

Abstract
Neural precursor cell expressed, developmentally downregulated 9 (NEDD9) supports oncogenic signaling in a number of solid and hematologic tumors. Little is known about the role of NEDD9 in ovarian carcinoma (OC), but available data suggest elevated mRNA and protein expression in advanced stage high grade cancers. We used a transgenic MISIIR-TAg mouse OC model combined with genetic ablation of Nedd9 to investigate its action in the development and progression of OC. A Nedd9−/− genotype delayed tumor growth rate, reduced incidence of ascites, and reduced expression and activation of signaling proteins including SRC, STAT3, E-cadherin and AURKA. Cell lines established from MISIIR-TAg;Nedd9−/− and MISIIR-TAg;Nedd9+/+ mice exhibited altered migration and invasion. Growth of these cells in a syngeneic allograft model indicated that systemic Nedd9 loss in the microenvironment had little impact on tumor allograft growth, but in a Nedd9 wild type background Nedd9−/− allografts exhibited significantly reduced growth, dissemination and oncogenic signaling compared to Nedd9+/+ allografts. Gene expression analysis revealed that Nedd9+/+ tumors exhibited more mesenchymal ‘stem-like’ transcriptional program, including increased expression of Aldh1a1 and Aldh1a2. Conversely, loss of Nedd9 resulted in increased expression of differentiation genes, including fallopian tube markers Foxj1, Ovgp1 and Pax8. Collectively, these data suggest that tumor cell-intrinsic Nedd9 expression promotes OC development and progression by broad induction of oncogenic protein signaling and stem/mesenchymal gene expression.
Keywords: NEDD9, ovarian carcinoma, metastasis, tumor microenvironment, gene expression
Introduction
Ovarian cancer is the fifth leading cause of cancer mortality and the deadliest gynecologic malignancy1. Most malignant ovarian tumors are epithelial in origin and broadly categorized as ovarian carcinoma (OC). OC is often diagnosed at advanced stages with five year survival below 40%1. The majority of patients respond to standard treatment (aggressive surgery and cytotoxic chemotherapy), but OC typically reemerges as incurable drug-resistant disease. A deeper understanding of the molecular and cellular mechanisms that contribute to disease development and progression are needed for meaningful improvements in patient outcomes.
Extensive molecular analyses of the most common and lethal subtype of OC, high-grade serous carcinoma (HGSC), reveals nearly ubiquitous mutation of TP53, a relatively small subset (10-15%) of cases with mutation of BRCA1 or BRCA2, and high degrees of genomic instability and tumor heterogeneity2–4. Mutation of other oncogenes and tumor suppressors occurs at low frequency, making identification of ‘actionable’ tumor vulnerabilities extremely challenging. Identification of alterations in functional or signaling pathways may be a more tractable way to group tumors by key functions. In this regard, tumor profiling has revealed gene expression signatures that predict outcomes5,6. An early study of advanced HGSC identified a unique signature of upregulated genes associated with regulation of motility and invasion, including NEDD9/HEF1/Cas-L7. NEDD9 is a non-catalytic Crk-associated substrate (CAS) family scaffolding protein that mediates the function of a large number of oncogenic proteins and is of particular interest because of its potential to coordinately regulate a number of metastatic signaling molecules (reviewed in8). Via its wide interactome, NEDD9 is involved in fundamental cellular processes, including adhesion, migration, invasion, cell cycle control and survival (reviewed in8). Studies performed in solid tumors, leukemias and lymphomas indicate NEDD9 typically promotes tumor growth, but in some tumor types is tumor suppressive (reviewed in8). The role of NEDD9 in OC is relatively unexplored, but two studies suggest a tumor-promoting role, with one study identifying elevated NEDD9 as part of a gene expression signature associated with advanced stage HGSC7 and a second study finding higher levels of NEDD9 protein expression in invasive OCs9.
To directly test the role of NEDD9 in OC, we employed genetically engineered mouse (GEM) models, including mice with targeted disruption of Nedd910 and a transgenic mouse model of spontaneous OC (MISIIR-TAg)11,12. MISIIR-TAg mice express the oncogenic SV40 large T (TAg) and small t (tag) antigen genes under control of the Müllerian inhibiting substance type II receptor (MISIIR) gene promoter. Expression of TAg results in functional inactivation of tumor suppressor proteins p53, RB and RB family proteins p107 & p130, and tag results in inactivation of PP2A, leading to activation of growth-promoting signaling cascades including PI3K/AKT13. These alterations are consistent with those most commonly identified human HGSCs, which exhibit nearly ubiquitous mutations in TP53 (96%), frequent RB pathway inactivation (67%) and elevated PI3K/AKT signaling (45%)4. Targeted disruption of Nedd9 does not affect fertility or viability, but mice exhibit impaired leukocyte adhesion, motility and trafficking via disruption of integrin and receptor signaling10. Hence, potential roles for NEDD9 in OC could be tumor cell-intrinsic or alternatively, due to its effects on the tumor microenvironment (TME): a point that has been minimally explored for any cancer. Several studies have demonstrated the presence of tumor infiltrating leukocytes in the OC immune microenvironment, with distinct subclasses having tumor-supportive or -inhibitory effects14–16. Thus, NEDD9 could plausibly influence OC development via effects on leukocyte presence and function in the TME. Using newly-established MISIIR-TAg;Nedd9−/− and MISIIR-TAg;Nedd9+/+ murine ovarian carcinoma (MOVCAR) cell lines and a related syngeneic orthotopic OC model17, we sought to parse the effects of tumor cell-intrinsic and TME-associated loss of Nedd9 in promoting OC growth in vivo. Lastly, as NEDD9 itself is transcriptionally upregulated in advanced OC7, we explored the global gene expression profiles of OC tumors isolated from MISIIR-TAg;Nedd9−/− and MISIIR-TAg;Nedd9+/+ mice to determine the effects of Nedd9 loss on gene expression. Together, our results show a tumor-promoting role for NEDD9 that is largely or entirely tumor cell-intrinsic and mediated by increased oncogenic signaling and a prominent shift toward more mesenchymal, stem-like gene expression.
Results
Deletion of Nedd9 delays development of ovarian tumors
To determine the effect of genetic ablation of Nedd9 in OC, transgenic MISIIR-TAg mice11,12 were crossed to Nedd9−/− mice10 and ovarian volume (tumor growth) was monitored and quantified in age-matched female MISIIR-TAg;Nedd9−/− (n=27) and MISIIR-TAg;Nedd9+/+ (n=24) mice by longitudinal in vivo magnetic resonance imaging (MRI)12,18. Tumor initiation begins early in MISIIR-TAg mice12, with TAg positive tumor cells present within normal sized ovaries of 4-8 week old mice (Supplementary Figure S1), and evidence of ovarian enlargement due to the presence of tumor beginning around 12 weeks of age. Thus, mice received baseline scans when the mean age was 11-12 weeks and 2-10 additional scans, with the endpoint defined as the point at which tumor volume or lack of animal wellness met humane criteria for euthanasia. Baseline MRI revealed a small difference in mean ovarian volume in age-matched mice (Fig. 1A and Table 1). Linear mixed-effects models with random intercepts were used to model longitudinal log-transformed volume data from the first three MRI scans, when all 51 mice were alive. Models included fixed effects for time, group and time by group interaction. The interaction of group and time, included to determine if the group effect varied significantly over time, was not statistically significant. In contrast, when the model was fit to the data without the interaction term, a significantly lower mean log-transformed volume was observed for MISIIR-TAg;Nedd9−/− mice compared to MISIIR-TAg;Nedd9+/+ controls at the third scan, when the mice were 15 weeks old (p=0.035, Fig. 1B and Table 1). Tumor growth slopes calculated for each individual mouse over the duration of the study revealed an overall delay in tumor growth rate in MISIIR-TAg;Nedd9−/− mice that closely approached significance (p=0.054, Fig. 1C). Endpoint data including age, final tumor volume, presence of ascites and gross and microscopic evidence of disease spread are summarized in Table 2. Abdominal ascites, a common feature of MISIIR-TAg mice11,18, was detected less frequently in MISIIR-TAg;Nedd9−/− mice (Fig. 1D and Table 2), but the trend did not reach significance. Histology, expression of TAg and common HGSC markers PAX8 or WT1 revealed no obvious genotype-associated differences at study endpoint or at early stage (Fig. 1E and Supplemental Figure S1A).
Figure 1. Spontaneous ovarian tumor growth in MISIIR-TAg;Nedd9+/+ and MISIIR-TAg;Nedd9−/− transgenic mice.
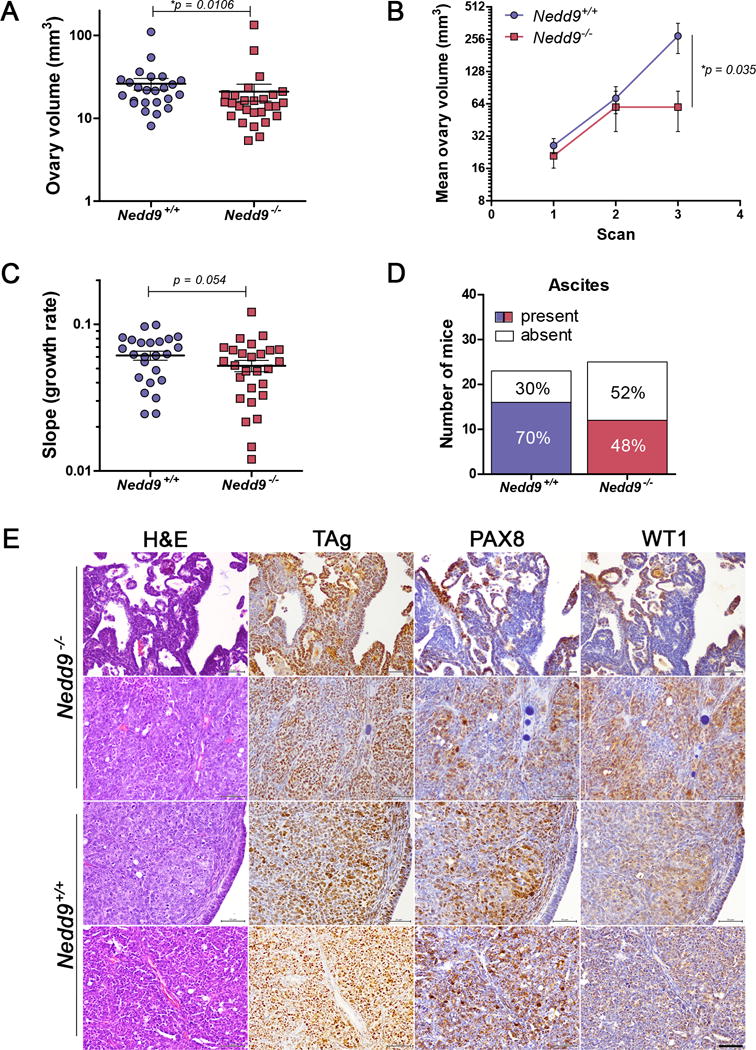
A) Initial ovarian tumor volume determined in baseline MRI scans of age matched female MISIIR-TAg;Nedd9+/+ (n=24, average age 80.96 days) and MISIIR-TAg;Nedd9−/− (n=27, average age 79.3 days) mice shows mean tumor volume is lower in MISIIR-TAg;Nedd9−/− mice. The numbers of randomly selected mice for each genotype were based on the known variability of the spontaneous tumor induction and latency of the MISIIR-TAg model. B) Tumor volumes calculated from the first three consecutive MRI datasets, when all mice in both cohorts were alive and of comparable mean age at each scan. By the third scan, MISIIR-TAg;Nedd9−/− mice show significantly decreased mean log-transformed tumor volume compared to MISIIR-TAg;Nedd9+/+ mice at comparable ages (n=24, average age 111.9 days vs. n=27 mice, average age 109.7 days). C) Longitudinal MRI datasets from all MISIIR-TAg;Nedd9+/+ (n=24) and MISIIR-TAg;Nedd9−/− (n=27) mice were used to determine the ovarian tumor volume at each scan for all mice on study. The growth rate (log-transformed growth slope) was calculated for each individual mouse and showed a delay in MISIIR-TAg;Nedd9−/− mice (mean slope=0.0521) compared to MISIIR-TAg;Nedd9+/+ controls (mean slope=0.0612). D) The number of mice with ascites detected at necropsy was reduced in the MISIIR-TAg;Nedd9−/− compared to MISIIR-TAg;Nedd9+/+ mice. E) Ovarian tumors from MISIIR-TAg;Nedd9−/− and MISIIR-TAg;Nedd9+/+ mice collected at study endpoint and paraffin embedded sections were stained with H&E, TAg, PAX8 and WT1. Representative images from tissues isolated from MISIIR-TAg;Nedd9−/− mice #129 (163 days) and 195 (204 days) and MISIIR-TAg;Nedd9+/+ mice #7509 (135 days) and 7431 (181 days) are shown (scale bar=50μm). Ovarian volumes at baseline scans were analyzed by the nonparametric two-tailed Wilcoxon-Mann-Whitney test. Tumor volume data were analyzed using linear mixed-effects models with random intercepts to model longitudinal log-transformed volume data from the first three time points. Individual tumor growth rates throughout the study were analyzed by the two-tailed Wilcoxon signed-rank test. Results are shown as mean ± s.e.m. Investigators were blinded to the genotype of the mice for MRI imaging and analysis of tumor volume.
Table 1.
Average age and tumor volume during early tumor development and at last scan in Nedd9+/+ and Nedd9−/− mice.
| Genotype | Baseline scan | 2nd scan | 3rd scan | FINAL Scan | ||||
|---|---|---|---|---|---|---|---|---|
| Mean age (days ± SD) |
Mean tumor volume (mm3± SD) |
Mean age (days ± SD) |
Mean tumor volume (mm3± SD) |
Mean age (days ± SD) | Mean tumor volume (mm3± SD) |
Mean age (days ± SD) | Mean tumor volume (mm3± SD) |
|
| MISIIR-TAg;Nedd9+/+ | 80.96 ± 17.34 | 26.16 ± 20.48 | 96.42 ± 16.71 | 68.90 ± 101.1 | 111.9 ± 18.39 | 274.5 ± 421.0 | 157.2 ± 23.3 | 1440.4 ± 617.8 |
| MISIIR-TAg;Nedd9−/− | 79.30 ± 12.88 | 21.00 ± 25.34 | 95.74 ± 16.55 | 61.34 ± 128.0 | 109.7 ± 16.59 | 135.1 ± 283.4 | 162.2 ± 24.0 | 1309.9 ± 843.5 |
Table 2.
Summary of endpoint data from longitudinal study of TgMISIIR-TAg;Nedd9+/+ and TgMISIIR-TAg;Nedd9−/− mice.
| MISIIR-TAg;Nedd9−/− mouse ID | Age at death (days) | Ovary (tumor) volume* | Ascites | Visible spread | Microscopic spread |
|---|---|---|---|---|---|
| 43 | 145 | 756.1 | − | + | + |
| 49 | 153 | 946.0 | − | − | + |
| 55 | 148 | 1208.9 | − | − | + |
| 63A | 182 | 1581.6 | + | + | + |
| 70 | 192 | 2284.1 | + | + | + |
| 94 | 130 | 122.7 | − | − | + |
| 115 | 141 | 691.4 | + | + | + |
| 128 | 151 | 35.4 | − | + | + |
| 129 | 163 | 1843.6 | + | + | + |
| 136 | 138 | 1255.0 | − | + | + |
| 143 | 188 | 730.3 | − | + | + |
| 144 | 163 | 1765.5 | + | + | + |
| 145 | 188 | 1889.7 | + | + | + |
| 146 | 193 | 530.5 | + | − | + |
| 156 | 138 | 1866.8 | + | + | + |
| 157 | 150 | 181.9 | n/a | n/a | n/a |
| 158 | 200 | 1748.6 | − | − | + |
| 159 | 158 | 1289.0 | − | + | + |
| 166 | 138 | 1680.5 | + | + | + |
| 168 | 150 | 1912.2 | − | + | + |
| 173 | 118 | 2563.2 | − | − | + |
| 192 | 183 | 2717.3 | + | − | + |
| 194 | 191 | 978.2 | + | + | + |
| 195 | 204 | 840.5 | − | − | + |
| 197 | 178 | 25.0 | − | − | + |
| 210 | 156 | 33.9 | n/a | n/a | n/a |
| 212 | 175 | 2870.9 | + | + | + |
|
| |||||
| MISIIR-TAg;Nedd9+/+ mouse ID | Age at death (days) | Ovary (tumor) volume | Ascites | Visible spread | Microscopic spread |
|
| |||||
| 7272 | 155 | 1237.0 | no | + | + |
| 7283 | 175 | 1615.8 | − | − | + |
| 7286 | 158 | 642.6 | − | − | + |
| 7429 | 186 | 947.4 | + | + | + |
| 7431 | 181 | 1635.0 | + | + | + |
| 7452 | 170 | 1015.1 | − | − | + |
| 7473 | 159 | 3125.8 | + | + | + |
| 7494 | 168 | 783.1 | − | + | + |
| 7496 | 128 | 33.3 | + | + | + |
| 7507 | 149 | 1527.9 | − | + | + |
| 7509 | 135 | 1921.6 | − | + | + |
| 7510 | 128 | 658.2 | + | + | + |
| 7516 | 209 | 2267.0 | + | + | + |
| 7518 | 136 | 1555.9 | + | − | + |
| 7523 | 212 | 426.4 | + | + | + |
| 7524 | 154 | 2225.6 | n/a | n/a | n/a |
| 7540 | 146 | 957.8 | − | − | + |
| 7547 | 157 | 1973.7 | − | + | + |
| 7549 | 183 | 1577.9 | + | + | + |
| 7555 | 132 | 1853.4 | − | + | + |
| 7575 | 137 | 889.3 | − | − | + |
| 7577 | 207 | 1356.4 | + | − | + |
| 7793 | 134 | 1424.4 | + | + | + |
| 7859 | 140 | 1227.3 | − | − | + |
| 7865 | 149 | 1725.9 | − | − | + |
| 7272 | 155 | 1237.0 | n/a | n/a | n/a |
| 7283 | 175 | 1615.8 | + | + | + |
| 7286 | 158 | 642.6 | − | + | + |
| 7429 | 186 | 947.4 | − | − | + |
| 7431 | 181 | 1635.0 | − | − | + |
measurement at last MRI scan
n/a = tissue not available for evaluation
There were no genotype-specific differences in tumor dissemination. All mice exhibited spread of tumor cells to omentum and over half showed evidence of infiltration of lung; invasive lesions in kidney, liver, intestine or pancreas were rare events in either genotype (Supplementary Tables S1 & S2, and Supplementary Fig. S1B&C). Similarly, no genotype-specific differences in the presence or extent of early lesions were observed; all young mice (4, 8, and 12 weeks old) exhibited oviductal (the murine equivalent of fallopian tube) epithelial atypia that increased in severity with age, and was similar to tubal intraepithelial carcinoma (TIC) and tubal carcinoma (TC) lesions detected in women with HGSC. There was an age-dependent increase in the presence of TAg+ tumor cells in the ovaries, and TAg+ tumor cells were occasionally observed in the hilum, the ovarian surface epithelium, and the epithelial cells lining paraovarian and/or ovarian inclusion cysts (Supplementary Table S3 and Supplementary Fig. S2). Similarly, an age-dependent increase in TAg and Nedd9 gene expression was observed in MISIIR-TAg mice (Supplementary Fig. S3C). Together, these results show that loss of Nedd9 delays early tumor growth rate and may limit the development of ascites, but suggest that compensatory mechanisms may overcome Nedd9 loss in this aggressive mouse model of OC.
Loss of Nedd9 disrupts oncogenic signaling in ovarian tumors
To understand the mechanisms that contribute to the delayed tumor growth in MISIIR-TAg;Nedd9−/− mice, expression and activation of a large panel of known NEDD9 interacting proteins were evaluated in tumor lysates of both genotypes (Fig. 2, Supplementary Fig. S4). Src activation was diminished in tumors from MISIIR-TAg;Nedd9−/− mice, as evidenced by the reduction of the activated (pSrcY416) and increase in auto-inhibited (pSrcY527) forms of Src. Levels of activated pFAKY397 and pFAKY861 were reduced in some cases, but this was less consistent (Supplementary Fig. S4). STAT3 activation (pSTAT3Y705), often elevated in aggressive OCs, was reduced in tumors from MISIIR-TAg;Nedd9−/− mice (Fig. 2). Tumors from MISIIR-TAg;Nedd9−/− mice also exhibited reduced levels of AURKA and E-cadherin (Fig. 2), proteins that support OC proliferation and survival19,20. In contrast, AKT, ERK, and N-cadherin showed no consistent differences in patterns of expression and/or activation (Supplementary Fig. S4). Induction of compensatory signaling mechanisms may support tumor growth in mice with deletion of Nedd9. An obvious candidate is p130Cas/BCAR1, a structural and functional paralogue of NEDD921; however, similar levels of p130Cas/BCAR1 were observed regardless of genotype (Fig. 2).
Figure 2. Expression and activation of oncogenic signaling in Nedd9 wild type and null ovarian tumors.
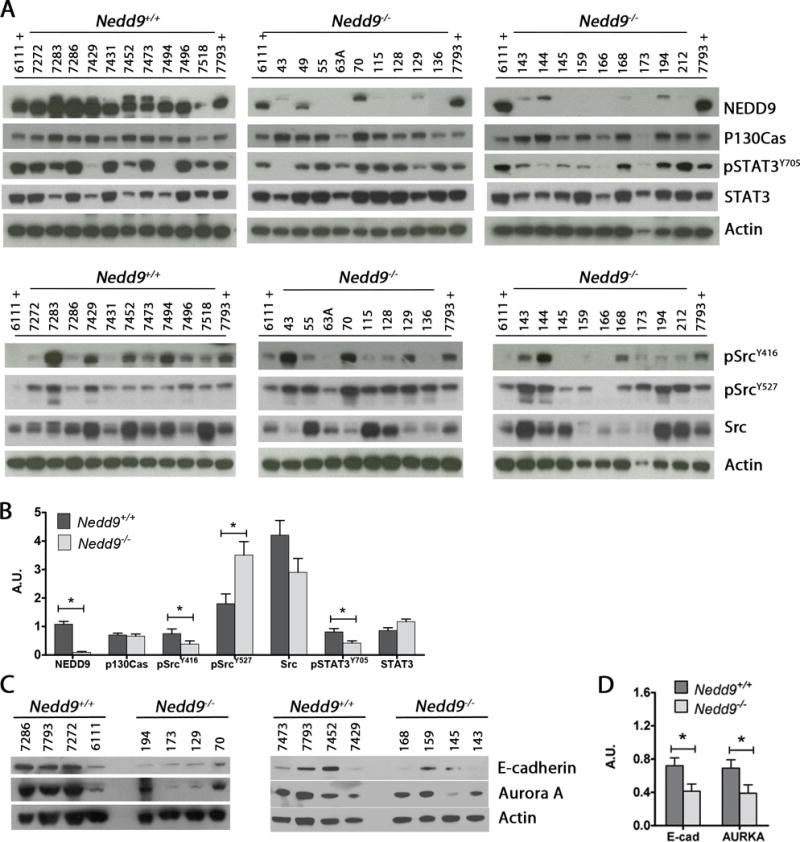
A) Western blot analysis of snap frozen tumor tissue lysates shows the level of expression and/or activation of indicated oncogenic proteins. Samples labeled with ‘+’ are reference specimens loaded on all gels for cross comparison. B) Densitometric analysis of the representative immunoblot images to show NEDD9 (upper band on the NEDD9 blot is non-specific), Src, pSRC, STAT3, pSTAT3, E-Cadherin and AURKA levels relative to β-actin. Representative western blots from 3 experiments (arbitrary number of samples). Densitometric analysis was performed using Image J and analyzed by the nonparametric two-tailed Wilcoxon-Mann-Whitney test. Bars labeled with asterisks are statistically significant (*P<0.05) and results are summarized as mean ± s.e.m.
Loss of Nedd9 alters in vitro migration and invasion of OC cells
To enable further study of the effects of loss of Nedd9 on properties of ovarian oncogenesis, murine ovarian carcinoma (MOVCAR) cell cultures were derived from malignant cells present in ascites collected from mice. Four independent MISIIR-TAg;Nedd9+/+ and MISIIR-TAg;Nedd9−/− tumor cell lines, whose OC origin was validated by TAg and cytokeratin expression and the presence/absence of NEDD9 protein (Supplementary Fig. S5A), were used to evaluate proliferation, adhesion, migration and invasion. No Nedd9 genotype-associated differences were observed in proliferation or in adhesion to type I collagen or fibronectin (Supplementary Fig. S5B-C). Real-time analysis of cell migration through transwell membranes showed an increased mean migration rate of Nedd9−/− cells in the absence of serum, but no differences in the presence of serum (Fig. 3A-B and Supplementary Fig. S5D). Additionally, Nedd9−/− cells invaded through 3D basement membrane significantly faster than controls, regardless of the presence of serum (Fig. 3C-D and Supplementary Fig. S5E). Wound closure and single cell motility analyses confirmed increased migration in Nedd9−/− cells compared to wild type controls (Fig. 3E-F and Supplementary Figs. S5F-H & S6). Depletion of wild type NEDD9 in MOVCAR and human OC cell lines (n=2 independent cell lines for each) showed results consistent with the Nedd9−/− ascites-derived MOVCAR cells, where shRNA-mediated depletion of NEDD9 significantly enhanced wound closure for all cell lines tested (Fig. 3G).
Figure 3. Effects of Nedd9 loss or depletion on in vitro migration and invasion of OC cells.
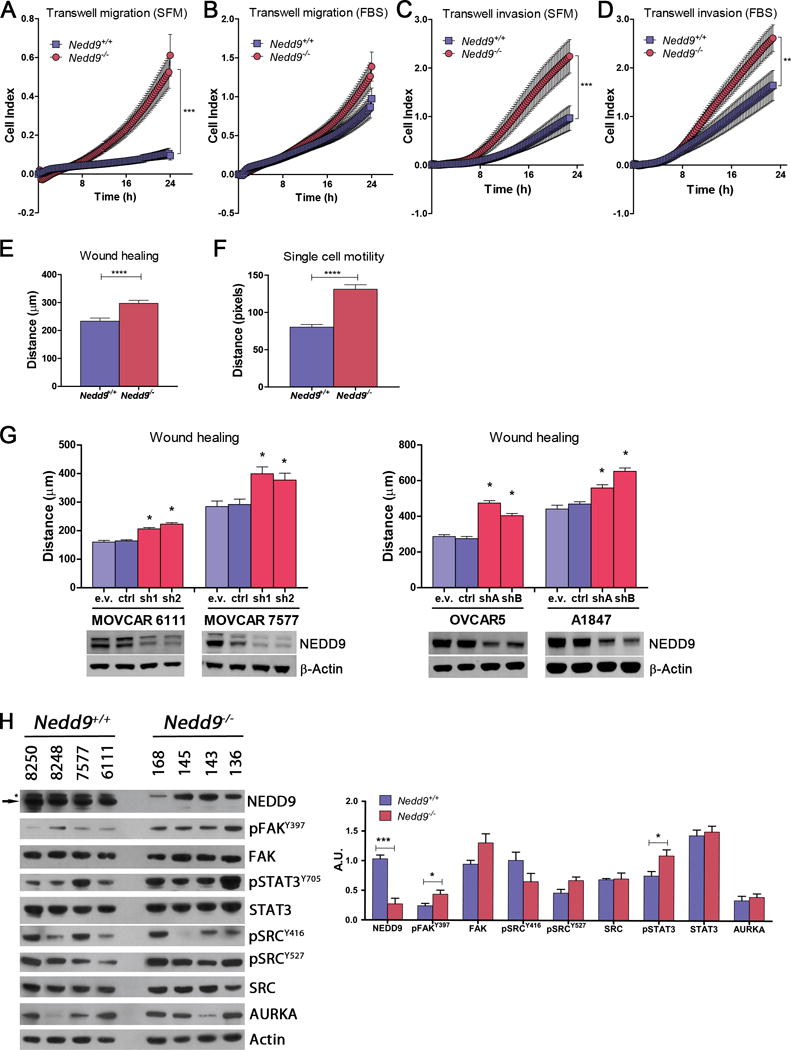
Four independent MOVCAR cell lines established from Nedd9+/+ (MOVCAR-6111, -7577, -8248 and -8250) and Nedd9−/− (MOVCAR-136, -143, -145 and -168) tumor bearing mice were assayed for migration and invasion using transwell migration, transwell invasion, wound healing and single cell motility assays and the mean values are shown for each assay. A) Average transwell migration rate was significantly increased in Nedd9−/− cell lines in the absence serum (SFM). B) No significant differences in transwell migration were detected in the presence of serum. C-D) transwell invasion through matrigel was increased in Nedd9−/− cell lines in the absence (C) and presence (D) of serum. E-F) Wound closure (E) and single cell motility (F) was also significantly increased in Nedd9−/− cell lines. G) Depletion NEDD9 in murine (MOVCAR-6111 and -7577) or human (OVCAR-5 and A1847) ovarian carcinoma cell lines by RNA interference (RNAi) with two independent NEDD9 targeting short hairpin RNAs (shRNA) resulted in more rapid would closure. Depletion of NEDD9 was confirmed by western blotting with anti-NEDD9 antibody and anti-β-Actin antibody as a loading control. All assays were performed in triplicate, analyzed by the nonparametric two-tailed Wilcoxon-Mann-Whitney test and bars labeled with asterisks are statistically significant (*P<0.05, ***P<0.001, ****P<0.0001, n.s. = not significant). H) Expression of a number of proteins interacting with NEDD9 was assessed by Western blot analysis and demonstrated elevation of pFAKY397 and pSTAT3Y705 in Nedd9−/− MOVCAR cells. The anti-NEDD9 antibody recognizes the 105 kDa NEDD9 protein (indicated by the arrow) as well as a non-specific higher molecular weight band (115-120 kDa) that is indicated by an asterisk. Charts represent mean values ± s.e.m.
The enhanced migration and invasion observed in cultured Nedd9−/− cells was somewhat paradoxical in light of the delay in early tumor growth observed in MISIIR-TAg;Nedd9−/− mice in vivo (Fig. 1). Interestingly, analysis of NEDD9 interacting proteins revealed key differences in cultured cells compared to tumors; pFAKY397and pSTAT3Y705 levels were significantly increased in 2D cultured Nedd9−/− cells compared to Nedd9+/+ control cells (Fig. 3H). Activation of these proteins could support the enhanced migration and invasion observed in cells in vitro.
Nedd9 in the microenvironment is not required for OC growth and dissemination
Nedd9−/− mice have defects in migration and trafficking of B and T lymphocytes to secondary lymphoid organs10. To determine if this or other NEDD9-mediated aspects of the TME contribute to OC we compared the effects of Nedd9 loss in the microenvironment on the growth of Nedd9+/+ MOVCAR cells in vivo in an immunocompetent orthotopic allograft model17. Female MISIIR-TAg-low mice do not develop tumors, but are permissive syngeneic hosts for growth of TAg-expressing MOVCAR cells due to low level TAg expression in the oviductal epithelium17. In contrast to the result with the MISIIR-TAg model, ovaries isolated from 4 and 14 week-old MISIIR-TAg-low mice exhibit similar levels of NEDD9 and no detectable TAg expression (Supplementary Fig. S3). MISIIR-TAg-low;Nedd9−/− and MISIIR-TAg-low;Nedd9+/+ mice received unilateral intrabursal implantation of luciferase-expressing Nedd9+/+ MOVCAR-5009-luc cells (Fig. 4A). Longitudinal bioluminescent imaging (BLI) showed no significant difference in luciferase expression from tumors in mice of either genotype (Fig. 4B). Mice from both groups reached humane criteria for euthanasia within the same time-frame (53.1±1.7 days for MISIIR-TAg-low;Nedd9+/+ vs. 52.8±1.9 days for MISIIR-TAg-low;Nedd9−/− mice), and no significant differences in primary tumor volume, the number or volume of tumor nodules, incidence of ascites or tumor histology (Fig. 4C-G). Protein lysates from MOVCAR-5009-luc tumors grown in MISIIR-TAg-low;Nedd9−/− and MISIIR-TAg-low;Nedd9+/+ hosts exhibited no consistent differences in expression or phosphorylation of oncogenic proteins analyzed (data not shown). Finally, detailed analysis of leukocyte populations detected in primary tumors, lymph nodes and peritoneal washes of MISIIR-TAg-low;Nedd9−/− and MISIIR-TAg-low;Nedd9+/+ hosts revealed significantly fewer CD45+NK1.1+CD3e+ natural killer cells in the peritoneal cavities of MISIIR-TAg-low;Nedd9−/− mice, but no significant differences in other leukocyte populations analyzed at any other site (Supplementary Table S4 and Fig. S7). Together, these results strongly suggest that the loss of Nedd9 in the host has no significant impact on leukocyte infiltration of OC tumors, the peritoneal environment, or other aspects of the TME that affect the growth or progression of orthotopic tumors. Thus, differences in spontaneous tumor growth in MISIIR-TAg;Nedd9−/− and MISIIR-TAg;Nedd9+/+ control mice are likely tumor cell-intrinsic.
Figure 4. Loss of Nedd9 in the tumor microenvironment does not impact ovarian tumor growth or dissemination.
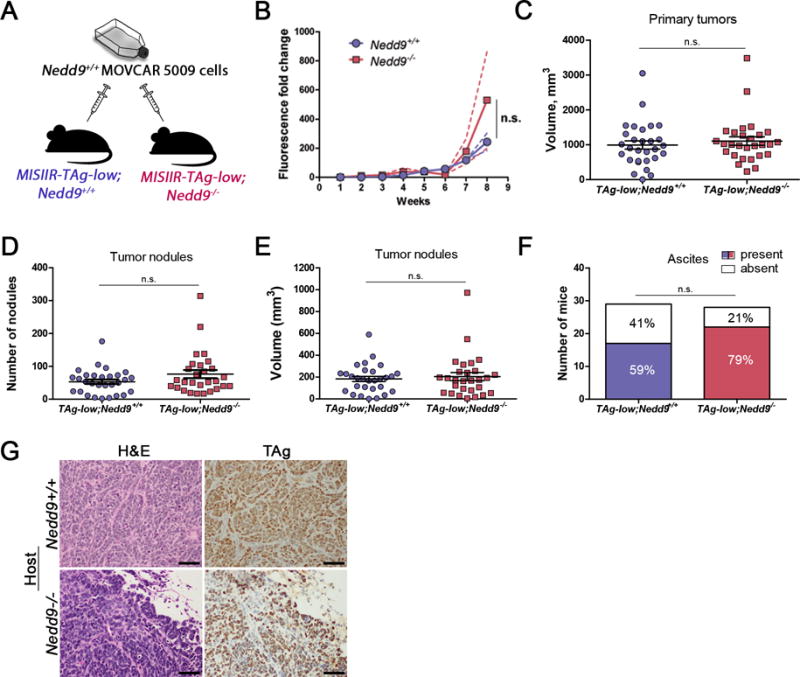
A) Schematic showing the experimental approach to understand the role of NEDD9 expressed in tumor microenvironment on OC development and progression: Nedd9+/+ and Nedd9−/− MISIIR-TAg-low mice were used as hosts for orthotopic (intrabursal) Nedd9+/+ MOVCAR cell allografts. Investigators were not blinded to the genotype of the allograft host. B) Longitudinal bioluminescent imaging (BLI) did not reveal significant effect of Nedd9-status on tumor growth rates. C-F) Necropsy analysis of tumor allografts grown in MISIIR-TAg-low;Nedd9+/+ and MISIIR-TAg-low;Nedd9−/− hosts showed no significant differences in volume of the primary tumor at the injection site (C) or the number (76.5 ± 12.4 versus 53.3 ± 6.7 tumor nodules) (D) or volume (E) of peritoneal tumor nodules or the incidence (F) of ascites detected. Three independent allograft implantation experiments were performed (n=29 MISIIR-TAg-low;Nedd9+/+ mice and n=28 MISIIR-TAg-low;Nedd9−/− mice in total) and results were analyzed by the nonparametric two-tailed Wilcoxon-Mann-Whitney test for significance. MISIIR-TAg-low;Nedd9+/+ and MISIIR-TAg-low;Nedd9−/− hosts mice were randomly selected for allograft implantation. Charts represent mean values ± s.e.m. G) H&E and TAg stained sections of MOVCAR-5009 allografts grown in Nedd9+/+ and Nedd9−/− MISIIR-TAg-low hosts (scale bar=50μm).
Tumor cell-intrinsic Nedd9 expression promotes OC growth and dissemination
To determine whether the aggressive characteristics of Nedd9−/− MOVCAR cells observed in vitro are maintained in vivo, we compared tumor growth and dissemination in TAg-Low;Nedd9+/+ hosts harboring orthotopic allografts of Nedd9−/− and Nedd9+/+ MOVCAR cells (Fig. 5A). Strikingly, unlike hyperaggressive tumor cell lines derived from Nedd9−/− mammary carcinomas22,23, allografts derived from Nedd9−/− cells grew more slowly than those derived from Nedd9+/+ cells (Fig. 5B-E). Moreover, there was a dramatic difference in tumor dissemination in Nedd9−/− mice, as evidenced by highly significant decreases in the average number of tumor nodules, ascites incidence and ascites volume in mice harboring Nedd9−/− compared to Nedd9+/+ controls (Fig. 5G-I). Western blot analysis performed on tumor cell lysates showed decreased expression and/or activation of oncogenic proteins in Nedd9−/− tumors (Fig. 5J), including total and activated FAK and STAT3 and E-cadherin. These results strongly support the initial observations in spontaneous tumors from transgenic mice, demonstrating prominent effects of loss of Nedd9 in OC cells, including delayed tumor growth and decreased tumor dissemination and ascites production.
Figure 5. Loss of Nedd9 in MOVCAR cells impairs OC dissemination.
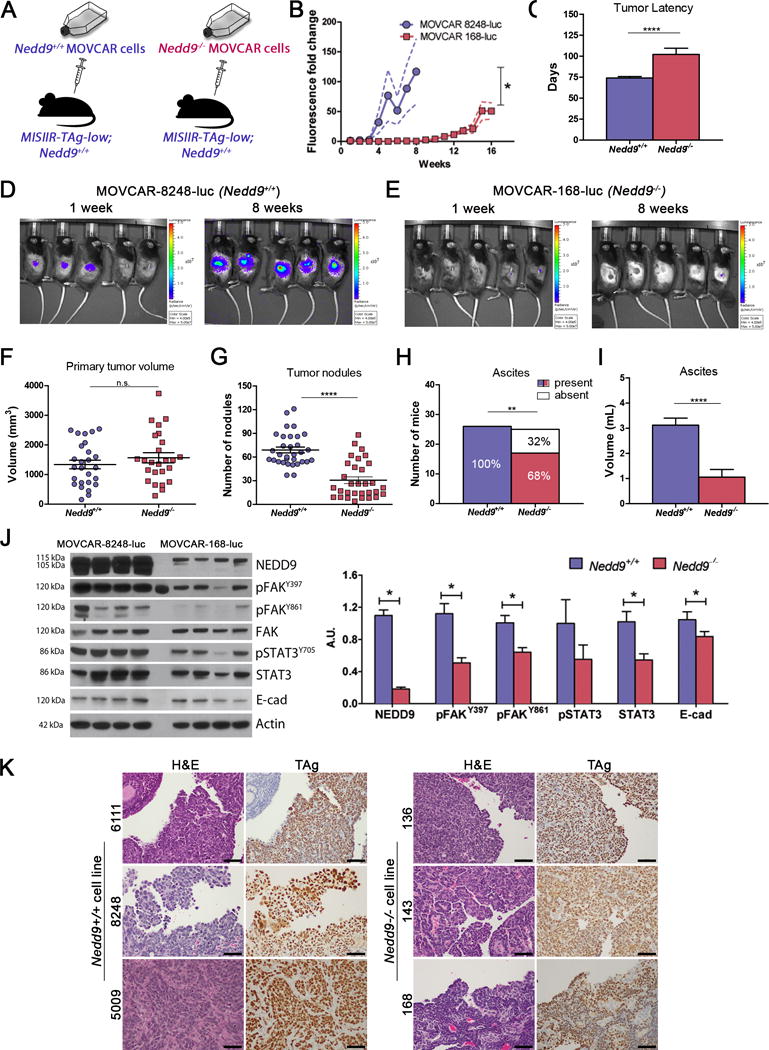
A) Schematic showing the experimental approach to confirm the tumor cell intrinsic role of NEDD9 on OC development and progression: MISIIR-TAg-low;Nedd9+/+ mice selected at random were used as hosts for orthotopic (intrabursal) Nedd9+/+ (MOVCAR-5009, -6111 and -8248) or Nedd9−/− (MOVCAR-136, -143 and -168) cell allografts. Investigators were not blinded to group identity. B) Growth of allografts monitored by weekly BLI showing delayed growth in MOVCAR 168-luc (Nedd9−/−) compared to MOVCAR-8248-luc (Nedd9+/+) allografts. C) Mice were sacrificed when they met humane criteria for euthanasia and showed significantly increased tumor latency of Nedd9−/− allografts compared to Nedd9+/+ allografts (102.2± 7.4 days versus 74.0 ± 1.8 days for controls, p=0.0004). D-E) Representative BLI images showing of MOVCAR-8248 (Nedd9+/+) allografts (D) compared to MOVCAR-168 (Nedd9−/−) controls (E). F) Necropsy analysis of tumor allografts grown in MISIIR-TAg-low;Nedd9+/+ hosts showed no significant difference in primary tumor at the injection site. G) The Nedd9−/− allografts exhibited significantly reduced mean number of peritoneal tumor nodules (30.7 ± 4.2 versus 69.0 ± 3.7, p<0.0001), and H) the incidence (17/25 [68%] versus 26/26 [100%], p<0.01) and I) mean volume (1.06 ml versus 3.12 ml, p<0.0001) of ascites detected. J) Immunoblot analysis of tumor lysates confirmed loss of NEDD9 (upper band is non-specific) and decreased activation of oncogenic signaling proteins FAK, STAT3 and E-cadherin in Nedd9−/− allografts compared to Nedd9+/+ controls. The results represent three independent experiments employing three independent Nedd9+/+ and Nedd9−/− MOVCAR cell lines each (n=10 mice/group/experiment). Data were analyzed by the nonparametric two-tailed Wilcoxon-Mann-Whitney test for significance of tumor latency, tumor and ascites volume, and tumor nodules, and the ascites fraction by the Fisher exact 2-sided test (*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001). Representative western blots from MOVCAR-8248 (Nedd9+/+) and MOVCAR-168 (Nedd9−/−) allograft tumors. Densitometric analysis was performed using Image J and analyzed by the nonparametric two-tailed Wilcoxon-Mann-Whitney test and bars labeled with asterisks are statistically significant (*P<0.05). Charts represent mean ± s.e.m. K) Representative H&E and TAg stained sections of allograft tumors from Nedd9+/+ and Nedd9−/− MOVCAR cells implanted into Nedd9+/+ MISIIR-TAg-low mice show similar morphology and TAg staining (scale bar=50μm).
Loss of Nedd9 alters gene expression
To further investigate the potential mechanisms of NEDD9 action in OC, we performed genome wide mRNA microarray analysis of tumors isolated from MISIIR-TAg;Nedd9+/+ and MISIIR-TAg;Nedd9−/− mice (n = 4 tumors of each genotype). Application of strict criteria of a 2-fold change and a false discovery rate (FDR) of 5% identified a subset of 46 genes as the most differentially regulated between the two groups (Fig. 6A and Supplementary Table S5). Among these, 36 genes were upregulated and 10 genes were downregulated in Nedd9−/− compared to Nedd9+/+ control tumors. Consistent with genotype, Nedd9 was among the most significantly downregulated genes in Nedd9−/− tumors (Fig. 6A). Expression of Nedd9 and a subset of seven genes were independently analyzed by qRT-PCR, validating differential expression for all but Atp2b2 (Fig. 6B and data not shown). Notable among the genes upregulated in Nedd9−/− tumors were Bmpr1b and Foxj1, both of which were previously shown to be downregulated in human OCs24,25. Expression of both genes was increased in individual Nedd9−/− tumors, and elevated protein expression was detected for FOXJ1 (Fig. 6B-C), a member of the forkhead box family of transcription factors.
Figure 6. Gene expression differences in Nedd9+/+ and Nedd9−/− tumors.
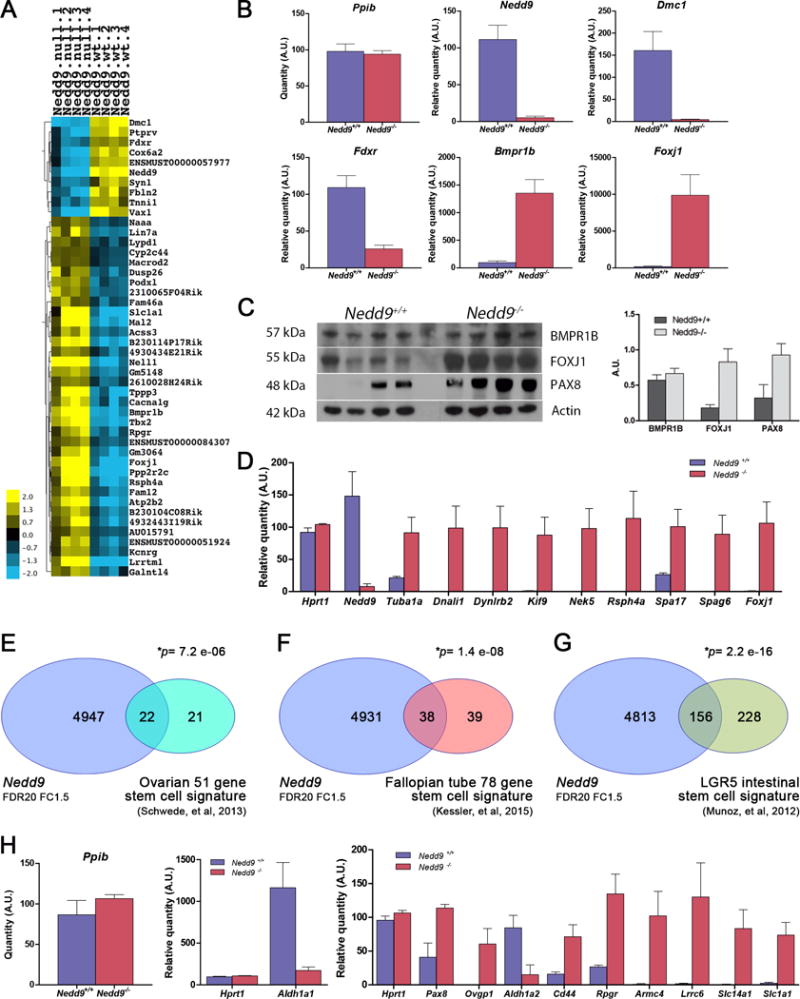
Global gene expression profiles were determined by RNA microarray analysis of Nedd9+/+ and Nedd9−/− tumor specimens, validated by quantitative RT-PCR and compared with publically available data sets. Gene expression data are accessible through Gene Expression Omnibus accession number GSE106911. A) Heat map showing 10 downregulated and 36 upregulated RNA probes in tumors isolated from Nedd9−/− mice compared to Nedd9+/+ controls (2-fold change, p<0.001 and FDR=5). B) Validation of differential gene expression of Nedd9, Dmc1, Fdxr, Bmpr1b and Foxj1 genes in individual tumors by qRT-PCR with expression of Ppib used as a normalizer. C) Western blot detection of expression of BMPR1B, FOXJ1 and PAX8 proteins in individual tumors. D) Validation of upregulation of FOXJ1 target genes regulating ciliogenesis: Tuba1a, Dnali1, Dynlrb2, Kif9, Nek5, Rsph4a, Spa17, Spag6 and Foxj1 in individual Nedd9−/− tumors by qRT-PCR with expression of Ppib and Hprt1 used as normalizer genes. Overlap of differentially expressed genes in the murine Nedd9+/+ and Nedd9−/− gene expression datasets (FDR20, FC1.5) to a 51-gene stem cell-like gene signature in human OC (E), a 78-gene stem cell-associated gene signature in fallopian tube (F) and a 384 gene stem cell signature in LGR5+ cells in murine intestinal crypt (G). H) qRT-PCR validation of individual overlapping genes identified from the above datasets in Nedd9+/+ and Nedd9−/− tumors. Ppib and Hprt were used as normalizer genes for each qRT-PCR experiment. Charts represent mean values ± s.e.m.
FOXJ1 is a potent regulator of the transcriptional program controlling the production of motile cilia, and a marker of ciliated cells of the fallopian tube epithelium (FTE)26. Upregulation of Foxj1 in tumors pointed to tissue-specific gene expression related to ciliated FTE differentiation. Consistent with this, analysis of the data set with more relaxed statistical cutoff (FDR<20%, 4-fold change cutoff, Supplementary Table S6) via the Gene Ontology (GO) database showed significant enrichment of genes with biological processes involved in the assembly and function of motile cilia, including axoneme and inner and outer dynein arm assembly, cilium assembly, movement, organization, morphogenesis and beat frequency (Supplementary Table S7). Differential expression of Foxj1 and eight known FOXJ1 transcriptional targets associated with motile ciliogenesis27, six of which were present in our extended list, was independently validated by qRT-PCR (Fig. 6D). Interestingly, analysis of the dataset also revealed significantly increased expression of Pax8 and Ovgp1, genes characteristically expressed in secretory FTE cells, the putative cell of origin of human HGSCs28–31 (Fig. 6C, D&H). Examination of normal oviducts in Nedd9−/− and Nedd9+/+ mice showed no apparent differences in the proportions of ciliated or secretory FTE (Supplementary Fig. S8). Therefore, the overexpression of several FTE-specific genes in Nedd9−/− tumors suggests that in the absence of Nedd9, ovarian tumors may exhibit gene expression profiles consistent with a more differentiated phenotype.
Recent work using Gene Set Enrichment Analysis (GSEA) identified a ‘stem cell-like’ gene expression signature in poorly differentiated aggressive tumors that is highly correlated with genes expressed in embryonic stem cells32. A subsequent study using this methodology defined a 51-gene ‘stem cell-like’ signature in HGSC33. Comparison of differentially expressed genes in the Nedd9−/− vs. Nedd9+/+ dataset (4969 significant genes (FDR20, FC1.5) among the total 24,345 known genes on the array) to the 51-gene stem cell-like gene signature showed significant overlap (Fisher’s exact test). Among the 51-gene human signature, a total of 43 homologous murine probes were identified, with 22/43 (51.1%, p=7.2 e-06) overlapping between the two datasets (Fig. 6E). To further explore the relationship of Nedd9 loss and FTE gene expression, we similarly compared our dataset with a 78-gene stem cell gene signature recently described in human fallopian tube (FT) organoids34. Maintenance of stem cell gene expression in FT organoids is Notch-dependent, with Notch inhibition decreasing expression of stem cell genes and increasing expression of genes associated with differentiation, including FOXJ134. Comparison of the murine Nedd9 dataset with the FT stem cell signature showed very significant overlap, with 38/77 (49.4%, p=1.4 e-08) homologous probes overlapping (Fig. 6F).
The 78-gene stem cell-associated signature for FT organoids was defined by identifying the subset of genes significantly enriched within a 384 gene stem cell signature previously defined in LGR5+ cells in murine intestinal crypt35. We also compared the overlap between the Nedd9 dataset and the murine LGR5+ intestinal crypt stem cell signature, and again found highly significant overlap of 156/384 (40.6%, p=2.2 e-16) genes (Fig. 6G). A subset of overlapping genes identified in these datasets, selected based on their known expression in FTE (Pax8 and Ovgp1), putative role as OC stem cell markers (Aldh1a1, Aldh1a2 and CD44), or presence in the OC stem cell signature (Slc14a1 and Scl1a1) or fallopian tube stem cell signature (Armc4 and Lrrc6), were validated by qRT-PCR (Fig. 6H). Together, these comparisons support the idea that loss of Nedd9 results in a shift in transcriptional program from a less differentiated more mesenchymal ‘stem cell-like’ profile to a more differentiated profile of gene expression in murine OC. These data also demonstrate relationship of gene expression profiles between murine MISIIR-TAg OCs and fallopian tube, a preferred site of origin of human HGSCs30.
Discussion
Elevated NEDD9 expression has been identified as a biomarker of aggressive human tumors, including melanoma, GBM, breast and lung cancers36–39 (and reviewed in8). While NEDD9 has generally been identified as a promoter of cancer cell aggressiveness, one study reported downregulation of NEDD9 as part of a gene expression signature predicting breast cancer cell metastasis to lung40. In a p210Bcr/Abl mouse model of chronic myelogenous leukemia (CML), loss of Nedd9 accelerated the development of CML/myeloproliferative neoplasm, increased infiltration of myeloid cells in several tissues and decreased overall survival41. These disparate observations suggest that the role of NEDD9 in cancer is likely tissue- and context-dependent. Analysis of OC clinical specimens and TCGA data (Supplementary Fig. S3) suggests overexpression of NEDD9 mRNA and protein is correlated with more advanced cancers and unfavorable prognosis4,42; however to date, the functional role of NEDD9 in OC has not been explored. The results of this study show for the first time the effects of genetic ablation of Nedd9 in an aggressive transgenic model of OC. Our results reveal that loss of Nedd9 did not abrogate tumor development, but resulted in a pronounced delay in tumor growth rate most apparent in young mice. Moreover, the frequency of malignant ascites development was reduced in mice lacking Nedd9. These observations led us to conclude that Nedd9 is not absolutely required for OC, but promotes OC growth and progression.
As Nedd9 has multiple functions, there are multiple avenues by which its loss of function can influence tumorigenesis. First, is via its well-established function as a scaffold protein that activates key oncogenic signaling proteins. In spontaneous tumors or allografts lacking Nedd9, expression or activation of Src, AURKA and STAT3, was reduced, and in Nedd9−/− allografts, FAK activation was also diminished. We and others have shown that each of these proteins support ovarian tumorigenesis18,19,43–46. The novel association between NEDD9 and STAT3 activation we report is consistent with tumor growth inhibition as our prior work demonstrated that targeted blockade of JAK/STAT3 in MISIIR-TAg mice inhibited OC development18. The reduction of E-cadherin in the absence of Nedd9 is different from most other tumor models8 and may seem inconsistent with the gene expression data suggesting a shift from more mesenchymal ‘stem cell-like’ toward more differentiated gene expression; however, in OC the production of malignant ascites and multicellular aggregates, key mechanisms of OC dissemination, are associated with high levels of E-cadherin expression20,47. Thus, reduction of E-cadherin levels in MISIIR-TAg;Nedd9−/− mice and in Nedd9−/− allografts in MISIIR-TAg-Low mice likely underlies the reduced ascites production and dissemination observed.
Interestingly, in mice that are Nedd9−/−, but lack the presence of a driver oncogene, the most notable observed defects were impaired lymphocyte motility and trafficking, with diminished chemotactic migration response of splenic B and T cells to CXCL1210. The importance of CXCL12 signaling axis in OC is well-established48, and analysis of this and other cytokine/chemokine expression in our models merits further study. The presence of tumor infiltrating lymphocytes (TILs) or T regulatory (TReg) cells in patient tumors is correlated with better and worse prognosis, respectively14–16. Notably, we have shown that blockade of JAK/STAT3 signaling in MISIIR-TAg mice inhibited OC development in part by reducing TReg cells in the peritoneal TME18. Thus, it was important to investigate the effects of loss of Nedd9 in the TME. However, our results provide convincing evidence that OC growth and dissemination in this model is not affected by loss of Nedd9 in the TME, instead supporting tumor-cell intrinsic effects.
Our in vivo findings are consistent with the MMTV-PyVmT mammary carcinoma model, where Nedd9 loss delayed tumor onset and decreased tumor multiplicity, but did not abrogate tumor development or progression22. In this model, loss of Nedd9 had prominent effects on NEDD9-regulated signaling proteins including SRC, FAK, Shc and AKT. Tumor-derived cell lines lacking Nedd9 showed increased anchorage-independent growth, 3D growth on matrices, and increased formation of orthotopic allografts and metastatic spread compared to Nedd9+/+ controls suggesting selection of cells with hyperaggressive characteristics23. The increased migration and invasion capacity observed in Nedd9−/− MOVCAR cells might similarly indicate hyperaggressive clonal outgrowths from Nedd9−/− OCs; however, when cells were grown in vivo, tumor latency increased and dissemination was significantly impaired. Thus, in vivo results confirm decreased tumorigenic potential in the absence of Nedd9. The basis for the difference in behavior in vitro and in vivo is unknown, but may be attributable to cell culture conditions49.
Accumulating evidence suggests that phenotypic behaviors (e.g., proliferation, drug response, migration, etc.) of cancer cells grown in 2D culture can be quite different from cells grown in 3D culture as spheroids, 3D organotypic cultures, or as tumors in vivo50. This may be particularly true for OC cells as these cells typically disseminate as clusters. Of critical importance to cell survival in vivo may be alterations in proteins and/or signaling pathways that protect cells from anoikis51. Notably, levels of E-cadherin, activated Src (pSRCY416) and FAK (pFAKY397), known to protect cells from anoikis, are diminished in primary Nedd9−/− tumors and/or Nedd9−/− MOVCAR allografts.
Loss of Nedd9 in the MMTV-Neu mammary cancer model had a more dramatic effect, limiting the incidence of tumors and increasing latency52. Unlike the MMTV-PyVmT or MISIIR-TAg models, there were no consistent effects on NEDD9-regulated signaling proteins/pathways, and also unlike our OC model, there was little difference in gene expression between Nedd9 wild type and null MMTV-Neu tumors22,52. The underlying defect in this model was a reduction in the number of luminal epithelial progenitors. The connection to progenitor cells is intriguing in the context of OC; however, this cannot be adequately studied as consensus regarding the progenitor cell of HGSCs in humans or mice is lacking. Together, observations in MISIIR-TAg and other mouse models lacking Nedd9 underscore the idea that its function is highly tissue- and context-dependent.
NEDD9 is associated with epithelial-to-mesenchymal transition in multiple tumor types (reviewed in8,53). Transcriptional profiling data showed significant differences between Nedd9 null and wild type tumors, with strong overlap of stem-cell-like signatures33,35,54, supporting an more mesenchymal ‘stem-like’ gene expression in Nedd9 wild type tumors and expression of genes associated with a differentiated phenotype in Nedd9 null tumors. Particularly striking was the downregulation of putative ovarian cancer stem cell markers (ALDH1A1, ALDH1A2 and CD44) and upregulation of common markers of FTE differentiation (PAX8, OVGP1 and FOXJ1) as well as a plethora of FOXJ1-regulated genes associated with ciliogenesis. Notably, altered expression of NANOG, a transcription factor required for stem cell renewal and pluripotency, has inverse effects on FOXJ1 expression in OC25. Over the past decade there has been a significant paradigm shift with the recognition that HGSC of the ovary likely arise in the FTE30. The altered expression of stem cell and FTE differentiation markers underscores the connection of NEDD9 in OC in the MISIIR-TAg model and provides in vivo evidence supporting a pro-tumorigenic role for NEDD9 in human HGSC.
Materials and methods
Mouse strains, breeding and tumor induction
Procedures involving mice were approved by the FCCC IACUC. C57BL/6J MISIIR-TAg and MISIIR-TAg-low mouse strains were developed in our laboratory and described previously11,12,17. MISIIR-TAg mice develop spontaneous bilateral ovarian tumors and MISIIR-TAg-low mice express TAg at low levels in the oviduct, do not develop ovarian tumors, and are permissive syngeneic hosts for allograft implantation of murine ovarian carcinoma (MOVCAR) cells isolated from MISIIR-TAg mice17. C57BL/6J Nedd9−/− mice were previously described10. Male MISIIR-TAg and MISIIR-TAg-low mice were crossed with female Nedd9−/− mice to obtain MISIIR-TAg/TAg-low;Nedd9+/− offspring and males were crossed with Nedd9−/− females to generate MISIIR-TAg;Nedd9−/− and MISIIR-TAg-low;Nedd9−/− mice. Tumor allografts were implanted in age-matched (13-15 week-old) MISIIR-TAg-low mice unilateral intrabursal injection of 3×105 luciferase expressing cells as described17,55.
Longitudinal in vivo tumor imaging
In vivo tumor imaging was performed in the FCCC Biological Imaging Facility and growth of spontaneous ovarian tumors in transgenic mice was monitored and quantified by magnetic resonance imaging (MRI) as described12,55. Mice received baseline images starting at approximately 10-12 weeks of age and were imaged biweekly (early stages) or weekly (later stages) for an additional 4-17 weeks. The study endpoint was set at the point at which individual mice reached IACUC-approved humane criteria for euthanasia, typically based on tumor volume or lack of wellness. Growth of orthotopic allografts was monitored by weekly bioluminescent imaging (BLI)17,55 with the same definition of study endpoint.
Tumor collection, tissue preparation and analysis
Mice were euthanized by CO2 inhalation and necropsied. Tumor volume (l × w2× 0.5) and the presence and number of tumor nodules was noted and affected tissues and portions of normal organs were collected and snap frozen or fixed in 10% neutral buffered formalin, paraffin embedded then sectioned for H&E staining and immunohistochemical detection of TAg, PAX8 and WT1 as described11.
Cell lines and culture conditions and antibodies
MOVCAR cell lines were derived from the ascites of MISIIR-TAg;Nedd9+/+ (MOVCAR-5009, -6111, -7577, -8248, -8250) and MISIIR-TAg;Nedd9−/− (MOVCAR-136, -143, -145, -168) mice in our laboratory as described12. Human OC cell lines OVCAR-5 and A1847 OC were obtained from the FCCC Cell Culture Facility (deposited by Dr. Thomas Hamilton) and maintained as described56. Cell lines were periodically checked for mycoplasma contamination OVCAR-5 cells were authenticated by STR profiling. Antibodies and commercial source: antibodies recognizing AKT (cat# 468S), pAKTS473 (cat# 3787S), ERK 1/2 (9102), pERK1/2T202-Y204 (cat# 9106S), Src (cat# 2108S), pSrcY416 (cat# 2101S), pSrcY527 (2105S), STAT3 (cat# 9132) and pSTAT3Y705 (cat# 9145S), pan-keratin (cat# 4545) (Cell Signaling Technology; Danvers, MA, USA), NEDD9 (cat# ab18056) (Abcam; Cambridge, MA, USA), SV40-TAg (cat# sc-147), β-actin (cat# sc-8432), E-cadherin (cat# sc-7870), N-cadherin (cat# sc-7939), p130Cas (cat# sc-860) (Santa Cruz Biotechnology; Santa Cruz, CA, USA), BMPR1B (cat# bs-6639R), and FOXJ1 (cat# 36887) (One World Lab; San Diego, CA, USA), and PAX8 (cat# 10336-I-AP) (Proteintech Group, Rosemont, IL, USA).
Immunoblot, migration and invasion assays
Cell and tumor tissue lysates, protein assays and Western analysis performed as described56. Transwell migration and invasion assays were performed using the xCELLigence system (Roche, Basel, Switzerland) for real-time cell analysis (RTCA) using Roche cellular invasion/migration (CIM)-plates according to manufacturer’s instructions and described in detail in Supplementary Methods.
In vitro knockdown of NEDD9
NEDD9 was depleted by lentiviral transduction of short hairpin RNA constructs targeting human (pGIPZ, GE Dharmacon; Lafayette, CO, USA) or murine (pLKO.1, GE Dharmacon) NEDD9 (shRNA sequences in Supplementary Table S8). Lentivirus was generated using HEK293T cells and Virapower Kit (Invitrogen, now Thermo Fisher Scientific; Waltham, MA, USA) and filtered medium used to infect target cells. Transduced cells were selected with puromycin. Empty vector was used as negative control and shRNAs targeting GAPDH and eGFP were used as non-specific controls for human and mouse lines respectively.
Gene expression analysis and quantitative RT-PCR
Tumor RNA was isolated and submitted to the FCCC Genomics Facility for gene expression analyses by microarray and quantitative RT-PCR as described in detail in18 and Supplementary Methods. Raw and normalized data are accessible through Gene Expression Omnibus accession number GSE106911. Differentially expressed genes were identified based on statistical significance (FDR<0.05) and biological significance using a fold-change cutoff of 2. Fisher’s exact test was used to test whether there is significant association between the murine NEDD9 essential genes from our study to those in previously published datasets33–35. Differential expression of individual genes was validated by qRT-PCR amplification as described18 using Power SYBR Green (Invitrogen, now Thermo Fisher Scientific) and gene specific-probes (Supplementary Table S9) or Taqman technology and probes (Invitrogen, now Thermo Fisher Scientific, Supplementary Table S10) with Ppib and Hprt1 as normalizer genes.
Statistical Analysis
Data were submitted to the Biostatics and Bioinformatics Facility for analysis. Wilcoxon two-sample two-sided tests were used to analyze the in vitro data. Specific analyses are also described in the results and figure legends. Multiple comparisons were not adjusted. For all analyses, p values of <0.05 were considered significant.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Drs. Zeng-jie Yang and David Schlaepfer for helpful discussions; Dr. Eric Ross and Ms. Ludmilla Demora of the FCCC Biostatistics and Bioinformatics Facility for helpful discussions and statistical analysis; Ms. Ellen Neulight for assistance with genotyping transgenic mice; Ms. Jane Miglo for technical assistance; and Mr. Xiang Hua in the Transgenic Mouse Facility for assistance with intrabursal injections. This work was supported by the FCCC Laboratory Animal, High Throughput Screening, Cell Culture, Biosample Repository, Biomedical Imaging, Histopathology, Transgenic Mouse and Biostatistics and Bioinformatics Facilities.
Financial Support: This work was supported by R01 CA136596 (to DCC); R01 CA63366 (to EAG); Ovarian SPORE P50 CA083638, the FCCC Core Grant NCI P30 CA006927, and charitable donations from the Roberta Dubrow Fund, the Teal Tea Foundation, and the Bucks County Board of Associates (to DCC).
Grant Support
This work was supported supported by R01 CA136596 (to DCC), R01 CA63366 (to EAG), and the FCCC Core Grant NCI P30 CA006927 as well as charitable donations (to D.C.C.’s laboratory) from the Roberta Dubrow Fund, the Teal Tea Foundation, the Bucks County Board of Associates and the Main Line Board of Associates.
Footnotes
Conflicts of interest: The authors have no conflicts of interest to declare.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: A Cancer Journal for Clinicians. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Bowtell DD, Bohm S, Ahmed AA, Aspuria PJ, Bast RC, Jr, Beral V, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer. 2015;15(11):668–79. doi: 10.1038/nrc4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McPherson A, Roth A, Laks E, Masud T, Bashashati A, Zhang AW, et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat Genet. 2016;48(7):758–67. doi: 10.1038/ng.3573. [DOI] [PubMed] [Google Scholar]
- 4.TCGA. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonome T, Levine DA, Shih J, Randonovich M, Pise-Masison CA, Bogomolniy F, et al. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res. 2008;68(13):5478–86. doi: 10.1158/0008-5472.CAN-07-6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verhaak RG, Tamayo P, Yang JY, Hubbard D, Zhang H, Creighton CJ, et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J Clin Invest. 2013;123(1):517–25. doi: 10.1172/JCI65833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Donninger H, Bonome T, Radonovich M, Pise-Masison CA, Brady J, Shih JH, et al. Whole genome expression profiling of advance stage papillary serous ovarian cancer reveals activated pathways. Oncogene. 2004;23(49):8065–77. doi: 10.1038/sj.onc.1207959. [DOI] [PubMed] [Google Scholar]
- 8.Shagisultanova E, Gaponova AV, Gabbasov R, Nicolas E, Golemis EA. Preclinical and clinical studies of the NEDD9 scaffold protein in cancer and other diseases. Gene. 2015;567(1):1–11. doi: 10.1016/j.gene.2015.04.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, Mu X, Zhou S, Zhang J, Dai J, Tang L, et al. NEDD9 overexpression is associated with the progression of and an unfavorable prognosis in epithelial ovarian cancer. Hum Pathol. 2014;45(2):401–8. doi: 10.1016/j.humpath.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 10.Seo S, Asai T, Saito T, Suzuki T, Morishita Y, Nakamoto T, et al. Crk-associated substrate lymphocyte type is required for lymphocyte trafficking and marginal zone B cell maintenance. J Immunol. 2005;175(6):3492–501. doi: 10.4049/jimmunol.175.6.3492. [DOI] [PubMed] [Google Scholar]
- 11.Connolly DC, Bao R, Nikitin AY, Stephens KC, Poole TW, Hua X, et al. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res. 2003;63(6):1389–97. [PubMed] [Google Scholar]
- 12.Hensley H, Quinn BA, Wolf RL, Litwin SL, Mabuchi S, Williams SJ, et al. Magnetic Resonance Imaging for Detection and Determination of Tumor Volume in a Genetically Engineered Mouse Model of Ovarian Cancer. Cancer Biol Ther. 2007;6(11) doi: 10.4161/cbt.6.11.4830. [DOI] [PubMed] [Google Scholar]
- 13.Colvin EK, Weir C, Ikin RJ, Hudson AL. SV40 TAg mouse models of cancer. Semin Cell Dev Biol. 2014;27:61–73. doi: 10.1016/j.semcdb.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 14.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 15.Facciabene A, Motz GT, Coukos G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res. 2012;72(9):2162–71. doi: 10.1158/0008-5472.CAN-11-3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. New England Journal of Medicine. 2003;348(3):203–13. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 17.Quinn BA, Xiao F, Bickel L, Martin L, Hua X, Klein-Szanto A, et al. Development of a syngeneic mouse model of epithelial ovarian cancer. J Ovarian Res. 2010;3:24. doi: 10.1186/1757-2215-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gritsina G, Xiao F, O’Brien SW, Gabbasov R, Maglaty MA, Xu RH, et al. Targeted Blockade of JAK/STAT3 Signaling Inhibits Ovarian Carcinoma Growth. Mol Cancer Ther. 2015;14(4):1035–47. doi: 10.1158/1535-7163.MCT-14-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Do TV, Xiao F, Bickel LE, Klein-Szanto AJ, Pathak HB, Hua X, et al. Aurora kinase A mediates epithelial ovarian cancer cell migration and adhesion. Oncogene. 2014;33(5):539–49. doi: 10.1038/onc.2012.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu S, Yang Yn, Dong L, Qiu W, Yang L, Wang X, et al. Construction and characteristics of an E-cadherin-related three-dimensional suspension growth model of ovarian cancer. Scientific reports. 2014;4:5646. doi: 10.1038/srep05646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nikonova AS, Gaponova AV, Kudinov AE, Golemis EA. CAS proteins in health and disease: An update. IUBMB Life. 2014;66(6):387–95. doi: 10.1002/iub.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Izumchenko E, Singh MK, Plotnikova OV, Tikhmyanova N, Little JL, Serebriiskii IG, et al. NEDD9 promotes oncogenic signaling in mammary tumor development. Cancer Res. 2009;69(18):7198–206. doi: 10.1158/0008-5472.CAN-09-0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh MK, Izumchenko E, Klein-Szanto AJ, Egleston BL, Wolfson M, Golemis EA. Enhanced genetic instability and dasatinib sensitivity in mammary tumor cells lacking NEDD9. Cancer Res. 2010;70(21):8907–16. doi: 10.1158/0008-5472.CAN-10-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma Y, Ma L, Guo Q, Zhang S. Expression of bone morphogenetic protein-2 and its receptors in epithelial ovarian cancer and their influence on the prognosis of ovarian cancer patients. J Exp Clin Cancer Res. 2010;29:85. doi: 10.1186/1756-9966-29-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siu MK, Wong ES, Kong DS, Chan HY, Jiang L, Wong OG, et al. Stem cell transcription factor NANOG controls cell migration and invasion via dysregulation of E-cadherin and FoxJ1 and contributes to adverse clinical outcome in ovarian cancers. Oncogene. 2013;32(30):3500–9. doi: 10.1038/onc.2012.363. [DOI] [PubMed] [Google Scholar]
- 26.Okada A, Ohta Y, Brody SL, Watanabe H, Krust A, Chambon P, et al. Role of foxj1 and estrogen receptor alpha in ciliated epithelial cell differentiation of the neonatal oviduct. J Mol Endocrinol. 2004;32(3):615–25. doi: 10.1677/jme.0.0320615. [DOI] [PubMed] [Google Scholar]
- 27.Thomas J, Morle L, Soulavie F, Laurencon A, Sagnol S, Durand B. Transcriptional control of genes involved in ciliogenesis: a first step in making cilia. Biol Cell. 2010;102(9):499–513. doi: 10.1042/BC20100035. [DOI] [PubMed] [Google Scholar]
- 28.Bowen NJ, Logani S, Dickerson EB, Kapa LB, Akhtar M, Benigno BB, et al. Emerging roles for PAX8 in ovarian cancer and endosalpingeal development. Gynecol Oncol. 2007;104(2):331–7. doi: 10.1016/j.ygyno.2006.08.052. [DOI] [PubMed] [Google Scholar]
- 29.Natraj U, Bhatt P, Vanage G, Moodbidri SB. Overexpression of monkey oviductal protein: purification and characterization of recombinant protein and its antibodies. Biol Reprod. 2002;67(6):1897–906. doi: 10.1095/biolreprod67.6.1897. [DOI] [PubMed] [Google Scholar]
- 30.Perets R, Drapkin R. It’s Totally Tubular…Riding The New Wave of Ovarian Cancer Research. Cancer Research. 2016;76(1):10–7. doi: 10.1158/0008-5472.CAN-15-1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu R, Zhai Y, Kuick R, Karnezis AN, Garcia P, Naseem A, et al. Impact of oviductal versus ovarian epithelial cell of origin on ovarian endometrioid carcinoma phenotype in the mouse. J Pathol. 2016;240(3):341–51. doi: 10.1002/path.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40(5):499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwede M, Spentzos D, Bentink S, Hofmann O, Haibe-Kains B, Harrington D, et al. Stem Cell-Like Gene Expression in Ovarian Cancer Predicts Type II Subtype and Prognosis. PLoS One. 2013;8(3):e57799. doi: 10.1371/journal.pone.0057799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kessler M, Hoffmann K, Brinkmann V, Thieck O, Jackisch S, Toelle B, et al. The Notch and Wnt pathways regulate stemness and differentiation in human fallopian tube organoids. Nat Commun. 2015;6:8989. doi: 10.1038/ncomms9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munoz J, Stange DE, Schepers AG, van de Wetering M, Koo BK, Itzkovitz S, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. Embo J. 2012;31(14):3079–91. doi: 10.1038/emboj.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ice RJ, McLaughlin SL, Livengood RH, Culp MV, Eddy ER, Ivanov AV, et al. NEDD9 depletion destabilizes Aurora A kinase and heightens the efficacy of Aurora A inhibitors: implications for treatment of metastatic solid tumors. Cancer Res. 2013;73(10):3168–80. doi: 10.1158/0008-5472.CAN-12-4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448(7155):807–10. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- 38.Kim M, Gans JD, Nogueira C, Wang A, Paik JH, Feng B, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125(7):1269–81. doi: 10.1016/j.cell.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 39.Natarajan M, Stewart JE, Golemis EA, Pugacheva EN, Alexandropoulos K, Cox BD, et al. HEF1 is a necessary and specific downstream effector of FAK that promotes the migration of glioblastoma cells. Oncogene. 2006;25(12):1721–32. doi: 10.1038/sj.onc.1209199. [DOI] [PubMed] [Google Scholar]
- 40.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436(7050):518–24. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seo S, Nakamoto T, Takeshita M, Lu J, Sato T, Suzuki T, et al. Crk-associated substrate lymphocyte type regulates myeloid cell motility and suppresses the progression of leukemia induced by p210Bcr/Abl. Cancer science. 2011;102(12):2109–17. doi: 10.1111/j.1349-7006.2011.02066.x. [DOI] [PubMed] [Google Scholar]
- 42.Wang Z, Shen M, Lu P, Li X, Zhu S, Yue S. NEDD9 may regulate hepatocellular carcinoma cell metastasis by promoting epithelial-mesenchymal-transition and stemness via repressing Smad7. Oncotarget. 2016;8(1) doi: 10.18632/oncotarget.13852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ratushny V, Pathak HB, Beeharry N, Tikhmyanova N, Xiao F, Li T, et al. Dual inhibition of SRC and Aurora kinases induces postmitotic attachment defects and cell death. Oncogene. 2012;31(10):1217–27. doi: 10.1038/onc.2011.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silver DL, Naora H, Liu J, Cheng W, Montell DJ. Activated signal transducer and activator of transcription (STAT) 3: localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004;64(10):3550–8. doi: 10.1158/0008-5472.CAN-03-3959. [DOI] [PubMed] [Google Scholar]
- 45.Ward KK, Tancioni I, Lawson C, Miller NL, Jean C, Chen XL, et al. Inhibition of focal adhesion kinase (FAK) activity prevents anchorage-independent ovarian carcinoma cell growth and tumor progression. Clinical & experimental metastasis. 2013;30(5):579–94. doi: 10.1007/s10585-012-9562-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wiener JR, Windham TC, Estrella VC, Parikh NU, Thall PF, Deavers MT, et al. Activated Src Protein Tyrosine Kinase Is Overexpressed in Late-Stage Human Ovarian Cancers. Gynecologic Oncology. 2003;88(1):73–9. doi: 10.1006/gyno.2002.6851. [DOI] [PubMed] [Google Scholar]
- 47.Sivertsen S, Berner A, Michael CW, Bedrossian C, Davidson B. Cadherin expression in ovarian carcinoma and malignant mesothelioma cell effusions. Acta cytologica. 2006;50(6):603–7. doi: 10.1159/000326027. [DOI] [PubMed] [Google Scholar]
- 48.Kulbe H, Chakravarty P, Leinster DA, Charles KA, Kwong J, Thompson RG, et al. A dynamic inflammatory cytokine network in the human ovarian cancer microenvironment. Cancer Res. 2012;72(1):66–75. doi: 10.1158/0008-5472.CAN-11-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhong J, Baquiran JB, Bonakdar N, Lees J, Ching YW, Pugacheva E, et al. NEDD9 stabilizes focal adhesions, increases binding to the extra-cellular matrix and differentially effects 2D versus 3D cell migration. PLoS One. 2012;7(4):e35058. doi: 10.1371/journal.pone.0035058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fuller E, Howell V. Culture Models to Define Key Mediators of Cancer Matrix Remodeling. Frontiers in Oncology. 2014;4(57) doi: 10.3389/fonc.2014.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buchheit CL, Weigel KJ, Schafer ZT. Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat Rev Cancer. 2014;14(9):632–41. doi: 10.1038/nrc3789. [DOI] [PubMed] [Google Scholar]
- 52.Little JL, Serzhanova V, Izumchenko E, Egleston BL, Parise E, Klein-Szanto AJ, et al. A requirement for Nedd9 in luminal progenitor cells prior to mammary tumorigenesis in MMTV-HER2/ErbB2 mice. Oncogene. 2014;33(4):411–20. doi: 10.1038/onc.2012.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beck TN, Chikwem AJ, Solanki NR, Golemis EA. Bioinformatic approaches to augment study of epithelial-to-mesenchymal transition in lung cancer. Physiological Genomics. 2014;46(19):699–724. doi: 10.1152/physiolgenomics.00062.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paik DY, Janzen DM, Schafenacker AM, Velasco VS, Shung MS, Cheng D, et al. Stem-like epithelial cells are concentrated in the distal end of the fallopian tube: a site for injury and serous cancer initiation. Stem cells. 2012;30(11):2487–97. doi: 10.1002/stem.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Connolly DC, Hensley HH. Xenograft and transgenic mouse models of epithelial ovarian cancer and non-invasive imaging modalities to monitor ovarian tumor growth in situ: applications in evaluating novel therapeutic agents. Curr Protoc Pharmacol. 2009 doi: 10.1002/0471141755.ph1412s45. Chapter 14:Unit14 2. [DOI] [PubMed] [Google Scholar]
- 56.Liu H, Xiao F, Serebriiskii IG, O’Brien SW, Maglaty MA, Astsaturov I, et al. Network Analysis Identifies an HSP90-Central Hub Susceptible in Ovarian Cancer. Clinical Cancer Research. 2013 doi: 10.1158/1078-0432. Published OnlineFirst July 30, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.