

Abstract
E-cadherin is a key component of the adherens junctions that are integral in cell adhesion and maintaining epithelial phenotype of cells. Homophilic E-cadherin binding between cells is important in mediating contact inhibition of proliferation when cells reach confluence. Loss of E-cadherin expression results in loss of contact inhibition and is associated with increased cell motility and advanced stages of cancer. In this review we discuss the role of E-cadherin and its downstream signaling in regulation of contact inhibition and the development and progression of cancer.
Introduction
E-cadherin is a member of the classical family of cadherins. Cadherins have been shown to play an important role in embryonic development and adult tissue homeostasis. These membrane spanning proteins mediate calcium dependent cell adhesion and cell junction formation, with E-cadherin being an integral component of the adherens junctions (AJs) and principal organizer of the epithelial phenotype (1, 2). The cytoplasmic tail of E-cadherin is associated with various catenins (α, β, p120) that link to the cytoskeleton and mediate down-stream signaling effects (3-5). These include the Hippo, Wnt, TGFβ, NF-κB and other growth factor signaling pathways (6-8). The loss of E-cadherin expression is associated with tumor progression and metastasis. Experimental studies show that re-expression of E-cadherin in cancer cells lacking it can prevent tumor progression and invasion making E-cadherin a classic tumor suppressor (9). This is explained in part due to E-cadherin’s adhesive function at the cell surface, which holds cells together, facilitates other cell–cell interactions and physically blocks the movement of cells (10-12). Further, E-cadherin homophilic binding can also lead to contact mediated inhibition of growth through modulation of growth inhibitory signals including the Hippo pathway, growth factor receptor tyrosine kinase (RTK) and Src family kinase signaling pathways (6, 13).
Unlike unicellular organisms, where cell growth and multiplication depend solely on the availability of nutrients in the surrounding environment, multicellular organisms have to sense adjoining cells and control their cell number and size. They have the ability to control cell growth and division mediated by contact between neighboring cells (14). The concept of contact inhibition refers to two different but related phenomena, contact inhibition of proliferation (CIP) and contact inhibition of locomotion (CIL) (15).
CIP refers to the phenomenon where cell proliferation is inhibited by cell density, often attributed to cell–cell contact. The basic properties of this phenomenon were established in the 1960s, along with the observation that such density-dependence of cell proliferation was also lost in transformed cells (14). CIP is important for development of normal differentiated tissues and is tightly regulated for proper tissue morphogenesis and organ development. Under normal circumstances, CIP is overcome in rapidly growing tissues during embryonic development, tissue regeneration, and wound healing. However, uncontrolled growth because of the loss of contact inhibition is a key step in the initiation and progression of several types of cancer. The underlying regulatory mechanisms of the contact inhibition of proliferation remain poorly understood, although cadherin-mediated cell–cell adhesion is thought to play an important role (Figure 1)(16, 17).
Figure 1. E-cadherin mediates contact inhibition of proliferation.
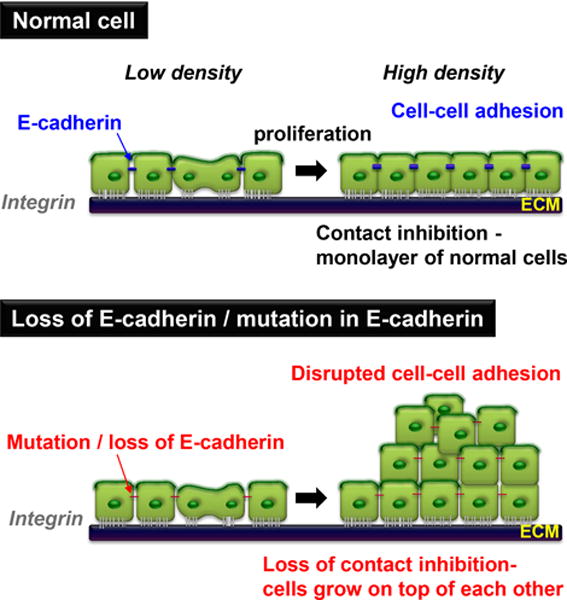
In culture, normal cells stop proliferating once they reach confluence upon homophillic E-cadherin binding, and subsequent formation of tight junctions. This results in mediating growth inhibitory signals and contact inhibition of proliferation (CIP). When cells either lose E-cadherin or E-cadherin is mutated, they continue proliferating, grow on top of each other and lose CIP.
On the other hand, CIL is the process through which cells cease moving after cell-cell contact, and it reduces migration of cells during development and malignant invasion of tumor cells. CIL was first observed by Abercrombie and Heaysman in the 1950s when they discovered that the direction of migration of chick heart fibroblasts was modified by their interaction with other cells (15, 18, 19). Even though E-cadherin has been implicated in regulation of CIL (20), in this review we will focus on the role of E-cadherin in contact inhibition of proliferation and cancer progression.
E-cadherin in contact inhibition of proliferation
Early observations found that membrane fractions slowed proliferation of sub-confluent cells and were important in initiating CIP (21, 22). Cell junction proteins that mediate cell-cell contact and a part of the membrane fractions were thus hypothesized to be key players in this process. Evidence for the role of E-cadherin in contact inhibition was obtained in cell culture when the absence of E-cadherin in cancer cell lines resulted in loss of CIP, which could be reversed by restoring E-cadherin expression (23). Additionally, when cells displayed CIP, disruption of E-cadherin binding between cells by treating them with E-cadherin blocking antibodies led to resumption of cell proliferation (24). The adhesion complex of E-cadherin with the catenins that link it to the actin cytoskeleton is important in this process as lung carcinoma cell lines that lack α-catenin have restored CIP upon its re-expression (25). E-cadherin could either affect CIP indirectly by bringing other cell surface receptors into contact or directly through homophilic ligation and downstream signaling. To demonstrate that the homophilic ligation of E-cadherin receptor was sufficient to regulate cell proliferation, independent of other cell interactions, Perrais et al applied E-cadherin coated beads to cells grown at low confluence and showed that E-cadherin ligation reduced cell proliferation, without affecting apoptosis (26). Recent studies have shown that this is through E-cadherin mediated regulation of the growth inhibitory Hippo signaling pathway which will be discussed in more detail in the following sections (6).
Another feature of CIP is the decrease in stimulation of cell proliferation in response to growth factors. Using micro-patterned substrata, Kim et al showed that the amount of EGF required to induce cell proliferation increased with an increase in cell density. A decrease in cell proliferation was observed in response to EGF treatment when E-cadherin was overexpressed and was reversed when E-cadherin was knocked down (27). This suggests that EGF induced cell proliferation depends upon E-cadherin mediated cell contact rather than just an increase in cell density.
Several mechanisms of how E-cadherin and AJs affect contact inhibition have been proposed. These include sequestration of the growth factor receptors, formation of junctional fences that prevent access to growth factors and alterations in downstream signaling events, and have been covered in several reviews (for review, see (28-31)). The cadherin complex can recruit the core components of the RNA-induced silencing complex (RISC) and multiple messenger RNAs and mature microRNAs via PLEKHA7 (32). They can also regulate Wnt/β-catenin, TGF-β, stem cell signaling and expression of MYC, JUN, and SOX2 mRNAs, thus playing an important role in maintenance of epithelial homeostasis (32). Other studies have unveiled that various mechanical cues, including actomyosin activity, individual cell areas, shape and stiffness, strain and topography of extracellular substrates affect proliferation of confluent epithelial cells (33). Although, it is incompletely understood how E-cadherin ligation and cytoskeletal tension cooperate to achieve contact inhibition of epithelial cell proliferation, many of these mechanical cues entail regulation of the growth inhibitory Hippo pathway discussed below.
The Hippo pathway and contact inhibition
The Hippo-Yap signaling pathway has been implicated in the contact inhibition of growth mediated by E-cadherin and other cell junction proteins (6, 34-37). The Hippo pathway is a highly regulated growth inhibitory pathway involved in organ size control, tissue development and regeneration, as well as in cancer initiation and progression. The core of the Hippo pathway consists of a kinase cascade, in association with several key scaffold proteins, which control YAP stability, nuclear localization, and transcriptional activation of its target genes (Figure 2) (38). Briefly, mammalian serine/threonine protein kinases MST1/2 (STK4/3; homologs of Drosophila Hippo) phosphorylate and activate large tumor suppressor 1/2 (LATS1/2; homologs of Drosophila Warts) which then phosphorylates the transcriptional activators Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ); homologs of Drosophila Yorkie (Yki). Phosphorylation of YAP/TAZ prevents their translocation to the nucleus and in some cell types it stimulates their degradation. Nuclear YAP and TAZ bind to the TEAD transcription factor family (homologs of Drosophila Scalloped [Sd]), and induce expression of a wide range of genes that enhance cell proliferation and survival.
Figure 2. Regulation of the Hippo signaling pathway.
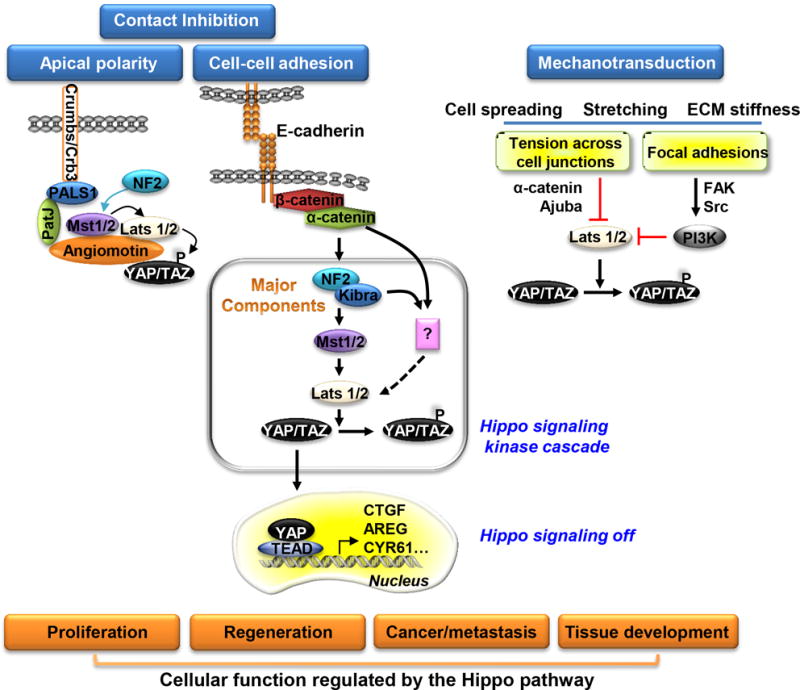
The Hippo signaling pathway is activated upon E-cadherin mediated cell adhesion, formation of tight junctions and apical polarity complexes while mechanical stress inhibits the pathway. Activation leads to growth inhibition upon cell contact. When activated, the pathway components form a complex at junctions where Mst phosphorylates Lats which then phosphorylates YAP. Phosphorylated YAP is retained in the cytoplasm, and cell growth is inhibited. When Hippo signaling is inactivated, the complex dissociates, preventing subsequent phosphorylation of Lats and YAP. YAP then translocates to the nucleus, binds TEADs and activates target gene expression and cell proliferation.
Activation of Hippo signaling by contact inhibition is readily observed as a decrease in the nuclear localization of YAP when cells are grown at high density in the absence of serum or growth factors. A direct role for E-cadherin-dependent cell contacts in the stimulation of the Hippo pathway has been demonstrated in some studies (6, 36). Also α-catenin has been found to regulate Hippo signaling (39-41), presumably due to its interaction with E-cadherin, although cadherin-independent activities of α-catenin have been proposed. E-cadherin stimulation of Hippo signaling depends on NF2/merlin (6, 36), which interacts with cadherins via α-catenin (42). NF2/merlin is a tumor suppressor protein that has long been implicated in contact inhibition of growth via several different mechanisms in addition to regulation of the Hippo-Yap pathway (43, 44). Tight junctions and associated apical polarity complexes also stimulate the Hippo pathway. Crumbs/Crb3 (mammalian Crumbs) and PatJ interact with NF2/merlin and angiomotin to stimulate the pathway (37, 45). E-cadherin-catenin and tight junction/polarity complexes likely function in parallel to stimulate Hippo signaling and mediate contact inhibition, acting to assess the integrity of the epithelial cell layer (34, 46).
The junctional localization of Lats plays an important role in regulation of the Hippo pathway. Lats (Warts) is the key kinase for the regulation of Yap/TAZ/Yorkie in the Hippo pathway; despite its name the Hippo pathway can sometimes be activated by upstream regulators independent of Hippo/Mst (47). Indeed, Lats interacts with several junction-associated proteins, which may control its activity more directly. In Drosophila Warts interacts predominantly with ajuba (Djub) and Expanded (and sometimes merlin) (39, 48), while in mammalian cells its main interactions with the membrane are mediate by NF2/merlin and angiomotins (no Drosophila homolog) (36, 37, 45). In one study, LATS1/2 and MST1/2 were found to be recruited by NF2/merlin and the scaffolding protein SAV1, respectively, to the plasma membrane where LATS1/2 are phosphorylated and activated by MST1/2 (48). In other recent Drosophila studies found that inactive Wts was localized at adherens junctions through interactions with ajuba and α-catenin, and relocation of Wts and Hippo to Crumbs–Expanded sub-apical membrane domain induced Wts phosphorylation and activation (39, 49). In mammals LATS1/2 is recruited to Crb3 at the apical membrane domain via NF2/merlin to induce YAP phosphorylation and thereby control airway cell differentiation (50). Angiomotin interacts with both NF2/merlin and Lats (as well as YAP) and its localization and phosphorylation state regulate the activity of the Hippo pathway in the early mouse embryo, which controls the differentiation of the trophectoderm versus inner cell mass (36). In the non-polarized inner cell mass E-cadherin recruits angiomotin via NF2/merlin to stimulate the pathway and inhibit nuclear accumulation of YAP. On the other hand, in the polarized outer trophectodermal cells, angiomotin is re-localized away from the E-cadherin contacts to the apical membrane, leading to inactivation of Lats and YAP nuclear accumulation. Thus, the assembly of complexes containing Lats at AJs and/or tight junctions mediates activation of the pathway, by both Hippo-dependent and Hippo independent mechanisms.
Aside from YAP phosphorylation, some studies have suggested that YAP is inactivated by physical sequestration at the membrane due to its direct binding to junctional/membrane proteins (40, 51). Although YAP can interact directly with some membrane-associated proteins, including angiomotin, it does not need to be membrane associated to remain out of the nucleus, and most of the nonnuclear YAP is present in the cytosol (6, 52). More likely, the association of YAP with membrane proteins is transient and control of its localization is a catalytic process resulting from its phosphorylation by Lats; and interaction of Lats with specific membrane complexes seems to control its catalytic activity.
Hippo-YAP signaling has also been shown to be regulated by mechanotransduction, responding to cell shape, cytoskeletal integrity, and tension across the cell or tissue, which has generated a great deal of excitement in the field (39, 53-56). Given the roles of cell junctions in the organization of the cytoskeleton and transmission of tension in tissues, this raises an important question about the relationship between contact inhibition and mechanotransduction. Importantly, they act reciprocally, with contact inhibition stimulating the Hippo pathway and reducing nuclear YAP while increased tension and/or actin cytoskeletal assembly leads to increased nuclear YAP. It has been proposed that the state of the actin cytoskeleton could regulate the pathway independent of cell junctions, but little is known about the potential mechanism (53, 54). One study claimed that a change in cell shape per-se is more important for regulation of YAP when cells grow to high density than the role of cell contacts (53). This phenomenon was proposed to regulate nuclear YAP independent of Lats or other Hippo activities; however most other studies have found that the regulation of YAP by the cytoskeleton depends on Lats (39, 55, 57, 58). Another possibility is that the state of the cytoskeleton influences the regulation of Lats by angiomotin, which has been shown to bind to actin filaments (45, 59). However, various cell junctions are major sites of actin cytoskeleton attachment to the membrane and represent the sites at which cytoskeletal tension is exerted, and therefore may be where the Hippo pathway is controlled.
Tension across cell junctions has indeed been shown to stimulate nuclear accumulation of YAP/yorkie via inhibition of Lats/Warts. Increasing tension at adherens junctions in Drosophila epithelia cause recruitment of ajuba to α-catenin associated with E-cadherin, which inhibits Warts activity resulting in inhibition of the pathway (39). A similar process may occur in mammalian epithelia, since stretching the cell monolayer results in YAP nuclear accumulation by a mechanism requiring intact AJs (56). Signaling at integrin-mediated adhesions with the extracellular matrix via Src kinase activation has also been found to stimulate nuclear accumulation of YAP in several systems (13, 60, 61) In one case Src was found to act directly on YAP independent of Lats (61), but in the others Src acted through PI3 kinase to inhibit Lats activity (13, 60), similar to the control of Lats and YAP by growth factor receptor signaling through PI3K (52). Control of YAP by integrin signaling depended on Focal Adhesion Kinase (FAK) (13), which is known to be dependent on tension at focal adhesions, indicating that this mechanism is a form of mechanotransduction.
These findings raise the question of whether stimulation of the Hippo pathway by the formation of cadherin- and tight junction-mediated contacts is mechanistically related to the inhibition of the pathway by actomyosin dependent tension across AJs. Formally, tension induced inhibition of the pathway at junctions could be the predominant signaling process, with high cell density and contact relieving the tension between cells. However, artificial simulation of E-cadherin contacts using E-cadherin coated beads, which presumably have little effect on tension between cells, is able to stimulate the pathway (6). Furthermore, it makes sense that activation of the Hippo pathway activity is required in order for it to become inhibited, leading to YAP nuclear accumulation. Formation of cell contacts may recruit Hippo pathway complexes and stimulate the pathway. Subsequent tension could overcome this process and inhibit the pathway. Whether the pathway inhibition by tension is an independent event or the reversal of pathway activation is unclear. The finding that ajuba recruitment to α-catenin in response to tension across cell junctions acts to inhibit Warts activity (39) suggests that mechanotransduction acts by additional steps. However, ajuba mediated inhibition of Warts at the AJ may act to prevent assembly and activation of the Hippo complex at the Crumbs-Apical polarity membrane (49). More work will be required to elucidate the relationship between these mechanisms.
E-cadherin and Cancer
Contact Inhibition and Cancer
CIP mimics normal tissue homeostasis contributing to organization of cells in normal tissues which is lost during the course of tumorigenesis and leads to abnormal tissue outgrowth. However several cancer cell lines and transformed cells overcome contact inhibition and continue to proliferate when they reach confluence, growing on top of each other and forming clumps unlike in a normally organized cell monolayer (Figure 1) (14). The loss of contact inhibition is considered one of the hallmarks of cancer and disrupts normal signal transduction pathways that result from cell contact and cell-cell interactions (16, 62).
To address whether loss of CIP played a role in tumor progression when E-cadherin was perturbed, Navarro et al re-expressed E-cadherin in cancer cell lines lacking it and observed a decrease in tumor size (9). This suggested that E-cadherin expression in these tumors could inhibit cell proliferation and contribute to CIP. Apart from CIP, loss of E-cadherin has been associated with advanced tumor stages and poor prognosis in patients with cancer. It is clear that E-cadherin and the cell-cell interactions it mediates play an important role in cancer progression and establishment of metastases. However, the molecular factors that relate cell junctions to tumor development and metastasis are still being uncovered. Here we discuss some of the roles of E-cadherin in cancer development and progression.
E-cadherin in cell migration and cancer metastasis
The loss of E-cadherin is a key characteristic of Epithelial to Mesenchymal transition (EMT) during which cells lose their epithelial phenotype and gain a more migratory mesenchymal phenotype (63). To form metastatic tumors, cancer cells must first detach from the primary tumor which can be facilitated by the EMT process (Figure 3). The functional loss of cell adhesion and cell junctions mediated by loss of E-cadherin homophilic binding enables cells to dissociate from the primary tumor, invade surrounding tissues and migrate to distant sites and establish metastatic tumors. Several transcriptional factors, including Snail, Slug, Twist, and Zeb1/2, are important for initiating EMT during embryogenesis, and are commonly mysregulated during cancer development (64). These transcription factors have been shown to directly inhibit E-cadherin gene expression, activate EMT and prevent E-cadherin mediated suppression of tumor cell motility and invasiveness (64). E-cadherin expression can also be altered through accumulation of mutations, loss of heterozygosity and epigenetic regulation of its expression resulting in promoter methylation. Loss of E-cadherin expression can promote tumor cell invasion and metastasis whereas increased expression of E-cadherin has been shown to reverse these phenotypes (65).
Figure 3. E-cadherin in cell migration and establishment of metastasis.
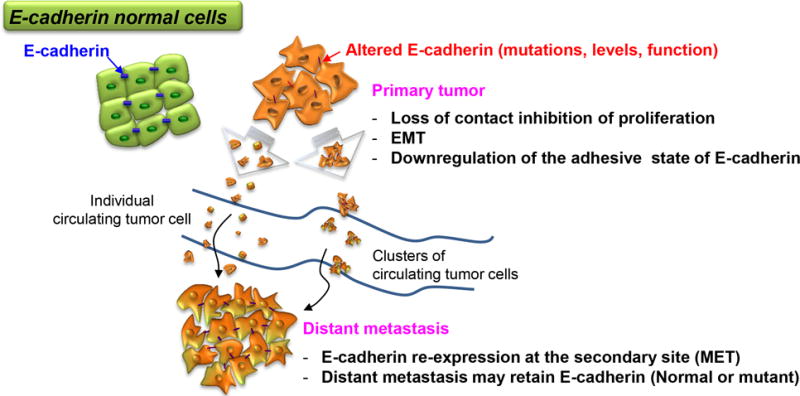
A primary tumor can generate either single cells or clusters that spread through the bloodstream to form distant metastases. Loss of, or mutations in E-cadherin facilitate dispersion of tumor cells through altered cell adhesion or EMT. However some dispersed cells retain E-cadherin expression particularly in clusters that are hypothesized to enhance survival in the blood and at metastatic sites. Alternatively, tumor cells might re-express E-cadherin at distant sites and lead to establishment of metastases. Thus regulation of E-cadherin at multiple levels can contribute to the development of metastatic tumors as discussed in more detail in the text.
EMT was originally thought to be necessary for epithelial cancer cells to acquire their migratory and invasive ability, and facilitate dissemination to distant tissues. However, recent studies have found that E-cadherin is often expressed in metastatic tumor cells (63, 66-68). One explanation is that the reverse phenomenon or Mesenchymal to Epithelial transition (MET) might take place after cells metastasize (67). Re-expression of E-cadherin in this setting is shown to increase binding to cells at the metastatic site and facilitate tumor cell survivability. However some tumors are further characterized by maintenance of a high level of E-cadherin throughout the metastatic process.
This observation that human cancers expressing an abundance of E-cadherin can actually metastasize without undergoing EMT poses the question of how they leave primary tumors. One hypothesis is that E-cadherin expressing tumor cells have the ability to down-regulate the adhesive activity of E-cadherin without affecting its expression at the cell surface. To test this hypothesis, Petrova et al. used tumor cells that retained E-cadherin expression even during metastasis and treated them with E-cadherin activating antibodies that increase E-cadherin adhesive activity on the cell surface (68). They found that stimulating the activity state of E-cadherin on the cell surface led to inhibition of metastatic progression despite high levels of E-cadherin expression. This finding is important as it suggests that E-cadherin can be allosterically regulated at the cell surface and adhesion can be regulated apart from cell surface expression.
The maintenance of E-cadherin expression for stable physical links between cells plays an essential role in this collective migration. These tumor cell clusters retain their epithelial phenotype and have been found to be more effective at establishing metastases than single cells (69, 70). It is suggested that cancer cell clusters can transition between distinct epithelial differentiation states to accomplish the proliferative versus migratory components of metastasis. E-cadherin has been shown to function in a positive feedback loop with Rac and the actin cytoskeleton to stabilize forward-directed protrusion and directionally persistent movement (71, 72). This mechanism could be important in several epithelial cancers that exhibit collective tumor cell invasion in culture.
E-cadherin in tumor initiation and progression
The role of E-cadherin as a tumor suppressor has been ascribed primarily as a result of its loss in EMT and/or regulation during establishment of metastasis. Apart from its role in metastasis, where its loss is associated with increased tumor cell migration and invasion, E-cadherin may also play a role in primary tumor development and progression. Changes in E-cadherin levels, adhesive strength and downstream signaling effects other than complete loss of expression at the cell surface may be important for these processes.
Analysis of the cancer genome atlas (TCGA) database shows that E-cadherin is frequently mutated in several types of cancers. Of particular interest is its role in hereditary diffuse gastric cancer (HDGC). Germline mutations in CDH1 (E-cadherin) are known to be a causal factor for HDGC, with 30% to 50% of all patients harboring such a mutation (73). Studying the HDGC mutations have provided important insights into the mechanisms of cadherin regulation. Though the majority of HDGC mutations are nonsense mutations leading to truncation or absence of the E-cadherin protein, about 20% are missense mutations leading to changes in the protein amino acid sequence. These mutations occur throughout E-cadherin protein including both the extracellular and cytoplasmic domains and mutated E-cadherin is still expressed on the cell surface. Though hypothesized to lack adhesive ability, interfere with E-cadherin calcium binding sites, or increase proteolytic degradation, some of these cancer causing mutations are not in residues known to mediate the homophilic binding. When expressed in tumor cells, mutated E-cadherin was shown to still retain adhesive activity with some differences in migratory behavior of tumor cells. This implies that mutations in E-cadherin can initiate tumor development and metastases through mechanisms other than through loss of cell-cell adhesion. One possibility is that these mutations allosterically regulate E-cadherin signaling analogous to integrin regulation (68, 74-76). This form of surface regulation has been previously shown to be involved in cell rearrangements and tissue morphogenesis in C-cadherin regulation during Xenopus gastrulation and could impact tumor development and progression (77).
E-cadherin signaling in cancer
E-cadherin mediated AJs are hubs of intracellular signaling that regulate cell proliferation, survival, invasion, and migration. Mutations in E-cadherin and the catenins that link it to the cytoskeleton, their protein levels or cellular localization have been implicated in several types of cancer. This could be in part due to alterations in E-cadherin mediated signaling. E-cadherin is involved in several oncogenic pathways including activation of Wnt signaling by nuclear localization of β-catenin, PI3K and MAPK in response to EGF ligands and growth factors, and more recently, the Hippo signaling pathway (Table 1) (4, 6, 28, 52). Here we discuss some of the molecular mechanisms underlying the role of the cadherin-catenin system in the regulation of cell proliferation, invasion, and intracellular signaling during cancer development and progression.
Table 1.
E-cadhein signaling in cancer
| Pathway | Effector molecules | Cancer phenotypes |
|---|---|---|
| Wnt | β-catenin, TCF, GSK | Stem cell phenotype, cell proliferation |
| Hippo | Mst, Lats, NF2, Amot, YAP | Cell proliferation, contact inhibition, anti-apoptosis |
| RTKs, Growth Factors | EGFR, ErbB2, Met, EGF, HGF, SF | EMT, cell motility, cell proliferation |
| GTPases | Rho A, Rac1,Cdc42 | Cell motility, cell proliferation |
Cadherin and regulation of the Wnt signaling pathway
β-catenin is the main nuclear effector of the Wnt signaling pathway. When Wnt signaling is activated, β-catenin is stabilized and translocates to the nucleus where it interacts with TCF to activate target gene expression (Figure 4). Apart from the Wnt pathway, it is important in other aspects of cadherin biology and cell signaling by mediating the functional interaction of cadherin with the actin cytoskeleton. This suggests a relationship between cadherins and Wnt signaling. Studies have shown that increased levels of cadherins on the cell surface can bind β -catenin and sequester it at the membrane, thereby antagonizing Wnt signaling preventing nuclear translocation of β-catenin (78-81). Also, reductions in cadherin levels can release β-catenin bound at the cell surface and enhance nuclear β-catenin signaling events in the presence of Wnt (82). This can be explained by the structural evidence that demonstrated β-catenin uses the same binding interface to interact with both TCF and cadherin ligands. Cadherins have a superior binding affinity to β-catenin and out compete the TCF- β-catenin interaction thereby preventing transcriptional activation of target genes (4). Taken together, these data have led to the common perception that cadherin loss promotes tumorigenesis by effectively releasing membrane-bound β-catenin into the cytosol, hence stimulating canonical Wnt signaling. However, although these experiments demonstrate that cadherin levels can affect β-catenin signaling, the idea that cadherin loss leads to nuclear translocation of free β-catenin and activation of target gene expression is not necessarily true. For example, analysis of breast cancer cell lines with transcriptional silencing of the E-cadherin gene does not support a correlation between loss of E-cad expression and activation of β-catenin signaling (83). According to several reports, gastric and pancreatic cancer cell lines that lack E-cadherin also do not manifest a corresponding upregulation of β-catenin signaling (83, 84). Moreover, depletion of E-cadherin in a mouse model for pancreatic cancer showed no activation of β-catenin signaling with the progression of these tumors (85). These findings suggest that β-catenin released from AJs is degraded under normal conditions with the Axin/APC degradation complex. Additional events, including Wnt activation, compromised proteasomal degradation of β-catenin, its tyrosine phosphorylation, and possibly a release from transcriptional inhibition, may be required to activate β-catenin signaling (10).
Figure 4. E-cadherin mediated regulation of the Wnt signaling pathway.

In the absence of Wnt, β-catenin is regulated by the combination of binding to the cytoplasmic domain of E-cadherin at the adherens junctions or degraded in the cytoplasm by the destruction complex (adenomatosis polyposis coli (APC), Axin, GSK3β). When Wnt signaling is activated, the destruction complex is inhibited, β-catenin is stabilized in the cytoplasm and translocates to the nucleus for activation of target gene expression. Loss of E-cadherin expression can thus free up β-catenin bound at the cell junctions which in the absence of Wnt signaling would likely be degraded by the destruction complex, or in the presence of Wnt enhance nuclear accumulation and target gene expression.
E-Cadherin and Hippo pathway in cancer
The activity of the Hippo pathway is frequently deregulated in several human cancers, though surprisingly the components of the pathway are not frequently mutated. Mutations occur in Merlin (NF2), a bona fide tumor suppressor gene particularly in schwannomas. Also an infrequent but highly penetrant activating mutation in YAP has been associated with lung cancer (86). Inhibition of the Hippo pathway through overexpression of YAP, or its constitutively active 5SA mutant lead to organ overgrowth and tumor development in mouse livers and mammary glands (38, 87). Similarly, altering other mediators of the pathway, such as downregulation of Lats1/2 activity has similar effects on tumor development. As discussed earlier E-cadherin is an important upstream regulator of activation of the Hippo signaling pathway and response to EGF growth factor signaling. E-cadherin re-expression in the MDA-MB-231 breast cancer cells lacking E-cadherin, leads to decreased response to growth factor stimulation, activation of the Hippo signaling pathway and reduced proliferation in these cells (6). In ErbB2/EGFR-transgenic mice, it was observed that YAP accumulated in nuclei of mammary glands, suggesting that EGFR signaling affects YAP in vivo similar to cell culture. The expression of dominant-negative Lats, which inhibits Hippo signaling lead to tumor formation in these ErbB2-transgenic mice, suggesting that Hippo signaling is involved in EGFR-induced mammary tumorigenesis (87).
However, little is known whether loss/mutations in E-cadherin regulate Hippo signaling and affect cancer development and progression. It is possible that some of the cancer promoting effects of loss of E-cadherin result in deregulation of Hippo signaling and nuclear localization of YAP. This could lead to activation of growth promoting and anti-apoptotic genes that allow tumor cells to overcome contact-inhibition, tumor growth and cancer progression. The role of E-cadherin in cell migration could be linked to the activation of the Hippo pathway by altering merlin localization. Recently, Das and colleagues found that during initiation of cell migration, E-cadherin mediated contractile pulling forces across the cell-cell boundary localize cortical merlin to the cytoplasm, facilitate Rac1 activation and lamellipodia formation (88). Survival of tumor cells that lose E-cadherin and detach from the primary tumor may also be affected by Hippo signaling. Inactivation of the Hippo pathway enables individual tumor cells to escape from induction of anoikis due to cytoskeletal re-organization, allowing enhanced survival while circulating in the bloodstream, thereby facilitating metastasis (89).
Cadherins and growth factor signaling
E-cadherin dependent regulation of growth factor signaling is crucial during development and in maintaining tissue homeostasis. In the Drosophila intestine an E-cad–Rho–EGFR cascade couples stem cell division to enterocyte apoptosis (90). E-cadherin adhesion controls secretion of EGF by inhibiting transcription of the EGF maturation factor rhomboid, thereby controlling stem cell proliferation and organ size. E-cadherin mediated tension can localize EGFR signaling for proper polarization of cells in epithelial barriers (91). Thus dysregulation of E-cadherin can lead to tissue dysmorphogenesis through altered growth factor signaling.
The role of E-cadherin on tumorigenesis by modulation of mitogenic signaling was first hypothesized when it was observed that cadherin adhesion resulted in contact inhibition and decreased cell proliferation in the PC9 lung carcinoma cell lines (25). Many epithelial cancers have elevated levels of EGF receptor which is implicated in cell proliferation, invasion and metastasis (92). E-cadherin co-accumulates with EGFR at cell contacts and can physically interact with EGF and other members of the ErbB receptor tyrosine kinase family (93). This is thought to modulate the accessibility of the receptor and inhibit cell responsiveness to EGF stimulation (8). E-cadherin ligation has also been shown to partially inhibit EGFR-mediated growth signaling by preventing the transphosphorylation of Tyr-845 of EGFR by Src family kinases and downstream inhibition of the Hippo pathway (6, 8, 26, 94). E-cadherin can interact with other receptors including Met (HGF receptor) at the AJs and mediate downstream signaling though HGF in breast cancer cells (95). The reduction in cell proliferation characteristic of dense cell cultures is thus mediated in part by cadherin-containing AJs that render the cells insensitive to growth factor stimulation.
While E-cadherin can influence tyrosine kinase signaling, the inverse is also true, growth factor signaling can also influence E-cadherin levels and cell-cell adhesion. Growth factors including EGF, HGF/scatter factor (SF), and FGF have been shown to initiate EMT (64, 96). They disrupt AJs and cause a dramatic switch in cell morphology by altering gene expression in which cells shift from an epithelial to a fibroblastic phenotype. Disruption of AJs by HGF stimulation involves the activation of PI3K, Src, MAPK and β-catenin-TCF transcriptional activity (96). EGF stimulation can lead to inhibition of the Hippo pathway through activation of the PI3K/ PDK pathway (52). These changes increase the migratory and invasive behavior of cells and facilitate tumor progression and metastasis.
Cadherins and Rho GTPases
Another family of signaling molecules that are modulated by E-cadherin binding at the cell surface is the Rho family of small GTPases (Rho, Rac, and Cdc42) (97). In their active GTP-bound form, these Rho GTPases interact with and activate target proteins that regulate actin polymerization, cell motility, and gene expression. They thus play an important role in the assembly and maintenance of the AJs and facilitate remodeling of the actin cytoskeleton in response to growth factors and mechanical stimuli (97). Deregulation of these small GTPases in transformed cells has been shown to interfere with cadherin function and facilitate tumorigenesis. For example, E-cadherin expression in Non-small cell lung cancers alters cell proliferation and migration by reducing levels of RhoA or Cdc42 (98). In Ras-transformed cells that are inefficient in the assembly of AJs, expression of Tiam1, a Rac activator, can restore AJ assembly and epithelial morphology reducing cell migration and invasion (99). Further these Rho GTPases have been implicated in regulation of the Hippo pathway in response to mechanical stress and this crosstalk could further be responsible for E-cadherin mediated tumorigenesis (100).
Conclusions and future perspectives
Much progress has been made in our understanding of the role of E-cadherin in CIP and cancer, yet there is no single answer as to how deregulation E-cadherin leads to tumor development and progression. As discussed above, E-cadherin is important in regulation of contact inhibition of proliferation through regulation of the Hippo signaling pathway. Other factors including cell shape, tension and size at high cell density have also been shown to be important in regulation of the Hippo pathway. Do these processes work together, or one initiates the other and are there ways to prevent inhibition of the pathway still need to be addressed. Loss of E-cadherin expression has been shown to be important in EMT and increased migratory and invasive behavior of tumor cells. But then again, E-cadherin expression was shown to promote collective cell migration and metastasis in several cancer models. Additionally, not much is known about how changes in surface expression of E-cadherin or the HDGC mutations can affect E-cadherin binding and down-stream signaling in cells. Allosteric regulation of the E-cadherin homophilic bond could lead to subtle changes in down-stream signaling effects that could impact tumor progression. Which signaling pathways are important, are they relevant in-vivo and how can we use this information to treat patients with cancer is still the universal question that has yet to be answered. Though we are making progress, there is still a lot to learn.
Acknowledgments
This work has been supported by the grant R01CA207115-02 awarded to Dr. Barry Gumbiner from the National Cancer Institute at the National Institutes of Health.
Footnotes
Conflict of Interest
The authors have no conflict of interest to disclose
Contributor Information
Alisha Mendonsa, Center for Developmental Biology and Regenerative Medicine, Seattle Children’s Research Institute.
Tae-Young Na, Center for Developmental Biology and Regenerative Medicine, Seattle Children’s Research Institute.
Barry M Gumbiner, Center for Developmental Biology and Regenerative Medicine, Seattle Children’s Research Institute; Department of Pediatrics, University of Washington School of Medicine; Department of Biochemistry, University of Washington School of Medicine.
References
- 1.Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nature reviews Molecular cell biology. 2005;6(8):622–34. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 2.Gumbiner B, Stevenson B, Grimaldi A. The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J Cell Biol. 1988;107(4):1575–87. doi: 10.1083/jcb.107.4.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thoreson MA, Anastasiadis PZ, Daniel JM, Ireton RC, Wheelock MJ, Johnson KR, et al. Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J Cell Biol. 2000;148(1):189–202. doi: 10.1083/jcb.148.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gottardi CJ, Gumbiner BM. Adhesion signaling: how beta-catenin interacts with its partners. Current biology : CB. 2001;11(19):R792–4. doi: 10.1016/s0960-9822(01)00473-0. [DOI] [PubMed] [Google Scholar]
- 5.Ozawa M, Baribault H, Kemler R. The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. The EMBO journal. 1989;8(6):1711–7. doi: 10.1002/j.1460-2075.1989.tb03563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim NG, Koh E, Chen X, Gumbiner BM. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc Natl Acad Sci U S A. 2011;108(29):11930–5. doi: 10.1073/pnas.1103345108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Padua D, Massague J. Roles of TGFbeta in metastasis. Cell research. 2009;19(1):89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]
- 8.Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. The EMBO journal. 2004;23(8):1739–48. doi: 10.1038/sj.emboj.7600136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Navarro P, Gomez M, Pizarro A, Gamallo C, Quintanilla M, Cano A. A role for the E-cadherin cell-cell adhesion molecule during tumor progression of mouse epidermal carcinogenesis. J Cell Biol. 1991;115(2):517–33. doi: 10.1083/jcb.115.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene. 2008;27(55):6920–9. doi: 10.1038/onc.2008.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392(6672):190–3. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- 12.Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochimica et biophysica acta. 1994;1198(1):11–26. doi: 10.1016/0304-419x(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 13.Kim NG, Gumbiner BM. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J Cell Biol. 2015;210(3):503–15. doi: 10.1083/jcb.201501025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine EM, Becker Y, Boone CW, Eagle H. Contact inhibition, macromolecular synthesis, and polyribosomes in cultured human diploid fibroblasts. Proc Natl Acad Sci U S A. 1965;53:350–6. doi: 10.1073/pnas.53.2.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ribatti D. A revisited concept: Contact inhibition of growth. From cell biology to malignancy. Experimental cell research. 2017;359(1):17–9. doi: 10.1016/j.yexcr.2017.06.012. [DOI] [PubMed] [Google Scholar]
- 16.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 17.Fagotto F, Gumbiner BM. Cell contact-dependent signaling. Developmental biology. 1996;180(2):445–54. doi: 10.1006/dbio.1996.0318. [DOI] [PubMed] [Google Scholar]
- 18.Abercrombie M, Heaysman JE. Observations on the social behaviour of cells in tissue culture. I. Speed of movement of chick heart fibroblasts in relation to their mutual contacts. Experimental cell research. 1953;5(1):111–31. doi: 10.1016/0014-4827(53)90098-6. [DOI] [PubMed] [Google Scholar]
- 19.Mayor R, Carmona-Fontaine C. Keeping in touch with contact inhibition of locomotion. Trends in cell biology. 2010;20(6):319–28. doi: 10.1016/j.tcb.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scarpa E, Szabo A, Bibonne A, Theveneau E, Parsons M, Mayor R. Cadherin Switch during EMT in Neural Crest Cells Leads to Contact Inhibition of Locomotion via Repolarization of Forces. Dev Cell. 2015;34(4):421–34. doi: 10.1016/j.devcel.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lieberman MA, Glaser L. Density-dependent regulation of cell growth: an example of a cell-cell recognition phenomenon. The Journal of membrane biology. 1981;63(1–2):1–11. doi: 10.1007/BF01969440. [DOI] [PubMed] [Google Scholar]
- 22.Whittenberger B, Glaser L. Inhibition of DNA synthesis in cultures of 3T3 cells by isolated surface membranes. Proc Natl Acad Sci U S A. 1977;74(6):2251–5. doi: 10.1073/pnas.74.6.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.St Croix B, Sheehan C, Rak JW, Florenes VA, Slingerland JM, Kerbel RS. E-Cadherin-dependent growth suppression is mediated by the cyclin-dependent kinase inhibitor p27(KIP1) J Cell Biol. 1998;142(2):557–71. doi: 10.1083/jcb.142.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Motti ML, Califano D, Baldassarre G, Celetti A, Merolla F, Forzati F, et al. Reduced E-cadherin expression contributes to the loss of p27kip1-mediated mechanism of contact inhibition in thyroid anaplastic carcinomas. Carcinogenesis. 2005;26(6):1021–34. doi: 10.1093/carcin/bgi050. [DOI] [PubMed] [Google Scholar]
- 25.Watabe M, Nagafuchi A, Tsukita S, Takeichi M. Induction of polarized cell-cell association and retardation of growth by activation of the E-cadherin-catenin adhesion system in a dispersed carcinoma line. J Cell Biol. 1994;127(1):247–56. doi: 10.1083/jcb.127.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perrais M, Chen X, Perez-Moreno M, Gumbiner BM. E-cadherin homophilic ligation inhibits cell growth and epidermal growth factor receptor signaling independently of other cell interactions. Mol Biol Cell. 2007;18(6):2013–25. doi: 10.1091/mbc.E06-04-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim JH, Kushiro K, Graham NA, Asthagiri AR. Tunable interplay between epidermal growth factor and cell-cell contact governs the spatial dynamics of epithelial growth. Proc Natl Acad Sci U S A. 2009;106(27):11149–53. doi: 10.1073/pnas.0812651106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kourtidis A, Lu R, Pence LJ, Anastasiadis PZ. A central role for cadherin signaling in cancer. Experimental cell research. 2017;358(1):78–85. doi: 10.1016/j.yexcr.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klezovitch O, Vasioukhin V. Cadherin signaling: keeping cells in touch. F1000Res. 2015;4:550. doi: 10.12688/f1000research.6445.1. F1000 Faculty Rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McClatchey AI, Yap AS. Contact inhibition (of proliferation) redux. Curr Opin Cell Biol. 2012;24(5):685–94. doi: 10.1016/j.ceb.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 31.Priya R, Yap AS. Active tension: the role of cadherin adhesion and signaling in generating junctional contractility. Curr Top Dev Biol. 2015;112:65–102. doi: 10.1016/bs.ctdb.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 32.Kourtidis A, Necela B, Lin WH, Lu R, Feathers RW, Asmann YW, et al. Cadherin complexes recruit mRNAs and RISC to regulate epithelial cell signaling. J Cell Biol. 2017;216(10):3073–85. doi: 10.1083/jcb.201612125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirata H, Samsonov M, Sokabe M. Actomyosin contractility provokes contact inhibition in E-cadherin-ligated keratinocytes. Sci Rep. 2017;7:46326. doi: 10.1038/srep46326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gumbiner BM, Kim NG. The Hippo-YAP signaling pathway and contact inhibition of growth. J Cell Sci. 2014;127(Pt 4):709–17. doi: 10.1242/jcs.140103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes Dev. 2013;27(4):355–71. doi: 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirate Y, Hirahara S, Inoue K, Suzuki A, Alarcon VB, Akimoto K, et al. Polarity-dependent distribution of angiomotin localizes hippo signaling in preimplantation embryos. Curr Biol. 2013;23(13):1181–94. doi: 10.1016/j.cub.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yi C, Troutman S, Fera D, Stemmer-Rachamimov A, Avila JL, Christian N, et al. A tight junction-associated Merlin-angiomotin complex mediates Merlin’s regulation of mitogenic signaling and tumor suppressive functions. Cancer Cell. 2011;19(4):527–40. doi: 10.1016/j.ccr.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130(6):1120–33. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rauskolb C, Sun S, Sun G, Pan Y, Irvine KD. Cytoskeletal tension inhibits Hippo signaling through an Ajuba-Warts complex. Cell. 2014;158(1):143–56. doi: 10.1016/j.cell.2014.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, et al. Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell. 2011;144(5):782–95. doi: 10.1016/j.cell.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silvis MR, Kreger BT, Lien WH, Klezovitch O, Rudakova GM, Camargo FD, et al. alpha-catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Sci Signal. 2011;4(174):ra33. doi: 10.1126/scisignal.2001823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gladden AB, Hebert AM, Schneeberger EE, McClatchey AI. The NF2 tumor suppressor, Merlin, regulates epidermal development through the establishment of a junctional polarity complex. Dev Cell. 2010;19(5):727–39. doi: 10.1016/j.devcel.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Curto M, Cole BK, Lallemand D, Liu CH, McClatchey AI. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J Cell Biol. 2007;177(5):893–903. doi: 10.1083/jcb.200703010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McClatchey AI, Yap AS. Contact inhibition (of proliferation) redux. Current opinion in cell biology. 2012;24(5):685–94. doi: 10.1016/j.ceb.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 45.Yi C, Kissil J. Merlin and Angiomotin in Hippo-Yap Signaling. In: Oren M, Aylon Y, editors. The Hippo Signaling Pathway and Cancer. New York: Springer Science+Business Media; 2013. [Google Scholar]
- 46.Yang CC, Graves HK, Moya IM, Tao C, Hamaratoglu F, Gladden AB, et al. Differential regulation of the Hippo pathway by adherens junctions and apical-basal cell polarity modules. Proc Natl Acad Sci U S A. 2015;112(6):1785–90. doi: 10.1073/pnas.1420850112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer cell. 2009;16(5):425–38. doi: 10.1016/j.ccr.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell. 2013;154(6):1342–55. doi: 10.1016/j.cell.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun S, Reddy BV, Irvine KD. Localization of Hippo signalling complexes and Warts activation in vivo. Nat Commun. 2015;6:8402. doi: 10.1038/ncomms9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szymaniak AD, Mahoney JE, Cardoso WV, Varelas X. Crumbs3-Mediated Polarity Directs Airway Epithelial Cell Fate through the Hippo Pathway Effector Yap. Dev Cell. 2015;34(3):283–96. doi: 10.1016/j.devcel.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13(4):246–57. doi: 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- 52.Fan R, Kim NG, Gumbiner BM. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc Natl Acad Sci U S A. 2013;110(7):2569–74. doi: 10.1073/pnas.1216462110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, et al. A Mechanical Checkpoint Controls Multicellular Growth through YAP/TAZ Regulation by Actin-Processing Factors. Cell. 2013;154(5):1047–59. doi: 10.1016/j.cell.2013.07.042. [DOI] [PubMed] [Google Scholar]
- 54.Halder G, Dupont S, Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev Mol Cell Biol. 2012;13(9):591–600. doi: 10.1038/nrm3416. [DOI] [PubMed] [Google Scholar]
- 55.Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012;26(1):54–68. doi: 10.1101/gad.173435.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Benham-Pyle BW, Pruitt BL, Nelson WJ. Cell adhesion. Mechanical strain induces E-cadherin-dependent Yap1 and beta-catenin activation to drive cell cycle entry. Science. 2015;348(6238):1024–7. doi: 10.1126/science.aaa4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sansores-Garcia L, Bossuyt W, Wada K, Yonemura S, Tao C, Sasaki H, et al. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. Embo J. 2011;30(12):2325–35. doi: 10.1038/emboj.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schroeder MC, Halder G. Regulation of the Hippo pathway by cell architecture and mechanical signals. Semin Cell Dev Biol. 2012;23(7):803–11. doi: 10.1016/j.semcdb.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 59.Mana-Capelli S, Paramasivam M, Dutta S, McCollum D. Angiomotins link F-actin architecture to Hippo pathway signaling. Mol Biol Cell. 2014;25(10):1676–85. doi: 10.1091/mbc.E13-11-0701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elbediwy A, Vincent-Mistiaen ZI, Spencer-Dene B, Stone RK, Boeing S, Wculek SK, et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development. 2016;143(10):1674–87. doi: 10.1242/dev.133728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li P, Silvis MR, Honaker Y, Lien WH, Arron ST, Vasioukhin V. alphaE-catenin inhibits a Src-YAP1 oncogenic module that couples tyrosine kinases and the effector of Hippo signaling pathway. Genes Dev. 2016;30(7):798–811. doi: 10.1101/gad.274951.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 63.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14(6):818–29. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 64.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews Molecular cell biology. 2014;15(3):178–96. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berx G, van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harbor perspectives in biology. 2009;1(6):a003129. doi: 10.1101/cshperspect.a003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell. 2017;168(4):670–91. doi: 10.1016/j.cell.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jolly MK, Ware KE, Gilja S, Somarelli JA, Levine H. EMT and MET: necessary or permissive for metastasis? Molecular oncology. 2017;11(7):755–69. doi: 10.1002/1878-0261.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Petrova YI, Schecterson L, Gumbiner BM. Roles for E-cadherin cell surface regulation in cancer. Mol Biol Cell. 2016;27(21):3233–44. doi: 10.1091/mbc.E16-01-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheung KJ, Ewald AJ. A collective route to metastasis: Seeding by tumor cell clusters. Science. 2016;352(6282):167–9. doi: 10.1126/science.aaf6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013;155(7):1639–51. doi: 10.1016/j.cell.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Friedl P, Noble PB, Walton PA, Laird DW, Chauvin PJ, Tabah RJ, et al. Migration of coordinated cell clusters in mesenchymal and epithelial cancer explants in vitro. Cancer research. 1995;55(20):4557–60. [PubMed] [Google Scholar]
- 72.Nabeshima K, Inoue T, Shimao Y, Kataoka H, Koono M. Cohort migration of carcinoma cells: differentiated colorectal carcinoma cells move as coherent cell clusters or sheets. Histology and histopathology. 1999;14(4):1183–97. doi: 10.14670/HH-14.1183. [DOI] [PubMed] [Google Scholar]
- 73.Hansford S, Kaurah P, Li-Chang H, Woo M, Senz J, Pinheiro H, et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA oncology. 2015;1(1):23–32. doi: 10.1001/jamaoncol.2014.168. [DOI] [PubMed] [Google Scholar]
- 74.Maiden SL, Petrova YI, Gumbiner BM. Microtubules Inhibit E-Cadherin Adhesive Activity by Maintaining Phosphorylated p120-Catenin in a Colon Carcinoma Cell Model. PLoS One. 2016;11(2):e0148574. doi: 10.1371/journal.pone.0148574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Petrova YI, Spano MM, Gumbiner BM. Conformational epitopes at cadherin calcium-binding sites and p120-catenin phosphorylation regulate cell adhesion. Mol Biol Cell. 2012;23(11):2092–108. doi: 10.1091/mbc.E11-12-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shashikanth N, Petrova YI, Park S, Chekan J, Maiden S, Spano M, et al. Allosteric Regulation of E-Cadherin Adhesion. J Biol Chem. 2015;290(35):21749–61. doi: 10.1074/jbc.M115.657098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhong Y, Brieher WM, Gumbiner BM. Analysis of C-cadherin regulation during tissue morphogenesis with an activating antibody. J Cell Biol. 1999;144(2):351–9. doi: 10.1083/jcb.144.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Heasman J, Crawford A, Goldstone K, Garner-Hamrick P, Gumbiner B, McCrea P, et al. Overexpression of cadherins and underexpression of beta-catenin inhibit dorsal mesoderm induction in early Xenopus embryos. Cell. 1994;79(5):791–803. doi: 10.1016/0092-8674(94)90069-8. [DOI] [PubMed] [Google Scholar]
- 79.Fagotto F, Funayama N, Gluck U, Gumbiner BM. Binding to cadherins antagonizes the signaling activity of beta-catenin during axis formation in Xenopus. J Cell Biol. 1996;132(6):1105–14. doi: 10.1083/jcb.132.6.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gottardi CJ, Gumbiner BM. Distinct molecular forms of beta-catenin are targeted to adhesive or transcriptional complexes. J Cell Biol. 2004;167(2):339–49. doi: 10.1083/jcb.200402153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gottardi CJ, Wong E, Gumbiner BM. E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J Cell Biol. 2001;153(5):1049–60. doi: 10.1083/jcb.153.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cox RT, Pai LM, Kirkpatrick C, Stein J, Peifer M. Roles of the C terminus of Armadillo in Wingless signaling in Drosophila. Genetics. 1999;153(1):319–32. doi: 10.1093/genetics/153.1.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.van de Wetering M, Barker N, Harkes IC, van der Heyden M, Dijk NJ, Hollestelle A, et al. Mutant E-cadherin breast cancer cells do not display constitutive Wnt signaling. Cancer research. 2001;61(1):278–84. [PubMed] [Google Scholar]
- 84.Caca K, Kolligs FT, Ji X, Hayes M, Qian J, Yahanda A, et al. Beta- and gamma-catenin mutations, but not E-cadherin inactivation, underlie T-cell factor/lymphoid enhancer factor transcriptional deregulation in gastric and pancreatic cancer. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research. 1999;10(6):369–76. [PubMed] [Google Scholar]
- 85.Herzig M, Savarese F, Novatchkova M, Semb H, Christofori G. Tumor progression induced by the loss of E-cadherin independent of beta-catenin/Tcf-mediated Wnt signaling. Oncogene. 2007;26(16):2290–8. doi: 10.1038/sj.onc.1210029. [DOI] [PubMed] [Google Scholar]
- 86.Chen Q, Zhang N, Xie R, Wang W, Cai J, Choi KS, et al. Homeostatic control of Hippo signaling activity revealed by an endogenous activating mutation in YAP. Genes Dev. 2015;29(12):1285–97. doi: 10.1101/gad.264234.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li H, Gumbiner BM. Deregulation of the Hippo pathway in mouse mammary stem cells promotes mammary tumorigenesis. Mammalian genome : official journal of the International Mammalian Genome Society. 2016;27(11–12):556–64. doi: 10.1007/s00335-016-9662-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Das T, Safferling K, Rausch S, Grabe N, Boehm H, Spatz JP. A molecular mechanotransduction pathway regulates collective migration of epithelial cells. Nat Cell Biol. 2015;17(3):276–87. doi: 10.1038/ncb3115. [DOI] [PubMed] [Google Scholar]
- 89.Dupont S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Experimental cell research. 2016;343(1):42–53. doi: 10.1016/j.yexcr.2015.10.034. [DOI] [PubMed] [Google Scholar]
- 90.Liang J, Balachandra S, Ngo S, O’Brien LE. Feedback regulation of steady-state epithelial turnover and organ size. Nature. 2017;548(7669):588–91. doi: 10.1038/nature23678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rubsam M, Mertz AF, Kubo A, Marg S, Jungst C, Goranci-Buzhala G, et al. E-cadherin integrates mechanotransduction and EGFR signaling to control junctional tissue polarization and tight junction positioning. Nat Commun. 2017;8(1):1250. doi: 10.1038/s41467-017-01170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yewale C, Baradia D, Vhora I, Patil S, Misra A. Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials. 2013;34(34):8690–707. doi: 10.1016/j.biomaterials.2013.07.100. [DOI] [PubMed] [Google Scholar]
- 93.Fedor-Chaiken M, Hein PW, Stewart JC, Brackenbury R, Kinch MS. E-cadherin binding modulates EGF receptor activation. Cell communication & adhesion. 2003;10(2):105–18. [PubMed] [Google Scholar]
- 94.Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, et al. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol. 2002;4(3):222–31. doi: 10.1038/ncb758. [DOI] [PubMed] [Google Scholar]
- 95.Ho-Yen CM, Jones JL, Kermorgant S. The clinical and functional significance of c-Met in breast cancer: a review. Breast cancer research : BCR. 2015;17:52. doi: 10.1186/s13058-015-0547-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A. The cadherin-catenin adhesion system in signaling and cancer. The Journal of clinical investigation. 2002;109(8):987–91. doi: 10.1172/JCI15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Citi S, Guerrera D, Spadaro D, Shah J. Epithelial junctions and Rho family GTPases: the zonular signalosome. Small GTPases. 2014;5(4):1–15. doi: 10.4161/21541248.2014.973760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Asnaghi L, Vass WC, Quadri R, Day PM, Qian X, Braverman R, et al. E-cadherin negatively regulates neoplastic growth in non-small cell lung cancer: role of Rho GTPases. Oncogene. 2010;29(19):2760–71. doi: 10.1038/onc.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Malliri A, van Es S, Huveneers S, Collard JG. The Rac exchange factor Tiam1 is required for the establishment and maintenance of cadherin-based adhesions. J Biol Chem. 2004;279(29):30092–8. doi: 10.1074/jbc.M401192200. [DOI] [PubMed] [Google Scholar]
- 100.Shi X, Yin Z, Ling B, Wang L, Liu C, Ruan X, et al. Rho differentially regulates the Hippo pathway by modulating the interaction between Amot and Nf2 in the blastocyst. Development (Cambridge, England) 2017;144(21):3957–67. doi: 10.1242/dev.157917. [DOI] [PubMed] [Google Scholar]