

Abstract
The pathophysiological mechanisms underlying the origin of several ovarian pathologies remain unclear. In addition to the genetic basis, developmental insults are gaining attention as a basis for the origin of these pathologies. Such early insults include maternal over or under nutrition, stress, and exposure to environmental chemicals. This chapter reviews the development and physiological function of the ovary, the known ovarian pathologies, the developmental check points of ovarian differentiation impacted by developmental insults, the role played by steroidal and metabolic factors as mediaries, the epigenetic mechanisms via which these mediaries induce their effects, and the knowledge gaps for targeting future studies to ultimately aid in the development of improved treatments.
1. Introduction
Ovary is the female gonad that is present as a pair in most animals and is responsible for generation of female gametes and production of hormones that regulate reproductive functions. Although the existence of the ovary was probably recognized long before the time of Aristotle, the first detailed descriptions came from Leonardo Da Vinci, Andreas Vesalius, Gabrielle Fallopio and others during the 16th century with Vesalius pointing to the existence of follicles within the ovary (Correia 1998). Later in the 17th century, William Harvey stated “ex ovo de vivum” pointing to everything coming from the egg or the ovum (Albertini 2015, Correia 1998). The concept put forth by Niels Stensen and Regnier de Graaf that the entire follicle was the egg that underwent fertilization and then released was later refuted by von Baer in the 19th century who described the ovum within the follicle as the fertilizing unit (Correia 1998, Pangas and Rajkovic 2015). Since then there has been an explosion of knowledge relative to ovarian structure, function and its role in sexuality and regeneration of life. Considering the key role played by the ovary in housing and supporting the oocyte - the germline of the next generation, and several ovarian pathologies contributing to reduced fecundity have been identified, it is imperative to recognize modifiable factors that contribute to the development of such pathologies.
i. Ovarian Structure
In most mammals, ovaries are present in the upper pelvic cavity on either side of the body held at place by a mesentery connected to a broad ligament. Structurally ovary is made of inner medulla, which contains vascular elements (blood and lymphatic vessels) and an outer cortex containing the ovarian follicles, the functional unit of the ovary. The cortex is enclosed in a capsule called tunica albuginea. The structure of the ovarian follicle varies depending on the stage of follicular differentiation (discussed in subsequent sections). A fully developed follicular unit is made up of the female gamete, ovum, and the surrounding somatic cells comprised of an avascular layer containing granulosa cells the site of estrogen production and theca cell layer, the androgen producing unit, where the nerve innervations and blood supply terminate.
ii. Establishment of the ovarian reserve/pool
Gonads develop in the urogenital ridge adjacent to the primitive embryonic kidney and exist initially in a bipotential or sexually undifferentiated state (Capel 2000). Presence or absence of testis-determining factor (TDF; also known as sex-determining region Y [SRY]) gene in the somatic cells (Capel 2000, Goodfellow and Lovell-Badge 1993) dictates whether the bipotential gonad differentiates into a testes or ovary, respectively. A ovary-determining gene (Z gene) has been postulated but is yet to be identified (Blecher and Erickson 2007). Although the dogma is ovarian development occurs by default or in a passive manner, gene expression studies point to active regulation of ovarian differentiation involving differential expression of various genes including Wnt4 (Yao 2005).
The bipotential primordial germ cells (PGCs), which form the ova in the females, originate from the endoderm of the yolk sac and migrate into the genital ridge and undergoes further proliferation (Hummitzsch, et al. 2015, Lin and Capel 2015). The coelomic epithelium also proliferates and infiltrates the mesenchyma of the genital ridge to form the primitive sex cords that surround the PGCs. The mesenchymal cells that are part of the mesonephros also migrate into the gonad at this time through the medulla (Smith, et al. 2014). Following sex determination, the bipotential gonad in the male immediately organizes into a testis, while the ovaries in the female show little structural differentiation. By the 7th week of gestation in human female fetus, primitive sex cords degenerate and are replaced by cortical sex cords (Knobil and Neill 1998). Around this time, the glomeruli and ductal cells that are part of the migrating mesonephros undergo regression and the remnant remains to form the ovarian rete (Smith, et al. 2014). Final stages of ovarian development involve involution of medulla and the development of cortex where sex cords split into clusters within which the germ cells develop into oocyte and the cord cells form the granulosa cells. The mesonephric rete cells that are initially located in the ovarian medulla increase in size through proliferation and migrate into the cortex. It has been suggested that theca cells originate from these mesenchymal mesonephric cells later on as follicular differentiation progresses (Smith, et al. 2014).
The PGCs first mitotically divide to form oogonia, which undergo first meiosis to form the primary oocyte, which occurs at around 9th week of fetal life in the human (Albertini 2015). Primary oocytes stay arrested at the diplotene stage until ovulation. The process of oocyte maturation is closely associated with follicular development. Upon formation of primary oocytes, they are surrounded by the cord-derived, epithelial origin, squamous cell layer called pregranulosa cells to form the primordial follicles. The ‘naked oocytes’ that are not surrounded by the pregranulosa cell layer undergoes atresia. In the humans, complete ovarian differentiation occurs in utero, but in altricial species such as rodents differentiation gets completed during postnatal life (McGee and Hsueh 2000). In the rodents, the PGCs migrate into gonadal ridge during late embryonic period and develop into oogonia such that at birth only sex cords and oogonia are present. Primordial follicles in the rodents develop between days 1–2 after birth in contrast to the humans where primordial follicles appear at 16 weeks of fetal life.
Unlike in the testes where spermatogonium undergoes mitotic renewal throughout reproductive life, the mitotic activity of the oogonia stops before or around birth depending on the species resulting in the number of oocytes formed in each species being set by the time of birth or early postnatal life. This premise has been challenged recently from studies in mice with the suggestion that oocyte renewal occurs even after formation of initial pool (Johnson, et al. 2004) and the stem cells that participate in this renewal are of bone marrow origin (Johnson, et al. 2005). Although other investigators have failed to confirm these findings (Zhang, et al. 2012), recent studies suggest presence of germline stem cells in the ovary (Wang, et al. 2014). The relevance of these findings as it relates to formation of functional ovarian follicles remains unclear (Hummitzsch, et al. 2015). As such, the prevailing dogma is that ovaries have a finite set of primordial follicles that constitutes the follicular pool or ovarian reserve (Figure 1) by birth to draw from throughout the lifespan of the female. Developmentally at about 20 weeks of gestation an estimated 6–7 million germ cells are present in the human ovaries reducing down to about 1–2 million follicles at birth and ~250,000 follicles at puberty. By 35 years of age only 25,000 follicles remain reducing further down to 2,500 follicles by 50 years of age (Gougeon, et al. 1994, Richardson, et al. 1987). By the time women are 65 years of age virtually no follicles are present in both ovaries (Costoff and Mahesh 1975). This reduction in follicular numbers has been shown to involve programmed cell death or apoptosis (Escobar, et al. 2011, Tilly 1996).
Figure 1:
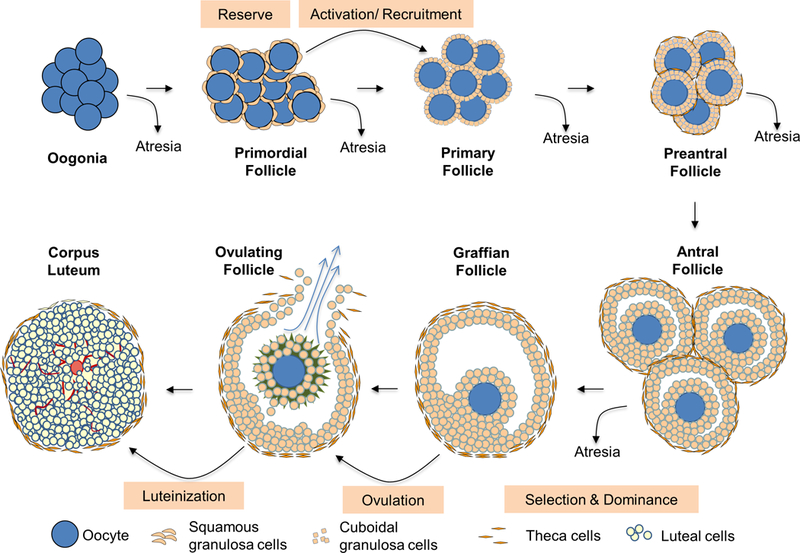
Schematic showing the different stages of follicular differentiation and the developmental checkpoints, potential targets for developmental insults.
iii. Follicular activation or recruitment
The vast majority of the primordial follicles from the follicular pool remain in a quiescent state with few follicles from this pool emerging to develop further during each reproductive cycle. The fate of those emerging from the pool during each cycle is continued differentiation or atresia (Figure 1). Follicular differentiation from primordial pool begins with follicular activation (also referred to as initial recruitment) and occurs in a gonadotropin-independent manner. Follicular activation is regulated by locally produced growth factors, hormones and cytokines [reviewed in (McLaughlin and McIver 2009)] such as kit ligand, fibroblast growth factor, transforming growth factor (TGF) α, leukemia inhibitory factor, bone morphogenetic protein (BMP) 4, anti-mullerian hormone (AMH), TGFβ and androgens (Durlinger, et al. 1999, Fortune 2003, Skinner 2005). While the exact mechanism of activation is not completely understood, gene knockout studies provide some insights. For instance, premature follicular activation following gene deletion of forkhead box O3a (FOXO3A) and protein kinase B (alpha serine/threonine-protein kinase, AKT) inhibitor, phosphatase and tensin homolog (Castrillon, et al. 2003, Reddy, et al. 2008) suggests that the primordial follicular pool is under inhibitory control and activation occur when the inhibitory factors decrease or stimulatory factors increase. As such, the balance between activators (kit ligand, fibroblast growth factor, TGFα, leukemia inhibitory factor and BMP4) and inhibitors (AMH and TGFβ) appear to drive follicular activation (Gougeon 2004). The activation process involves intracellular signaling pathways such as phosphoinositide 3-kinase (PI3K), AKT and mammalian target of rapamycin [reviewed in (Sobinoff, et al. 2013)] and leads to formation of primary follicles that is characterized by cuboidal shaped-granulosa cell layer.
iv. Follicular selection and dominance
Further differentiation of primary follicle into preantral follicle involves acquisition of theca layer around the granulosa cell layer and granulosa cell division through mitosis to form multiple layers of granulosa cells (the theca layer remains as a single layer at this stage) (Figure 1). The primary follicles are evident in the human fetal ovary at 20 weeks and antral follicles by 32 weeks of gestation. In contrast, primary follicles appear by day 3–5 and antral follicles by day 15–17 after birth in the rodents (McGee and Hsueh 2000).
The follicular growth from primary to preantral stage is considered to be gonadotropin independent. Identification of follicle-stimulating hormone (FSH) receptors during early stages of follicular development (Touraine, et al. 1999) and the finding that loss of FSH affects preantral follicle development (McGee and Hsueh 2000) goes against this premise and suggests that gonadotropins do influence preantral follicular development. The non-gonadotropin players that influence follicular growth during this transition are TGFβ, growth differentiation factor (GDF) 9, BMP family members, estrogens, androgens, insulin and insulin-like growth factor (IGF) 1 (Zeleznik 2004). As the preantral follicle acquires multiple granulosa cell layers, they become less sensitive to oocyte-secreted growth factors and transition from the gonadotropin-independent to gonadotropin-dependent state. Furthermore, there is a reduction in the amount of AMH produced by the growing follicles resulting in increased sensitivity of granulosa cells to FSH (Dewailly, et al. 2016) consistent with them becoming more gonadotropin responsive. In the humans, constant pools of preantral follicles are always present such that it has been referred to as the second reserve of follicles (Monniaux, et al. 2014).
Preantral follicles differentiate into antral follicles (Figure 1) with the appearance of antrum, a cavity filled with follicular fluid containing granulosa cell secretions and diffusions from blood vessels in the theca layer. At this stage, theca layer also differentiates into inner layer of theca interna and the outer layer of theca externa. Within the follicle, oocyte gets surrounded by granulosa cells called corona radiata and is suspended in the antral fluid held by a stalk of granulosa cells called the cumulus oophorus (Figure 1). Following the onset of menses, a rise in FSH secretion leads to cyclic recruitment (McGee and Hsueh 2000) of a small cohort of antral follicles that gets selected to grow further.
Antral follicular development was believed to occur either continuously or through a single recruitment process where the privileged follicle(s) continue to develop further (Baerwald, et al. 2012). However, advancement in non-invasive ultrasound monitoring technology has shown that follicular development in the human and domestic animals occur in waves, which involves synchronous growth of a group of antral follicles occurring at regular intervals during the menstrual / estrous cycle (Baerwald, et al. 2012). The ovulatory follicle originates from the final wave with all other follicles undergoing demise through atretic process, although exceptions have been reported (Baerwald, et al. 2003).
Depending on ovulatory quota of the species one or more follicles emerge to become dominant to ovulate during the ovulatory wave. The rest of the follicles from the cohort termed non-dominant or subordinate undergo atresia (Zeleznik 2001). The dominant follicles due to their increased sensitivity to withstand the low FSH milieu, a consequence of negative feedback from increasing levels of estradiol produced by the growing follicles, continue their growth. The members of the IGF family and their binding proteins have a role in determining follicular selection and / or dominance (Fortune, et al. 2004).
Follicular growth is also associated with increased secretion of steroidal hormones, androgens and estrogens by theca and granulosa cells, respectively, as suggested by the two-cell two-gonadotropin hypothesis (Liu and Hsueh 1986). Increasing estradiol levels promote expression of luteinizing hormone (LH) receptors in granulosa cells thus allowing them to switch their dependence to LH from FSH thereby preparing the follicle, now known as preovulatory or Graafian follicle, to respond to the ovulatory LH surge.
Ovulation involves expulsion of the oocyte (Figure 1) that undergoes final stages of maturation and resumption of meiosis and germinal vesicle breakdown. The follicular somatic cells left behind after ovulation transforms into the endocrine structure called corpus luteum through a process called luteinization (Figure 1). This involves neovascularization of the granulosa cell layer which along with theca cells invade the antrum where they undergo differentiation into luteal cells. Luteinization is also associated with switch in the major hormone production from estradiol to progesterone. Both the ovulatory process and luteinization are intricately regulated by members of various gene families such as proteases that breakdown follicular wall to allow oocyte expulsion, epidermal growth factors that promote cumulus granulosa cell expansion and proteases that breakdown follicular wall to allow oocyte expulsion (Conti, et al. 2012, Richards 2007, Russell and Robker 2007, Stouffer, et al. 2007).
v. Corpus Luteum
Corpus luteum is a transient endocrine structure that produces progesterone required to support uterine development, implantation and pregnancy. The corpus luteum ceases function and structurally involutes through the process known as luteolysis either at the end of a non-fecund reproductive ovarian cycle or when placenta develops to take over its function in the human or at the end of pregnancy in other species. The process of luteolysis may depend on the species as in some species they undergo functional and structural luteolysis and involutes into connective tissue laden corpus albicans while in other species such as rodents, they only undergo functional luteolysis leading to many corpora lutea from multiple cycles being present in the ovary (Smith and Meidan 2014, Stouffer, et al. 2013).
2). Ovarian Pathologies
Ovarian dysfunctions underlie many infertility disorders. There are several ovarian pathologies some of genetic origin and others stemming from genetic susceptibility and environmental interactions. Ovarian pathologies include primary ovary insufficiency (POI), Turner’s syndrome, fragile X syndrome, polycystic ovary syndrome (PCOS), ovarian cysts, ovarian torsion, hyperthecosis, luteomas and ovarian cancers. Although a case can be made for some of these to have extra-ovarian origin, this chapter will discuss conditions that have major changes within the ovary and discuss briefly the influence of extra-ovarian factors in the pathogenesis of the ovarian condition. These conditions emerge due to defects in one or more check points of follicular developmental trajectory such as ovarian reserve, activation, selection, dominance, ovulation or corpus luteum formation (Table 1).
Table 1:
Common ovarian pathologies and the stage of follicular development affected.
| Stages in Ovarian Follicular Development | Phenotype | |||||||
|---|---|---|---|---|---|---|---|---|
| Ovarian Pathologies |
Reserve Establishment |
Reserve Depletion |
Activation/ Recruitment |
Selection | Dominance | Ovulation | Luteal Phase |
|
| Turner’s Syndrome |
↓ | ? | Loss of oocytes | |||||
| Fragile X Syndrome |
? | ↑ | B* | B* | B* | B* | B* | Reduced primordial follicle pool; reduced follicular development |
| Primary Ovarian Insufficiency |
? | ↓ | ↑ | Reduced primordial follicle pool: Premature loss of follicle pool |
||||
| Polycystic Ovary Syndrome |
↑ | B* | B* | B* | B* | Multifollicular appearance; Absence of preovulatory follicles; Absence of corpus luteum |
||
| Follicular Cysts |
B | B | Large diameter cyst | |||||
| Luteal Cysts | B* | B | Fluid filled corpus luteum | |||||
| Luteomas | ↑* | Brownish-yellow nodular mass |
||||||
| Hyperreactio Luteinalis |
↑* | Bilateral cystic ovarian enlargement |
||||||
| Germ Cell Tumors |
? | Ovarian cancer | ||||||
| Stromal Cell Tumors |
? | ? | ? | Ovarian cancer | ||||
↑ Increase; ↓ Decrease; ? Unknown; B Blocked;
Predicted based the ovarian phenotype observed
i. Premature ovarian insufficiency
The POI condition is characterized by decline in ovarian function due to premature depletion of follicles that result in an earlier than average menopause affecting approximately 1 to 2% of women under 40 years-of-age (Monniaux, et al. 2014). This could be the result of chromosomal abnormalities such as mutation in the fragile X mental retardation 1 (FMR1) gene (Qin, et al. 2015), autoimmune disorders (Silva, et al. 2014), or outcome of chemo / radiotherapy (Thomakos, et al. 2015). Additionally, environmental xenobiotic exposures can cause premature menopause potentially due to POI (Grindler, et al. 2015, Hoyer and Keating 2014). The POI in autoimmune disorders may also be associated with endocrine disorders such as hypoparathyroidism and hypoadrenalism (Silva, et al. 2014, Wemeau, et al. 2013). The POI may be the result of increased atresia of early follicular stages or premature activation of the follicular pool leading to its depletion.
ii. Turner’s Syndrome
Turner syndrome, also known as gonadal dysgenesis, is a genetic disorder arising from chromosomal abnormalities containing a missing X chromosome (45,X karyotype) (Shah, et al. 2003). It occurs with a frequency of 0.02% of livebirths and is characterized by gonadal dysgenesis with presence of gonadal streaks, lack of pubertal development, amenorrhea, and hypergonadotropic hypogonadism along with premature ovarian failure (Venkatesh, et al. 2014). Within the ovary, accelerated loss of ovarian follicles by apoptosis begins from 18 weeks of gestation and the rate of depletion varies such that 45,X karyotype phenotypes range from complete loss of follicles to mosaic karyotypes showing follicular development (Novak, et al. 1995).
iii. Fragile X Syndrome
Fragile X syndrome (FXS), also known at Martin-Bell syndrome, characterized by developmental delays, learning disabilities, and behavioral problems also manifest an ovarian phenotype of POI. In these subjects, POI is caused by either a decrease in initial pool of follicles or through an accelerated rate of atresia (Wittenberger, et al. 2007). Other factors that contribute to decreased ovarian function in FXS cannot be ruled out as evidenced from the decreased levels of circulating inhibin B, inhibin A, and progesterone seen in these patients many years preceding POI (Welt, et al. 2004).
iv. Polycystic ovary syndrome
PCOS is the most common infertility disorder affecting about 5.5% to 19.9% of women of reproductive age (Lizneva, et al. 2016). It is a characterized by functional hyperandrogenism (clinical and biochemical), menstrual irregularities, chronic anovulation, polycystic ovaries, and reduced fertility (Conway, et al. 2014, Dumesic, et al. 2015). Majority of patients with PCOS are at increased risk of developing type 2 diabetes mellitus (T2DM) and cardiovascular diseases (Diamanti-Kandarakis and Dunaif 2012, Moran, et al. 2015). The etiology of PCOS remains unknown. Multiple factors including genetics and early developmental exposures have been implicated (Puttabyatappa, et al. 2016). While high familial aggregation (Legro, et al. 1998) and the observations among Dutch twins that heritability for PCOS is 0.79 (Vink, et al. 2006) provide evidence in support of genetic susceptibility, studies with congenital adrenal hyperplasia patients (Barnes, et al. 1994) and animal models (Abbott, et al. 2008, Maliqueo, et al. 2014, Padmanabhan and Veiga-Lopez 2013a) implicate prenatal androgen excess in the development of PCOS.
The ovarian phenotype of PCOS subjects is the multifollicular appearance, stemming from accumulation of antral follicles, which could originate from enhanced recruitment and / or decreased depletion. While longitudinal studies characterizing developmental ontogeny of ovarian defects in PCOS subjects is lacking, cross-sectional studies showing a decrease in number of primordial follicles and a corresponding increase in number of growing follicles are supportive of enhanced recruitment (Webber, et al. 2003). The prevailing endocrine milieu, namely hyperandrogenic and hyperinsulinemic status of PCOS subjects, either by increasing FSH action and / or decreasing the negative regulators of follicular activation might contribute to the enhanced follicular recruitment (Dumesic and Richards 2013). Another contributing factor for development of multifollicular ovarian morphology is follicular arrest that may involve failure to select follicle pool to differentiate further (Franks, et al. 2008), low FSH to stimulate follicular growth (Rebar, et al. 1976) and / or reduced rate of atresia (Webber, et al. 2003). While follicular arrest has been postulated (Franks, et al. 2000), serial ultrasonographic studies are yet to be undertaken in these subjects to prove or disprove this premise.
v. Ovarian cysts
Ovarian cysts are a common problem in women of reproductive age (Kozak, et al. 2005) and domestic animals such as dairy cattle (Kesler and Garverick 1982). In US alone a quarter million women per year have been diagnosed with ovarian cysts (Kozak, et al. 2005). Commonly observed ovarian cysts include functional cysts, luteinized unruptured follicles (LUF), and enlarged follicles. Functional ovarian cysts are observed during the menstrual / estrous cycle and include follicular and corpus luteum cysts. Follicular cysts are ovarian follicles that fail to ovulate, persist for more than 2 cycles and develop 30mm or more in diameter. They usually regress within days or weeks of their development. Hemorrhagic cysts are follicular cysts that are filled with blood through extravasation. Although defects in hypothalamic and pituitary secretions primarily contribute to the follicular cyst development, intra-ovarian contributors cannot be ruled out (Ortega, et al. 2015a). Corpus luteum cysts forms when the follicular remnant after ovulation reseals a fluid filled cavity without infiltration of the luteal cells. LUFs are ovarian cysts where the follicle that reached preovulatory stage, fails to ovulate, entraps the oocyte with rest of the follicle undergoing luteinization. Enlarged follicles are similar to follicular cysts but fail to persist for more than 2 cycles.
vi. Luteomas and hyperreactio luteinalis
These are the two most common ovarian pathologies that are apparent during pregnancy and frequently cause hyperandrogenic state and induce maternal virilization (Joshi and Dunaif 1995, Phelan and Conway 2011). Pregnancy luteomas usually appear as a solid ovarian mass that are benign and in up to 50% of cases present in both ovaries. These appear as brownish-yellow nodular mass made of lutein cells with internal hemorrhagic deposits. This condition is common among Afro-Caribbean ethnic subgroup and other predisposing factors are maternal age, multiparity, and PCOS (Joshi and Dunaif 1995). Growth in these luteomas is promoted by human chorionic gonadotropin (hCG) and they usually regress after childbirth very rarely requiring surgical interventions (Wang, et al. 2005).
Hyperreactio luteinalis (HL) is a relatively rare condition characterized by bilateral cystic ovarian enlargement and excessive androgen production during pregnancy (Joshi and Dunaif 1995). The causes for this condition is not known but similar to luteoma arise due to increased responsiveness to hCG with the cyst made up mostly of theca lutein cells (Bradshaw, et al. 1986). Conditions with elevated levels of hCG such as multiple pregnancy, trophoblastic diseases or chronic kidney disease, can induce the development of HL (Phelan and Conway 2011).
vii. Ovarian cancers
According to the National Cancer Institute (NCI), ovarian cancer is the fifth leading cause of cancer death in women (Al-Alem and Curry 2015). Ovarian cancers can originate in different cell types and include epithelial, sex cord stromal, and germ cell tumors (Bast, et al. 2009). Among these the epithelial cell cancers that originate from ovarian surface or reproductive tract epithelium accounts for about 90% of the ovarian cancers (Al-Alem and Curry 2015). Risk factors include genetics and increased number of ovulatory cycles from nulliparity and early menarche with late menopause (Rossing, et al. 1994). The ovarian surface epithelial cell tumor can develop due to “incessant ovulation” leading to inflammatory reaction, oxidative stress, and release of growth factors that damage the ovarian surface epithelium and promote their growth culminating in the development of tumor (Fathalla 1971).
Gonadal stromal tumors comprise a heterogeneous group of benign and malignant cancers that include granulosa cell tumor (GCT) and Sertoli-Leydig cell tumour (SLCT) (Karnezis, et al. 2017). There are juvenile and adult types of GCT depending on the affected age group and involve somatic missense mutation in the forkhead box L2 gene. Majority of these tumors are estrogenic although androgenic or progestogenic varieties have been reported (Foulkes, et al. 2016). The SLCTs observed in young to middle-aged women are rare tumors involving dicer 1, ribonuclease III (DICER1) mutations that promote Sertoli and Leydig cell differentiation from granulosa cells and often present androgenic phenotypes (Karnezis, et al. 2017).
Ovarian germ cell tumors (OGCT) are derived from primordial germ cells and comprised of a diverse group of histologic subtypes that can be either benign or malignant. Malignant OGCTs, which include dysgerminomas and nondysgerminomas are rare, accounting for only 1% of all ovarian tumors with an average age of onset of age < 25 years (Cools, et al. 2011).
viii. Ovarian torsion
Adnexal torsion is one of a few gynecologic surgical emergencies and refers to rotation of the ovary at its pedicle and consequent occlusion of the ovarian artery and/or vein. Misdiagnosis or delay in treatment can have permanent sequelae including loss of an ovary with effect on future fertility, peritonitis, and even death (Sasaki and Miller 2014). Adnexal torsion commonly occurs in the pregnant women while other predisposing causes are benign ovarian cysts (follicular, luteal or theca lutein cysts), cystadenomas, mature dermoid cysts, malignant tumor, pelvic inflammatory disease, or endometriosis (Ricci, et al. 2016). Torsion of a normal ovary is uncommon but can occur in adolescents (Spinelli, et al. 2015).
3). Basis of ovarian pathologies
Originally, most of the non-infectious disease states were thought to be due to aberrant changes in the organism’s genetic information (DNA sequence) resulting from mutations and deletions or gene fusion, amplification or tandem duplication (Tang and Ho 2007) thus providing the genetic basis for the origin of the disease. In recent years, developmental exposure to adverse insults during critical periods of organ differentiation is also getting attention as one of the leading causes. In the context of ovarian pathologies, as discussed above, the critical period of ovarian differentiation occurs during prenatal life in precocial species (human, monkeys, sheep, cow) and extends to postnatal life in altricial species (rats and mouse) (Padmanabhan and Veiga-Lopez 2013b). Insults occurring during these critical windows result in adaptive changes that could have either positive or negative consequences, a concept known as the developmental origins of health and disease (DoHAD) (Barker 2004). The concept was first proposed based on epidemiological data from the 1944–1945 Dutch famine cohort showing maternal starvation during gestation correlates with an increased risk for cardiovascular and metabolic diseases in the offspring (Ravelli, et al. 1976). Since then it has become quite apparent that varying developmental insults such as maternal undernutrition, overnutrition, stress, lifestyle, disease states and exposures to environmental chemicals contribute to the development of adult reproductive and metabolic pathologies (Padmanabhan, et al. 2016). The genetic and environmental bases for the development of various ovarian pathologies are addressed below.
i. Genetic
Although the genetically determined ovarian disorders such as Turner syndrome and FXS are relatively infrequent, other ovarian conditions such as ovarian cancers, POI, and PCOS that are more frequent appear to also have a genetic contribution (Franks and McCarthy 2004). The genetic basis for ovarian defects for the most part stem from chromosomal aberrations (Turners and FXS) although polymorphism and phenotypes with multifactorial inheritance (POI, PCOS and ovarian cancers) have been reported (Brugh, et al. 2003, Franks and McCarthy 2004).
Turner syndrome characterized by POI stems from a missing X chromosome (45,X karyotype) (Shah, et al. 2003). In most cases the single X chromosome is of maternal origin (Hassold, et al. 1988). 47,XXX is another condition with chromosomal abnormality that has an extra X chromosome of maternal origin and is associated with early onset of menopause (Hassold, et al. 1996). Additionally cases with four or more X chromosomes have been reported where the severity of the symptoms increases with the number of X chromosomes (Shah, et al. 2003). These chromosomal abnormalities involving extra X chromosomes are also characterized by POI suggestive of a strong genetic basis. Gene mutations in the FMR1 gene are shown to be responsible for the POI in FXS patients (Qin, et al. 2015). Gene mutations and gene polymorphisms are implicated in considering the genetic basis for the origin of PCOS and ovarian cancers and techniques such as genome wide association studies have mapped various susceptibility genes for PCOS (Dunaif 2016, Puttabyatappa, et al. 2016, Zhao, et al. 2016) and ovarian cancers (Longacre, et al. 2016).
ii. Developmental insults and programming agents
The impact of early developmental insults as they relate to various ovarian developmental check points that stem from maternal undernutrition, overnutrition, stress and exposure to environmental chemicals are discussed below and summarized in Table 2.
Table 2:
Developmental insults and the stage of follicular development affected.
| Stages in Ovarian Follicular Development | Remarks | |||||||
|---|---|---|---|---|---|---|---|---|
| Maternal Insult | Reserve Establishment |
Reserve Depletion |
Activation/ Recruitment |
Selection | Dominance | Ovulation | Luteal Phase |
|
| Undernutrition | ↓ Chan et al., 2005 Bernal et al., 2010 |
↑* Mossa et al., 2013 |
B* Khorram et al., 2015 |
B* Chan et al., 2005 Khorram et al., 2015 |
Increased oocyte apoptosis; Decrease in primordial and increase in other follicle classes; appearance of cysts |
|||
| Overnutrition | ↓* Tsoulis et al., 2016 |
↑* Cheong et al., 2014 |
B* Cheong et al., 2014 |
B* Cheong et al., 2014 |
Increase in oocyte apoptosis; Decrease in primordial and increase in other follicle classes; follicular developmental defects |
|||
| Stress | ↓* Barra et al., 2014 Ashworth et al., 2016 |
↑* Barra et al., 2014 Ashworth et al., 2016 Hulas-Stasiak et al., 2015 Ristic et al., 2008 |
Increased oocyte apoptosis; Decrease in primordial and increase in other follicle classes |
|||||
| EDC | ↓ | ↑ | ↑ | B | B | B | See below for specific EDC examples |
|
| -Bisphenol A | ↓* Karowan and Pepling 2012 Adewale et al., 2009 |
↓* | ↓* Karowan and Pepling 2012 |
B* Newbold et al., 2007 Markey et al., 2003 |
B* Newbold et al., 2007 Markey et al., 2003 |
B* Newbold et al., 2007 Markey et al., 2003 |
Reduced germ nest breakdown; increased primordial follicles at early age with increased antral and cystic follicles at older age |
|
| -Methoxychlor | ↑* | ↑* Armenti et al., 2008 |
B* Armenti et al., 2008 |
B* Armenti et al., 2008 |
B* Armenti et al., 2008 |
Increased number of preantral and antral follicles; reduced corpus luteum; numerous cystic follicles |
||
| -Diethylstilbesterol | ↓ Kim et al., 2009 |
↓* | ↓* Karavan & Pepling 2012 Yamamoto et al., 2003 |
B* Newbold et al., 1983 |
B* Newbold et al., 1983 |
Reduced germ cell nest breakdown; increased primordial follicles at early age with increased antral and cystic follicles at older age |
||
| -Phthalates | ↓ Zhang et al., 2014 |
↑* | ↑ Moyer et al., 2012 |
B* Moyer et al., 2012 |
B* Moyer et al., 2012 |
Reduced germ cell nest breakdown; increased preantral and antral follicles |
||
↑ Increase; ↓ Decrease; B Blocked;
Indicates the affected follicular developmental stage is predicted based on the phenotype observed
a. Maternal Undernutrition
The concept that maternal undernutrition through either caloric or protein reduction leads to in utero growth restriction (IUGR) and adult onset metabolic and cardiovascular diseases (Barker and Osmond 1988), also extends to ovarian pathologies. In general, clinical and experimental studies provide evidence that nutritional deficiency during early-life is associated with a decline in ovarian follicular reserve and changes in ovulation rates (Chan, et al. 2015). For instance, one study in rats found maternal undernutrition that lasted throughout the pregnancy and lactation resulted in reduced ovarian reserve (Bernal, et al. 2010). Similarly, undernourishment during the first trimester in cows, a precocial species as human, also resulted in reduced ovarian reserve in the offspring, based on their reduction in their antral follicle count (Mossa, et al. 2013). Another study in rodents found maternal undernutrition confined to just the second half of the pregnancy resulted in large ovarian cysts, reduced number of corpora lutea, and elevated circulating testosterone (T) (Khorram, et al. 2015). The differences in phenotypic outcomes in the two rodent studies (Bernal, et al. 2010, Khorram, et al. 2015) may relate to differences in the timing of undernutrition. To what extent such outcomes are a function of the IUGR induced by maternal undernutrition is unclear.
b. Maternal Overnutrition
Overnutrition, the opposite end of the nutritional spectrum from undernutrition, and consequent obesity in women is also associated with increased reproductive risks manifested as irregular menstrual cycles and anovulation and PCOS (Naderpoor, et al. 2015). Obesity is also a risk factor for abnormal ovarian development in the offspring. For instance, neonatal offspring of rats fed high fat diet became obese, had fewer oocytes at embryonic day 20 and showed reduced AMH signaling at postnatal day 4 that favor increased follicular activation (Tsoulis, et al. 2016). As adults these offspring manifested increased follicular atresia, reduced FSH responsiveness and high AMH levels in antral follicles indicative of a compromised follicular environment. Maternal high fat diet also produced deleterious effects in mice evidenced as obesity, reduced primordial, antral, and Graafian follicular numbers along with increased expression levels of FOXO3A and GDF9 (Cheong, et al. 2014). Similar studies with precocial species that focus on ovarian pathologies are lacking.
Considering maternal obesity also leads to offspring obesity (Zambrano and Nathanielsz 2013), it is unclear to what extent the ovarian disruptions seen in the offspring are a function of the obese phenotype of the offspring themselves, which is likely to replicate the maternal environment of the obese mothers. Reduced adipokine and increased inflammatory markers are hallmarks of obese mothers (Jungheim, et al. 2012), aspects that are likely to be detrimental to ovarian function (Boots and Jungheim 2015, Campos, et al. 2015). If so, the impact of maternal obesity on oocyte metabolism, meiosis and maturity will also be seen in the obese F1 generation, setting up a self-perpetuating vicious cycle. The effect of obesity on the ovary may be reflected as lipid accumulation in oocytes (Robker, et al. 2009), a detriment to oocyte competence (Wu, et al. 2011). For instance, increases in follicular fluid free fatty acid concentrations in women undergoing IVF have been found to be associated with poor oocyte quality (Jungheim, et al. 2011) and saturated free fatty acids have been shown to induce apoptosis of human granulosa cells (Mu, et al. 2001). Multi-generational studies focusing on the impact of maternal obesity on offspring ovarian reserve and function along with offspring metabolic phenotype are required to determine if ovarian dysfunctions are a reflection of repetitive obese phenotype reflecting the prevailing metabolic environment or transgenerationally programmed.
c. Maternal Stress
Maternal stress can arise from major life events bereavement, catastrophic events, depression or anxiety (Dunkel Schetter 2011) involving classic endocrine response of increased glucocorticoid secretion that could underlie abnormal ovarian programming. Programming by maternal undernutrition discussed above, may also be secondary to the stress response they manifest (Whirledge and Cidlowski 2010). Stress can also elevate sympathetic neurotransmitters such as norepinephrine (Nugent, et al. 2012) and influence ovarian function (Lara, et al. 2002).
Animal models of maternal stress have provided evidence supportive of maladaptive programming of offspring ovarian differentiation and function. For instance, gestational exposure of rats to cold stress was found to reduce primordial follicle formation in the offspring either through reduced germinal nest breakdown or increase in oocyte apoptosis indicative of reduced ovarian reserve as well as decrease in the number of growing follicles suggestive of defects in the follicular activation / recruitment process (Barra, et al. 2014). Likewise prenatal social stress in pigs (Ashworth, et al. 2011) and rats (Ashworth, et al. 2016) resulted in fewer primordial and more primary follicles, respectively, stressing the impact of maternal stress in programming offspring ovarian reserve and follicular activation. To what extent the impact of other stressors such as heat and bright light involve ovarian developmental defects in delaying vaginal opening and increasing estrus cycle length in the offspring (Politch and Herrenkohl 1984) remains to be ascertained.
Because glucocorticoid secretion is the main endocrine response during stress, animal models administered glucocorticoids during gestation help address the effects of stress hormones on the ovary. For instance, prenatal dexamethasone, a synthetic glucocorticoid, treatment resulted in a decrease in primordial follicular number in spiny mouse (Hulas-Stasiak, et al. 2015) and rat (Ristic, et al. 2008). Because maternal stress also leads to maternal and fetal androgen excess as evidenced from increases in ano-genital distance (Barrett and Swan 2015, Herrenkohl and Scott 1984) and elevations in circulating levels of T (Barrett and Swan 2015, Takahashi, et al. 1998), another well-established programming agent (Hakim, et al. 2016), to what extent androgens are involved in mediating the ovarian response in stress models remains to be addressed.
d. Environmental Chemical Exposures
Targeted studies in animals have provided enough evidence that developmental exposure to endocrine disrupting chemicals (EDCs), chemicals with endocrine like functions (Diamanti-Kandarakis, et al. 2009, Gore, et al. 2015), affect offspring ovarian development and function. Maternal exposure to common EDCs such as methoxychlor (MXC), diethylstilbestrol (DES), bisphenol A (BPA), phthalates and genistein have an impact on the ovary in various animal models culminating in reduced ovarian reserve, POI, and / or development of cystic follicles (Crain, et al. 2008, Hannon and Flaws 2015, Zama and Uzumcu 2010). For instance, perinatal (gestational day 19 to postnatal day 7) exposure to MXC in the rat and gestational (days 9–16 or 9–20) exposure to BPA in mice increased the number of preantral and antral follicles, induced cystic follicles, and reduced the number of corpus luteum (Armenti, et al. 2008, Markey, et al. 2003, Newbold, et al. 2009), indicative of defects at the level of follicular activation / recruitment, selection, dominance and ovulation. Other studies found neonatal exposure to diethylstilbestrol (DES) in mice (Kim, et al. 2009) and BPA in rats (Adewale, et al. 2009) lead to the development of multioocyte follicles indicative of defects in the establishment of the ovarian reserve. Amongst the phthalates, neonatal diethylhexyl phthalate (DEHP) treatment affected germ cell nest breakdown (Zhang, et al. 2014), while gestational (days 17–19) mono-2-ethylhexylphthalate (MEHP) exposure increased number of preantral and antral follicles in the offspring (Moyer and Hixon 2012) indicating that the former negatively affected the establishment of ovarian reserve and the latter activation / recruitment process suggestive of early depletion of ovarian reserve and potential for premature reproductive aging. These and other observations that are reviewed elsewhere (Hannon and Flaws 2015, Peretz, et al. 2014, Zama and Uzumcu 2010) point to the profound impact of perinatal EDC exposure in programming ovarian defects (Nilsson and Skinner 2015).
Although association studies point to detrimental effects of EDC exposure on ovarian function in humans (Bhattacharya and Keating 2012, Craig, et al. 2011, Hoyer and Keating 2014), the impact of developmental exposures to EDCs on ovarian function in the offspring have not been studied. Findings that 1) EDCs are present in the human maternal circulation (Inoue, et al. 2004, Mazdai, et al. 2003, Peretz, et al. 2014, Sathyanarayana, et al. 2008), 2) EDCs such as BPA are associated with excess androgen - a well-documented developmental programming agent (Zhou, et al. 2008), 3) observations of reduced pregnancy rates in daughters of mothers with high serum dichlorodiphenyltrichloroethane (DDT) compounds (Cohn, et al. 2003), and 4) a steady decline in fertility in the developed world with increase in the manufacture and use of chemicals (Woodruff 2011) indicate that developmental exposures to EDC are a threat to female reproductive function. To what extent this is manifested at the ovarian level, is a research gap to focus future studies upon.
4). Mechanism of programmed disruptions
Although the types of ovarian defects induced by the different developmental insults (undernutrition, overnutrition, stress, EDCs) reflect the timing of exposure as it relates to critical periods of ovarian differentiation, common mediators and mechanisms appear to be involved (Padmanabhan, et al. 2016). The common programming mediators include steroidal and metabolic factors.
i. Steroidal and metabolic factors in programming ovarian defects
Hormones, both steroidal and metabolic, play an important role in organizing the structure / function of various organ systems including the ovary. Among the steroidal hormones estradiol, T and glucocorticoids of gonadal and / or adrenal origin are the major programmers of reproductive dysfunctions. The fact that the undifferentiated gonad develops into a testes or ovary depending on the presence or absence of T during the critical period of gonadal differentiation - former driving the development of testes and the latter formation of ovaries (Capel 2000, Jost 1983) - highlight the importance of steroid hormones as programming agents of ovarian differentiation and dysfunctions in offspring.
The role played by abnormal maternal exposure to steroids in the development of ovarian pathologies in the offspring is evident from animal studies involving administration of agonist, receptor antagonist, steroid synthesis inhibitors, or EDCs with steroidogenic potential during the developmental window (Table 3). For instance creation of a hypoestrogenic milieu in fetal baboon by maternal treatment with letrozole - an aromatase inhibitor - from mid to late gestation leads to a reduction in primordial follicular numbers that was prevented by estradiol co-treatment (Zachos, et al. 2002), thus stressing the role played by estradiol in setting the ovarian reserve. In altricial species such as mice, where this window occurs during postnatal life, administration of native (Chen, et al. 2007) or synthetic (Karavan and Pepling 2012) estradiol during neonatal life also reduces ovarian reserve. Treatment with DES, a potent synthetic estrogen, from days 7–21 of gestation increases the percentages of primary and secondary ovarian follicles in the pups (Yamamoto, et al. 2003) and induces inflammation, medullary tubule-like structures in the interstitial compartment and intra- and para-ovarian cysts in the 10 month offspring (Newbold, et al. 1983).
Table 3:
Steroidal influence on the stage of follicular development.
| Animal Model | Reference | Stages in Ovarian Follicular Development | Remarks | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Species | Treatment | Reserve Establishment |
Reserve Depletion |
Activation /Recruitment |
Selection | Dominance | Ovulation | Luteal Phase |
||
| Rhesus Macaques |
Testosterone GD 40- 55/65/75 (term 164) |
Abbott et al 2005 |
B* | B* | B* | Anovulation; polyfollicular ovarian morphology |
||||
| Rhesus Macaques |
Testosterone GD 100/115- 124/139 (term 164) |
Abbott et al 2005 |
B* | B* | B* | Anovulation; polyfollicular ovarian morphology |
||||
| Baboon | Letrozole GD 100–164 (term 184) |
Zachos et al., 2002 |
↓ | ↓ | Ovarian outer cortex has fewer follicles and more interfollicular nests in fetuses. Adult ovarian phenotype not assessed |
|||||
| Sufflock Sheep |
Testosterone GD 30–90 (term 147) |
Padmanabhan & Veiga-Lopez 2013b |
↑ | ↑ | B | B | B | B | Multifollicular phenotype; reduced primordial and increase in other follicular classes; anovulation |
|
| Sufflock Sheep |
Testosterone GD 60–90 (term 147) |
Steckler et al., 2007 Veiga-Lopez et al, 2016 |
↓* | Altered intrafollicular steroids; Reduced fecundity |
||||||
| Sufflock Sheep |
DHT GD 30–90 (term 147) |
Padmanabhan & Veiga-Lopez 2013b |
↑ | ↑ | Decrease in percentage of primordial follicles with corresponding increase in other follicular classes in fetal ovary |
|||||
| Scottish Greyface x Texel Sheep |
Testosterone GD 62–102 (term 147) |
Hogg et al., 2011 Hogg et al., 2012 |
No changes in ovarian morphology or follicular counts at fetal and 11 months of age |
|||||||
| Rat (Sprague Dawley) |
Testosterone GD 16–19 (term 21) |
Wu et al., 2010 | ↑* | B* | B* | Increase in preantral and antral follicles; fewer preovulatory follicles and corpus luteum |
||||
| Rat (Sprague Dawley) |
DHT GD 16–19 (term 21) |
Wu et al., 2010 | ↑* | B* | B* | Increase in preantral and antral follicles; fewer preovulatory follicles and corpus luteum |
||||
| Mouse | DHT GD 16–18 (term 20) |
Rolland et al., 2010 |
B* | Longer estrous cycle | ||||||
| Rat# (Wistar) |
DHT 21–111d PN |
Manneras et al., 2007 |
B* | B* | B* | Multiple Cystic follicles | ||||
| Rat# (Wistar) |
Letrozole 21–111d PN |
Manneras et al., 2007 |
B* | B* | B* | Multiple Cystic follicles | ||||
| Rat# (Wistar) |
Letrozole 34–55d PN |
Kafali et al 2004 |
B* | B* | B* | Multiple Cystic follicles | ||||
| Rat# (Wistar) |
Letrozole 56–77d PN |
Baravalle et al 2006 |
B* | B* | B* | Multiple Cystic follicles | ||||
| Mouse# | Letrozole 28–63d PN |
Kauffman et al., 2015 |
B* | B* | B* | Multiple Cystic follicles | ||||
| Rat# (Wistar) |
Estradiol Valerate 14d PN |
Rosa-E-Silva et al., 2003 | B* | B* | B* | Multiple Cystic follicles | ||||
| Mouse | Estradiol 1–4d PN |
Karowan and Pepling 2012 Chen et al 2007 |
↓ | ↓ | ↓ | Impaired germ cell nest breakdown; increased primordial follicle numbers |
||||
| Mouse (CD1) |
Ethinyl estradiol 1–4d PN |
Karowan and Pepling 2012 |
↓ | ↓ | ↓ | Impaired germ cell nest breakdown and increased primordial follicle numbers |
||||
GD = Gestational day; DHT = Dihydrotestosterone; PN = postnatal. ↑ Increase; ↓ Decrease; B Blocked;
Indicates the affected follicular developmental stage is predicted based on the phenotype observed;
Indicates animal models with treatments outside the ovarian developmental window.
While studies testing the effects of maternal exposure to excess estradiol or deficiency at the ovarian level are few, a large number of studies involving both altricial and precocial models have investigated the effects of maternal exposure to T in the context of developmental origin of PCOS (Abbott, et al. 2008, Maliqueo, et al. 2014, Padmanabhan and Veiga-Lopez 2013a). Amongst the animal models studied, Suffolk sheep model of gestational T exposure is the only model that has undertaken serial ultrasonographic studies and extensively investigated the ovary at the organismal, tissue and molecular level to relate phenotypic changes with underlying molecular changes (Cardoso, et al. 2015, Padmanabhan and Veiga-Lopez 2013b). T excess from days 30 to 90 of gestation in sheep doubled the number of primordial follicles in day 90 T-treated fetuses, supportive of advancement in follicular differentiation (Comim, et al. 2015), produced a multifollicular ovarian morphology due to accumulation of antral follicles, increased recruitment, induced follicular persistence, and induced ovulatory and corpus luteum dysfunctions (Cardoso, et al. 2015, Padmanabhan and Veiga-Lopez 2013b), defects also reported in women with PCOS (Dumesic and Richards 2013, Padmanabhan and Veiga-Lopez 2013b).
Serial ultrasonographic studies in this sheep model found multifollicular appearance to be the result of increased follicular recruitment and follicular persistence (Cardoso, et al. 2015). Of relevance, a multifollicular phenotype is also evident in prenatal T-treated monkeys (Abbott, et al. 2008), although serial ultrasonographic studies are lacking to assess contribution from recruitment and persistence. As opposed to the arrested follicular phenotype of women with PCOS (Franks, et al. 2008), prenatal T-treated monkeys (Abbott, et al. 2008) and sheep (Padmanabhan and Veiga-Lopez 2013b), the follicles in prenatal T-treated rats appear to be cystic in nature (Maliqueo, et al. 2014).
Examination of key mediators of ovarian function at the tissue level in the 30–90 day T-treated sheep model of PCOS has yielded mechanistic insights (Figure 2) linking molecular / protein changes with functional changes. Importantly, use of ovarian sections from the same set of animals for assessment of various molecular / protein markers has helped obtain an integrative picture of mechanisms underlying the ovarian pathology. These investigations indicate that increased androgen receptor expression (which promotes follicular activation) (Ortega, et al. 2009), in the absence of changes in B-cell lymphoma 2 (BCL2, which instills resistance to atresia) and reduced BCL2-associated X protein (Salvetti, et al. 2012) in early growing follicles are the likely contributors to the enhanced follicular recruitment. On the contrary, increased expression levels of follistatin, reduced activin βB (West, et al. 2001) and increased AMH protein expression (Veiga-Lopez, et al. 2012) in preantral and antral follicles likely creates a net negative FSH milieu, thus contributing to the follicular arrest and follicular persistence. In addition, reduced adiponectin - an insulin sensitizer, (Ortega, et al. 2010), alterations in (1) the apoptotic machinery manifested as decreased BCL2 and caspase 3 in granulosa / theca cells of antral follicles (Salvetti, et al. 2012), (2) intrafollicular androgen / estrogen ratio the result of reduced granulosa aromatase (CYP19A1) and thecal 17α hydroxylase (CYP17A1) (Padmanabhan, et al. 2014), (3) steroid receptor balance expressed as an increase in estrogen receptor α (ESR1) and decrease in ESR 2 coupled with increased androgen receptor expression in granulosa cells of antral follicle (Ortega, et al. 2009) and (4) ratio of metalloproteases to its inhibitor (Puttabyatappa, et al. 2017) in antral follicles, as well as disruptions in ovarian stromal vasculature (Ortega, et al. 2015b) are all consistent with follicular arrest and /or block in the progression of preovulatory follicle development.
Figure 2:
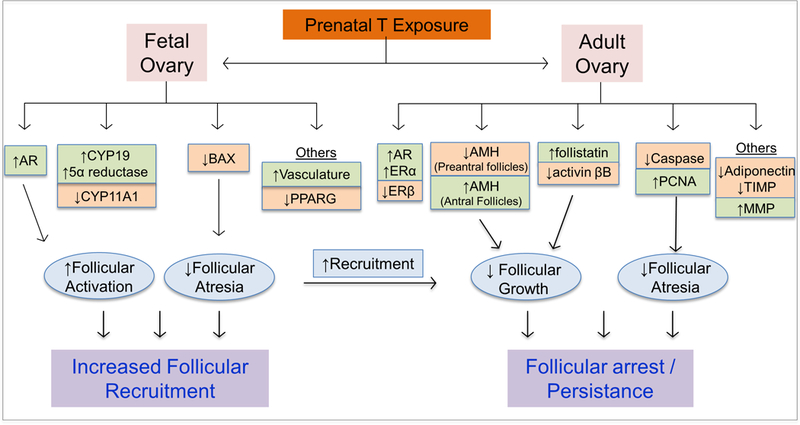
Fetal and adult ovarian changes that account for enhanced follicular recruitment and follicular persistence evidenced in prenatal T-treated sheep. Increases in gene / protein markers are denoted in green and decreases in orange.
While ovarian stimulation studies found exogenous gonadotropins are capable of rescuing ovulatory function in prenatal T-treated (days 30–90 of fetal life) sheep (Steckler, et al. 2008), similar studies using an even less severe phenotype (T treatment from days 60–90 of fetal life) found ovarian defects continue to persist in the developing preovulatory follicles and involves impairment in the final maturation step and intrafollicular steroid milieu (Veiga-Lopez, et al. 2016). Disruptions in intrafollicular steroidal milieu accompanied by a reduction in oocyte competence have also been evidenced in prenatal T-treated monkeys (Dumesic, et al. 2003), a finding that could potentially explain the reduced fecundity observed in prenatal T-treated sheep (Steckler, et al. 2007).
Other studies with Scottish Greyface x Texel sheep found T treatment from gestational days 60 to 90 or 62 to102 did not produce multifollicular ovarian phenotype (Hogg, et al. 2012) indicating that the critical window for programming this phenotype spans from days 30–60 of gestation, when gonadal differentiation occurs. However, in vitro studies of follicles from these prenatal T-treated sheep provided evidence in support of increased thecal androgen production (Hogg, et al. 2012). In addition these investigators found that a single injection of T directly into the fetus on day 60 altered the expression pattern of key genes involved in steroid biosynthesis and action (Hogg, et al. 2011). The functional significance of the findings from the direct fetal T administration and late gestational Scottish Greyface x Texel T exposure models are unclear, as adult reproductive phenotypes remain yet to be characterized.
As T can be aromatized to estradiol and gestational T treatment increases maternal and fetal T and estradiol concentrations (Abi Salloum, et al. 2015), the ovarian changes detailed above could be programmed by androgenic and estrogenic pathways. Comparative studies with prenatal dihydrotestosterone (DHT, a non-aromatizable form of T) treated sheep suggest that the programming of follicular recruitment is androgen-dependent while the follicular persistence and multifollicular appearance is estrogen-dependent (Padmanabhan and Veiga-Lopez 2011). However, because DHT can be metabolized to estrogen agonist 3β-diol (Handa, et al. 2008) and exert its action via estrogen receptor beta, one needs to exert caution in interpreting these findings relative to the type of steroid serving as programming agent.
As opposed to the information available on prenatal programming of ovarian structure / function by gonadal steroids, the role played by glucocorticoids in the developmental programming of offspring ovarian defects are yet to be elucidated. However, because dexamethasone has been shown to increase oocyte apoptosis in fetal ovaries in vitro (Poulain, et al. 2012), the potential for stress-induced increases in glucocorticoids having an effect in establishing the ovarian reserve cannot be overlooked.
Relating the findings from prenatal T and estradiol-treated models to undernutrition, overnutrition, stress and EDC exposure models discussed in section 3, there are commonalities in phenotypic outcomes. For instance, the impact of prenatal undernourishment, overnourishment, or stress on follicular activation / recruitment etc. parallel that seen after prenatal exposure to excess T and thus may relate to the elevated androgen levels seen also in these models (Barrett and Swan 2015, Mossa, et al. 2013, Whyte, et al. 2007).
Similarly, the impact of developmental exposure to EDCs on ovarian function may relate to their potential to serve as steroidomimetics and / or alter steroid biosynthesis (Diamanti-Kandarakis, et al. 2009, Gore 2008). EDCs with steroidomimetic capability include 1) phytoestrogens, dioxins, MXC, dichlorodiphenyltrichloroethane (DDT), and endosulfan that affect estrogen signaling pathways, 2) vinclozolin and DDT metabolites that affect androgen receptor actions, and 3) BPA, phthalates and polychlorinated biphenyl (PCB) that affect both pathways (Cooper and Kavlock 1997).
In stark contrast to the data available relative to steroidal programming of ovarian function, information relative to metabolic programming of offspring ovarian perturbations is limiting. Clearly maternal undernutrition (Fowden and Forhead 2004) and overnutrition (Holemans, et al. 2004) leads to metabolic alterations manifested as hyperinsulinemia, hyperleptinemia, hyperglycemia, dyslipidemia, etc. It needs to be recognized that some of the changes observed in prenatal T-treated sheep model discussed above may have also been subject to programming influences via maternal hyperinsulinemia induced by gestational T treatment (Abi Salloum, et al. 2015). Because hyperinsulinemic and hyperandrogenic states often co-exist during pregnancy in other disorders such as PCOS, obesity, and preeclampsia, it is difficult to pinpoint the specific metabolic / steroidal contributions programming the ovarian pathology. Because insulin is required for establishment of ovarian reserve (Isik, et al. 2012) metabolic contributions to programming of ovarian dysfunctions are likely to exist. The programing effects of other factors associated with metabolic disorders such as adipokines, chemokines, amino acids, fatty acids and glucose (Heerwagen, et al. 2010, Padmanabhan, et al. 2016), that have been shown to have direct effects at the ovarian level, have not been investigated in the context of developmental origin of ovarian pathologies. Furthermore, elevated reactive oxygen species generated through oxidative stress in metabolic disorders (Dong, et al. 2013) due to their potential to damage cell components, such as proteins, lipids, and DNA (Valko, et al. 2007), can also have an impact on the developing ovary. Interventional studies targeting the metabolic factor in question, such as gestational insulin sensitizer treatment and follow up of ovarian function in offspring are very much needed to address the role played by metabolic factors in programming ovarian dysfunctions in the offspring.
ii. Epigenetic mechanisms
The mechanisms by which steroidal and metabolic mediators program the ovarian developmental trajectory likely involve epigenetic modulations. Epigenetic changes involve heritable changes in the gene function without altering the DNA sequence. These occur through methylation of DNA, histone modifications, and noncoding RNA expression. Methylation of DNA occurs at cytosine residues in CpG dinucleotides (Ehrlich and Lacey 2013). Histone modification involves acetylation, methylation, phosphorylation, palmitoylation and ubiquitination of amino acid residues (Turner 2011). Noncoding RNAs include micro, small and long RNA that do not code for any proteins but regulate gene expression of target genes through post-transcriptional processing (Amaral and Mattick 2008). These epigenetic modifications can reduce (or silence) expression or degrade mRNA post-transcription (Heard and Martienssen 2014) of keys genes involved in programming ovarian function.
Relatively little is known in terms of epigenetic programming of ovarian disorders with most studies pointing to associations than causality. For instance, the programming of ovarian dysfunction in the prenatal T-treated sheep model discussed above involves epigenetic alterations as evidenced from increased expression of microRNAs in fetal day 65 ovaries, several of which are predicted to target ESR1, CYP19 and Sry-related high-mobility-group box (SOX) family (Luense, et al. 2011), genes of importance to gonadal development. Further evidence that epigenetic alterations are involved in programming ovarian disorders comes from studies with EDCs (Huo, et al. 2015, Zama and Uzumcu 2010). For instance, gestational BPA treatment was found to increase expression of microRNAs that regulate SOX family genes, kit ligand and insulin-related genes that regulate ovarian follicular differentiation and insulin sensitivity (Veiga-Lopez, et al. 2013). Because silencing kit ligand, a growth factor that maintains primordial follicular pool, would negatively affect the ovarian reserve (Gougeon 2004), the ovarian defects observed in gestational (9–16 days) BPA exposed mice (Newbold, et al. 2009) may be secondary to the impact on the microRNA that regulates Kit ligand expression. Similarly, exposures to MXC, BPA and DEHP have been found to alter DNA methyl transferase (DNMT), an enzyme involved in methylating gene promoters (Cruz, et al. 2014). In other studies with ovaries from perinatal MXC-exposed rats, targeted genome-wide methylation analysis revealed hypermethylation of multiple genes involved in estrogen receptor, phosphatase and tensin homolog (PTEN) and insulin-mediated cell signaling along with increase in DNMT expression (Zama and Uzumcu 2013). The accelerated follicle loss observed in these rats (Zama and Uzumcu 2013) may therefore result from silencing of genes involved in PTEN signaling such as Foxo3, likely due to removal of the inhibitory action on AKT (Sobinoff, et al. 2013). Because BPA exposure has been shown to reduce histone methylation in the mouse follicles in vitro (Trapphoff, et al. 2013), the involvement of histone modifications in the developmental programming of ovarian dysfunctions by BPA and other EDCs cannot be ruled out.
Importantly, epigenetic changes induced by developmental insults can be transferred to subsequent generations (Miska and Ferguson-Smith 2016). A role for epigenetics in the etiology of ovarian disease and transgenerational transmission is evident at the level of epigenome and transcriptome in the F3 granulosa cells following ancestral exposure to fungicide, pesticide mixture, plastic mixture, dioxin and a hydrocarbon mixture spanning the window of embryonic gonadal sex determination (Skinner 2014). These changes appear to underlie the disease phenotypes manifested as increase in ovarian cysts and reduction in primordial follicle pool in F1 and F3 offspring (Nilsson and Skinner 2015). In addition, crossing the ancestrally MXC exposed F3 females with wild types male rats produced similar ovarian defects (reduction in primordial pools and polycystic ovaries) in F4 females indicating that the epigenetic transfer occurs through female germline (Manikkam, et al. 2014). These emerging data indicates genome wide epigenetic alterations indeed occurs in response to developmental insults and point to the need for developing intervention strategies such as use of dietary supplements (methyl donors like folic acid) to prevent epigenetic alterations and development of ovarian pathologies.
5). Conclusions
As discussed above, in addition to genetic basis, developmental insults also contribute to the establishment of many ovarian pathologies. These insults, albeit of varying origin, target several developmental check points such as establishment of ovarian reserve, activation / recruitment, selection, dominance, ovulation and formation of corpus luteum with the final phenotypic outcome likely dictated by the developmental time point of insult. All these developmental insults work via common mediaries such as steroidal and metabolic factors to induce gene-specific epigenetic changes that alter the expression pattern of genes and consequently the ovarian developmental trajectory culminating in adult pathologies. With many such insults, it is difficult to distinguish the contribution from steroidal and metabolic mediaries in programming the pathologies as they often coexist. Interventions targeting the specific steroidal or metabolic factors are necessary to identify the specific mediator or determine if both metabolic and steroidal factors synergize in the programming of the pathology.
Although the focus of this chapter has been on the impact of developmental insults, recent evidence suggests that a subsequent insult (a second hit) has the potential to amplify or unmask defects induced by the first insult (Puttabyatappa, et al. 2016). For instance, postnatal obesity in sheep amplifies the reproductive defects (Steckler, et al. 2009) and postnatal estradiol increases the incidences of subluteal cycles (Veiga-Lopez, et al. 2014) in prenatal T-treated sheep. Such studies are lacking at the ovarian level. Considering the potential for interaction between developmental insults and subsequent changes in lifestyle, diet, and environmental exposures, this is an important avenue for future investigation. In addition, because susceptibility to a given insult is likely to differ from individual to individual, focus on gene x environment interactions should be taken into consideration in probing these pathologies.
It also needs to be recognized that for the most part impacts of the various developmental insults at the level of the ovary have come mostly from altricial species. While this has been crucial for addressing underlying mechanisms such as transgenerational transfer of pathology to subsequent generations and testing intervention strategies, similar studies are required in precocial species such as sheep, cow and non-human primates for effective human translation. This is important as ovarian differentiation in these species gets completed in utero similar to humans, as opposed to them getting completed postnatally in altricial models.
Since targeted mechanistic studies are not possible in human at the ovarian level, there is also a need for development of biomarkers for recognizing early onset of developmentally programmed ovarian pathologies. Currently, AMH and antral follicular count are widely used to assess recruitment defects and declining ovarian reserve in patients undergoing assisted reproductive technologies (ART) procedures (Tal and Seifer 2017). Noninvasive ultrasonography tools and circulating biomarkers are very much needed for early detection of ovarian dysfunctions in ‘at risk’ individuals.
Effective intervention strategies are required to prevent ovarian pathologies originating or amplified by developmental insults. Because use of pharmacological agents that block common endocrine programmers such as T and insulin have the potential to serve as programming agents themselves, intervention strategies need to focus on lifestyle changes. When environmental endocrine disrupting chemicals are identified as the culprit, focus needs to be not only on lifestyle changes but also at the regulatory level to reduce exposures.
Acknowledgments
This work was supported by National Institutes of Health grant P01 HD44232.
Contributor Information
Muraly Puttabyatappa, Department of Pediatrics, University of Michigan, Ann Arbor, MI.
Vasantha Padmanabhan, Department of Pediatrics, University of Michigan, Ann Arbor, MI.
References
- Abbott DH, Barnett DK, Bruns CM & Dumesic DA 2005. Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome? Hum Reprod Update 11 357–374. [DOI] [PubMed] [Google Scholar]
- Abbott DH, Barnett DK, Levine JE, Padmanabhan V, Dumesic DA, Jacoris S, and Tarantal AF 2008. Endocrine antecedents of polycystic ovary syndrome in fetal and infant prenatally androgenized female rhesus monkeys. Biol Reprod 79 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abi Salloum B, Veiga-Lopez A, Abbott DH, Burant CF, and Padmanabhan V 2015. Developmental programming: exposure to testosterone excess disrupts steroidal and metabolic environment in pregnant sheep. Endocrinology 156 2323–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adewale HB, Jefferson WN, Newbold RR, and Patisaul HB 2009. Neonatal bisphenol-a exposure alters rat reproductive development and ovarian morphology without impairing activation of gonadotropin-releasing hormone neurons. Biol Reprod 81 690–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexanderson C, Eriksson E, Stener-Victorin E, Lystig T, Gabrielsson B, Lonn M & Holmang A 2007. Postnatal testosterone exposure results in insulin resistance, enlarged mesenteric adipocytes, and an atherogenic lipid profile in adult female rats: comparisons with estradiol and dihydrotestosterone. Endocrinology 148 5369–5376. [DOI] [PubMed] [Google Scholar]
- Al-Alem L, and Curry TE Jr 2015. Ovarian cancer: involvement of the matrix metalloproteinases. Reproduction 150 R55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertini DF 2015. Chapter 2 - The Mammalian Oocyte, Knobil and Neill’s Physiology of Reproduction (Fourth Edition), pp. 59–97. San Diego: Academic Press. [Google Scholar]
- Amaral PP, and Mattick JS 2008. Noncoding RNA in development. Mamm Genome 19 454–492. [DOI] [PubMed] [Google Scholar]
- Armenti AE, Zama AM, Passantino L, and Uzumcu M 2008. Developmental methoxychlor exposure affects multiple reproductive parameters and ovarian folliculogenesis and gene expression in adult rats. Toxicol Appl Pharmacol 233 286–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth CJ, George SO, Hogg CO, Lai YT, and Brunton PJ 2016. Sex-specific prenatal stress effects on the rat reproductive axis and adrenal gland structure. Reproduction 151 709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth CJ, Hogg CO, Hoeks CW, Donald RD, Duncan WC, Lawrence AB, and Rutherford KM 2011. Pre-natal social stress and post-natal pain affect the developing pig reproductive axis. Reproduction 142 907–914. [DOI] [PubMed] [Google Scholar]
- Baerwald AR, Adams GP, and Pierson RA 2003. Characterization of ovarian follicular wave dynamics in women. Biol Reprod 69 1023–1031. [DOI] [PubMed] [Google Scholar]
- Baerwald AR, Adams GP, and Pierson RA 2012. Ovarian antral folliculogenesis during the human menstrual cycle: a review. Hum Reprod Update 18 73–91. [DOI] [PubMed] [Google Scholar]
- Baravalle C, Salvetti NR, Mira GA, Pezzone N & Ortega HH 2006. Microscopic characterization of follicular structures in letrozole-induced polycystic ovarian syndrome in the rat. Arch Med Res 37 830–839. [DOI] [PubMed] [Google Scholar]
- Barker D, and Osmond C 1988. Low birth weight and hypertension. BMJ: British Medical Journal 297–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ 2004. The developmental origins of adult disease. J Am Coll Nutr 23 588S–595S. [DOI] [PubMed] [Google Scholar]
- Barnes RB, Rosenfield RL, Ehrmann DA, Cara JF, Cuttler L, Levitsky LL, and Rosenthal IM 1994. Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab 79 1328–1333. [DOI] [PubMed] [Google Scholar]
- Barra R, Cruz G, Mayerhofer A, Paredes A, and Lara HE 2014. Maternal sympathetic stress impairs follicular development and puberty of the offspring. Reproduction 148 137–145. [DOI] [PubMed] [Google Scholar]
- Barrett ES, and Swan SH 2015. Stress and Androgen Activity During Fetal Development. Endocrinology 156 3435–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bast RC Jr., Hennessy B, and Mills GB 2009. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer 9 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal AB, Vickers MH, Hampton MB, Poynton RA, and Sloboda DM 2010. Maternal undernutrition significantly impacts ovarian follicle number and increases ovarian oxidative stress in adult rat offspring. PLoS One 5 e15558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya P, and Keating AF 2012. Impact of environmental exposures on ovarian function and role of xenobiotic metabolism during ovotoxicity. Toxicol Appl Pharmacol 261 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blecher SR, and Erickson RP 2007. Genetics of sexual development: a new paradigm. Am J Med Genet A 143A 3054–3068. [DOI] [PubMed] [Google Scholar]
- Boots CE, and Jungheim ES 2015. Inflammation and Human Ovarian Follicular Dynamics. Semin Reprod Med 33 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw KD, Santos-Ramos R, Rawlins SC, MacDonald PC, and Parker CR Jr 1986. Endocrine studies in a pregnancy complicated by ovarian theca lutein cysts and hyperreactio luteinalis. Obstet Gynecol 67 66S–69S. [DOI] [PubMed] [Google Scholar]
- Brugh VM 3rd, Maduro MR, and Lamb DJ 2003. Genetic disorders and infertility. Urol Clin North Am 30 143–152. [DOI] [PubMed] [Google Scholar]
- Campos DB, Albornoz M, Papa PC, Palin MF, Bordignon V, and Murphy BD 2015. Relationship between adiponectin and fertility in the female pig. Reprod Fertil Dev 27 458–470. [DOI] [PubMed] [Google Scholar]
- Capel B 2000. The battle of the sexes. Mech Dev 92 89–103. [DOI] [PubMed] [Google Scholar]
- Cardoso RC, Puttabyatappa M, and Padmanabhan V 2015. Steroidogenic versus Metabolic Programming of Reproductive Neuroendocrine, Ovarian and Metabolic Dysfunctions. Neuroendocrinology 102 226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrillon DH, Miao L, Kollipara R, Horner JW, and DePinho RA 2003. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science 301 215–218. [DOI] [PubMed] [Google Scholar]
- Chan KA, Tsoulis MW, and Sloboda DM 2015. Early-life nutritional effects on the female reproductive system. J Endocrinol 224 R45–62. [DOI] [PubMed] [Google Scholar]
- Chen Y, Jefferson WN, Newbold RR, Padilla-Banks E, and Pepling ME 2007. Estradiol, progesterone, and genistein inhibit oocyte nest breakdown and primordial follicle assembly in the neonatal mouse ovary in vitro and in vivo. Endocrinology 148 3580–3590. [DOI] [PubMed] [Google Scholar]
- Cheong Y, Sadek KH, Bruce KD, Macklon N, and Cagampang FR 2014. Diet-induced maternal obesity alters ovarian morphology and gene expression in the adult mouse offspring. Fertil Steril 102 899–907. [DOI] [PubMed] [Google Scholar]
- Cohn BA, Cirillo PM, Wolff MS, Schwingl PJ, Cohen RD, Sholtz RI, Ferrara A, Christianson RE, van den Berg BJ, and Siiteri PK 2003. DDT and DDE exposure in mothers and time to pregnancy in daughters. Lancet 361 2205–2206. [DOI] [PubMed] [Google Scholar]
- Comim FV, Hardy K, Robinson J, and Franks S 2015. Disorders of follicle development and steroidogenesis in ovaries of androgenised foetal sheep. J Endocrinol 225 39–46. [DOI] [PubMed] [Google Scholar]
- Conti M, Hsieh M, Zamah AM, and Oh JS 2012. Novel signaling mechanisms in the ovary during oocyte maturation and ovulation. Mol Cell Endocrinol 356 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway G, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Franks S, Gambineri A, Kelestimur F, Macut D, Micic D, Pasquali R, Pfeifer M, Pignatelli D, Pugeat M, Yildiz BO, and EPSI Group 2014. The polycystic ovary syndrome: a position statement from the European Society of Endocrinology. Eur J Endocrinol 171 P1–29. [DOI] [PubMed] [Google Scholar]
- Cools M, Wolffenbuttel KP, Drop SL, Oosterhuis JW, and Looijenga LH 2011. Gonadal development and tumor formation at the crossroads of male and female sex determination. Sex Dev 5 167–180. [DOI] [PubMed] [Google Scholar]
- Cooper RL, and Kavlock RJ 1997. Endocrine disruptors and reproductive development: a weight-of-evidence overview. J Endocrinol 152 159–166. [DOI] [PubMed] [Google Scholar]
- Correia CP 1998. The ovary of Eve egg and sperm and preformation, xxiii, 396 p. Chicago, Ill.: University of Chicago Press. [Google Scholar]
- Costoff A, and Mahesh VB 1975. Primordial follicles with normal oocytes in the ovaries of postmenopausal women. J Am Geriatr Soc 23 193–196. [DOI] [PubMed] [Google Scholar]
- Craig ZR, Wang W, and Flaws JA 2011. Endocrine-disrupting chemicals in ovarian function: effects on steroidogenesis, metabolism and nuclear receptor signaling. Reproduction 142 633–646. [DOI] [PubMed] [Google Scholar]
- Crain DA, Janssen SJ, Edwards TM, Heindel J, Ho SM, Hunt P, Iguchi T, Juul A, McLachlan JA, Schwartz J, Skakkebaek N, Soto AM, Swan S, Walker C, Woodruff TK, Woodruff TJ, Giudice LC, and Guillette LJ Jr 2008. Female reproductive disorders: the roles of endocrine-disrupting compounds and developmental timing. Fertil Steril 90 911–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz G, Foster W, Paredes A, Yi KD, and Uzumcu M 2014. Long-term effects of early-life exposure to environmental oestrogens on ovarian function: role of epigenetics. J Neuroendocrinol 26 613–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaan KC, Berger LL, Kesler DJ, McKeith FK, Faulkner DB & Cmarik GF 1988. Effect of prenatal androgenization on growth performance and carcass characteristics of steers and heifers. J Anim Sci 66 1864–1870. [DOI] [PubMed] [Google Scholar]
- Dewailly D, Robin G, Peigne M, Decanter C, Pigny P, and Catteau-Jonard S 2016. Interactions between androgens, FSH, anti-Mullerian hormone and estradiol during folliculogenesis in the human normal and polycystic ovary. Hum Reprod Update 22 709–724. [DOI] [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, and Gore AC 2009. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev 30 293–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, and Dunaif A 2012. Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocr Rev 33 981–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong M, Zheng Q, Ford SP, Nathanielsz PW, and Ren J 2013. Maternal obesity, lipotoxicity and cardiovascular diseases in offspring. J Mol Cell Cardiol 55 111–116. [DOI] [PubMed] [Google Scholar]
- Dumesic DA, Oberfield SE, Stener-Victorin E, Marshall JC, Laven JS, and Legro RS 2015. Scientific Statement on the Diagnostic Criteria, Epidemiology, Pathophysiology, and Molecular Genetics of Polycystic Ovary Syndrome. Endocr Rev 36 487–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumesic DA, and Richards JS 2013. Ontogeny of the ovary in polycystic ovary syndrome. Fertil Steril 100 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumesic DA, Schramm RD, Bird IM, Peterson E, Paprocki AM, Zhou R, and Abbott DH 2003. Reduced intrafollicular androstenedione and estradiol levels in early-treated prenatally androgenized female rhesus monkeys receiving follicle-stimulating hormone therapy for in vitro fertilization. Biol Reprod 69 1213–1219. [DOI] [PubMed] [Google Scholar]
- Dunaif A 2016. Perspectives in Polycystic Ovary Syndrome: From Hair to Eternity. J Clin Endocrinol Metab 101 759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkel Schetter C 2011. Psychological science on pregnancy: stress processes, biopsychosocial models, and emerging research issues. Annu Rev Psychol 62 531–558. [DOI] [PubMed] [Google Scholar]
- Durlinger AL, Kramer P, Karels B, de Jong FH, Uilenbroek JT, Grootegoed JA, and Themmen AP 1999. Control of primordial follicle recruitment by anti-Mullerian hormone in the mouse ovary. Endocrinology 140 5789–5796. [DOI] [PubMed] [Google Scholar]
- Ehrlich M, and Lacey M 2013. DNA methylation and differentiation: silencing, upregulation and modulation of gene expression. Epigenomics 5 553–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar M, Vázquez-Nin G , and Echeverría O 2011. Follicular Cells, Cell Death in Mammalian Ovary, 185–200. Springer; Netherlands. [Google Scholar]
- Fathalla MF 1971. Incessant ovulation--a factor in ovarian neoplasia? Lancet 2 163. [DOI] [PubMed] [Google Scholar]
- Fortune J 2003. The early stages of follicular development: activation of primordial follicles and growth of preantral follicles. Animal Reproduction Science 78 135–163. [DOI] [PubMed] [Google Scholar]
- Fortune JE, Rivera GM, and Yang MY 2004. Follicular development: the role of the follicular microenvironment in selection of the dominant follicle. Anim Reprod Sci 82–83 109–126. [DOI] [PubMed] [Google Scholar]
- Foulkes WD, Gore M, and McCluggage WG 2016. Rare non-epithelial ovarian neoplasms: Pathology, genetics and treatment. Gynecologic Oncology 142 190–198. [DOI] [PubMed] [Google Scholar]
- Fowden AL, and Forhead AJ 2004. Endocrine mechanisms of intrauterine programming. Reproduction 127 515–526. [DOI] [PubMed] [Google Scholar]
- Franks S, Mason H, and Willis D 2000. Follicular dynamics in the polycystic ovary syndrome. Mol Cell Endocrinol 163 49–52. [DOI] [PubMed] [Google Scholar]
- Franks S, and McCarthy M 2004. Genetics of ovarian disorders: polycystic ovary syndrome. Rev Endocr Metab Disord 5 69–76. [DOI] [PubMed] [Google Scholar]
- Franks S, Stark J, and Hardy K 2008. Follicle dynamics and anovulation in polycystic ovary syndrome. Hum Reprod Update 14 367–378. [DOI] [PubMed] [Google Scholar]
- Goodfellow PN, and Lovell-Badge R 1993. SRY and sex determination in mammals. Annu Rev Genet 27 71–92. [DOI] [PubMed] [Google Scholar]
- Gore AC 2008. Developmental programming and endocrine disruptor effects on reproductive neuroendocrine systems. Front Neuroendocrinol 29 358–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore AC, Chappell VA, Fenton SE, Flaws JA, Nadal A, Prins GS, Toppari J, and Zoeller RT 2015. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr Rev er20151010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gougeon A 2004. Dynamics of human follicular growth: morphologic, dynamic, and functional aspects. The ovary 2 25–43. [Google Scholar]
- Gougeon A, Ecochard R, and Thalabard JC 1994. Age-related changes of the population of human ovarian follicles: increase in the disappearance rate of non-growing and early-growing follicles in aging women. Biol Reprod 50 653–663. [DOI] [PubMed] [Google Scholar]
- Grindler NM, Allsworth JE, Macones GA, Kannan K, Roehl KA, and Cooper AR 2015. Persistent organic pollutants and early menopause in U.S. women. PLoS One 10 e0116057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim C, V Padmanabhan, and Vyas AK 2016. Gestational Hyperandrogenism in Developmental Programming. Endocrinology en20161801. [DOI] [PMC free article] [PubMed]
- Handa RJ, Pak TR, Kudwa AE, Lund TD, and Hinds L 2008. An alternate pathway for androgen regulation of brain function: activation of estrogen receptor beta by the metabolite of dihydrotestosterone, 5alpha-androstane-3beta,17beta-diol. Horm Behav 53 741–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon PR, and Flaws JA 2015. The effects of phthalates on the ovary. Front Endocrinol (Lausanne) 6 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassold T, Abruzzo M, Adkins K, Griffin D, Merrill M, Millie E, Saker D, Shen J, and Zaragoza M 1996. Human aneuploidy: incidence, origin, and etiology. Environ Mol Mutagen 28 167–175. [DOI] [PubMed] [Google Scholar]
- Hassold T, Benham F, and Leppert M 1988. Cytogenetic and molecular analysis of sex-chromosome monosomy. Am J Hum Genet 42 534–541. [PMC free article] [PubMed] [Google Scholar]
- Heard E, and Martienssen RA 2014. Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerwagen MJ, Miller MR, Barbour LA, and Friedman JE 2010. Maternal obesity and fetal metabolic programming: a fertile epigenetic soil. Am J Physiol Regul Integr Comp Physiol 299 R711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrenkohl LR, and Scott S 1984. Prenatal stress and postnatal androgen: effects on reproduction in female rats. Experientia 40 101–103. [DOI] [PubMed] [Google Scholar]
- Hogg K, McNeilly AS, and Duncan WC 2011. Prenatal androgen exposure leads to alterations in gene and protein expression in the ovine fetal ovary. Endocrinology 152 2048–2059. [DOI] [PubMed] [Google Scholar]
- Hogg K, Young JM, Oliver EM, Souza CJ, McNeilly AS, and Duncan WC 2012. Enhanced thecal androgen production is prenatally programmed in an ovine model of polycystic ovary syndrome. Endocrinology 153 450–461. [DOI] [PubMed] [Google Scholar]
- Holemans K, Caluwaerts S, Poston L , and Van Assche FA 2004. Diet-induced obesity in the rat: a model for gestational diabetes mellitus. American journal of obstetrics and gynecology 190 858–865. [DOI] [PubMed] [Google Scholar]
- Hoyer PB, and Keating AF 2014. Xenobiotic effects in the ovary: temporary versus permanent infertility. Expert Opin Drug Metab Toxicol 10 511–523. [DOI] [PubMed] [Google Scholar]
- Hulas-Stasiak M, Dobrowolski P, and Tomaszewska E 2015. Prenatally administered dexamethasone impairs folliculogenesis in spiny mouse offspring. Reprod Fertil Dev [DOI] [PubMed]
- Hummitzsch K, Anderson RA, Wilhelm D, Wu J, Telfer EE, Russell DL, Robertson SA, and Rodgers RJ 2015. Stem cells, progenitor cells, and lineage decisions in the ovary. Endocr Rev 36 65–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo X, Chen D, He Y, Zhu W, Zhou W, and Zhang J 2015. Bisphenol-A and Female Infertility: A Possible Role of Gene-Environment Interactions. Int J Environ Res Public Health 12 11101–11116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Okada F, Ito R, Kato S, Sasaki S, Nakajima S, Uno A, Saijo Y, Sata F, Yoshimura Y, Kishi R, and Nakazawa H 2004. Perfluorooctane sulfonate (PFOS) and related perfluorinated compounds in human maternal and cord blood samples: assessment of PFOS exposure in a susceptible population during pregnancy. Environ Health Perspect 112 1204–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isik S, Ozcan HN, Ozuguz U, Tutuncu YA, Berker D, Alimli AG, Akbaba G, Karademir MA, and Guler S 2012. Evaluation of ovarian reserve based on hormonal parameters, ovarian volume, and antral follicle count in women with type 2 diabetes mellitus. J Clin Endocrinol Metab 97 261–269. [DOI] [PubMed] [Google Scholar]
- Johnson J, Bagley J, Skaznik-Wikiel M, Lee HJ, Adams GB, Niikura Y, Tschudy KS, Tilly JC, Cortes ML, Forkert R, Spitzer T, J Iacomini, Scadden DT, and Tilly JL 2005. Oocyte generation in adult mammalian ovaries by putative germ cells in bone marrow and peripheral blood. Cell 122 303–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J, Canning J, Kaneko T, Pru JK, and Tilly JL 2004. Germline stem cells and follicular renewal in the postnatal mammalian ovary. Nature 428 145–150. [DOI] [PubMed] [Google Scholar]
- Joshi R, and Dunaif A 1995. Ovarian disorders of pregnancy. Endocrinol Metab Clin North Am 24 153–169. [PubMed] [Google Scholar]
- Jost A 1983. Genetic and hormonal factors in sex differentiation of the brain. Psychoneuroendocrinology 8 183–193. [DOI] [PubMed] [Google Scholar]
- Jungheim ES, Macones GA, Odem RR, Patterson BW, Lanzendorf SE, Ratts VS, and Moley KH 2011. Associations between free fatty acids, cumulus oocyte complex morphology and ovarian function during in vitro fertilization. Fertil Steril 95 1970–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungheim ES, Travieso JL, Carson KR, and Moley KH 2012. Obesity and reproductive function. Obstet Gynecol Clin North Am 39 479–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafali H, Iriadam M, Ozardali I & Demir N 2004. Letrozole-induced polycystic ovaries in the rat: a new model for cystic ovarian disease. Arch Med Res 35 103–108. [DOI] [PubMed] [Google Scholar]
- Karavan JR, and Pepling ME 2012. Effects of estrogenic compounds on neonatal oocyte development. Reproductive Toxicology 34 51–56. [DOI] [PubMed] [Google Scholar]
- Karnezis AN, Cho KR, Gilks CB, Pearce CL, and Huntsman DG 2017. The disparate origins of ovarian cancers: pathogenesis and prevention strategies. Nat Rev Cancer 17 65–74. [DOI] [PubMed] [Google Scholar]
- Kauffman AS, Thackray VG, Ryan GE, Tolson KP, Glidewell-Kenney CA, Semaan SJ, Poling MC, Iwata N, Breen KM, Duleba AJ, Stener-Victorin E, Shimasaki S, Webster NJ & Mellon PL 2015. A Novel Letrozole Model Recapitulates Both the Reproductive and Metabolic Phenotypes of Polycystic Ovary Syndrome in Female Mice. Biol Reprod 93 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesler DJ, and Garverick HA 1982. Ovarian cysts in dairy cattle: a review. J Anim Sci 55 1147–1159. [DOI] [PubMed] [Google Scholar]
- Khorram O, Keen-Rinehart E, Chuang TD, Ross MG, and Desai M 2015. Maternal undernutrition induces premature reproductive senescence in adult female rat offspring. Fertil Steril 103 291–298 e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Nakajima T, Hayashi S, Chambon P, Watanabe H, Iguchi T, and Sato T 2009. Effects of diethylstilbestrol on programmed oocyte death and induction of polyovular follicles in neonatal mouse ovaries. Biol Reprod 81 1002–1009. [DOI] [PubMed] [Google Scholar]
- Knobil E, and Neill JD 1998. Encyclopedia of reproduction San Diego: Academic Press. [Google Scholar]
- Kozak LJ, Owings MF, and Hall MJ 2005. National Hospital Discharge Survey: 2002 annual summary with detailed diagnosis and procedure data. Vital Health Stat 13 1–199. [PubMed] [Google Scholar]
- Lara HE, Dorfman M, Venegas M, Luza SM, Luna SL, Mayerhofer A, Guimaraes MA, Rosa ESAA, and Ramirez VD 2002. Changes in sympathetic nerve activity of the mammalian ovary during a normal estrous cycle and in polycystic ovary syndrome: Studies on norepinephrine release. Microsc Res Tech 59 495–502. [DOI] [PubMed] [Google Scholar]
- Legro RS, Driscoll D, Strauss JF 3rd, Fox J, and Dunaif A 1998. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci U S A 95 14956–14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YT, and Capel B 2015. Cell fate commitment during mammalian sex determination. Curr Opin Genet Dev 32 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YX, and Hsueh AJ 1986. Synergism between granulosa and theca-interstitial cells in estrogen biosynthesis by gonadotropin-treated rat ovaries: studies on the two-cell, two-gonadotropin hypothesis using steroid antisera. Biol Reprod 35 27–36. [DOI] [PubMed] [Google Scholar]
- Lizneva D, Suturina L, Walker W, Brakta S, Gavrilova-Jordan L, and Azziz R 2016. Criteria, prevalence, and phenotypes of polycystic ovary syndrome. Fertil Steril 106 6–15. [DOI] [PubMed] [Google Scholar]
- Longacre M, Snyder NA, Housman G, Leary M, Lapinska K, Heerboth S, Willbanks A, and Sarkar S 2016. A Comparative Analysis of Genetic and Epigenetic Events of Breast and Ovarian Cancer Related to Tumorigenesis. Int J Mol Sci 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luense LJ, Veiga-Lopez A, Padmanabhan V, and Christenson LK 2011. Developmental programming: gestational testosterone treatment alters fetal ovarian gene expression. Endocrinology 152 4974–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maliqueo M, Benrick A, and Stener-Victorin E 2014. Rodent models of polycystic ovary syndrome: phenotypic presentation, pathophysiology, and the effects of different interventions. Semin Reprod Med 32 183–193. [DOI] [PubMed] [Google Scholar]
- Manikkam M, Haque MM, Guerrero-Bosagna C, Nilsson EE, and Skinner MK 2014. Pesticide methoxychlor promotes the epigenetic transgenerational inheritance of adult-onset disease through the female germline. PLoS One 9 e102091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manneras L, Cajander S, Holmang A, Seleskovic Z, Lystig T, Lonn M & Stener-Victorin E 2007. A new rat model exhibiting both ovarian and metabolic characteristics of polycystic ovary syndrome. Endocrinology 148 3781–3791. [DOI] [PubMed] [Google Scholar]
- Markey CM, Coombs MA, Sonnenschein C, and Soto AM 2003. Mammalian development in a changing environment: exposure to endocrine disruptors reveals the developmental plasticity of steroid-hormone target organs. Evolution & development 5 67–75. [DOI] [PubMed] [Google Scholar]
- Mazdai A, Dodder NG, Abernathy MP, Hites RA, and Bigsby RM 2003. Polybrominated diphenyl ethers in maternal and fetal blood samples. Environ Health Perspect 111 1249–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee EA, and Hsueh AJ 2000. Initial and cyclic recruitment of ovarian follicles. Endocr Rev 21 200–214. [DOI] [PubMed] [Google Scholar]
- McLaughlin EA, and McIver SC 2009. Awakening the oocyte: controlling primordial follicle development. Reproduction 137 1–11. [DOI] [PubMed] [Google Scholar]
- Miska EA, and Ferguson-Smith AC 2016. Transgenerational inheritance: Models and mechanisms of non-DNA sequence-based inheritance. Science 354 59–63. [DOI] [PubMed] [Google Scholar]
- Monniaux D, Clement F, Dalbies-Tran R, Estienne A, Fabre S, Mansanet C, and Monget P 2014. The ovarian reserve of primordial follicles and the dynamic reserve of antral growing follicles: what is the link? Biol Reprod 90 85. [DOI] [PubMed] [Google Scholar]
- Moran LJ, Norman RJ, and Teede HJ 2015. Metabolic risk in PCOS: phenotype and adiposity impact. Trends Endocrinol Metab 26 136–143. [DOI] [PubMed] [Google Scholar]
- Mossa F, Carter F, Walsh SW, Kenny DA, Smith GW, Ireland JL, Hildebrandt TB, Lonergan P, Ireland JJ, and Evans AC 2013. Maternal undernutrition in cows impairs ovarian and cardiovascular systems in their offspring. Biol Reprod 88 92. [DOI] [PubMed] [Google Scholar]
- Moyer B, and Hixon ML 2012. Reproductive effects in F1 adult females exposed in utero to moderate to high doses of mono-2-ethylhexylphthalate (MEHP). Reprod Toxicol 34 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu YM, Yanase T, Nishi Y, Tanaka A, Saito M, Jin CH, Mukasa C, Okabe T, Nomura M, Goto K, and Nawata H 2001. Saturated FFAs, palmitic acid and stearic acid, induce apoptosis in human granulosa cells. Endocrinology 142 3590–3597. [DOI] [PubMed] [Google Scholar]
- Naderpoor N, Shorakae S, Joham A, Boyle J, De Courten B, and Teede HJ 2015. Obesity and polycystic ovary syndrome. Minerva Endocrinol 40 37–51. [PubMed] [Google Scholar]
- Newbold RR, Bullock BC, and Mc Lachlan JA 1983. Exposure to diethylstilbestrol during pregnancy permanently alters the ovary and oviduct. Biol Reprod 28 735–744. [DOI] [PubMed] [Google Scholar]
- Newbold RR, Jefferson WN, and Padilla-Banks E 2009. Prenatal exposure to bisphenol a at environmentally relevant doses adversely affects the murine female reproductive tract later in life. Environ Health Perspect 117 879–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson EE, and Skinner MK 2015. Environmentally Induced Epigenetic Transgenerational Inheritance of Reproductive Disease. Biol Reprod 93 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak AZ, Kokai GK, Popovic VP, Ludoski MD, and Jurukovski VA 1995. Interphase cytogenetics on paraffin-embedded sections of ovary for detection of genomic constitution in a patient with Turner’s syndrome and chromosomal mosaicism. Hum Genet 95 293–298. [DOI] [PubMed] [Google Scholar]
- Nugent BM, Tobet SA, Lara HE, Lucion AB, Wilson ME, Recabarren SE, and Paredes AH 2012. Hormonal programming across the lifespan. Horm Metab Res 44 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega HH, Marelli BE, Rey F, Amweg AN, Diaz PU, Stangaferro ML, and Salvetti NR 2015a. Molecular aspects of bovine cystic ovarian disease pathogenesis. Reproduction 149 R251–264. [DOI] [PubMed] [Google Scholar]
- Ortega HH, Rey F, Velazquez MM, and Padmanabhan V 2010. Developmental programming: effect of prenatal steroid excess on intraovarian components of insulin signaling pathway and related proteins in sheep. Biol Reprod 82 1065–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega HH, Salvetti NR, and Padmanabhan V 2009. Developmental programming: prenatal androgen excess disrupts ovarian steroid receptor balance. Reproduction 137 865–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega HH, Veiga-Lopez A, Sreedharan S, del Lujan Velazquez MM, Salvetti NR, and Padmanabhan V 2015b. Developmental Programming: Does Prenatal Steroid Excess Disrupt the Ovarian VEGF System in Sheep? Biol Reprod 93 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan V, Cardoso RC, and Puttabyatappa M 2016. Developmental Programming, a Pathway to Disease. Endocrinology 157 1328–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan V, Salvetti NR, Matiller V, and Ortega HH 2014. Developmental Programming: Prenatal Steroid Excess Disrupts Key Members of Intraovarian Steroidogenic Pathway in Sheep. Endocrinology 155 3649–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan V, and Veiga-Lopez A 2011. Developmental origin of reproductive and metabolic dysfunctions: androgenic versus estrogenic reprogramming. Semin Reprod Med 29 173–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan V, and Veiga-Lopez A 2013a. Animal models of the polycystic ovary syndrome phenotype. Steroids 78 734–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan V, and Veiga-Lopez A 2013b. Sheep models of polycystic ovary syndrome phenotype. Mol Cell Endocrinol 373 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangas SA, and Rajkovic A 2015. Chapter 21 - Follicular Development: Mouse, Sheep, and Human Models, Knobil and Neill’s Physiology of Reproduction (Fourth Edition), pp. 947–995. San Diego: Academic Press. [Google Scholar]
- Peretz J, Vrooman L, Ricke WA, Hunt PA, Ehrlich S, Hauser R, Padmanabhan V, Taylor HS, Swan SH, VandeVoort CA, and Flaws JA 2014. Bisphenol a and reproductive health: update of experimental and human evidence, 2007–2013. Environ Health Perspect 122 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan N, and Conway GS 2011. Management of ovarian disease in pregnancy. Best Pract Res Clin Endocrinol Metab 25 985–992. [DOI] [PubMed] [Google Scholar]
- Politch JA, and Herrenkohl LR 1984. Effects of prenatal stress on reproduction in male and female mice. Physiol Behav 32 95–99. [DOI] [PubMed] [Google Scholar]
- Poulain M, Frydman N, Duquenne C, N’Tumba-Byn T, Benachi A, Habert R, Rouiller-Fabre V, and Livera G 2012 Dexamethasone induces germ cell apoptosis in the human fetal ovary. J Clin Endocrinol Metab 97 E1890–1897. [DOI] [PubMed] [Google Scholar]
- Puttabyatappa M, Burns A, Martin JD, Mesquita M, Veiga-Lopez A, and Padmanabhan V 2017. Developmental Programming: Gestational Exposure to Excess Testosterone Alters Expression of Ovarian Matrix Metalloproteases and Their Target Proteins. Reprod Sci 10.1177/1933719117697127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttabyatappa M, Cardoso RC, and Padmanabhan V 2016. Effect of maternal PCOS and PCOS-like phenotype on the offspring’s health. Mol Cell Endocrinol 435 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Jiao X, Simpson JL, and Chen ZJ 2015. Genetics of primary ovarian insufficiency: new developments and opportunities. Hum Reprod Update 21 787–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravelli GP, Stein ZA, and Susser MW 1976. Obesity in young men after famine exposure in utero and early infancy. N Engl J Med 295 349–353. [DOI] [PubMed] [Google Scholar]
- Rebar R, Judd HL, Yen SS, Rakoff J, Vandenberg G, and Naftolin F 1976. Characterization of the inappropriate gonadotropin secretion in polycystic ovary syndrome. J Clin Invest 57 1320–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P, Liu L, Adhikari D, Jagarlamudi K, Rajareddy S, Shen Y, Du C, Tang W, Hamalainen T, Peng SL, Lan ZJ, Cooney AJ, Huhtaniemi I, and Liu K 2008. Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science 319 611–613. [DOI] [PubMed] [Google Scholar]
- Ricci ZJ, Oh SK, Stein MW, Kaul B, Flusberg M, Chernyak V, Rozenblit AM, and Mazzariol FA 2016. Solid organ abdominal ischemia, part I: clinical features, etiology, imaging findings, and management. Clin Imaging 40 720–731. [DOI] [PubMed] [Google Scholar]
- Richards JS 2007. Genetics of ovulation. Semin Reprod Med 25 235–242. [DOI] [PubMed] [Google Scholar]
- Richardson SJ, Senikas V, and Nelson JF 1987. Follicular depletion during the menopausal transition: evidence for accelerated loss and ultimate exhaustion. J Clin Endocrinol Metab 65 1231–1237. [DOI] [PubMed] [Google Scholar]
- Ristic N, Nestorovic N, Manojlovic-Stojanoski M, Filipovic B, Sosic-Jurjevic B, Milosevic V, and Sekulic M 2008. Maternal dexamethasone treatment reduces ovarian follicle number in neonatal rat offspring. J Microsc 232 549–557. [DOI] [PubMed] [Google Scholar]
- Robker RL, Akison LK, Bennett BD, Thrupp PN, Chura LR, Russell DL, Lane M, and Norman RJ 2009. Obese women exhibit differences in ovarian metabolites, hormones, and gene expression compared with moderate-weight women. J Clin Endocrinol Metab 94 1533–1540. [DOI] [PubMed] [Google Scholar]
- Roland AV, Nunemaker CS, Keller SR & Moenter SM 2010. Prenatal androgen exposure programs metabolic dysfunction in female mice. J Endocrinol 207 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa ESA, Guimaraes MA, Padmanabhan V & Lara HE 2003. Prepubertal administration of estradiol valerate disrupts cyclicity and leads to cystic ovarian morphology during adult life in the rat: role of sympathetic innervation. Endocrinology 144 4289–4297. [DOI] [PubMed] [Google Scholar]
- Rossing MA, Daling JR, Weiss NS, Moore DE, and Self SG 1994. Ovarian tumors in a cohort of infertile women. N Engl J Med 331 771–776. [DOI] [PubMed] [Google Scholar]
- Russell DL, and Robker RL 2007. Molecular mechanisms of ovulation: co-ordination through the cumulus complex. Hum Reprod Update 13 289–312. [DOI] [PubMed] [Google Scholar]
- Salvetti NR, Ortega HH, Veiga-Lopez A, and Padmanabhan V 2012. Developmental programming: impact of prenatal testosterone excess on ovarian cell proliferation and apoptotic factors in sheep. Biol Reprod 87 22, 21–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki KJ, and Miller CE 2014. Adnexal torsion: review of the literature. J Minim Invasive Gynecol 21 196–202. [DOI] [PubMed] [Google Scholar]
- Sathyanarayana S, Calafat AM, Liu F, and Swan SH 2008. Maternal and infant urinary phthalate metabolite concentrations: are they related? Environ Res 108 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K, Sivapalan G, Gibbons N, Tempest H, and DK Griffin 2003. The genetic basis of infertility. Reproduction 126 13–25. [DOI] [PubMed] [Google Scholar]
- Silva CA, Yamakami LY, Aikawa NE, Araujo DB, Carvalho JF, and Bonfa E 2014. Autoimmune primary ovarian insufficiency. Autoimmun Rev 13 427–430. [DOI] [PubMed] [Google Scholar]
- Skinner MK 2005. Regulation of primordial follicle assembly and development. Human Reproduction Update 11 461–471. [DOI] [PubMed] [Google Scholar]
- Skinner MK 2014. Endocrine disruptor induction of epigenetic transgenerational inheritance of disease. Mol Cell Endocrinol 398 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GW, and Meidan R 2014. Ever-changing cell interactions during the life span of the corpus luteum: relevance to luteal regression. Reprod Biol 14 75–82. [DOI] [PubMed] [Google Scholar]
- Smith P, Wilhelm D, and Rodgers RJ 2014. Development of mammalian ovary. J Endocrinol 221 R145–161. [DOI] [PubMed] [Google Scholar]
- Sobinoff AP, Sutherland JM, and McLaughlin EA 2013. Intracellular signalling during female gametogenesis. Mol Hum Reprod 19 265–278. [DOI] [PubMed] [Google Scholar]
- Spinelli C, Piscioneri J, and Strambi S 2015. Adnexal torsion in adolescents: update and review of the literature. Curr Opin Obstet Gynecol 27 320–325. [DOI] [PubMed] [Google Scholar]
- Steckler T, Roberts E, Doop D, Lee T, and Padmanabhan V 2007. Developmental programming in sheep: administration of testosterone during 60–90 days of pregnancy reduces breeding success and pregnancy outcome. Theriogenology 67 459–467. [DOI] [PubMed] [Google Scholar]
- Steckler TL, Herkimer C, Dumesic DA, and Padmanabhan V 2009. Developmental programming: excess weight gain amplifies the effects of prenatal testosterone excess on reproductive cyclicity—implication for polycystic ovary syndrome. Endocrinology 150 1456–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steckler TL, Lee JS, Ye W, Inskeep EK, and Padmanabhan V 2008. Developmental programming: exogenous gonadotropin treatment rescues ovulatory function but does not completely normalize ovarian function in sheep treated prenatally with testosterone. Biol Reprod 79 686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouffer RL, Bishop CV, Bogan RL, Xu F, and Hennebold JD 2013. Endocrine and local control of the primate corpus luteum. Reprod Biol 13 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouffer RL, Xu F, and Duffy DM 2007. Molecular control of ovulation and luteinization in the primate follicle. Front Biosci 12 297–307. [DOI] [PubMed] [Google Scholar]
- Takahashi LK, Turner JG, and Kalin NH 1998. Prolonged stress-induced elevation in plasma corticosterone during pregnancy in the rat: implications for prenatal stress studies. Psychoneuroendocrinology 23 571–581. [DOI] [PubMed] [Google Scholar]
- Tal R, and Seifer DB 2017. Ovarian reserve testing: A user’s guide. American Journal of Obstetrics and Gynecology [DOI] [PubMed]
- Tang WY, and Ho SM 2007. Epigenetic reprogramming and imprinting in origins of disease. Rev Endocr Metab Disord 8 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomakos N, Trachana SP, Koutroumpa I, Rodolakis A, and Gavalas NG 2015. Molecular aspects and clinical methods for preserving ovarian reserves in women receiving cancer treatment. Clin Exp Obstet Gynecol 42 416–425. [PubMed] [Google Scholar]
- Tilly JL 1996. The molecular basis of ovarian cell death during germ cell attrition, follicular atresia, and luteolysis. Front Biosci 1 d1–11. [DOI] [PubMed] [Google Scholar]
- Touraine P, Beau I, Gougeon A, Meduri G, A Desroches, Pichard C, Detoeuf M, Paniel B, Prieur M, Zorn JR, Milgrom E, Kuttenn F, and Misrahi M 1999 New natural inactivating mutations of the follicle-stimulating hormone receptor: correlations between receptor function and phenotype. Mol Endocrinol 13 1844–1854. [DOI] [PubMed] [Google Scholar]
- Trapphoff T, Heiligentag M, El Hajj N, Haaf T, and Eichenlaub-Ritter U 2013. Chronic exposure to a low concentration of bisphenol A during follicle culture affects the epigenetic status of germinal vesicles and metaphase II oocytes. Fertil Steril 100 1758–1767 e1751. [DOI] [PubMed] [Google Scholar]
- Tsoulis MW, Chang PE, Moore CJ, Chan KA, Gohir W, Petrik JJ, Vickers MH, Connor KL, and Sloboda DM 2016. Maternal High-Fat Diet-Induced Loss of Fetal Oocytes Is Associated with Compromised Follicle Growth in Adult Rat Offspring. Biol Reprod 94 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BM 2011. Environmental sensing by chromatin: an epigenetic contribution to evolutionary change. FEBS Lett 585 2032–2040. [DOI] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, and Telser J 2007. Free radicals and antioxidants in normal physiological functions and human disease. The international journal of biochemistry & cell biology 39 44–84. [DOI] [PubMed] [Google Scholar]
- Veiga-Lopez A, Luense LJ, Christenson LK, and Padmanabhan V 2013 Developmental programming: gestational bisphenol-A treatment alters trajectory of fetal ovarian gene expression. Endocrinology 154 1873–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Lopez A, J Moeller, Abbott DH, and Padmanabhan V 2016. Developmental programming: rescuing disruptions in preovulatory follicle growth and steroidogenesis from prenatal testosterone disruption. J Ovarian Res 9 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Lopez A, Wurst AK, Steckler TL, Ye W, and Padmanabhan V 2014. Developmental programming: postnatal estradiol amplifies ovarian follicular defects induced by fetal exposure to excess testosterone and dihydrotestosterone in sheep. Reprod Sci 21 444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Lopez A, Ye W, and Padmanabhan V 2012. Developmental programming: prenatal testosterone excess disrupts anti-Müllerian hormone expression in preantral and antral follicles. Fertility and sterility 97 748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh T, Suresh PS, and Tsutsumi R 2014. New insights into the genetic basis of infertility. Appl Clin Genet 7 235–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink JM, Sadrzadeh S, Lambalk CB, and Boomsma DI 2006. Heritability of polycystic ovary syndrome in a Dutch twin-family study. J Clin Endocrinol Metab 91 2100–2104. [DOI] [PubMed] [Google Scholar]
- Wang H, Jiang M, Bi H, Chen X, He L, Li X, and Wu J 2014. Conversion of female germline stem cells from neonatal and prepubertal mice into pluripotent stem cells. J Mol Cell Biol 6 164–171. [DOI] [PubMed] [Google Scholar]
- Wang YC, Su HY, Liu JY, Chang FW, and Chen CH 2005. Maternal and female fetal virilization caused by pregnancy luteomas. Fertil Steril 84 509. [DOI] [PubMed] [Google Scholar]
- Webber LJ, Stubbs S, Stark J, Trew GH, Margara R, Hardy K, and Franks S 2003. Formation and early development of follicles in the polycystic ovary. Lancet 362 1017–1021. [DOI] [PubMed] [Google Scholar]
- Welt CK, Smith PC, and Taylor AE 2004. Evidence of Early Ovarian Aging in Fragile X Premutation Carriers. The Journal of Clinical Endocrinology & Metabolism 89 4569–4574. [DOI] [PubMed] [Google Scholar]
- Wemeau JL, Proust-Lemoine E, Ryndak A, and Vanhove L 2013. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens) 12 39–45. [DOI] [PubMed] [Google Scholar]
- West C, Foster DL, Evans NP, Robinson J, and Padmanabhan V 2001. Intra-follicular activin availability is altered in prenatally-androgenized lambs. Mol Cell Endocrinol 185 51–59. [DOI] [PubMed] [Google Scholar]
- Whirledge S, and Cidlowski JA 2010. Glucocorticoids, stress, and fertility. Minerva Endocrinol 35 109–125. [PMC free article] [PubMed] [Google Scholar]
- Whyte J, Alexenko A, Davis A, Ellersieck M, Fountain E, and Rosenfeld C 2007. Maternal diet composition alters serum steroid and free fatty acid concentrations and vaginal pH in mice. Journal of Endocrinology 192 75–81. [DOI] [PubMed] [Google Scholar]
- Wittenberger MD, Hagerman RJ, Sherman SL, McConkie-Rosell A, Welt CK, Rebar RW, Corrigan EC, Simpson JL, and Nelson LM 2007. The FMR1 premutation and reproduction. Fertil Steril 87 456–465. [DOI] [PubMed] [Google Scholar]
- Woodruff TJ 2011. Bridging epidemiology and model organisms to increase understanding of endocrine disrupting chemicals and human health effects. J Steroid Biochem Mol Biol 127 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LL, Norman RJ, and Robker RL 2011. The impact of obesity on oocytes: evidence for lipotoxicity mechanisms. Reprod Fertil Dev 24 29–34. [DOI] [PubMed] [Google Scholar]
- Wu XY, Li ZL, Wu CY, Liu YM, Lin H, Wang SH & Xiao WF 2010. Endocrine traits of polycystic ovary syndrome in prenatally androgenized female Sprague-Dawley rats. Endocr J 57 201–209. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Shirai M, Sugita K, Nagai N, Miura Y, Mogi R, Yamamoto K, Tamura A, and Arishima K 2003. Effects of maternal exposure to diethylstilbestrol on the development of the reproductive system and thyroid function in male and female rat offspring. J Toxicol Sci 28 385–394. [DOI] [PubMed] [Google Scholar]
- Yao HH 2005. The pathway to femaleness: current knowledge on embryonic development of the ovary. Mol Cell Endocrinol 230 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos NC, Billiar RB, Albrecht ED, and Pepe GJ 2002. Developmental regulation of baboon fetal ovarian maturation by estrogen. Biol Reprod 67 1148–1156. [DOI] [PubMed] [Google Scholar]
- Zama AM, and Uzumcu M 2010. Epigenetic effects of endocrine-disrupting chemicals on female reproduction: an ovarian perspective. Front Neuroendocrinol 31 420–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zama AM, and Uzumcu M 2013. Targeted genome-wide methylation and gene expression analyses reveal signaling pathways involved in ovarian dysfunction after developmental EDC exposure in rats. Biol Reprod 88 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano E, and Nathanielsz PW 2013. Mechanisms by which maternal obesity programs offspring for obesity: evidence from animal studies. Nutr Rev 71 Suppl 1 S42–54. [DOI] [PubMed] [Google Scholar]
- Zeleznik AJ 2001. Follicle selection in primates: “many are called but few are chosen”. Biol Reprod 65 655–659. [DOI] [PubMed] [Google Scholar]
- Zeleznik AJ 2004. The physiology of follicle selection. Reprod Biol Endocrinol 2 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zheng W, Shen Y Adhikari D, Ueno H, and Liu K 2012. Experimental evidence showing that no mitotically active female germline progenitors exist in postnatal mouse ovaries. Proc Natl Acad Sci U S A 109 12580–12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Li L, Qin XS, Zhou Y, Zhang XF, Wang LQ, De Felici M, Chen H, Qin GQ, and Shen W 2014. Di-(2-ethylhexyl) phthalate and bisphenol A exposure impairs mouse primordial follicle assembly in vitro. Environ Mol Mutagen 55 343–353. [DOI] [PubMed] [Google Scholar]
- Zhao H, Lv Y, Li L, and Chen ZJ 2016. Genetic Studies on Polycystic Ovary Syndrome. Best Pract Res Clin Obstet Gynaecol 37 56–65. [DOI] [PubMed] [Google Scholar]
- Zhou W, Liu J, Liao L, Han S, and Liu J 2008. Effect of bisphenol A on steroid hormone production in rat ovarian theca-interstitial and granulosa cells. Mol Cell Endocrinol 283 12–18. [DOI] [PubMed] [Google Scholar]