

Abstract
In genome-wide association studies (GWAS) for thousands of phenotypes in large biobanks, most binary traits have substantially fewer cases than controls. Both of the widely used approaches, linear mixed model and the recently proposed logistic mixed model, perform poorly - producing large type I error rates - in the analysis of unbalanced case-control phenotypes. Here we propose a scalable and accurate generalized mixed model association test that uses the saddlepoint approximation to calibrate the distribution of score test statistics. This method, SAIGE, provides accurate p-values even when case-control ratios are extremely unbalanced. It utilizes state-of-art optimization strategies to reduce computational cost, and hence is applicable to GWAS for thousands of phenotypes by large biobanks. Through the analysis of UK Biobank data of 408,961 white British European-ancestry samples for >1400 binary phenotypes, we show that SAIGE can efficiently analyze large sample data, controlling for unbalanced case-control ratios and sample relatedness.
Introduction
Decreases in genotyping cost allow for large biobanks to genotype all participants, enabling genome-wide scale phenome-wide association studies (PheWAS) in hundreds of thousands of samples. In a typical genome-wide PheWAS, GWAS for tens of million variants are performed for thousands of phenotypes constructed from Electronic Health Records (EHR) and/or survey questionnaires from participants in large cohorts1,2. For binary traits based on disease/condition status in PheWAS, cases are typically defined as individuals with specific International Classification of Disease (ICD) codes within the EHR. Controls are usually all participants without the same or other related conditions1,2. Due to the low prevalence of many conditions/diseases, case-control ratios are often unbalanced (case:control=1:10) or extremely unbalanced (case:control<1:100). The scale of data and the unbalanced nature of binary traits pose substantial challenges for genome-wide PheWAS in biobanks.
Population structure and relatedness are major confounders in genetic association studies and also need to be controlled in PheWAS. Linear mixed models (LMM) are widely used to account for these issues in GWAS for both binary and quantitative traits3–8. However, since LMM is not designed to analyze binary traits, it can have inflated type I error rates, especially in the presence of unbalanced case-control ratios. Recently, Chen, H. et al. have proposed to use logistic mixed models and developed a score test called the generalized mixed model association test (GMMAT)9. GMMAT assumes that score test statistics asymptotically follow a Gaussian distribution to estimate asymptotic p-values. Although GMMAT test statistics are more robust than the LMM based approaches, it can also suffer type I error rate inflation when case-control ratios are unbalanced, because unbalanced case-control ratios invalidate asymptotic assumptions of logistic regression10. In addition, since GMMAT requires O(MN2) computation and O(N2) memory space, where M is the number of genetic variants to be tested and N is the number of individuals, it cannot handle data with hundreds of thousands of samples.
Here, we propose a novel method to allow for analysis of very large samples, for binary traits with unbalanced case-control ratios, which also infers and accounts for sample relatedness. Our method, Scalable and Accurate Implementation of GEneralized mixed model (SAIGE), uses the saddlepoint approximation (SPA)11,12 to calibrate unbalanced case-control ratios in score tests based on logistic mixed models. Since SPA uses all the cumulants, and hence all the moments, it is more accurate than using the Gaussian distribution, which uses only the first two moments. Similar to BOLT-LMM8, the large sample size method for linear mixed models, our method utilizes state-of-art optimization strategies, such as the preconditioned conjugate gradient (PCG) approach13 for solving linear systems for large cohorts without requiring a pre-computed genetic relationship matrix (GRM). The overall computation cost of this proposed method is O(MN), which is substantially lower than the computation cost of GMMAT9 and many popular LMM methods, such as GEMMA7. In addition, we reduce the memory use by compactly storing raw genotypes instead of calculating and storing the GRM.
We have demonstrated that SAIGE controls for the inflated type I error rates for binary traits with unbalanced case-control ratios in related samples through simulation and the UK Biobank data of 408,961 white British samples14,15. By evaluating its computation performance, we demonstrate the feasibility of SAIGE for large-scale PheWAS.
RESULTS
Overview of Methods
The SAIGE method contains two main steps: 1. Fitting the null logistic mixed model to estimate variance component and other model parameters. 2. Testing for association between each genetic variant and phenotypes by applying SPA to the score test statistics. Step 1 iteratively estimates the model parameters using the computational efficient average information restricted maximum likelihood (AI-REML) algorithm16, which is also used in GMMAT9. Several optimization strategies have been applied in step 1 to make fitting the null logistic mixed model practical for large data sets, such as the UK Biobank14,15. First, the spectral decomposition has been replaced by the PCG to solve linear systems without inversing the GRM13 (as in BOLT-LMM8). The PCG method iteratively finds solutions of the linear system in a computation and memory-efficient way. Thus, instead of requiring a pre-computed GRM, which costs a significant amount of time to calculate when sample sizes are large, SAIGE uses the raw genotypes as input. The computation time is about O(M1N) times the number of iterations for the conjugate gradient to converge, where M1 is a number of variants to be used for constructing GRM. Second, to further reduce the memory usage during the model fitting, the raw genotypes are stored in a binary vector and elements of GRM are calculated when needed rather than being stored, so the memory usage is M1N/4 bytes (as in BOLT-LMM8 and GenABEL17). For example, for the UK Biobank data with M1 = 93,511 and N = 408,961 (white British participants), the memory usage drops from 669 Gigabytes(Gb) for storing the GRM with float numbers to 9.56 Gb for the raw genotypes in a binary vector.
After fitting the null logistic mixed model, the estimate of the random effects for each individual is obtained. The ratio of the variances of the score statistics with and without incorporating the variance components for the random effects is calculated using a subset of randomly selected genetic variants, similar to BOLT-LMM8 and GRAMMAR-Gamma18. This ratio has been previously suggested to be constant for score tests based on LMMs18. We have shown that the ratio is also approximately constant for all genetic variants with MAC ≥ 20 in the scenario of the logistic mixed models through analytic derivation and simulations (Supplementary Note and Supplementary Figure 1).
In step 2, for each variant, the variance ratio is used to calibrate the score statistic variance that does not incorporate variance components for random effects. Since GRM is no longer needed for this step, the computation time to obtain the score statistic for each variant is O(N). SAIGE next approximates the score test statistics using the SPA to obtain more accurate p-values than the normal distribution. A faster version of the SPA test, similar to the fastSPA method in the SPAtest R package that we recently developed12, is used to further improve the computation time, which exploits the sparsity in low frequency or rare variants to reduce the computation cost.
Computation and Memory Cost
The key features of SAIGE compared to other existing methods are presented in Table 1, showing that SAIGE is the only mixed-model association method that is able to account for the unbalanced case-control ratios while remaining computationally practical for large data sets. To further evaluate the computational performance of SAIGE, we randomly sampled subsets from the 408,458 white British UK Biobank participants who are defined as either coronary artery disease (CAD) cases (31,355) or controls (377,103) based on the PheWAS Code 4112,14,15 followed by benchmarking association tests using SAIGE and other existing methods on 200,000 genetic markers randomly selected out of the 71 million with imputation info 0.3. The non-genetic covariates sex, birth year, and principal components 1 to 4 were adjusted in all tests. The log10 of the memory usage and projected computation time for testing the full set of 71 million genetic variants are plotted against the sample size as shown in Supplementary Figure 2 and Supplementary Table 1. Although SAIGE and BOLT-LMM have the same order of computational complexity (Table 1), SAIGE was slower than BOLT-LMM across all sample sizes (ex. 517 vs 360 CPU hours when N=408,458). This is due to the fact that fitting logistic mixed model requires more iterative steps than linear mixed model, and applying SPA requires additional computation. SAIGE requires slightly less memory than BOLT-LMM (10 to 11 Gb when N=408,458) and the low memory usage makes both methods feasible for the large data set. In contrast, GMMAT and GEMMA requires substantially more computation time and memory usage. For example, when N=400,000, projected memory usages of both GMMAT and GEMMA are more than 600 Gb. The actual computation time and memory usage of association tests for the full UK Biobank data for CAD are given in Table 1. SAIGE required 517 CPU hours and 10.3 Gb memory to analyze 71 million variants that have imputation info 0.3 for 408,458 samples, which indicates that the analysis will be done in ~26 hours with 20 CPU cores.
Table 1.
Comparison of different methods for GWAS with mixed effect models
| Method Features | Algorithm Complexity | Benchmarks for UK Biobank Data Coronary Artery Disease (PheCode 411) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Does not require pre- computed genetic relationship matrix |
Feasible for large sample sizes |
Developed for binary traits |
Accounts for unbalanced case- control ratio |
Tests quantitative traits |
Time complexity | Memory usage (Gbyte) |
Time CPU hrs |
Memory | ||||
| Step 1 | Step 2 | Step 1 | Step 2 | |||||||||
| Logistic mixed model | SAIGE | ✓ | ✓ | ✓ | ✓ | ✓ | O(PM1N1.5) * | O(MN) | M1N/4 | N | 517 | 10.3G |
| GMMAT | ✓ | ✓ | O(PN3) | O(MN2) | F N2 | F N2 | NA | NA | ||||
| Linear mixed model | BOLT-LMM | ✓ | ✓ | ✓ | O(PM1N1.5)* | O(MN) | M1N/4 | N | 360 | 10.9G | ||
| GEMMA | ✓ | O(N3) | O(MN2) | F N2 | FN2 | NA | NA | |||||
N: number of samples
P: number of iterations required to reach convergence
M1: number of markers used to construct the kinship matrix;
M: total number of markers to be tested
F: Byte for floating number
Number of iterations in PCG is assumed as O(N0.5)8
Association analysis of binary traits in UK Biobank data
We applied SAIGE to several randomly selected binary traits defined by the PheWAS Codes (PheCode) of UK Biobank2,14,15 and compared the association results with those obtained from the method based on linear mixed models, BOLT-LMM8, and SAIGE without the saddlepoint approximation (SAIGE-NoSPA), which is asymptotically equivalent to GMMAT9. Due to computation and memory cost, the current GMMAT method cannot analyze the UK Biobank data. We restrict our analysis to markers directly genotyped or imputed by the Haplotype Reference Consortium (HRC)19 panel due to quality control issues of non-HRC markers reported by the UK BioBank. Approximately 28 million markers with minor allele counts (MAC) ≥ 20 and imputation info score 0.3 were used in the analysis. Among 408,961 white British participants in the UK Biobank, 132,179 have at least one up to the third degree relative among the genotyped individuals14,15. We used 93,511 high quality genotyped variants to construct the GRM. In the UK Biobank data, most binary phenotypes based on PheCodes (1,431 out of 1,688; 84.8%) have case-control ratio lower than 1:100 (Supplementary Figure 3) and would likely demonstrate problematic inflation of association test statistics without SPA.
Association results of four exemplary binary traits that have various case-control ratios are plotted in Manhattan plots shown in Figure 1 and in the quantile-quantile (QQ) plots stratified by minor allele frequency (MAF) shown in Figure 2. The four binary traits are coronary artery disease (PheCode 411) with 31,355 cases and 377,103 controls (1:12), colorectal cancer (PheCode 153) with 4,562 cases and 382,756 controls (1:84), glaucoma (PheCode 365) with 4,462 cases and 397,761 controls (1:89), and thyroid cancer (PheCode 193) with 358 cases and 407,399 controls (1:1138). In the Manhattan plots in Figure 1, each locus that contains any variant with p-value < 5×10−8 is highlighted as blue or green to indicate whether this locus has been reported by previous studies or not. Supplementary Table 2 presents the number of all significant loci and those that have not been previously reported by each method for each trait and Supplementary Table 3 lists all significant loci identified by SAIGE. A significant locus is defined to be potentially novel if it is located outside 500kb of any previously reported ones.
Figure 1.
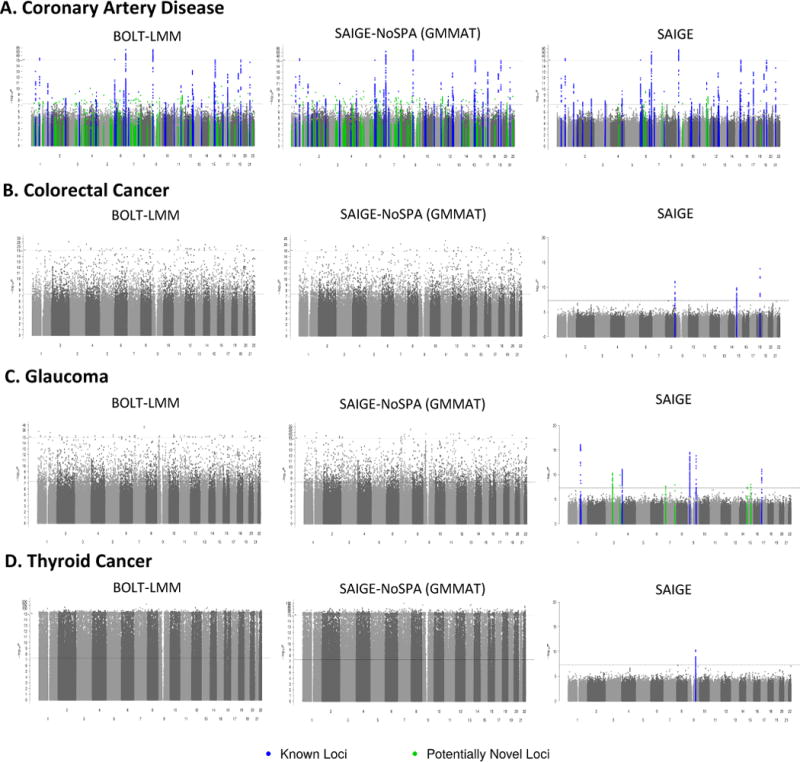
Manhattan plots of GWAS results for four binary phenotypes with various case-control ratios in the UK Biobank.
GWAS results from SAIGE, SAIGE-NoSPA(asymptotically equivalent to GMMAT) and BOLT-LMM are shown for A. coronary artery disease (PheCode 411, case:control = 1:12, N = 408,458), B. colorectal cancer (PheCode 153, case:control = 1:84, N = 387,318), C. glaucoma (PheCode 365, case: control = 1:89, N = 402,223), and D. thyroid cancer (PheCode 193, case:control=1:1138, N = 407,757). N: sample size. Blue: loci with association p-value < 5×10−8, which have been previously reported, Green: loci that have association p-value < 5×10−8 and have not been reported before. Since results from SAIGE-noSPA and BOLT-LMM contain many false positive signals for colorectal cancer, glaucoma, and thyroid cancer, the significant loci are not highlighted. The upper dashed line marks the break point for the different scales of the y axis and the lower dashed line marks the genome-wide significance (p-value = 5×10−8).
Figure 2.
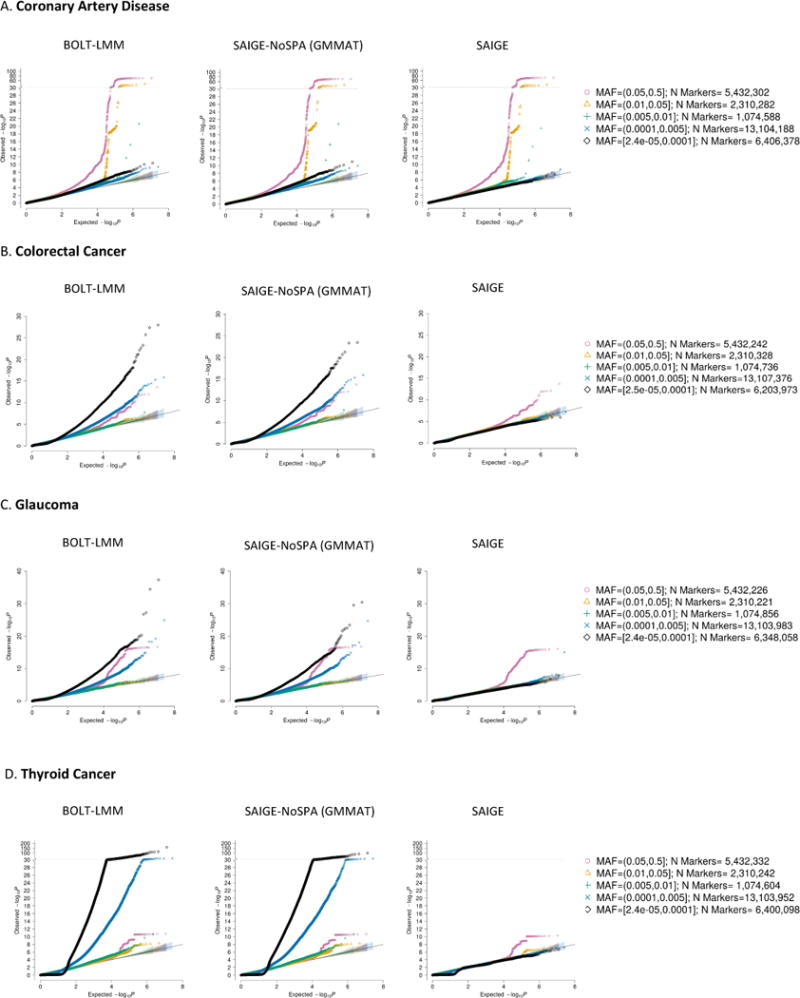
Quantile-quantile plots of GWAS results for four binary phenotypes with various case-control ratios in the UK Biobank.
GWAS results from SAIGE, SAIGE-NoSPA (asymptotically equivalent to GMMAT) and BOLT-LMM are shown for A. coronary artery disease (PheCode 411, case: control = 1:12, N = 408,458), B. colorectal cancer (PheCode 153, case: control = 1:84, N = 387,318), C. glaucoma (PheCode 365, case: control = 1:89, N = 402,223), and D. thyroid cancer (PheCode 193, case: control=1:1138, N = 407,757). N: sample size.
Both Manhattan and QQ plots show BOLT-LMM and SAIGE-NoSPA have greatly inflated type I error rates. The inflation problem is more severe as case-control ratios become more unbalanced and the MAF of the tested variants decreases. The genomic inflation factors (λ) at the 0.001, 0.01 p-value percentiles are shown for several MAF categories in Supplementary Table 4. For the colorectal cancer GWAS which has case-control ratio 1:84, λ at the 0.001 p-value percentile is 1.68 and 1.71 for variants with MAF by SAIGE-NoSPA and BOLT-LMM, while λ is 0.99 by SAIGE. The inflation is even more severe for the test results by SAIGE-NoSPA and BOLT-LMM for the thyroid cancer, which has case-control ratio 1:1138, with the λ at the 0.001 p-value percentile around 4 to 5 for variants with MAF and all variants, respectively. With the unbalanced case-control ratio accounted for in SAIGE, the λ is again very close to 1.
We have generated summary statistics for all 1,403 PheCode-derived binary traits in 408,961 UK Biobank white British European-ancestry samples using SAIGE software and made them available in a public repository (see Supplementary Note for URL).
Simulation Studies
We investigated the type I error control and power of two logistic mixed model approaches, SAIGE and GMMAT, and the linear mixed model method BOLT-LMM that computes mixed model association statistics under the infinitesimal and non-infinitesimal models through simulation studies. We followed the steps described in the Online Methods section to simulate genotypes for 1,000 families, each with 10 family members (N=10,000), based on the pedigree shown in Supplementary Figure 4.
Type I error rates
The type I error rates for SAIGE, SAIGE-NoSPA, GMMAT, and BOLT-LMM have been evaluated based on the association tests performed on 109 simulated genetic variants. The variants were simulated using the same MAF spectrum of the UK Biobank HRC imputation data with case-control ratio 1:99, 1:9, and 1:1. Two different values of variance component parameter =1 and 2 were considered, which correspond to the liability scale heritability 0.23 and 0.38, respectively. The empirical type I error rates at the α = 5×10−4 and α = 5×10−8 are shown in the Supplementary Table 5. SAIGE-NoSPA, GMMAT, and BOLT-LMM have greatly inflated type I error rates when the case-control ratios are moderately or extremely unbalanced and slightly deflated type I error rates when the case-control ratios are balanced. This is expected as previous studies have suggested inflation of the score tests in the presence of the unbalanced case-control ratios and deflation in balanced studies10,12. Unlike GMMAT and BOLT-LMM, SAIGE has no inflation when case-control ratios are unbalanced. When the case-control ratios are balanced, SAIGE has correct type I error rates when =1 and slightly deflated type I error rates when =2. We also observed that GMMAT score test statistics do not follow the normal distribution when MAF is low and case-control is unbalanced (Supplementary Figure 5).
To further investigate the type I error rates by MAF and case-control ratios, we carried out additional simulations. Supplementary Figure 6 shows QQ plots of 1,000,000 rare variants (MAF = 0.005) with various case-control ratios (1:1, 1:9, and 1:99) and Supplementary Figure 7 shows QQ plots of 1,000,000 variants with different MAF (0.005, 0.01, and 0.3) when case-control ratio was 1:99. Consistent to what has been observed in the real data study, GMMAT and SAIGE-NoSPA is more inflated for less frequent variants with more unbalanced case-control ratios. In contrast, SAIGE has successfully corrected this problem.
To evaluate whether SAIGE can control type I error rates in the presence of population stratification, we have simulated two subpopulations with Fst 0.013, which corresponds to the average Fst between Finnish and non-Finnish Europeans20. We assumed that subpopulations have different disease prevalences (0.01 for subpopulation 1 and 0.02 for subpopulation 2, 0.1 for subpopulation 1 and 0.2 for subpopulation 2, and 0.5 for subpopulation 1 and 0.4 for subpopulation 2). Both subpopulations have 1,000 families, each with 10 family members based on the pedigree shown in Supplementary Figure 4. Association tests were performed on 10 million simulated markers and the first four principle components were included as covariates (Supplementary Figure 8). QQ plots (Supplementary Figure 9) show that the test statistics were well calibrated regardless of the variance component parameter and prevalence. This simulation result demonstrates that SAIGE produces well-calibrated p-values in the presence of population stratification.
Power
Next, we evaluated empirical power. Since power simulation requires re-estimating a variance component parameter for each variant to test, to reduce computational burden, we used SAIGE-NoSPA instead of the original GMMAT software. Due to the inflated type I error rates of BOLT-LMM and GMMAT (and SAIGE-NoSPA), for a fair comparison, we estimated power at the test-specific empirical levels that yield type I error rate α = 5×10−8 (Supplementary Table 6). Supplementary Figure 10 shows the power curve by odds ratios for variants with MAF 0.05, 0.1 and 0.2 when =1. When the case-control ratio is balanced, the power of SAIGE, SAIGE-NoSPA and BOLT-LMM were nearly identical. For studies with moderately unbalanced case-control ratio (case:control=1:9), SAIGE has higher power than SAIGE-NoSPA and BOLT-LMM, which is due to very small empirical α for SAIGE-NoSPA and BOLT-LMM resulted from type I error inflation. The power gap is much larger when the case-control ratios are extremely unbalanced. Power results for =2 yielded the same conclusion regarding the methods comparison (data not shown).
Overall simulation studies show that SAIGE can control type I error rates even when case-control ratios are extremely unbalanced and can be more powerful than GMMAT and BOLT-LMM. In contrast, GMMAT and BOLT-LMM suffer type I error inflation, and the inflation is especially severe with low MAF and unbalanced case-control ratios.
DISCUSSION
In this paper, we have presented a method to perform the association tests for binary traits in large cohorts in the presence of sample relatedness, which provides accurate p-value estimates for even extremely unbalanced case-control settings (with a prevalence < 0.1%). The dramatic decrease of the genotyping cost over the last decade allows more and more large biobanks to genotype all of their participants followed by genome-wide PheWAS, in which GWASs are performed for all thousands of diseases/conditions characterized based on EHR and/or survey questionnaires to identify genetic risk factors across different phenotypes1,2,21. Several challenges exist for PheWAS studies by large cohorts. Statistically, inflated type I error rates caused by unbalanced case-control ratios and sample relatedness need to be corrected. Computationally, most of existing mixed model association methods are not feasible for large sample sizes. Our method, SAIGE, uses logistic mixed model to account for the sample relatedness and applies the saddlepoint approximation (SPA) to correct the inflation caused by the unbalanced case-control ratio in the score tests based on logistic mixed models.
SAIGE successfully corrects the inflation of type I error rates of low-frequency variants with binary traits that have unbalanced case-control ratios while also accounting for the relatedness among samples. Furthermore, our method uses several optimization strategies that are similar to those used by BOLT-LMM to improve its computational feasibility for large cohorts. For example, the preconditioned conjugate gradient algorithm is used to solve linear systems instead of the Cholesky decomposition method so that the time complexity for fitting the null logistic model is decreased from O(N3) to approximately O(M1N1.5), where M1 is the number of pruned markers used for estimating the genetic relationship matrix and the N is the sample size.
In the selection of genetic markers (M1) for estimating the kinship matrix and the variance component, trade off exists between computational cost and performance of adjusting for sample relatedness. Increasing the number of markers used for that step linearly increases the computation time and memory. On the other hand, using too few markers may not be sufficient to account for all subtle sample relatedness. For example, Yang et al. have shown that using a few thousand markers is not sufficient to yield correct type I error control22. In the UK Biobank data analysis, we used 93,511 independent, high quality genotyped variants, which were used by the UK Biobank data group to estimate the kinship coefficients between samples15. We carried out a sensitivity analysis by increasing the number of markers to 340,447 (Supplementary Note Section 2.3). Using more markers to estimate the kinship matrix for the UK Biobank data analysis produced generally similar association p-values but with lambdas closer to 1.
Using genome-wide genetic markers to adjust for sample relatedness tends to have the proximal contamination problem, which can reduce association test power6,8,22,23. To avoid it, the leave-one-chromosome-out (LOCO) scheme can be used. We implemented the LOCO option in SAIGE. A sensitivity analysis (Supplementary Note Section 1.2.5) on the four exemplary binary phenotypes in the UK Biobank suggested that the proximal contamination in GWAS for diseases with relatively low prevalence, such as thyroid cancer, glaucoma, and colorectal cancer, is not as substantial as for more common diseases, e.g. coronary artery disease.
Given the inflation of type I errors of linear mixed model for rare variants (MAF < 0.5%) with unbalanced case-control phenotypes, current GWAS studies address the problem by excluding rare variants from the analysis. However, this practice can lead to false negative results if associated rare variants are simply excluded rather than analyzed properly. For example, after using SAIGE to analyze rare variants in the UK Biobank, we identified a nonsense variant in MYOC (MAF = 0.14%) that was significantly associated with glaucoma. In our preliminary analysis of UK Biobank data of 1,283 non-sex specific phenotypes, we observed 1,609 genetic variants, including variants in the same locus, with minor allele frequency < 0.5% with SAIGE p-values < 5×10−8 (Supplementary Note Section 2.4). The method as implemented in SAIGE can control for type I error rates regardless of MAF and case-control ratios and will facilitate identification of rare disease-associated variants.
There are several limitations in SAIGE. First, the time for algorithm convergence may vary among phenotypes and study samples given different heritability levels and sample relatedness. Second, SAIGE has been observed to be slightly conservative when case-control ratios are extremely unbalanced (Supplementary Table 5). Third, the accurate odds ratio estimation requires fitting the model under the alternative and is not computational efficient. Similar to several other mixed model methods3,8,18, SAIGE estimates odds ratios for genetic markers using the parameter estimates from the null model. Fourth, SAIGE assumes that the effect sizes of genetic markers are normally distributed with a mean of zero and standard deviation of one, which follows an infinitesimal architecture. With this assumption, SAIGE may sacrifice power to detect genetic signals whose genetic architecture is non-infinitesimal. Last, the variance component estimates from SAIGE is biased and hence it should not be used to estimate the heritability (Supplementary Note Section 2.1). This is because SAIGE uses penalized quasi-likelihood (PQL) to estimate . However, as shown in our simulation studies and elsewhere9, PQL-based approaches works well to adjust for sample relatedness. In future, we plan to extend the current single variant test to gene- or region-based multiple variant test to improve power for identifying disease susceptibility rare variants.
With the emergence of large-scale biobank, PheWAS will be an important tool to identify genetic components of complex traits. Here we describe a scalable and accurate method, SAIGE, for the analysis of binary phenotypes in genome-wide PheWAS. Currently, SAIGE is the only available approach to adjust for both case-control imbalance and family relatedness, which are commonly observed in PheWAS datasets. In addition, the optimization approaches used in SAIGE make it scalable for the current largest (UK Biobank) and future much larger datasets. Through simulation and real data analysis, we have demonstrated that our method can efficiently analyze a dataset with 400,000 samples and adjust for type I error rates even when the case-control ratios are extremely unbalanced. Our method will provide an accurate and scalable solution for large scale biobank data analysis and ultimately contribute to identify genetic mechanism of complex diseases.
ONLINE METHODS
Generalized linear mixed model for binary traits
In a case-control study with sample size , we denote the status of the ith individual using or 0 for being a case or a control. Let the vector represent covariates including the intercept and represent the allele counts ( ) for the variant to test. The logistic mixed model can be written as
where is the probability for the ith individual being a case given the covariates and genotypes as well as the random effect, which is denoted as . The random effect is assumed to be distributed as (0, τ ), where is an genetic relationship matrix (GRM) and is the additive genetic variance. The is a coefficient vector of fixed effects and is a coefficient of the genetic effect.
Estimate variance component and other model parameters (Step 1)
To fit the null model, , penalized quasi-likelihood (PQL) method1 and the AI-REML algorithm2 are used to iteratively estimate , ). At iteration k, let , ) be estimated , ), the estimated mean of , , and = be an matrix of the variance of working vector )/ . To obtain log quasi-likelihood and average information at each iteration, the current GMMAT approach calculates the inverse of . Since it is computationally too expensive for large N, we use the preconditioned conjugate gradient (PCG)3,4, which allows calculating quasi-likelihood and average information without calculating (See Supplementary Note for details). PCG is a numerical method to find solutions of linear system. It is particularly useful when the system is very large. BOLT-LMM5 successfully used it to estimate variance component in linear mixed model.
A score test statistics for Ho: is where G and Y are genotype and phenotype vectors, respectively, and is the estimated mean of Y under the null hypothesis, and is the covariate adjusted genotype vector. The variance of T, Var(T) , where . For each variant, given , calculation of Var(T) requires O(N2) computation. In addition, since our approach does not calculate , and hence , obtaining Var(T) requires applying PCG for each variant, which can be computationally very expensive. To reduce computation cost, we use the same approximation approach used in BOLT-LMM and GRAMMAR-GAMMAR6, in which we estimate a variance of T with assuming that true random effect is given, and then calculate ratio between these two variance. Suppose Var(T)* = , which is a variance estimate of T assuming is given. Let r = Var(T)/Var(T)* ratio of these two different types of variance estimates. In Supplementary Note, we have shown that the ratio is approximately constant for all variants. Using this fact, we can estimate r using a relatively small number of variants. In all the numerical studies in this paper, we used 30 variants to estimate r.
Score test with SPA (Step 2)
Suppose is the estimated ratio (i.e. r) in Step 1. Now the variance adjusted test statistics is
which has mean zero and variance under the null hypothesis. The computation of requires O(N) computation. The traditional score tests assume that T (and hence Tadj) asymptotically follows a Gaussian distribution under Ho: , which is using only the first two moments (mean and variance). When the case-control ratios are unbalanced and variants have low MAC, the underlying distribution of Tadj can be substantially different from Gaussian distribution. To obtain accurate p-values, we use Saddlepoint approximation method (SPA)7–9, which approximates distribution using the entire cumulant generating function (CGF). A fast version of SPA (fastSPA)9 has recently been developed and applied to PheWAS, and provides accurate p-values even when case-control ratios are extremely unbalanced (ex. case:control=1:600).
To apply fastSPA to Tadj we need to obtain CGF of Tadj first. To do this, we use the fact that given , Tadj is a weighted sum of independent Bernoulli random variables. The approximated cumulant generating function is
where the constant c=Var*(T) −1/2. Let and are first and second derivatives of K with respect to t. To calculate the probability that , where q is an observed test statistic, we use the following formula7
where , and is the solution of the equation . As fastSPA9, we exploit the sparsity of genotype vector when MAF of variants are low. In addition, since normal approximation works well when the test statistic is close to the mean, we use the normal distribution when the test statistic is within two standard deviation of the mean.
Data simulation
We carried out a series of simulations to evaluate and compare the performance of SAIGE to GMMAT. We randomly simulated a set of 1,000,000 base-pair “pseudo” sequences, in which variants are independent to each other. Alleles for each variant were randomly drawn from Binomial(n = 2, p = MAF). Then we performed the gene-dropping simulation10 using these sequences as founder haplotypes that were propagated through the pedigree of 10 family members shown in Supplementary Figure 4. Binary phenotypes were generated from the following logistic mixed model
where Gi is the genotype value, is the genetic log odds ratio, is the random effect simulated from . Two covariates, X1 and X2, were simulated from Bernoulli(0.5) and N(0,1), respectively. The intercept was determined by given prevalence (i.e. case-control ratios).
To evaluate the type I error rates at genome-wide α=5×10−8, 10 million markers along with 100 sets of phenotypes with different random seeds for case-control ratios 1:99, 1:9, and 1:1 were simulated with . Given and 2, the liability scale heritability is 0.23 and 0.38, respectively11 (Supplementary Note Section 2.1). Association tests were performed on the 10 million genetic markers for each of the 100 sets of phenotypes using SAIGE, GMMAT, and BOLT-LMM, therefore in total 109 tests were performed. To have a realistic MAF spectrum, MAFs were randomly sampled from the MAF spectrum in UK Biobank data (Supplementary Figure 11). Additional type I error simulations were carried out for five different MAFs (0.005, 0.01, 0.05, 0.1 and 0.3) to evaluate type I error rates by MAFs and in the presence of population stratification (Supplementary Note Section 2.2).
For the power simulation, phenotypes were generated under the alternative hypothesis . For each of the MAF 0.05 and 0.2, we simulated 1,000 datasets, and power was evaluated at test-specific empirical α, which yields nominal α=5×10−8. The empirical α was estimated from the previous type I error simulations. As the same as type I error simulations, three different case-control ratios (1:99, 1:9, and 1:1) were considered.
Note that since we evaluated the empirical type I error rates and power based on genetic variants that were simulated independently, the LD Score regression12 calibration and the leave-one-chromosome-out (LOCO) scheme were not applied in BOLT-LMM or SAIGE.
Performance evaluation of approaches used to obtain computational scalability
We evaluated the numerical stability and convergence for numerical and asymptotic approximations that we use to achieve the computational scalability: PCG (Supplementary Figure 12), randomized trace estimator (Supplementary Figure 13), variance ratio estimation (Supplementary Figure 14), LOCO (Supplementary Figure 15), and variance component parameter estimation (Supplementary Table 7 and Supplementary Figure 16). In addition, we compared the results of UK Biobank data analysis using a GRM constructed from a larger number of markers (M1 = 340,447) (Supplementary Table 8 and Supplementary Figure 17 and 18).
Phenotype definition in UK Biobank
We used a previously published scheme to defined disease-specific binary phenotypes by combining hospital ICD-9 codes into hierarchical PheCodes, each representing a more or less specific disease group13.
ICD-10 codes were mapped to PheCodes using a combination of available maps through the Unified Medical Language System(https://www.nlm.nih.gov/research/umls/) and other sources, string matching, and manual review. Study participants were labeled a PheCode if they had one or more of the PheCode-specific ICD codes. Cases were all study participants with the PheCode of interest and controls were all study participants without the PheCode of interest or any related PheCodes. Gender checks were performed, so PheCodes specific for one gender could not mistakenly be assigned to the other gender.
Genome build
All genomic coordinates are given in NCBI Build 37/UCSC hg19.
Statistical analysis
With the assumption of the additive genetic model, we performed GWAS using SAIGE (version 0.13) on 28 million genetic markers for 1,403 binary phenotypes of 408,961 white British participants, who passed the quality control in the UK Biobank14. In the logistic mixed model by SAIGE, the first four principal components, sex and birth year were included as the non-genetic covariates. To evaluate the performance of SAIGE to account for sample relatedness and unbalanced case-control ratio for binary phenotypes, GWAS results from SAIGE were compared to those from SAIGE-NoSPA(asymptotically equivalent to GMMAT version 0.96) and BOLT-LMM (version 2.3) for four exemplary binary phenotypes with various case-control ratios. The numbers of samples used for analysis are included in the legend of each Figure. The genomic inflation factors (λ) were calculated as the ratio of observed and expected chi-square statistic at the 0.001 and 0.01 p-value percentiles.
Reporting Summary
Further information on study design is available in the Nature Research Reporting Summary linked to this article.
Code and data availability
SAIGE is implemented as an open-source R package available at https://github.com/weizhouUMICH/SAIGE/. The GWAS results for 1403 binary phenotypes with the PheCodes13 constructed based on ICD codes in UK Biobank using SAIGE are currently available for public download at https://www.leelabsg.org/resources. Information for all phenotypes can be found in the website above. We also display the results in the Michigan PheWeb http://pheweb.sph.umich.edu/UKBiobank, which consists of Manhattan plots, Q-Q plots, and regional association plots for each phenotype as well as the PheWAS plots for every genetic marker.
Supplementary Material
Acknowledgments
This research has been conducted using the UK Biobank Resource under application number 24460. SL and RD were supported by NIH R01 HG008773. CJW was supported by NIH R35 HL135824. WZ was supported by the University of Michigan Rackham Predoctoral Fellowship. JBN was supported by the Danish Heart Foundation and the Lundbeck Foundation. JCD, AG, LB, and WQW was supported by NIH R01 LM010685 and U2C OD023196.
Footnotes
- Michigan PheWeb, http://pheweb.sph.umich.edu/UKBiobank;
- GWAS Summary Statistics for > 1400 binary phenotypes in the UK Biobank by SAIGE, https://www.leelabsg.org/resources;
AUTHOR CONTRIBUTIONS
W.Z. C.W. and S.L. designed experiments. W.Z. and S.L. performed experiments. J.B., L.G.F., A.G., L.A.B, W-Q. W, J.C.D constructed phenotypes for the UK Biobank data. W.Z., J.L., S.A.G., H.M.K., C.W., S.L. and G.R.A. analyzed UK Biobank data. P.V. created the PheWeb. M.E.G. and K.H. provided data. W.Z., J.B., A.G., J.C.D., R.D., C.W. and S.L. wrote the manuscript.
COMPETING FINANCIAL INTERESTS STATEMENT
The authors declare no competing financial interests.
References
- 1.Bush WS, Oetjens MT, Crawford DC. Unravelling the human genome-phenome relationship using phenome-wide association studies. Nat Rev Genet. 2016;17:129–145. doi: 10.1038/nrg.2015.36. [DOI] [PubMed] [Google Scholar]
- 2.Denny JC, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol. 2013;31:1102–1110. doi: 10.1038/nbt.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang HM, et al. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42:348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Z, et al. Mixed linear model approach adapted for genome-wide association studies. Nat Genet. 2010;42:355–360. doi: 10.1038/ng.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lippert C, et al. FaST linear mixed models for genome-wide association studies. Nat Methods. 2011;8:833–835. doi: 10.1038/nmeth.1681. [DOI] [PubMed] [Google Scholar]
- 7.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44:821–824. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loh PR, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet. 2015;47:284–290. doi: 10.1038/ng.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen H, et al. Control for Population Structure and Relatedness for Binary Traits in Genetic Association Studies via Logistic Mixed Models. Am J Hum Genet. 2016;98:653–666. doi: 10.1016/j.ajhg.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma C, Blackwell T, Boehnke M, Scott LJ, GoT2D investigators Recommended Joint and Meta-Analysis Strategies for Case-Control Association Testing of Single Low-Count Variants. Genet Epidemiol. 2013;37:539–550. doi: 10.1002/gepi.21742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuonen D. Saddlepoint approximations for distributions of quadratic forms in normal variables. Biometrika. 1999;4:7. [Google Scholar]
- 12.Dey R, Schmidt EM, Abecasis GR, Lee S. A fast and accurate algorithm to test for binary phenotypes and its application to PheWAS. Am J Hum Genet. 2017;101:37–49. doi: 10.1016/j.ajhg.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaasschieter EF. Preconditioned conjugate gradients for solving singular systems. J Comput Appl Math. 1988;24:265–275. [Google Scholar]
- 14.Sudlow C, et al. UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age. PLOS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bycroft C, et al. Genome-wide genetic data on ~500,000 UK Biobank participants. bioRxiv. 2017;166298 doi: 10.1101/166298. [DOI] [Google Scholar]
- 16.Gilmour AR, Thompson R, Cullis BR. Average Information REML: An Efficient Algorithm for Variance Parameter Estimation in Linear Mixed Models. Biometrics. 1995;51:1440. [Google Scholar]
- 17.Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 18.Svishcheva GR, Axenovich TI, Belonogova NM, van Duijn CM, Aulchenko YS. Rapid variance components–based method for whole-genome association analysis. Nat Genet. 2012;44:1166–1170. doi: 10.1038/ng.2410. [DOI] [PubMed] [Google Scholar]
- 19.McCarthy S, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–1283. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelis M, et al. Genetic structure of Europeans: a view from the North-East. PLoS One. 2009;4:e5472. doi: 10.1371/journal.pone.0005472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shameer K, et al. A genome- and phenome-wide association study to identify genetic variants influencing platelet count and volume and their pleiotropic effects. Hum Genet. 2014;133:95–109. doi: 10.1007/s00439-013-1355-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J, Zaitlen NA, Goddard ME, Visscher PM, Price AL. Advantages and pitfalls in the application of mixed-model association methods. Nat Genet. 2014;46:100–106. doi: 10.1038/ng.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Listgarten J, et al. Improved linear mixed models for genome-wide association studies. Nat Methods. 2012;9:525–526. doi: 10.1038/nmeth.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS-ONLY REFERENCES
- 1.Breslow NE, Clayton DG. Approximate Inference in Generalized Linear Mixed Models. J Am Stat Assoc. 1993;88:9. [Google Scholar]
- 2.Gilmour AR, Thompson R, Cullis BR. Average Information REML: An Efficient Algorithm for Variance Parameter Estimation in Linear Mixed Models. Biometrics. 1995;51:1440. [Google Scholar]
- 3.Kaasschieter EF. Preconditioned conjugate gradients for solving singular systems. J Comput Appl Math. 1988;24:265–275. [Google Scholar]
- 4.Hestenes MR, Eduard S. Methods of conjugate gradients for solving linear systems. Vol. 49. NBS; 1952. [Google Scholar]
- 5.Loh PR, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet. 2015;47:284–290. doi: 10.1038/ng.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Svishcheva GR, Axenovich TI, Belonogova NM, van Duijn CM, Aulchenko YS. Rapid variance components–based method for whole-genome association analysis. Nat Genet. 2012;44:1166–1170. doi: 10.1038/ng.2410. [DOI] [PubMed] [Google Scholar]
- 7.Kuonen D. Saddlepoint approximations for distributions of quadratic forms in normal variables. Biometrika. 1999;4:7. [Google Scholar]
- 8.Imhof JP. Computing the Distribution of Quadratic Forms in Normal Variables. Biometrika Trust. 1961;48:8. [Google Scholar]
- 9.Dey R, Schmidt EM, Abecasis GR, Lee S. A fast and accurate algorithm to test for binary phenotypes and its application to PheWAS. Am J Hum Genet. 2017;101:37–49. doi: 10.1016/j.ajhg.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 11.de Villemereuil P, Schielzeth H, Nakagawa S, Morrissey M. General Methods for Evolutionary Quantitative Genetic Inference from Generalized Mixed Models. Genetics. 2016;204:1281–1294. doi: 10.1534/genetics.115.186536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bulik-Sullivan BK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Denny JC, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol. 2013;31:1102–1110. doi: 10.1038/nbt.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sudlow C, et al. UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age. PLOS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.