).
Plasma levels of VWF and FVIII can be modified by both genetic and environmental factors, and the heritability of VWF plasma levels is estimated at approximately 65% (13–17). Polymorphisms in the VWF gene and ABO blood group locus collectively account for 25%–35% of this variation, indicating that additional genetic modifiers of this quantitative trait exist. The Cohorts for Heart and Aging Research in Genome Epidemiology (CHARGE) genome-wide association study (GWAS) was the first to identify novel VWF quantitative trait loci including STAB2, SCARA5, CLEC4M, STXBP2, STX2, and TC2N (18). Both the STAB2 and CLEC4M genes encode endocytic receptors expressed by liver sinusoidal endothelial cells (LSECs), a component of the reticuloendothelial system that facilitates the removal of apoptotic cells and aged plasma proteins (19). While we have previously characterized the ability of CLEC4M to bind and internalize VWF (20), the ability of LSECs to regulate VWF-FVIII clearance has not been described.
STAB2 encodes stabilin-2, a class H scavenger receptor expressed on the sinusoidal endothelial cells of the liver, spleen, and lymphatics that functions as a clearance receptor for hyaluronic acid and other glycosaminoglycans (21–23). Stabilin-2 is composed of repetitive extracellular fasciclin and EGF-like domains with a single X-link domain that is C-type lectin–like, located near its transmembrane region. Subsequent to the CHARGE study, additional GWAS-based publications have independently confirmed that common STAB2 variants associate with plasma levels of VWF and/or FVIII in normal individuals or subjects with low VWF levels (18, 24). Additionally, a study of low-frequency variants demonstrated that the STAB2 p.E2377K variant associates with a 33.6% and 26.8% increase in VWF and FVIII levels, respectively (25), suggesting that stabilin-2 is a significant regulator of plasma VWF-FVIII levels. Based on its known function and association with plasma VWF levels, stabilin-2 may act as a clearance receptor for VWF and/or FVIII; however, the mechanistic basis for this relationship has not been assessed.
In addition to facilitating protein clearance, binding of VWF-FVIII to endocytic receptors can regulate interactions with cells in the immune system. The development of FVIII neutralizing antibodies (termed inhibitors) in response to infused FVIII replacement therapy occurs in approximately 30% of patients with severe hemophilia A, while anti-VWF antibodies have been reported in approximately 5% of type 3 VWD patients treated with VWF-FVIII concentrates (26, 27). Emerging evidence suggests that VWF plays an important role in regulating the immune response to FVIII (28). This effect may involve influencing FVIII biodistribution and its interactions with endocytic receptors expressed on antigen-presenting cells (APCs). Internalization of VWF-FVIII by APCs leads to lysosomal degradation of the protein complex or to presentation of VWF and FVIII peptides on MHC class II to cognate CD4+ T cells (26). However, little is currently known about the receptors that mediate VWF-FVIII clearance or the APC subsets involved in this process. To date, no single endocytic receptor has been shown to influence the immune response to VWF-FVIII in vivo.
In these studies, we assess the ability of LSECs to regulate the clearance of VWF-FVIII using in vivo and ex vivo methodologies. We further characterize the role of stabilin-2 as an endocytic receptor for VWF-FVIII that modifies the in vivo half-life of this complex. We assess the ability of single-nucleotide variants (SNVs) in the STAB2 gene to modify the VWF-binding ability of stabilin-2, or to associate with plasma VWF levels in type 1 VWD patients. Finally, we determine the influence of stabilin-2 deficiency on the immune response to exogenous VWF and FVIII.
Results
LSECs contribute to the clearance of VWF and FVIII in vivo.
To visually assess the contribution of LSECs to the clearance of VWF in vivo, we coated 100-nm fluorescent polystyrene microspheres with plasma-derived VWF (pdVWF; FVIII-free). Microsphere coating with VWF was assessed using cell and solid-phase binding assays (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI96400DS1). In this conformation, VWF has been previously shown to bind macrophage LRP-1 in a shear-dependent manner (29). Using VWF-KO mice, LSECs (CD31) and Kupffer cells (F4/80) were labeled using fluorescent antibodies, and the mouse liver was exteriorized by midline laparotomy. Microspheres were then delivered i.v. via jugular catheter, and uptake by liver cells was imaged with intravital spinning disc confocal microscopy. The ability of the VWF coating to influence the cellular distribution of polystyrene microspheres was quantified by image analysis using ImageJ software. All microspheres were observed binding to cells in the liver within 3 minutes of infusion. When uncoated polystyrene microspheres or microspheres coated with glycine were infused into VWF-deficient mice, removal was mediated predominantly by Kupffer cells (Figure 1, A and B), likely through a scavenger receptor or complement C3 pathways (30, 31). However, when microspheres were coated with bovine serum albumin (BSA), a known ligand for LSECs (23), clearance was mediated by both Kupffer cells and LSECs (P < 0.0001) (Figure 1C). Similarly, VWF-coated polystyrene microspheres were internalized by Kupffer cells and LSECs (P < 0.0001) (Figure 1, D–F, Supplemental Figure 2, and Supplemental Videos 1 and 2).
Figure 1. In vivo uptake of VWF by endothelial cells in the murine liver and spleen.

Association of coated fluorescent microspheres (green) with LSECs (anti-CD31, blue) and Kupffer cells (KCs) (anti-F4/80, red) was visualized in real time with spinning disc confocal microscopy. (A–E) Intravital fluorescent images of uncoated microspheres (A), glycine-coated microspheres (B), BSA-coated microspheres (C), and VWF-coated microspheres (D and E). Scale bars: 50 μm. Images are representative of n = 3–5 independent experiments. (F) Quantitative analysis was performed on multiple still images from n = 3–4 independent experiments ± SEM by t test; *P < 0.05. See Supplemental Figures 1 and 2 and Supplemental Videos 1 and 2 for characterization of coated microspheres. (G–N) VWF-KO mice were infused with human pdVWF for 30 minutes, and visualization of VWF localization in the liver and spleen was performed with IHC and immunofluorescence (IF). Images are representative of n = 3 independent experiments. (G) Association of human pdVWF with cells in the murine liver using IHC (scale bar: 100 μm). (H) Association of LSECs (CD31, red) and VWF (green) by IF (scale bar: 20 μm). (I) Association of human pdVWF (green) with KCs (F4/80, red) by IF (scale bar: 20 μm). (J) Association of human pdVWF with cells in the murine spleen using IHC (scale bar: 200 μm). (K) Association of splenic endothelial cells (CD31, red) and VWF (green) by IHC (scale bar: 20 μm). (L) Association of human pdVWF (green) with KCs (F4/80, red) by IHC. (M and N) Super-resolution imaging of VWF associating with CD31 (M) and F4/80 (N) (scale bars: 2.5 μm) in the liver. Images are representative of n = 3 independent experiments. For all images, blue indicates DAPI.
To confirm our intravital observations, we performed immunohistochemistry (IHC) on liver and spleen sections from VWF-KO mice 30 minutes after tail vein injection of human pdVWF (200 U/kg). We observed that the majority of human VWF was associated with CD31-expressing sinusoidal endothelial cells or F4/80-expressing macrophages (Figure 1, G–I, M, and N for super-resolution images), consistent with our intravital observations. Similarly, infused human pdVWF associated with cells in the marginal zone of the spleen that expressed CD31 and F4/80 (Figure 1, J–L). We also took super-resolution images of infused VWF associating with LSECs and Kupffer cells in the liver (Figure 1, M and N). 3D image reconstructions of VWF associating with CD31- or F4/80-expressing cells in the murine liver can be found in Supplemental Figure 3 and Supplemental Videos 3 and 4. Overall, the staining of infused VWF throughout the liver was variable (Supplemental Figure 4), which may be related to the pattern of expression of endocytic VWF receptors as well as differential exposure of VWF to shear as it circulates throughout the liver.
To quantitatively assess the influence of LSECs on mediating VWF-FVIII clearance, we pretreated normal C57BL/6 mice with cyclophosphamide, which has been previously reported to disrupt the sinusoidal endothelium but not affect Kupffer cell function (32, 33). In normal mice, plasma VWF:Ag and FVIII:C levels were significantly increased (Figure 2, A and B), while VWF propeptide/VWF:Ag ratio (used as a surrogate marker for VWF clearance) was significantly decreased, indicating reduced VWF clearance (Figure 2C). The lower magnitude of FVIII:C increase compared with VWF may be related to impaired FVIII synthesis as a result of cyclophosphamide-induced sinusoidal endothelial cell toxicity. VWF and CD31 staining was less intense in the livers of cyclophosphamide-treated mice that were infused with human pdVWF (Figure 2, D–F), suggesting that cyclophosphamide treatment was partially cytotoxic to LSECs. We confirmed the involvement of LSECs in regulating clearance of human VWF and FVIII by performing half-life studies using cyclophosphamide-treated mice. VWF-KO mice that were pretreated with cyclophosphamide 24 hours before infusion with human pdVWF had a significantly increased half-life (P < 0.0001) relative to untreated controls (Figure 2G). Similarly, FVIII-KO mice treated with cyclophosphamide before infusion with human recombinant FVIII (rFVIII) had a significantly increased half-life relative to untreated controls (P < 0.0001) (Figure 2H), which may be related to increased circulating endogenous VWF:Ag in response to cyclophosphamide treatment. The ability of CD31-expressing LSECs to endocytose murine pdVWF is shown in Supplemental Figure 5.
Figure 2. Cyclophosphamide-induced LSEC cytotoxicity increases the plasma levels and half-life of VWF-FVIII.

(A–C) Normal C57BL/6 mice were treated with cyclophosphamide to induce LSEC cytotoxicity, and plasma levels of VWF:Ag (A), FVIII:C (B), and VWFpp/VWF:Ag ratio (C) were measured (n = 10–14 animals per condition). (D and E) Association of infused human pdVWF (green) with LSECs (CD31, red) in untreated VWF-KO (D) and cyclophosphamide-treated VWF-KO (E) mice. Images are representative of n = 3 independent experiments; scale bars: 20 μm. (F) Quantitative analysis of VWF, CD31, and F4/80 staining from n = 3 independent experiments by t test; *P < 0.05. (G and H) Influence of cyclophosphamide treatment on VWF half-life in VWF-KO mice (n = 12 per treatment condition) (G) and FVIII half-life in FVIII-KO mice (n = 12 per treatment condition) (H). See statistical summary of half-life studies in Table 1.
LSECs are strongly regulated by their microenvironment and rapidly lose their phenotype in cell culture (19). We were unable to detect the expression of either stabilin-2 or CLEC4M by primary cultures of commercially available human LSECs (data not shown). Therefore, to demonstrate the association between LSECs, their surface receptors stabilin-2 and CLEC4M, and VWF, we performed IHC on normal human liver tissue (Figure 3A). VWF staining was strongest in vascular endothelial cells, and weaker and punctate in the liver sinusoids (Figure 3, B and C). Both sinusoidal and vascular endothelial cells express CD31, while only sinusoidal endothelial cells express stabilin-2 and CLEC4M. In the liver sinusoids, VWF associated with CD68-expressing Kupffer cells (Figure 3D) and CLEC4M-, stabilin-2–, and CD31-expressing LSECs (Figure 3, E–G). VWF in the sinusoids partially colocalized with EEA-1, a marker of early endosomes, and LAMP2, a marker of late endosomes/lysosomes (Figure 3, H and I). In the human spleen, VWF associated weakly with CD68-expressing macrophages and stabilin-2–expressing and CD31-expressing splenic endothelial cells (Figure 3, J–L).
Figure 3. VWF associates with endocytic endothelial cells in the human liver and spleen.

Association of VWF with cells in the normal human liver was characterized by IF. (A) H&E stain of normal human liver (scale bar: 100 μm). (B and C) VWF (green) was found within the vascular endothelium (B) and the sinusoids (C). (D) Association of VWF (green) with Kupffer cells (CD68, red). (E–G) Association of VWF (green) with the LSEC markers stabilin-2 (E, red), CLEC4M (F, red), and CD31 (G, red). (H and I) Association of VWF (green) with early endosomes (EEA1, red) (H) and late endosomes (LAMP2, red) (I) in the liver sinusoids. Figure depicts representative images from a total of n = 3 sections from 1 case; scale bars: 20 μm. (J–L) Association of VWF (green) with splenic macrophages (CD68, red) (J) and CD31-expressing (red) (K) and stabilin-2–expressing (red) (L) endothelial cells in the spleen (scale bar: 20 μm). Figure depicts representative images from a total of n = 3 sections from 2 cases. For all images, blue indicates DAPI.
The LSEC receptor stabilin-2 binds and internalizes human VWF-FVIII complex.
Murine LSECs express a series of endocytic receptors that may regulate VWF-FVIII clearance, including SIGNR1 (a homolog of the human receptor CLEC4M) (34–36) and stabilin-2 (21, 37). We first investigated the influence of SIGNR1 on VWF-FVIII clearance in VWF/SIGNR1 double-knockout (DKO) mice (Supplemental Figure 6), and observed no difference in endogenous VWF:Ag or FVIII:C or change in half-life of human pdVWF or FVIII in SIGNR1-deficient mice compared with C57BL/6 controls.
We next investigated the role of stabilin-2 in regulating VWF-FVIII clearance in vivo. Human pdVWF infused into VWF-KO mice associated with stabilin-2–expressing cells in the liver and spleen (Figure 4, A and D), and stabilin-2 deficiency altered the VWF staining intensity and cellular localization in both organs (Figure 4, B, C, E, and F). LSECs from VWF-KO mice were next isolated by MACS and cultured for 24 hours ex vivo. Characterization using flow cytometry and immunofluorescence demonstrated that these cells were CD146+, CD31+, stabilin-2+, and F4/80– and capable of endocytosing acetylated LDL (Figure 4G and data not shown). When exposed to VWF and FVIII products, we observed that these cells were able to bind and endocytose human recombinant VWF (rVWF) and pdVWF, murine rVWF, and human plasma-derived FVIII (pdFVIII) that was VWF-bound (Figure 4, H–L). For Figure 4I, an orthogonal view of a stabilin-2–expressing LSEC is inset to demonstrate internalization of VWF. LSECs interacted weakly with human rFVIII in the absence of VWF (data not shown).
Figure 4. Stabilin-2–expressing cells bind and internalize VWF in vivo and in vitro.

(A–F) The influence and association between stabilin-2 and VWF were characterized in the murine liver and spleen. (A) Association of infused human pdVWF (green) with stabilin-2–expressing cells (red) in the liver of a VWF-KO mouse (scale bar: 20 μm). (B and C) Association of human pdVWF (green) with LSECs (CD31, red) in VWF-KO (B) and VWF/STAB2 DKO (C) mice (scale bars: 100 μm). (D) Association of infused human pdVWF (green) with stabilin-2–expressing cells (red) in the murine spleen (scale bar: 20 μm). (E and F) Association of VWF (green) with splenic endothelial cells (CD31, red) in VWF-KO (E) and VWF/STAB2 DKO (F) mice (scale bars: 20 μm). All images are representative of n = 3 independent experiments. (G–L) IF of isolated VWF-KO murine LSECs cultured ex vivo. (G) Expression of CD31 (green) and stabilin-2 (red). (H–L) IF of LSECs exposed to 2 U/ml VWF and/or FVIII for 1 hour. (H) Binding of human pdVWF (hpdVWF, green) to CD31-expressing LSECs (red). (I–K) Binding of hpdVWF (I) (orthogonal view inset), human recombinant VWF (hrVWF) (J), and murine recombinant VWF (mrVWF, red) (K) to stabilin-2–expressing LSECS (red). (L) Binding of human pdFVIII (+VWF) [hpdFVIII (+VWF)] (green) to stabilin-2–expressing LSECs (red). Scale bars: 20 μm. (M–R) IF of HEK 293 cells transfected with the human and murine stabilin-2 cDNA and exposed to 2 U/ml VWF and/or FVIII for 1 hour. (M and N) Binding of hpdVWF (M, green) and hpdFVIII (+VWF) (N, green) to human stabilin-2–expressing (red) HEK 293 cells. (O) Colocalization of VWF (green) and FVIII (red) on stabilin-2–expressing HEK 293 cells. (P–R) Binding of hpdVWF (green) (P), hrVWF (green) (Q), and mrVWF (green) (R) to murine stabilin-2–expressing (red) HEK 293 cells. For all images, blue indicates DAPI; and yellow, colocalization. Scale bars: 20 μm. All images are representative of n ≥ 3 independent experiments.
To confirm the ability of stabilin-2 to mediate VWF endocytosis by LSECs, we transfected HEK 293T cells with human and murine stabilin-2 cDNAs, and assessed the ability of these cells to bind and internalize VWF-FVIII. HEK 293 cells transfected with empty vector backbone did not interact with VWF or FVIII (data not shown). Human stabilin-2–expressing HEK 293T cells were able to bind and internalize human pdVWF and FVIII in a VWF-dependent manner (Figure 4, M and N), and VWF and FVIII partially colocalized on stabilin-2–expressing cells (Figure 4O), suggesting that the receptor interacts with these ligands as a complex. Human rFVIII, in the absence of VWF, interacts weakly with human stabilin-2–expressing cells (data not shown). HEK 293T cells expressing murine stabilin-2 demonstrated binding of both human rVWF and pdVWF but not murine rVWF (Figure 4, P–R). Analysis confirming the colocalization of VWF and stabilin-2 on stabilin-2–expressing cells using Coloc-2 can be found in Supplemental Figure 7.
We next characterized the influence of stabilin-2 deficiency on plasma levels and half-life of VWF-FVIII in a mouse model. No significant differences in endogenous plasma VWF:Ag levels were observed in STAB2-KO C57BL/6 mice (38) compared with age-matched normal C57BL/6 control mice (Figure 5A). No significant differences in the half-life of murine rVWF and pdVWF in VWF/STAB2 DKO mice relative to VWF-KO controls (Supplemental Figure 6) were observed. As stabilin-2–expressing HEK 293 cells interact with human but not murine VWF, we next evaluated the influence of murine stabilin-2 deficiency on the half-life of human VWF. In VWF/STAB2 DKO mice, the half-life of human rVWF (Figure 5B) and human pdVWF (Wilate) (Figure 5C) was significantly increased (P < 0.0001) in comparison with VWF-KO mice (summary statistics in Table 1). We also assessed the influence of macrophage depletion by clodronate liposomes or LSEC cytotoxicity induced by cyclophosphamide treatment on VWF half-life in VWF/STAB2 DKO mice. Clodronate liposome treatment increased VWF half-life in VWF/STAB2 DKO mice compared with liposome-treated controls (P = 0.018) (Figure 5D), and, notably, cyclophosphamide treatment also increased VWF half-life in VWF/STAB2 DKO mice (P = 0.0008) (Figure 5E).
Figure 5. Stabilin-2 deficiency increases the half-life of human VWF-FVIII.

(A) Plasma levels of VWF:Ag were measured in age-matched normal C57BL/6 mice compared with stabilin-2–deficient (STAB2-KO) C57BL/6 mice (n = 26–38 animals per condition by t test). (B and C) Influence of stabilin-2 deficiency on the half-life of infused human rVWF (n = 12 animals per condition) (B) and human pdVWF (FVIII-bound 1:1) (n = 16 animals per condition) (C). (D) The combined influence of macrophage depletion using clodronate liposomes and stabilin-2 deficiency was assessed using human pdVWF (FVIII-free) (n = 8 animals per condition). (E) The combined influence of cyclophosphamide-induced LSEC cytotoxicity and stabilin-2 deficiency was assessed using human pdVWF (FVIII-free) (n = 8–16 animals per condition). (F) The influence of stabilin-2 deficiency on the half-life of infused human pdFVIII (+VWF) (n = 16 animals per condition) was measured in VWF-KO compared with VWF/STAB2 DKO mice. See statistical summary of half-life studies in Table 1.
Table 1. Summary statistics for VWF-FVIII half-life studies.

We also assessed the influence of stabilin-2 deficiency on FVIII clearance. When we reanalyzed samples from the VWF-FVIII complex infusion studies for FVIII:Ag (using an ELISA that does not detect murine FVIII), we observed that there was a prolongation of the slow phase of FVIII clearance in VWF/STAB2 DKO mice relative to VWF-KO controls (Figure 5F). As VWF and FVIII exist in a dynamic equilibrium with high rates of association and disassociation, the competition of endogenous murine FVIII for the FVIII binding site on human VWF may result in displacement of human FVIII, resulting in its accelerated proteolysis and clearance when compared with the half-life of human VWF. In the absence of human or murine VWF, there was no influence of stabilin-2 on the half-life of human rFVIII (Supplemental Figure 6) in comparison with VWF-deficient controls. This result confirms our observation that stabilin-2 interacts weakly with human FVIII in the absence of human VWF, and that human FVIII is cleared by stabilin-2 only in the presence of human VWF.
Genetic variability and N-linked glycans modulate the interaction between human VWF and the clearance receptor stabilin-2.
As VWF may interact with its clearance receptors through both peptide sequences and N-linked glycans, and stabilin-2 contains an X-link domain with C-type lectin capabilities, we assessed the influence of VWF N-linked glycans to mediate the interaction between VWF and stabilin-2. We first removed the N-linked glycans on human pdVWF and confirmed effective deglycosylation by concanavalin A lectin solid-phase ELISA (data not shown). We then exposed the de–N-glycosylated VWF to HEK 293T cells expressing human stabilin-2. Interestingly, stabilin-2–expressing cells demonstrated enhanced uptake of de–N-glycosylated human VWF as compared with control VWF (P < 0.0001) (Figure 6, A–C). We also characterized the influence of N-linked glycans on stabilin-2–mediated VWF clearance in vivo. The half-life of de–N-glycosylated VWF was significantly longer in VWF/STAB2 DKO mice than in VWF-KO mice (P < 0.0001) (Figure 6D). Finally, we demonstrated that competitive inhibition using known stabilin-2 ligands could decrease binding and internalization of VWF by stabilin-2–expressing cells (Figure 6I).
Figure 6. Genetic and biochemical regulation of VWF interactions with stabilin-2.

HEK 293 cells were transiently transfected with the human stabilin-2 cDNA and incubated with 2 U/ml human pdVWF or 2 U/ml human pdVWF pretreated with PNGase F to remove its N-linked glycans (de-N VWF). (A) Quantification of de-N VWF binding to stabilin-2–expressing cells. (B and C) Human pdVWF and de–N-glycosylated VWF (green) binding to human stabilin-2–expressing (red) cells. Images are representative of n = 6 independent experiments. Scale bars: 40 μm. (D) The influence of VWF N-linked glycans on the clearance of VWF by stabilin-2 was characterized in vivo (n = 8 animals per condition). See statistical summary of half-life studies in Table 1. The stabilin-2 variant p.E2377K was introduced into the human stabilin-2 cDNA by site-directed mutagenesis. (E and F) HEK 293 cells were transiently transfected with the stabilin-2 cDNA p.E2377K 1:1 with WT (E, red) or p.E2377K stabilin-2 alone (F, red) and incubated with 2 U/ml human pdVWF (green). Images are representative of n = 4 independent experiments; scale bars: 40 μm. For all experiments, blue indicates DAPI; and yellow, colocalization. (G) Quantification of VWF binding to HEK 293 cells expressing the p.E2377K stabilin-2 variant. (H) Flow cytometric analysis of p.E2377K stabilin-2 expression. Pathogenicity assessment of the stabilin-2 p.E2377K variant is described in Table 2. To demonstrate competitive binding to stabilin-2, known stabilin-2 ligands were preincubated with HEK 293 cells expressing murine stabilin-2 for 15 minutes and then incubated with 2 U/ml human pdVWF for 1 hour. (I) Quantitative IF was performed to compare untreated VWF binding to cells pretreated with stabilin-2 ligands (n = 3–4 independent experiments). HA, hyaluronic acid; DS, dermatan sulfate; UFH, unfractionated heparin. Throughout figure, *P < 0.05, **P < 0.001 as determined by t test.
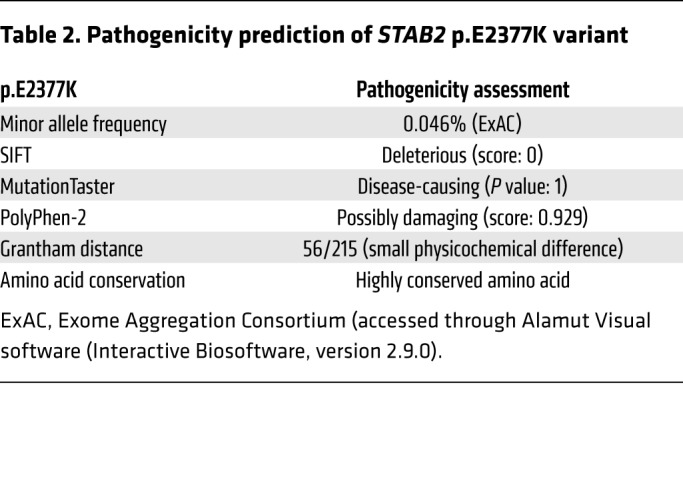
The rare stabilin-2 variant p.E2377K (rs141041254) has been previously shown to associate with plasma levels of VWF and FVIII in normal individuals (β: 26.81 and 33.65, respectively) (25). In silico analysis of the stabilin-2 p.E2377K variant using PolyPhen-2, SIFT, and MutationTaster (data accessed through Alamut Visual software [Interactive Biosoftware, version 2.9.0]) suggests that this variant is damaging to protein expression and/or function (Table 2). To understand the influence of the p.E2377K variant on the ability of stabilin-2 to interact with VWF, we generated this variant in the human stabilin-2 cDNA using site-directed mutagenesis and transiently transfected it into HEK 293T cells. Equivalent transfection efficiency of each plasmid was confirmed by TaqMan quantitative PCR analysis (Supplemental Figure 8). Compared with WT stabilin-2, the p.E2377K variant demonstrated a decreased total number of stabilin-2–expressing cells (WT 12.4% vs. p.E2377K 1.2% positive) (Figure 6H). For the cells that did express detectable levels of the p.E2377K stabilin-2 variant, the total amount of stabilin-2 expressed was decreased (48% geometric mean) relative to WT stabilin-2. Using immunofluorescence, we observed that for HEK 293T cells transfected with the p.E2377K stabilin-2 variant, VWF binding was significantly reduced (P < 0.0001) in comparison with cells transfected with the WT stabilin-2 (Figure 6, E–G).
Table 2. Pathogenicity prediction of STAB2 p.E2377K variant.
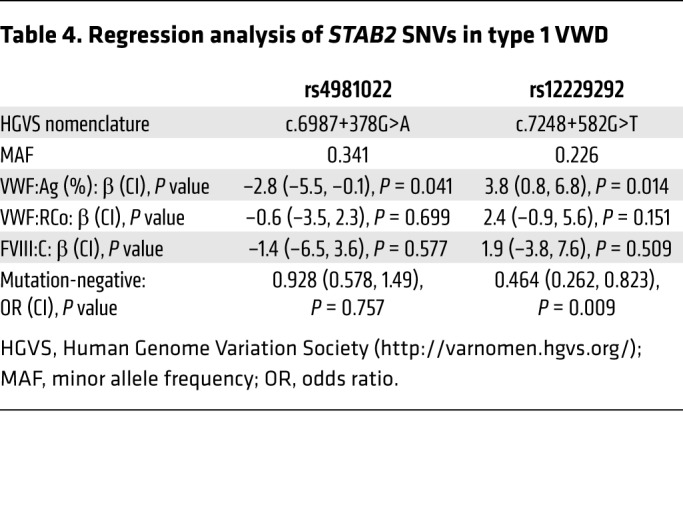
The common STAB2 SNVs rs12229292 and rs4981022 associate with plasma levels of VWF:Ag and/or FVIII:C in normal individuals (18). We hypothesized that they may also contribute to variability in plasma VWF:Ag and/or FVIII:C in individuals with low VWF levels and type 1 VWD, characterized by a partial quantitative deficiency of VWF:Ag. We genotyped 165 type 1 VWD patients (VWF:Ag 0.05–0.5 IU/ml) (see Table 3 for patient characteristics) for these SNVs and performed linear regression in order to characterize the influence of these SNVs on plasma levels of VWF:Ag and FVIII:C (Figure 7). STAB2 rs12229292 associated with elevated VWF:Ag (β: 3.8%, P = 0.014) and FVIII:C (β: 1.9, P = 0.509) and STAB2 rs4981022 associated with decreased VWF:Ag (β: –2.8%, P = 0.041) and FVIII:C (β: –1.4%, P = 0.577) in these patients, consistent with the proposal that STAB2 variability influences plasma levels of VWF and FVIII (Table 4).
Table 3. Population characteristics of type 1 VWD cohort.
Figure 7. Common SNVs in STAB2 influence plasma VWF:Ag levels in type 1 VWD patients.

One hundred sixty-five type 1 VWD index cases were genotyped for the STAB2 variants rs4981022 and rs12229292 to assess the contribution of STAB2 genetic diversity to plasma VWF:Ag and FVIII:C. (A and B) Association between STAB2 rs4981022 genotype and VWF:Ag (A) and FVIII:C (B) in type 1 VWD patients. (C and D) Association between STAB2 rs12229292 genotype and VWF:Ag (C) and FVIII:C (D) in type 1 VWD patients. Throughout figure, % = mean VWF:Ag or FVIII:C. Patient phenotypic data are found in Table 3. Linear regression and statistical analysis are found in Table 4.
Table 4. Regression analysis of STAB2 SNVs in type 1 VWD.
Stabilin-2 is an immunoregulatory receptor for human VWF-FVIII.
In addition to influencing the half-life of endogenous human VWF-FVIII, stabilin-2 may also play a role in regulating the intracellular fate of cleared exogenous VWF-FVIII used as replacement therapy for the inherited bleeding disorders hemophilia A and VWD. The most frequent and severe complication of this treatment in hemophilia A is the development of neutralizing anti-FVIII antibodies (termed FVIII inhibitors). In addition, anti-VWF antibodies have been reported in type 3 VWD cases. The internalization, proteolysis, and presentation of VWF-FVIII peptides by APCs on MHC II results in either tolerance or immunity to the protein antigens. However, the mechanisms by which VWF-FVIII is internalized by these receptors are largely uncharacterized, and details concerning the fate of the internalized proteins are also lacking. We hypothesized that associations might exist between mechanisms that regulate VWF-FVIII clearance and those that contribute to its immunogenic response. In this context, we next addressed the potential of stabilin-2 to influence the immunogenicity of these proteins.
Here, we intravenously infused normal and STAB2-KO mice with 4 weekly doses of 2 IU human pdFVIII (80 IU/kg) containing approximately 4.8 IU human pdVWF, and assessed the plasma for anti–human VWF and anti–human FVIII IgG antibodies 4 weeks after the initial exposure (Figure 8A). We observed a substantial reduction in the titer of total anti–human VWF IgG antibodies in STAB2-KO mice compared with normal mice (Figure 8B; P = 0.003), although the incidence of anti-VWF antibodies in both groups was 100% (data not shown). In parallel, we observed a significant decrease in the incidence of anti–human FVIII antibodies (Figure 8C; P = 0.02), as well as a significant decrease in the magnitude of the anti–human FVIII response (Figure 8D; P = 0.003). When the same immune challenge protocol was repeated in VWF-KO and VWF/STAB2-DKO mice, we observed a similar influence of stabilin-2 deficiency on the titer of anti-VWF IgG. However, no anti-FVIII IgG immune response was observed in VWF KO mice (Supplemental Figure 10). To demonstrate that STAB2-KO mice are capable of mounting a robust immune response against human VWF and FVIII, we infused 2 IU pdFVIII together with 1 μg lipopolysaccharide (LPS) for 2 weeks, and collected blood 4 weeks after the initial infusion (Figure 8A). We observed a substantial increase in the titer of anti-VWF antibodies (Figure 8B), as well as an increase in the incidence of anti-FVIII antibodies (Figure 8C; P = 0.003) and their titers (Figure 8D; P = 0.0001).
Figure 8. Stabilin-2 deficiency modifies the immune response to human pdVWF-FVIII.

(A) Normal or STAB2-KO C57BL/6 mice received intravenously 2 IU human FVIII (either rFVIII or pdFVIII complexed with pdVWF at a 1:2.4 ratio) weekly for 4 weeks (n = 10 for all conditions). For the immune challenge protocol, mice received 1 μg LPS i.p. for the first 2 weeks of treatment with pdFVIII (n = 6). On week 5, blood was collected and assayed for anti-VWF and anti-FVIII IgG. (B) Titer of anti-VWF IgG in mice receiving pdFVIII (by Mann-Whitney U). (C and D) Anti-FVIII IgG incidence (C) and titer (D) in mice receiving pdFVIII. (E and F) Anti-FVIII IgG incidence (E) and titer (F) in mice receiving rFVIII. Throughout figure, *P < 0.05 as determined by Fisher’s exact test or Mann-Whitney U.
Stabilin-2 is a clearance receptor for human VWF but not murine VWF. To demonstrate that the influence of stabilin-2 on FVIII immune response was human VWF–dependent, we performed similar experiments using human rFVIII. Upon infusion of 4 weekly doses of human rFVIII, we observed a modest but not statistically significant (P = 0.08) decrease in the incidence of anti–human FVIII IgG in STAB-KO mice compared with normal mice (Figure 8E). Similarly, the titers of anti-FVIII antibodies were decreased, but to a lesser extent than with the VWF-containing FVIII concentrates (Figure 8F).
To examine the potential of stabilin-2 as a therapeutic target for reducing the FVIII immune response, we intravenously administered VWF-containing human pdFVIII with 100 μg of the stabilin-2 ligand hyaluronic acid into WT C57BL/6 mice (Figure 9A). Following 4 weekly coinfusions, we did not observe a significant difference in the immune response against human VWF (Figure 9B). In contrast, we observed a substantial reduction in the titer of FVIII-specific IgG (Figure 9C; P < 0.01) and overall incidence of FVIII-specific IgG (Figure 9C; P < 0.001).
Figure 9. The stabilin-2 ligand hyaluronic acid modifies the immune response to human pdVWF-FVIII.

(A) Normal C57BL/6 mice received intravenously 2 IU human pdFVIII (complexed with pdVWF at a 1:2.4 ratio) with 100 μg hyaluronic acid (HA) weekly for 4 weeks (n = 10 for all conditions). On week 5, blood was collected and assayed for anti-VWF and anti-FVIII IgG. (B) Titer of anti-VWF IgG in mice receiving pdFVIII with or without hyaluronic acid. (C and D) Anti-FVIII IgG titer (C) and incidence (D) in mice receiving pdFVIII with or without hyaluronic acid. Throughout figure, *P < 0.05, **P < 0.001 as determined by Fisher’s exact test or Mann-Whitney U.
Discussion
Receptor-mediated endocytosis of the VWF-FVIII complex can influence the pathobiological conditions associated with their procoagulant or immunogenic properties. The clearance and immunogenicity of molecules can be affected not only by the expression of endocytic cell surface receptors, but by the proteolytic capabilities of the cell, expression of MHC II and costimulatory molecules, and the tissue microenvironment (26, 39, 40). However, there is much that is still unknown about mechanisms that regulate the removal of the VWF-FVIII protein complex from the circulation, and the intracellular fate of these ligands involving either lysosomal degradation or antigen presentation. The clearance of VWF and FVIII is a complex and semiselective process, involving a number of ligand-receptor interactions including the hepatocyte- and macrophage-expressed asialoglycoprotein receptor, SIGLEC5, SR-A1, and members of the LDL receptor family (12, 29, 41–44). In immunological studies, FVIII has been shown to associate in part with both dendritic cells and marginal zone macrophages in the mouse spleen (10, 11), while the binding of FVIII to dendritic cell receptors has been described in vitro (45). However, to date, no endocytic receptor has been implicated in regulating the immune response to VWF-FVIII in vivo. Importantly, macrophage depletion attenuates VWF clearance approximately 2-fold, and decreases but does not fully inhibit the FVIII immune response, suggesting that alternative pathways exist that regulate both processes (10, 11, 46, 47).
LSECs have the highest endocytic capacity of any cell in the body, and are reported to express a series of endocytic receptors including stabilin-2, CLEC4M, the mannose receptor, SR-A1, SR-B1, CD36, stabilin-1, FcγRIIb2, LSECtin, LRP-1, and LYVE-1. Both stabilin-2 and CLEC4M have been identified by GWAS as associating with plasma VWF or FVIII levels in normal individuals (18, 24, 25). In these studies, we characterize the scavenger receptor stabilin-2 as a novel clearance and immunomodulatory receptor for the human VWF-FVIII complex. We first demonstrate that stabilin-2/CD31–expressing LSECs and splenic endothelial cells are capable of endocytosing VWF using a combination of intravital microscopy, IHC, and ex vivo cell culture (Figures 1–4). Collectively, we observed that both human and murine VWF can be endocytosed and cleared by LSECs (Figures 1 and 2 and Supplemental Figure 4), but that stabilin-2 functions as a clearance receptor for only human and not murine VWF (Figures 4 and 5 and Supplemental Figure 6). Interestingly, treatment of VWF/STAB2 DKO mice with cyclophosphamide increased the half-life of human pdVWF (Figure 5E), and reduced but did not abolish the association between VWF and CD31-expressing cells, suggesting that cyclophosphamide does not ablate the entire hepatic endothelium, and that additional clearance receptor(s) for human and murine VWF are expressed by murine LSECs. We confirmed that the murine SIGNR1 receptor, which is homologous to the human CLEC4M receptor, does not regulate the half-life of human or murine VWF (Supplemental Figure 6).
Recognition of VWF by its clearance receptors can involve binding to a specific amino acid sequence, or to its glycans, and may also be influenced by the 3D conformation of the VWF molecule. The mechanistic basis by which stabilin-2 regulates the clearance of human but not murine VWF is not yet known, although it likely does not involve expression of N-linked glycans (Figure 6). As human and murine VWF share an approximately 80% amino acid identity, it may involve interactions between human or murine stabilin-2 and an amino acid sequence in human VWF that is not conserved in murine VWF. Interestingly, we have observed that murine stabilin-2 can regulate the clearance of the murine VWF propeptide, which is composed of repeating D1 and D2 domains with homology to the D′D3 and D4 regions found in the mature VWF molecule (48). Future studies will be aimed at better understanding the structural basis of this interaction.
We next assessed the influence of VWF–stabilin-2 interactions on quantitative VWF pathologies. Type 1 VWD is characterized by partial VWF quantitative deficiency, and exhibits both incomplete penetrance and variable expressivity. We genotyped a cohort of 165 type 1 VWD patients (Table 3) for common STAB2 SNVs that have previously associated with either VWF and/or FVIII levels in GWASs of normal subjects (18). We observed that both STAB2 rs4981022 and rs12229292 SNVs associated with VWF:Ag levels in our type 1 VWD population, with a magnitude of effect and directionality that was congruous with results observed in the CHARGE GWAS (Figure 7 and Table 4). In contrast, a previous study did not find an association between rs4981022 and VWF:Ag levels in a cohort of Dutch type 1 VWD patients (49). An increased median patient age, differences in ethnicity, inclusion/exclusion criteria related to VWF:Ag levels, and a lower proportion of VWF mutation–negative patients in the Dutch population may explain these differences. Consistent with our results, however, a GWAS found that the STAB2 SNV rs4981022 associates with low VWF plasma levels (24). Further studies on the influence of rare and common STAB2 variants on the type 1 VWD phenotype are required to better understand this relationship.
Epidemiological studies have demonstrated that elevated levels of VWF-FVIII are associated with venous and arterial thrombosis, and variants within the VWF gene that increase VWF:Ag or risk for thrombosis have been described (50–52). Additionally, variants in STAB2 have been shown to associate with either elevated levels of VWF-FVIII, or increased risk for thrombosis in population-based studies (25, 53, 54). While the CHARGE STAB2 SNV rs4981022 was not associated with an increased risk for venous thrombosis in a case-controlled study (52), a pedigree linkage analysis and GWAS identified the STAB2 SNV rs1593812 as a modifier for the incidence of venous thromboembolism (VTE) (55). Additionally, whole exome sequencing of VTE patients demonstrated an increased incidence of rare STAB2 variants in comparison with healthy controls (54). Further, in normal individuals the rare stabilin-2 variant p.E2377K (rs141041254) is associated with significantly elevated VWF and FVIII levels (β: 33.6% and 26.8%, respectively). In silico and in vitro analysis of the p.E2377K stabilin-2 variant indicates that it is damaging to stabilin-2 protein function or expression by decreasing stabilin-2 expression in transfected HEK 293 cells (Table 2 and Figure 6). While the exact mechanistic basis by which this variant modifies stabilin-2 expression is not yet known, in silico CentroidFold (http://rtools.cbrc.jp/centroidfold/) analysis predicts that the rs141041254 SNV alters the stabilin-2 mRNA secondary structure (Supplemental Figure 8), which might influence mRNA stability and translation efficiency (56). Thus, this variant appears to mediate its influence on VWF levels by decreasing the expression of stabilin-2, suggesting that haploinsufficiency of stabilin-2 leads to increased plasma levels of VWF-FVIII, which may influence the subsequent risk for thrombosis. However, as stabilin-2 is a scavenger receptor that regulates the clearance of other plasma constituents, the influence of stabilin-2 variants on altered clearance of other inflammatory regulators, coagulation factors, or apoptotic blood cells cannot be excluded.
We finally assessed the influence of stabilin-2 on the immune response to VWF-FVIII. Previous studies have demonstrated that human rFVIII in the presence of endogenous murine VWF associates with marginal zone macrophages in the murine spleen and that these APCs thus presumably contribute to the early immune response to FVIII (11). However, the addition of human VWF can block the interaction between FVIII and blood monocyte-derived dendritic cells in vitro (47). Therefore, the characterization of the association between human FVIII and cells in the spleen in the absence of human VWF might be incomplete in light of our observations that stabilin-2 interacts with human but not murine VWF. In this study, we demonstrate that physiological concentrations of infused human VWF associate in part with CD31- and stabilin-2–expressing cells in the red pulp of the spleen (Figure 1 and Figure 4). Moreover, stabilin-2 deficiency decreases the association between human VWF and CD31-expressing cells in the murine spleen. Using stabilin-2–expressing cells in vitro, we demonstrate that human FVIII binds stabilin-2 in a VWF-dependent manner, and that VWF and FVIII partially colocalize on the cell surface, indicating that they are endocytosed as a complex (Figure 4). We also demonstrate that stabilin-2 deficiency only alters FVIII half-life when it is coinfused with human pdVWF, suggesting that VWF and FVIII are cleared by stabilin-2 as a complex (Figure 5).
We then assessed the influence of stabilin-2 on the immune response to VWF-FVIII. We treated either normal C57BL/6 or STAB2-KO C57BL/6 mice with equimolar concentrations of 2 different preparations of human FVIII: a human pdVWF-FVIII concentrate or human rFVIII (human VWF-free) (Figure 8). We observed that when FVIII was complexed with human VWF, stabilin-2 deficiency significantly attenuated the immune response to VWF and FVIII, which was restored when mice were immune-challenged with low-dose LPS. In contrast, when human rFVIII was administered in the absence of human VWF, the immune response to rFVIII was not statistically different (P = 0.08), although a similar pattern was observed. Taken together, these studies suggest that stabilin-2 deficiency influences the immune response to VWF-FVIII, and that the response to FVIII is largely VWF-dependent, consistent with the effect observed in the clearance studies (Supplemental Table 1). We finally assessed the ability of stabilin-2 competitive inhibition to modulate the immune response to VWF-FVIII. Our in vitro assays demonstrate that preincubation of stabilin-2–expressing cells with the known stabilin-2 ligand hyaluronic acid can attenuate the binding and internalization of VWF by stabilin-2 (Figure 6I), while in vivo, coinfusion of hyaluronic acid with human VWF-FVIII decreased the immune response to FVIII in normal C57BL/6 mice (Figure 9).
The mechanistic basis by which stabilin-2 deficiency regulates the immune response to VWF-FVIII is incompletely understood and is currently subject to further investigation using models that address the issue of using human proteins in a murine system. The spleen has been the long-standing focus as the site of the immune response to FVIII owing to the tolerogenic environment in the liver and evidence that splenectomized mice have a decreased but not absent immune response to FVIII (11). As LSECs express stabilin-2, MHC II, and costimulatory molecules, although at relatively low levels, there is a potential that they may function as APCs capable of presenting VWF-FVIII peptides to cognate CD4+ T cells (57, 58). However, it is more likely that stabilin-2 expressed on the splenic sinusoidal endothelium acts as a key mediator in the directed shuttling of FVIII antigen in a VWF-dependent manner through marginal zone macrophage and marginal B cell compartments of the spleen (11, 59, 60). Furthermore, engagement of stabilin-2 initiates signaling through extracellular signal–regulated kinases 1 and 2 (ERK1/2), and can induce NF-κB activation of proinflammatory gene expression that can, in turn, enhance the likelihood of an immune response (61). As evidence for this hypothesis, the cotreatment of STAB2-KO mice with NF-κB–activating LPS and pdVWF-FVIII stimulated a robust anti-FVIII immune response (Figure 8), and different splenic cytokine profiles have been observed when FVIII-KO mice are treated with human pdVWF-FVIII compared with human rFVIII (62).
It is also possible that an antiinflammatory/immunotolerant phenotype may be related to altered plasma expression of pro-/antiinflammatory factors related to stabilin-2 deficiency. Although STAB2-KO mice have not been reported to have altered cytokine profiles compared with normal controls, they have highly elevated levels of the ligand hyaluronic acid (38, 63). While hyaluronic acid on its own has been reported to have immunomodulatory influences, including cell signaling through Toll-like receptors, inflammation, and T cell recruitment via interactions with CD44 (64), its rapid in vivo half-life of 3 to 5 minutes (65) suggests that at least some of its influence on the FVIII immune response may be mediated via a competitive binding mechanism involving stabilin-2. In addition to its interactions with stabilin-2, hyaluronic acid could also alter engagement of VWF-FVIII with other receptors including CD44, RHAMM, layilin, and LYVE-1.
These studies used a C57BL/6 mouse model that expresses normal levels of murine VWF-FVIII, where the immune response is elicited in response to an infused xenoprotein, human VWF and/or FVIII. As expected, the immune response to human FVIII is decreased in magnitude compared with what is typically observed in FVIII-KO animals, as evidenced by Bethesda analysis (Supplemental Figure 9). It is likely that in normal mice exposed to human VWF-FVIII there is no significant increase in FVIII neutralizing antibodies owing to the conservation of FVIII epitopes between human and murine proteins that are crucial for protein activity. Thus, characterization of the VWF-FVIII immune response in normal mice more closely models the development of FVIII antibodies in mild/moderate hemophilia A patients, where endogenous FVIII cross-reactive material is present and FVIII antibodies incompletely inactivate FVIII (66). In contrast, epidemiological studies have shown that an increased incidence in the immune response to FVIII associates with the severity of the F8 gene mutation; similarly, FVIII-KO mice exhibit an almost 100% inhibitor response to exogenous FVIII (67, 68). The influence of stabilin-2 deficiency on FVIII immunogenicity in a mouse model of severe hemophilia A remains to be assessed.
Genetic association studies have demonstrated an interaction between variants in the STAB2 locus and plasma levels of VWF-FVIII in normal individuals. Here we determine the mechanistic basis of this association by characterizing stabilin-2 as a clearance receptor for VWF-FVIII. Our studies have also characterized the role of VWF–stabilin-2 interactions in thrombosis and type 1 VWD, pathobiological conditions involving quantitative VWF abnormalities. Moreover, we demonstrate that stabilin-2 regulates the immune response to VWF-FVIII concentrates, and that competitive blockade with the stabilin-2 ligand hyaluronic acid can mitigate the immune response to FVIII. These studies may improve prediction of individuals with elevated VWF:Ag levels at risk for thrombosis, or development of a molecular diagnostic algorithm for type 1 VWD patients who do not have a pathogenic variant in their VWF gene. Therapeutically, competitive inhibition of stabilin-2 may also allow for the development of coagulation factor replacement products for hemophilia A or VWD with improved pharmacokinetic profiles and decreased immunogenicity.
Methods
Materials
Human plasma-derived VWF-FVIII (2.4:1, Humate-P) was from CSL Behring, human plasma-derived VWF-FVIII (1:1, Wilate) was from Octapharma, human recombinant FVIII (Advate) was from Baxalta/Shire, and human plasma-derived VWF (FVIII-free) was provided by Biotest. Murine and human recombinant VWF was produced by HEK 293 cells stably expressing the VWF cDNA. Murine pdVWF was generated by hydrodynamic tail vein injection of the murine WT VWF cDNA into VWF/FVIII DKO mice; plasma was collected 4 days after hydrodynamic injection and pooled. C57BL/6 mice were from The Jackson Laboratory. VWF-KO (69), FVIII-KO (70), and STAB2-KO (38) mice originated as previously described.
Animal models
Intravital microscopy.
VWF-KO mice were anesthetized by an i.p. injection of ketamine/xylazine (200/10 mg/kg), and the right internal jugular vein was cannulated with PE10 polyethylene tubing for i.v. delivery of microspheres and anesthesia. The murine liver was exteriorized through a midline laparotomy, followed by removal of the abdominal skin and muscle, and dissection of the falciform ligament to facilitate detachment of the liver from the diaphragm, which allowed for more stable image capture. Mice were then placed on a customized stage with an in-built coverslip that allowed for externalization of a small hepatic segment. LSECs and Kupffer cells were visualized by infusion of anti-CD31 (MEC 13.3, Becton Dickinson) and anti-F4/80 (BM8, eBioscience), respectively. Plasma-derived VWF (FVIII-free), glycine (Sigma-Aldrich), or BSA (Sigma-Aldrich) was coated onto 100 nm fluorescent polystyrene microspheres (Fluoresbrite YG Microspheres, Polysciences Inc.) in accordance with the manufacturer’s protocols and administered via catheter. Imaging was performed within the liver distal to where the hepatic artery and portal vein provide the blood supply to the sinusoids using an inverted Quorum Wave FX Spinning Disc confocal microscope (Quorum Technologies Inc.) and a Hamamatsu Orca EMCCD camera. Livers were imaged for up to 5 minutes after infusion.
Clearance studies.
STAB2-KO mice on a C57BL/6 background were crossed with C57BL/6 VWF-KO mice (3–11 generations) to generate VWF/STAB2 double-knockout (DKO) mice. VWF or FVIII products were administered to mice at 200 U/kg by tail vein injection. Blood was collected into 10% buffered citrate through the retro-orbital plexus. Samples were centrifuged at 10,000 g for 10 minutes to collect platelet-poor plasma and stored at –80°C until analysis. To induce LSEC cytotoxicity, mice received an i.p. injection of 200 mg/kg cyclophosphamide (Sigma-Aldrich) 24 hours prior to VWF half-life study (33). To transiently deplete macrophages including Kupffer cells, VWF/STAB2 DKO mice received a tail vein i.v. injection of 200 μl of anionic clodronate liposome or control liposomes (FormuMax Scientific Inc.) 48 hours prior to VWF half-life study.
Immunogenicity studies.
Naive normal C57BL/6 mice from our colony or STAB2-KO mice (38) received 4 weekly i.v. infusions of 2 IU of either plasma-derived FVIII (pdFVIII), containing a 2.4:1 ratio of VWF to FVIII, or human recombinant FVIII (rFVIII) (Figure 8A). Terminal samples were collected via the inferior vena cava into one-tenth volume sodium citrate 28 days after the initial injection. For the immune challenge protocol, mice received 1 μg LPS (InvivoGen) with VWF-FVIII complex for the first 2 injections. For the hyaluronic acid (Abcam) coinfusion studies, 100 μg hyaluronic acid was administered with each pdFVIII injection (Figure 9A).
Cell culture
Isolation and culture of LSECs.
LSECs were isolated from VWF-KO mice by positive magnetic cell sorting using CD146 microbeads (Miltenyi Biotec). Briefly, livers were mechanically and enzymatically separated using collagenase D (Roche) and strained to ensure a single-cell suspension. An LSEC-rich cell population was generated by centrifugation using HistoDenz (Sigma-Aldrich), and then labeled with anti-CD146 microbeads. Cells were flowed over a magnetic column and washed, and magnetically retained CD146+ cells were eluted. LSECs were seeded onto coverslips coated with poly-l-lysine (Sigma-Aldrich) and cultured for 24 hours in mouse endothelial cell medium (Cell Biologics).
Stabilin-2–expressing HEK 293 cells.
A HEK 293 (American Type Culture Collection) stable cell line expressing the murine stabilin-2 (mSTAB2) cDNA in the pEF vector was maintained in αMEM supplemented with 10% FBS (Life Technologies), 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, 250 ng/ml fungizone, 100 μg/ml hygromycin, and 50 μg/ml blasticidin (Thermo Fisher Scientific). Cells were routinely more than 70% stabilin-2–positive by flow cytometry. Human stabilin-2–expressing cells were generated transiently in HEK 293 T cells by Lipofectamine transfection (LTX, Invitrogen) according to the manufacturer’s protocols. The human STAB2 rs141041254 (c.7129G>A, p.E2377K) variant was generated in the human STAB2 cDNA by site-directed mutagenesis (Q5 Site-Directed Mutagenesis Kit, New England Biolabs).
Exposure of LSECs or stabilin-2–expressing HEK 293 T cells to VWF-FVIII.
Cells were washed with HBSS (Invitrogen) and exposed to 2 U/ml VWF-FVIII in binding buffer (10 mM HEPES pH 4, 135 mM NaCl, 10 mM KCl, 5 mM CaCl2, 2 mM MgSO4) at 37°C for 1 hour unless otherwise described. For the de–N-glycosylated VWF studies, VWF N-linked glycans were removed by PNGase F treatment (New England Biolabs) according to the manufacturer’s protocols. Effective de–N-glycosylation was confirmed by lectin binding to concanavalin A (Vector Laboratories) as previously described (20). For the ligand binding inhibition studies, murine stabilin-2–expressing cells were preincubated with the known stabilin-2 ligands hyaluronic acid (Abcam), dermatan sulfate (Sigma-Aldrich), BSA (Sigma-Aldrich), and mannan (Sigma-Aldrich) for 15 minutes before exposure to 1.5 U/ml human pdVWF for 1 hour.
Imaging studies
Antibodies.
Antibodies used for imaging studies included sheep anti–human FVIII (SAF8C-AP, Affinity Biologicals), rabbit anti-VWF (A0082, DAKO), sheep anti-VWF (ab11173, Abcam), mouse anti–stabilin-2 clone 3.1 (71), rat anti–mouse stabilin-2 (D317-3, MBL), rabbit anti–human stabilin-2 (121893, Abcam), mouse anti–human LAMP2 (NBP2-22217, Novus), mouse anti–human EEA1 (sc-365652, Santa Cruz Biotechnology), rat anti–mouse CD31 (DIA-310, Dianova), mouse anti–human CD31 (M0823, DAKO), rabbit anti–human CD31 (ab28364, Abcam), rabbit anti–human CD68 (HPA048982, Sigma-Aldrich), rabbit anti–mouse F4/80 (MBP2-12506, Novus), and mouse anti–human CLEC4M (120604, R&D Systems).
Immunohistochemistry.
IHC analysis was performed on frozen tissue sections to image murine stabilin-2–expressing cells in the liver and spleen. Briefly, murine liver and spleen were snap-frozen in OCT compound (VWR) using an acetone-dry ice bath. Ten-micrometer sections were soaked in PBS to remove OCT and fixed using Histochoice (Sigma-Aldrich). All other IHC analyses were performed on formalin-fixed paraffin-embedded tissues. Seven-micrometer sections were rehydrated with toluene/ethanol/water washes before incubation at 95°C in Target Retrieval Solution pH 6.0 (DAKO). All tissue sections were blocked in a humid chamber for 10 minutes in serum-free protein block (DAKO), 20 minutes in 0.1% Sudan Black B in 70% ethanol, and washed with TBST. The primary antibody diluted in TBST/1% BSA was applied overnight at 4°C, slides were washed, and secondary antibody plus DAPI was applied for 1 hour at room temperature. Slides were coverslipped with fluorescent mounting media (DAKO).
Immunocytochemistry.
Cells were washed, fixed with Cytofix/Cytoperm (Becton Dickinson), permeabilized with 0.1% Triton X-100, and blocked with protein block (DAKO). Cells were incubated with primary antibody in PBS/1% BSA overnight at 4°C. Cells were washed and incubated with appropriate secondary antibodies (Invitrogen), stained with DAPI, and mounted using fluorescent mounting media (DAKO).
Microscopy.
Slides were imaged with a Quorum Wave FX Spinning Disc confocal microscope and Hamamatsu Orca high-resolution camera or Nikon Eclipse NiE microscope in-built with camera (Nikon). Nanoscopy was performed on an SP8 Tandem Scanning White light laser (WLL) confocal platform (Leica Microsystems) equipped with stimulated emission depletion (STED) with depletion lasers at 592 nm and 660 nm. Images were deconvolved using Hyugens deconvolution software and analyzed using ImageJ or FIJI software (NIH).
VWF:Ag, FVIII:Ag, and FVIII:C
Human and murine VWF:Ag was measured using DAKO antibodies (72). Factor VIII antigen ELISA (Affinity Biologicals) was performed according to the manufacturer’s protocols with modifications. After sample incubation, plates were incubated with biotinylated sheep anti–factor VIII for 1 hour followed by a 1-hour incubation with streptavidin poly-HRP (Pierce/Thermo Fisher Scientific). FVIII:C was measured using the Chromogenix Coatest assay (Diapharma) according to the manufacturer’s directions. Murine VWF propeptide (VWFpp) was measured as previously described (72) using 349.2 and 349.3 antibodies provided by S. Haberichter (Blood Research Institute, BloodCenter of Wisconsin, Milwaukee, Wisconsin, USA).
Genotyping
Patient characteristics.
Patients were derived from the Canadian and Milwaukee type 1 VWD cohorts as previously described (20, 73, 74). Inclusion criteria included a bleeding diathesis, plasma levels of VWF:Ag between 0.05 and 0.50 U/ml, plasma levels of VWF:RCo ≤ 0.6 U/ml, and normal multimers. Laboratory tests for VWF:Ag, VWF:RCo, and FVIII:C were performed either at the source clinic attended by the patient (Canadian cohort) or by the Zimmerman project (Milwaukee cohort).
Genotyping and statistical analysis.
The STAB2 SNVs rs12229292 and rs4981022 were genotyped by TaqMan assay (Thermo Fisher Scientific). Patient characteristics are described in Table 3. Linear regression analysis was performed to quantify differences in VWF:Ag, VWF:RCo, and FVIII:C in type 1 VWD patients for each SNV, and logistic regression was used for mutation-negative patient analysis. All models were adjusted for age and ABO blood group. Regression analysis was performed using IBM-SPSS version 24.0 for Windows 2016.
FVIII/VWF antibody and inhibitor assays
Total FVIII- or VWF-specific IgG was detected by ELISA. In brief, human rFVIII or human pdVWF (FVIII-free) was adsorbed at 1 μg/ml to Nunc PolySorp (Thermo Fisher Scientific) plates overnight. Wells were blocked with 1% gelatin, and sample plasma was subsequently added in serial dilutions starting at 1:40. An HRP-conjugated polyclonal goat anti-mouse IgG (Southern Biotech) was used for detection. Positive titers exhibited optical density values of greater than the blank + 0.3 at 490 nm. FVIII inhibitors were measured by 1-stage activated partial thromboplastin time (APTT) clotting assay using a Siemens BCS XP automated coagulometer. Sample plasma was incubated at 56°C for 30 minutes to inactivate residual FVIII. Plasma was then serially diluted in human FVIII-deficient plasma (Precision Biologics) and incubated with normal human pooled plasma for 2 hours at 37°C. Positive inhibitor samples were defined as >0.6 Bethesda units. Statistical differences were calculated using Fisher’s exact test for antibody/inhibitor incidence, and the Mann-Whitney U test for comparisons of antibody/inhibitor titer using GraphPad Prism 7.0.
Statistics
Unless specified, t tests (2-tailed) or 1-way ANOVA was performed on experiments with n ≥ 4 using GraphPad InStat software version 3.06. Plasma half-lives were calculated using GraphPad Prism 7.0. Values are expressed as mean ± SEM. Asterisks in figures denote *P < 0.05 and **P < 0.001.
Study approval
All animal experiments were approved by the Queen’s University Animal Care Committee (Kingston, Ontario, Canada). Patient studies were approved by the Children’s Hospital of Wisconsin (Milwaukee, Wisconsin, USA) Institutional Review Board and the Queen’s University Health Sciences Research Ethics Board (Kingston, Ontario, Canada). Informed consent was obtained from all participants in accordance with the Declaration of Helsinki.
Author contributions
LLS and JDL designed and performed experiments, analyzed and interpreted data, and wrote the manuscript. JM and CN performed experiments and analyzed and interpreted the data. IG, ASP, and KN performed experiments. KS, CG, JK, and SG provided STAB2-KO mice and other reagents. WH performed statistical analyses. PDJ and RRM provided patient samples. DL designed experiments, analyzed and interpreted data, and edited the manuscript.
Supplementary Material
Acknowledgments
We thank S. Haberichter for the murine VWFpp antibodies. We thank L. Casey, S. Nanji, and N. Renwick (Queen’s University, Kingston, Ontario, Canada) for the healthy liver and spleen samples. We thank S. Tinlin, L. Boudreau, M. Gordon, P. Christopherson, and C. Dwyer for additional technical assistance. LLS was supported by postdoctoral fellowships from the Heart and Stroke Foundation of Canada and the Canadian Institutes for Health Research. DL is a Canada Research Chair in Molecular Hemostasis. This study was supported by funds from the Canadian Institutes of Health Research (MOP-97849) and from the NIH for the Zimmerman Program (HL081588).
Version 1. 08/20/2018
Electronic publication
Version 2. 08/31/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(9):4057–4073.https://doi.org/10.1172/JCI96400.
Contributor Information
Colleen Notley, Email: c.notley@queensu.ca.
Ilinca Georgescu, Email: 8ig4@queensu.ca.
Jeff Mewburn, Email: mewburnj@queensu.ca.
Kate Nesbitt, Email: kate.nesbitt@queensu.ca.
Kai Schledzewski, Email: kai.schledzewski@medma.uni-heidelberg.de.
Cyrill Géraud, Email: cyrill.geraud@umm.de.
Julia Kzhyshkowska, Email: Julia.Kzhyshkowska@umm.de.
Sergij Goerdt, Email: Sergij.Goerdt@umm.de.
Wilma Hopman, Email: hopmanw@kgh.kari.net.
Robert R. Montgomery, Email: bob.montgomery@bcw.edu.
David Lillicrap, Email: david.lillicrap@queensu.ca.
References
- 1.Pipe SW, Montgomery RR, Pratt KP, Lenting PJ, Lillicrap D. Life in the shadow of a dominant partner: the FVIII-VWF association and its clinical implications for hemophilia A. Blood. 2016;128(16):2007–2016. doi: 10.1182/blood-2016-04-713289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chauhan AK, Kisucka J, Lamb CB, Bergmeier W, Wagner DD. von Willebrand factor and factor VIII are independently required to form stable occlusive thrombi in injured veins. Blood. 2007;109(6):2424–2429. doi: 10.1182/blood-2006-06-028241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brill A, et al. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117(4):1400–1407. doi: 10.1182/blood-2010-05-287623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nichols TC, et al. The roles of von Willebrand factor and factor VIII in arterial thrombosis: studies in canine von Willebrand disease and hemophilia A. Blood. 1993;81(10):2644–2651. [PubMed] [Google Scholar]
- 5.Golder M, Mewburn J, Lillicrap D. In vitro and in vivo evaluation of the effect of elevated factor VIII on the thrombogenic process. Thromb Haemost. 2013;109(1):53–60. doi: 10.1160/TH12-05-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan J, et al. Patterns of expression of factor VIII and von Willebrand factor by endothelial cell subsets in vivo. Blood. 2016;128(1):104–109. doi: 10.1182/blood-2015-12-684688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Do H, Healey JF, Waller EK, Lollar P. Expression of factor VIII by murine liver sinusoidal endothelial cells. J Biol Chem. 1999;274(28):19587–19592. doi: 10.1074/jbc.274.28.19587. [DOI] [PubMed] [Google Scholar]
- 8.Fahs SA, Hille MT, Shi Q, Weiler H, Montgomery RR. A conditional knockout mouse model reveals endothelial cells as the principal and possibly exclusive source of plasma factor VIII. Blood. 2014;123(24):3706–3713. doi: 10.1182/blood-2014-02-555151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Everett LA, Cleuren AC, Khoriaty RN, Ginsburg D. Murine coagulation factor VIII is synthesized in endothelial cells. Blood. 2014;123(24):3697–3705. doi: 10.1182/blood-2014-02-554501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Schooten CJ, et al. Macrophages contribute to the cellular uptake of von Willebrand factor and factor VIII in vivo. Blood. 2008;112(5):1704–1712. doi: 10.1182/blood-2008-01-133181. [DOI] [PubMed] [Google Scholar]
- 11.Navarrete A, et al. Splenic marginal zone antigen-presenting cells are critical for the primary allo-immune response to therapeutic factor VIII in hemophilia A. J Thromb Haemost. 2009;7(11):1816–1823. doi: 10.1111/j.1538-7836.2009.03571.x. [DOI] [PubMed] [Google Scholar]
- 12.Grewal PK, et al. The Ashwell receptor mitigates the lethal coagulopathy of sepsis. Nat Med. 2008;14(6):648–655. doi: 10.1038/nm1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orstavik KH, Magnus P, Reisner H, Berg K, Graham JB, Nance W. Factor VIII and factor IX in a twin population. Evidence for a major effect of ABO locus on factor VIII level. Am J Hum Genet. 1985;37(1):89–101. [PMC free article] [PubMed] [Google Scholar]
- 14.Desch KC, et al. Linkage analysis identifies a locus for plasma von Willebrand factor undetected by genome-wide association. Proc Natl Acad Sci U S A. 2013;110(2):588–593. doi: 10.1073/pnas.1219885110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Lange M, Snieder H, Ariëns RAS, Spector TD, Grant PJ. The genetics of haemostasis: a twin study. Lancet. 2001;357(9250):101–105. doi: 10.1016/S0140-6736(00)03541-8. [DOI] [PubMed] [Google Scholar]
- 16.Bladbjerg EM, de Maat MP, Christensen K, Bathum L, Jespersen J, Hjelmborg J. Genetic influence on thrombotic risk markers in the elderly — a Danish twin study. J Thromb Haemost. 2006;4(3):599–607. doi: 10.1111/j.1538-7836.2005.01778.x. [DOI] [PubMed] [Google Scholar]
- 17.Souto JC, et al. Genome-wide linkage analysis of von Willebrand factor plasma levels: results from the GAIT project. Thromb Haemost. 2003;89(3):468–474. doi: 10.1055/s-0037-1613375. [DOI] [PubMed] [Google Scholar]
- 18.Smith NL, et al. Novel associations of multiple genetic loci with plasma levels of factor VII, factor VIII, and von Willebrand factor: the CHARGE (Cohorts for Heart and Aging Research in Genome Epidemiology) Consortium. Circulation. 2010;121(12):1382–1392. doi: 10.1161/CIRCULATIONAHA.109.869156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elvevold K, Smedsrød B, Martinez I. The liver sinusoidal endothelial cell: a cell type of controversial and confusing identity. Am J Physiol Gastrointest Liver Physiol. 2008;294(2):G391–G400. doi: 10.1152/ajpgi.00167.2007. [DOI] [PubMed] [Google Scholar]
- 20.Rydz N, et al. The C-type lectin receptor CLEC4M binds, internalizes, and clears von Willebrand factor and contributes to the variation in plasma von Willebrand factor levels. Blood. 2013;121(26):5228–5237. doi: 10.1182/blood-2012-10-457507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Falkowski M, Schledzewski K, Hansen B, Goerdt S. Expression of stabilin-2, a novel fasciclin-like hyaluronan receptor protein, in murine sinusoidal endothelia, avascular tissues, and at solid/liquid interfaces. Histochem Cell Biol. 2003;120(5):361–369. doi: 10.1007/s00418-003-0585-5. [DOI] [PubMed] [Google Scholar]
- 22.Politz O, et al. Stabilin-1 and -2 constitute a novel family of fasciclin-like hyaluronan receptor homologues. Biochem J. 2002;362(pt 1):155–164. doi: 10.1042/0264-6021:3620155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Connolly MK, et al. In hepatic fibrosis, liver sinusoidal endothelial cells acquire enhanced immunogenicity. J Immunol. 2010;185(4):2200–2208. doi: 10.4049/jimmunol.1000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Loon J, et al. Genome-wide association studies identify genetic loci for low von Willebrand factor levels. Eur J Hum Genet. 2016;24(7):1035–1040. doi: 10.1038/ejhg.2015.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huffman JE, et al. Rare and low-frequency variants and their association with plasma levels of fibrinogen, FVII, FVIII, and vWF. Blood. 2015;126(11):e19–e29. doi: 10.1182/blood-2015-02-624551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai JD, Georgescu MT, Hough C, Lillicrap D. To clear or to fear: an innate perspective on factor VIII immunity. Cell Immunol. 2016;301:82–89. doi: 10.1016/j.cellimm.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.James PD, Lillicrap D, Mannucci PM. Alloantibodies in von Willebrand disease. Blood. 2013;122(5):636–640. doi: 10.1182/blood-2012-10-462085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peyvandi F, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. 2016;374(21):2054–2064. doi: 10.1056/NEJMoa1516437. [DOI] [PubMed] [Google Scholar]
- 29.Rastegarlari G, et al. Macrophage LRP1 contributes to the clearance of von Willebrand factor. Blood. 2012;119(9):2126–2134. doi: 10.1182/blood-2011-08-373605. [DOI] [PubMed] [Google Scholar]
- 30.Ogawara K, et al. Mechanisms of hepatic disposition of polystyrene microspheres in rats: effects of serum depend on the sizes of microspheres. J Control Release. 1999;61(3):241–250. doi: 10.1016/S0168-3659(99)00121-2. [DOI] [PubMed] [Google Scholar]
- 31.Palecanda A, et al. Role of the scavenger receptor MARCO in alveolar macrophage binding of unopsonized environmental particles. J Exp Med. 1999;189(9):1497–1506. doi: 10.1084/jem.189.9.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malhi H, et al. Cyclophosphamide disrupts hepatic sinusoidal endothelium and improves transplanted cell engraftment in rat liver. Hepatology. 2002;36(1):112–121. doi: 10.1053/jhep.2002.33896. [DOI] [PubMed] [Google Scholar]
- 33.van Til NP, et al. Kupffer cells and not liver sinusoidal endothelial cells prevent lentiviral transduction of hepatocytes. Mol Ther. 2005;11(1):26–34. doi: 10.1016/j.ymthe.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 34.Powlesland AS, Ward EM, Sadhu SK, Guo Y, Taylor ME, Drickamer K. Widely divergent biochemical properties of the complete set of mouse DC-SIGN-related proteins. J Biol Chem. 2006;281(29):20440–20449. doi: 10.1074/jbc.M601925200. [DOI] [PubMed] [Google Scholar]
- 35.Parent SA, et al. Molecular characterization of the murine SIGNR1 gene encoding a C-type lectin homologous to human DC-SIGN and DC-SIGNR. Gene. 2002;293(1–2):33–46. doi: 10.1016/s0378-1119(02)00722-9. [DOI] [PubMed] [Google Scholar]
- 36.Tanne A, et al. A murine DC-SIGN homologue contributes to early host defense against Mycobacterium tuberculosis. J Exp Med. 2009;206(10):2205–2220. doi: 10.1084/jem.20090188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lalor PF, Lai WK, Curbishley SM, Shetty S, Adams DH. Human hepatic sinusoidal endothelial cells can be distinguished by expression of phenotypic markers related to their specialised functions in vivo. World J Gastroenterol. 2006;12(34):5429–5439. doi: 10.3748/wjg.v12.i34.5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schledzewski K, et al. Deficiency of liver sinusoidal scavenger receptors stabilin-1 and -2 in mice causes glomerulofibrotic nephropathy via impaired hepatic clearance of noxious blood factors. J Clin Invest. 2011;121(2):703–714. doi: 10.1172/JCI44740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lai JD, et al. Concurrent influenza vaccination reduces anti-FVIII antibody responses in murine hemophilia A. Blood. 2016;127(26):3439–3449. doi: 10.1182/blood-2015-11-679282. [DOI] [PubMed] [Google Scholar]
- 40.Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science. 2005;307(5715):1630–1634. doi: 10.1126/science.1108003. [DOI] [PubMed] [Google Scholar]
- 41.Pegon JN, et al. Factor VIII and von Willebrand factor are ligands for the carbohydrate-receptor Siglec-5. Haematologica. 2012;97(12):1855–1863. doi: 10.3324/haematol.2012.063297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bovenschen N, Mertens K, Hu L, Havekes LM, van Vlijmen BJ. LDL receptor cooperates with LDL receptor-related protein in regulating plasma levels of coagulation factor VIII in vivo. Blood. 2005;106(3):906–912. doi: 10.1182/blood-2004-11-4230. [DOI] [PubMed] [Google Scholar]
- 43.Bovenschen N, Rijken DC, Havekes LM, van Vlijmen BJ, Mertens K. The B domain of coagulation factor VIII interacts with the asialoglycoprotein receptor. J Thromb Haemost. 2005;3(6):1257–1265. doi: 10.1111/j.1538-7836.2005.01389.x. [DOI] [PubMed] [Google Scholar]
- 44.Wohner N, et al. Macrophage scavenger receptor SR-AI contributes to the clearance of von Willebrand factor. Haematologica. 2018;103(4):728–737. doi: 10.3324/haematol.2017.175216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dasgupta S, et al. A role for exposed mannosylations in presentation of human therapeutic self-proteins to CD4+ T lymphocytes. Proc Natl Acad Sci U S A. 2007;104(21):8965–8970. doi: 10.1073/pnas.0702120104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rawley O, et al. von Willebrand factor arginine 1205 substitution results in accelerated macrophage-dependent clearance in vivo. J Thromb Haemost. 2015;13(5):821–826. doi: 10.1111/jth.12875. [DOI] [PubMed] [Google Scholar]
- 47.Dasgupta S, et al. VWF protects FVIII from endocytosis by dendritic cells and subsequent presentation to immune effectors. Blood. 2007;109(2):610–612. doi: 10.1182/blood-2006-05-022756. [DOI] [PubMed] [Google Scholar]
- 48.Rawley O, Nesbitt K, Swystun L, Lillicrap D. Scavenger-receptor stabilin-2 is a major regulator of mouse VWF propeptide clearance. Blood Transfus. 2017;15(suppl 3):s484–s485. [Google Scholar]
- 49.Sanders YV, et al. CLEC4M and STXBP5 gene variations contribute to von Willebrand factor level variation in von Willebrand disease. J Thromb Haemost. 2015;13(6):956–966. doi: 10.1111/jth.12927. [DOI] [PubMed] [Google Scholar]
- 50.van der Meer IM, et al. Genetic variability of von Willebrand factor and risk of coronary heart disease: the Rotterdam Study. Br J Haematol. 2004;124(3):343–347. doi: 10.1046/j.1365-2141.2003.04776.x. [DOI] [PubMed] [Google Scholar]
- 51.van Schie MC, et al. Variation in the von Willebrand factor gene is associated with von Willebrand factor levels and with the risk for cardiovascular disease. Blood. 2011;117(4):1393–1399. doi: 10.1182/blood-2010-03-273961. [DOI] [PubMed] [Google Scholar]
- 52.Smith NL, et al. Genetic variation associated with plasma von Willebrand factor levels and the risk of incident venous thrombosis. Blood. 2011;117(22):6007–6011. doi: 10.1182/blood-2010-10-315473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Desch KC, et al. Exome sequencing studies identify mutations in STAB2 as a genetic risk for venous thromboembolic disease. Blood. 2017;130(suppl 1):457 [Google Scholar]
- 54.Desch KC, et al. Exome sequencing in venous thromboembolic disease identifies excess mutation burden in PROS1, PROC, SERPINC1, STAB2. Blood. 2016;128(22):3794 [Google Scholar]
- 55.Antoni G, et al. A multi-stage multi-design strategy provides strong evidence that the BAI3 locus is associated with early-onset venous thromboembolism. J Thromb Haemost. 2010;8(12):2671–2679. doi: 10.1111/j.1538-7836.2010.04092.x. [DOI] [PubMed] [Google Scholar]
- 56.Manning KS, Cooper TA. The roles of RNA processing in translating genotype to phenotype. Nat Rev Mol Cell Biol. 2017;18(2):102–114. doi: 10.1038/nrm.2016.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Limmer A, et al. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat Med. 2000;6(12):1348–1354. doi: 10.1038/82161. [DOI] [PubMed] [Google Scholar]
- 58.Lohse AW, et al. Antigen-presenting function and B7 expression of murine sinusoidal endothelial cells and Kupffer cells. Gastroenterology. 1996;110(4):1175–1181. doi: 10.1053/gast.1996.v110.pm8613007. [DOI] [PubMed] [Google Scholar]
- 59.Zerra PE, et al. Marginal zone B cells are critical to factor VIII inhibitor formation in mice with hemophilia A. Blood. 2017;130(23):2559–2568. doi: 10.1182/blood-2017-05-782912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lai JD, et al. Early cellular interactions and immune transcriptome profiles in human factor VIII-exposed hemophilia A mice. J Thromb Haemost. 2018;16(3):533–545. doi: 10.1111/jth.13936. [DOI] [PubMed] [Google Scholar]
- 61.Pandey MS, Weigel PH. A hyaluronan receptor for endocytosis (HARE) link domain N-glycan is required for extracellular signal-regulated kinase (ERK) and nuclear factor-κB (NF-κB) signaling in response to the uptake of hyaluronan but not heparin, dermatan sulfate, or acetylated low density lipoprotein (LDL) J Biol Chem. 2014;289(32):21807–21817. doi: 10.1074/jbc.M114.565846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qadura M, et al. Recombinant and plasma-derived factor VIII products induce distinct splenic cytokine microenvironments in hemophilia A mice. Blood. 2009;114(4):871–880. doi: 10.1182/blood-2008-09-174649. [DOI] [PubMed] [Google Scholar]
- 63.Harris EN, et al. N-glycans on the link domain of human HARE/Stabilin-2 are needed for hyaluronan binding to purified ecto-domain, but not for cellular endocytosis of hyaluronan. Glycobiology. 2010;20(8):991–1001. doi: 10.1093/glycob/cwq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiang D, Liang J, Noble PW. Hyaluronan as an immune regulator in human diseases. Physiol Rev. 2011;91(1):221–264. doi: 10.1152/physrev.00052.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fraser JR, Laurent TC, Pertoft H, Baxter E. Plasma clearance, tissue distribution and metabolism of hyaluronic acid injected intravenously in the rabbit. Biochem J. 1981;200(2):415–424. doi: 10.1042/bj2000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Witmer C, Young G. Factor VIII inhibitors in hemophilia A: rationale and latest evidence. Ther Adv Hematol. 2013;4(1):59–72. doi: 10.1177/2040620712464509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qadura M, et al. Immunoglobulin isotypes and functional anti-FVIII antibodies in response to FVIII treatment in Balb/c and C57BL/6 haemophilia A mice. Haemophilia. 2011;17(2):288–295. doi: 10.1111/j.1365-2516.2010.02397.x. [DOI] [PubMed] [Google Scholar]
- 68.Gouw SC, et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic review and meta-analysis. Blood. 2012;119(12):2922–2934. doi: 10.1182/blood-2011-09-379453. [DOI] [PubMed] [Google Scholar]
- 69.Denis C, et al. A mouse model of severe von Willebrand disease: defects in hemostasis and thrombosis. Proc Natl Acad Sci U S A. 1998;95(16):9524–9529. doi: 10.1073/pnas.95.16.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH. Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10(1):119–121. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 71.Martens JH, et al. Differential expression of a gene signature for scavenger/lectin receptors by endothelial cells and macrophages in human lymph node sinuses, the primary sites of regional metastasis. J Pathol. 2006;208(4):574–589. doi: 10.1002/path.1921. [DOI] [PubMed] [Google Scholar]
- 72.Pruss CM, et al. Pathologic mechanisms of type 1 VWD mutations R1205H and Y1584C through in vitro and in vivo mouse models. Blood. 2011;117(16):4358–4366. doi: 10.1182/blood-2010-08-303727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.James PD, et al. The mutational spectrum of type 1 von Willebrand disease: Results from a Canadian cohort study. Blood. 2007;109(1):145–154. doi: 10.1182/blood-2006-05-021105.. [DOI] [PubMed] [Google Scholar]
- 74.Flood VH, et al. Clinical and laboratory variability in a cohort of patients diagnosed with type 1 VWD in the United States. Blood. 2016;127(20):2481–2488. doi: 10.1182/blood-2015-10-673681. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.