

Abstract
Ca2+ flux across the inner mitochondrial membrane (IMM) regulates cellular bioenergetics, intra-cellular cytoplasmic Ca2+ signals, and various cell death pathways. Ca2+ entry into the mitochondria occurs due to the highly negative membrane potential (ΔΨm) through a selective inward rectifying MCU channel. In addition to being regulated by various mitochondrial matrix resident proteins such as MICUs, MCUb, MCUR1 and EMRE, the channel is transcriptionally regulated by upstream Ca2+ cascade, post transnational modification and by divalent cations. The mode of regulation either inhibits or enhances MCU channel activity and thus regulates mitochondrial metabolism and cell fate.
Graphical abstract
Ca2+ ion is a versatile second messenger essential for a variety of kinetically different cellular processes from fertilization to cell death [1]. While some processes like endocytosis occur in seconds, other processes such as gene transcription take up to hours and how Ca2+ regulates these diverse processes is a question still being studied [2]. A rise in cytosolic Ca2+ (cCa2+) can occur either by Ca2+ entry through plasma membrane channels (voltage gated/receptor mediated/second messenger mediated channels/store operated channels) or release from intracellular Ca2+ stores [3]. One ubiquitous mode of receptor-regulated Ca2+ entry is capacitative calcium entry, first proposed by Putney [4]. At any given point, resting cCa2+ is kept low (~ 100nM) and this is achieved by Ca2+ efflux mechanisms of pumps (PMCA), exchangers (NCX) and organelles such as ER and the mitochondria that act as Ca2+ sinks. cCa2+ transients are defined and shaped by the mitochondria. The first observation of mitochondrial Ca2+ (mCa2+) uptake was evidenced five decades ago when several groups witnessed isolated mitochondria to buffer Ca2+ [5]. Since then, mCa2+ has been extensively studied. Three main roles have been attributed to Ca2+ uptake by the mitochondria: 1) Ca2+ is utilized by the dehydrogenases of the TCA cycle (pyruvate dehydrogenase, iso-citrate dehydrogenase and α-ketoglutarate dehydrogenase) for ATP generation. 2) To maintain cCa2+ dynamics. 3) Activation of various cell death pathways of apoptosis and necrosis [6–10].
Ca2+ entry into the mitochondria is due to the high electrochemical gradient (~ −180 mV) and occurs without the transport of any other ions hence making it a uniporter. Studies on isolated mitochondria estimated a flux of more than 10,000 Ca2+ ions per second through the channel [5]. Although the properties of the uniporter were established, its molecular identity remained elusive. The identification of the mitochondrial calcium uniporter (MCU) was made possible with the advances in integrative genomics [11, 12]. Progress in the field has been made in identifying and characterizing MCU and its regulatory molecules.
Here, in this review we summarize the architecture of MCU channel, its mitochondrial resident regulators, regulation of the channel by ions and redox molecules and go onto explore the activity of the channel in pathological conditions along with therapeutic insights.
Architecture of MCU
Early work on mCa2+ uptake revealed that Ca2+ uptake into the mitochondria was ΔΨm dependent with no transport of anions hence making it a uniporter. The transport of Ca2+ followed second order kinetics and was sensitive to Ruthenium Red (RR) (an inorganic cationic dye that binds acidic mucopolysaccharides and phospholipids)[13, 14]. Electrophysiological studies defined the channel to be an inward rectifying current with high specificity for divalent cations Ca2+ ~Sr2+ ≫ Mn2+ ~ Ba2+ and to be inhibited at Nano molar concentrations of RR and its analog Ru360 [15]. Although the biophysical properties of the channel were characterized as being 1. Electrogenic dependent 2. Selective and 3. Low affinity for Ca2+, it was not until 2011 when two independent groups discovered the molecular identity of the channel. Whole genome phylogenetic profiling, RNA co-expression analysis and organelle wide protein co-expression analysis revealed an RR sensitive transmembrane protein resident of the IMM that is part of a large complex called the MCU [11, 12]. Reconstitution of MCU in planar lipid bilayer produced channel recordings with conductance similar to previously findings. Additionally, presence of RR failed to produce Ca2+-permeable channel activity indicating sensitivity to ruthenium, a characteristic of the mCa2+ transport [12]. Consistent with the finding that all vertebrate mitochondria take up Ca2+, the expression of MCU is conserved across eukaryotes except in yeast. In contrast to their evolutionary sister group Amoebozoa that have a single homolog of MCU and MICU1, yeast do not exhibit uniporter activity with no homologs of the uniporter components [16]. Because of this property yeast serve as an excellent heterologous expression system. Reconstitution of DdMCU (MCU from Dictyostelium discoideum) in yeast was sufficient to evoke a mCa2+ uptake response that was otherwise absent, further suggesting that MCU was the pore-forming subunit of the uniporter complex. MCU, the pore forming channel has been identified to be a two transmembrane, Ca2+ selective, ruthenium sensitive channel with its selectivity filter in the inter-membrane space (IMS) and N and C terminal domains resting in the mitochondrial matrix [16].
The structure of N-terminal domain (NTD) of MCU spanning exons 3 and 4 adopts a β-grasp like fold that entails an α–helix and six β-strands that form the central core with two highly conserved leucine rich loops [17]. Atomic resolution structure of NTD revealed a cluster of negatively charged residues called the MCU-regulating acidic patch (MRAP) in the β-grasp fold domain that binds divalent cations. Either interaction of Ca2+ or Mg2+ with the MRAP domain or mutations in the MRAP domain destabilizes MCU and shifts the self-association equilibrium to monomer with a loss of mCa2+ uptake. This study for the first time shows that like most Ca2+ channels that are regulated by Ca2+ feedback mechanisms, MCU is autoregulated by matrix Ca2+ and Mg2+ binding to the MRAP domain in the NTD of MCU [18]. Expression of MCUΔNTD localizes to the mitochondria, and forms MCU oligomers with intact ΔΨm. Deletion of the NTD showed a significant reduction in mCa2+ uptake with intact cCa2+ dynamics. Co-immuno precipitation assay revealed that loss of NTD had diminished interaction with MCUR1 (a positive regulator of MCU) but not MICU1 and MICU2 [17]. The C-terminal domain (CTD) harbors two transmembrane domains and two coiled-coiled domains that are required for interaction with its regulators. Nuclear magnetic resonance (NMR) in combination with electron microscopy revealed the architecture of the MCU channel to be a pentamer. The second transmembrane (TM2) and the coiled-coil helix (CCH) form the inner cores of the pentamer that are wrapped by the first TM helix and CCH respectively. TM1 and TM2 domains are connected by a loop consisting of conserved acidic residues called the DIME motif loop that forms the pentameric barrel at the mouth of the pore. Asp240 and Glu243 are positioned inside the barrel to form two carboxylate rings that make up the selectivity filter of the channel [19]. Although this finding for the first time reported the structure of the C.elegans MCU pore, no proof of channel activity was performed in the pentameric state.
Regulators of MCU channel
MCU exists as part of a heteromeric complex that consist of MICU1, MICU2, MCUR1, EMRE, MCUb and SLC25A23 [20–24]. Sequence analysis of MCU identified a gene with 50% similarity to MCU called MCUb. MCUb is conserved across all vertebrates and absent in species of plants, kinetoplastids, Nematoda, and Arthropoda where MCU is present. Immuno-precipitation and foster resonance energy transfer (FRET) analysis revealed interaction of MCU and MCUb. However, electrophysiological studies of purified MCUb inserted into planar lipid bilayers showed no channel activity. While RNAi mediated silencing of MCUb in HeLa cells demonstrated an increase in mCa2+ uptake upon stimulation with histamine, over-expression of MCUb resulted in a marked decrease in mCa2+ uptake. These pieces of data indicate that MCUb functions as a dominant negative MCU sub-unit [25].
The activity of the channel is determined by its negative regulator MICU1 and its positive regulator MCUR1. Like most ion channels that open upon agonist stimulation, MCU channel opens only when cCa2+ rises above 1–2 μM. The threshold is set by its gatekeeper MICU1. The EF hands of MICU1 sense the rise in cCa2+ (> 3 μM) and physically dissociate from the channel thus transforming MCU to its open confirmation [26]. MICU1 was the first MCU uniplex component to be identified using targeted RNAi screen based on clues from comparative physiology, evolutionary genomics and organelle proteomics. MICU1 was found to be an IMS resident protein with its EF hand domains facing the cytosol. It was also shown that silencing MICU1 abrogated mCa2+ uptake without dissipating ΔΨm [21, 27]. Contrary to the previous findings that MICU1 is an IMS resident protein, authentic biochemical and confocal validations from our laboratory reveal MICU1 to be a soluble matrix protein that senses rise in cCa2+ or mCa2+ with its EF hands and opens MCU channel. Serial mutation analysis revealed that the evolutionary conserved poly basic region of MICU1 (99 –110 aa) directly interacts with the coiled-coiled domains of MCU. Mutations in polybasic region but not EF hand domain results in loss of interaction with MCU [28]. Structural analysis of MICU1 reveals that under Ca2+-free conditions MICU1 forms a hexamer that interacts with MCU and inhibits its activity through the c-helix [29]. Complementary to our findings, genetic deletion of MICU1 exhibits increased Ca2+ uptake at low cCa2+ with a concomitant mCa2+ overload. This matrix overload resulted in altered mitochondrial morphology particularly in the skeletal muscles [30]. Autosomal recessive mutations in MICU1 presents with proximal skeletal muscle weakness with neurological features of chorea, tremors or ataxia [31, 32]. MICU1−/− animals also exhibit similar abnormalities to that seen in humans. Crispr-Cas9 mediated deletion of Micu1 yielded significant perinatal lethality suggesting that MICU1 is dispensable for embryonic development but essential post-natal [30]. Along these lines it was also observed that MICU1 plays a key role in liver regeneration post partial hepatectomy (PHx). Loss of MICU1 results in elevated inflammatory state post PHx with failure of the hepatocytes from entering the cell cycle due to mCa2+ overload mediated necrosis of hepatocytes [33].
Bio-informatic analysis revealed two paralogues of MICU1 that evolved in the vertebrates possibly by a gene duplication event. MICU2 and MICU3 were found to contain conserved domains as MICU1 along with two canonical EF hand domains with different expression patterns. While MICU1 and MICU2 were expressed in almost all tissues, MICU3 was specific to the central nervous system. The first study that identified the protein demonstrated mitochondrial localization of MICU2 and interaction with MICU1. RNAi mediated silencing of MICU2 revealed a reduced rate of mCa2+ uptake and premature release of matrix calcium. Hence it was concluded that MICU2 played a role in mCa2+ uptake [22]. Subsequently, gene knock out studies using transcription activator-like effector nuclease (TALEN) technology was utilized to generate MICU1 and MICU2 KO in HEK293T cells to functionally distinguish the roles of the two proteins. Addition of 1μM bolus of Ca2+ revealed a rapid uptake by MICU1 KO but not control mitochondria due to the loss of gate-keeping effect. MICU2 KO cells also exhibited similar gate-keeping characteristic albeit with reduced rate. Importantly, the impaired calcium handling was rescued upon re-expression of the ablated protein signifying that the phenotype is not because of off-target effects of gene KO [34]. It is now accepted that under low cCa2+, MICU1 and MICU2 function as gate-keepers of MCU. Controversy in the role of MICU1 and MICU2 under conditions of high cCa2+ exists and further studies are warranted. Genetic deletion of Micu2 in animals produced off-springs at Mendelian ratio with comparable sizes to wild-type littermates that survived > 18 months. Liver mitochondria from MICU2−/− animals took up low-regime Ca2+ which otherwise was inhibited by liver mitochondrial from wild-type animals, indicating gate-keeping effect of the protein. On the other hand, 25 μM Ca2+ pulse was taken up at a slower rate, a phenotype attributed to the reduction in MCU complex proteins [35]. In a recent finding, MICU1-MICU2 hetero-dimers were shown to exhibit steep co-operativity at sub-micromolar cCa2+, thus functioning as ON/OFF switches for MCU-mediated mCa2+ uptake [36].
A positive regulator of the channel, MCU regulator 1 (MCUR1) was identified in an RNAi screen of 45 mitochondrial proteins. Of the 45 genes, only one RNAi resulted in the inhibition of mitochondrial calcium uptake. MCUR1 directly interacts with the channel and promotes Ca2+-uptake into the mitochondria. Silencing of this ubiquitously expressed protein resulted in blunted mCa2+ uptake under basal and activated conditions, with no changes in cCa2+ dynamics. The reduced mCa2+- uptake then resulted in perturbed bioenergetics in MCUR1 KD cells [20, 37]. Findings from another laboratory suggest that MCUR1 mediates assembly of complex IV cytochrome oxidase (COX) and fibroblasts from patient mutations in COX exhibit similar phenotype to loss of MCUR1. Their study defines MCUR1 as an assembly factor of complex IV, thus loss of which results in impaired oxidation phosphorylation (Ox-phos) leading to reduced ΔΨm [38]. A decrease in ΔΨm, decreases the driving force for Ca2+ and hence reduced mCa2+ uptake in the absence of MCUR1. Later in vivo studies in MCUR1 addressed this controversy. To understand the role of MCUR1 in high and moderate ox-phos demanding tissues, heart (MCUR1fl/flα-MHCCre; cMCUR1 KO) and vasculature (MCUR1fl/flVE-Cad-Cre; MCUR1ΔEC) specific deletion of MCUR1 were generated [39]. While germ line deletion of MCUR1 in endothelial cells (ECs) did not show any discernable phenotype, cMCUR1 KO animals were smaller and died 3 weeks after birth. Electrophysiological and biochemical studies from tissue specific KOs demonstrated a significant decrease in MCU current due to the disruption of MCU super complex in the absence of MCUR1. Blunting of MCU current when the transmembrane voltage is clamped suggests a direct effect of MCUR1 on mCa2+ uptake and not an indirect effect of decreased ΔΨm. MCUR1’s ability to serve as MCU scaffolding factor stems from i) it being a transmembrane protein with coiled-coil domains essential for protein interaction ii) failure of MCU complex to assemble in the absence of MCUR1 and iii) considerable reduction in IMCU [39]. The role of MCUR1 in mCa2+ uptake may not be consistent for all species. Drosophila cells do not show a difference in mCa2+ uptake in the absence of MCUR1 [40].
MCU proteome analysis revealed essential MCU regulator (EMRE) in addition to MICU1, MICU2 and MCUb. EMRE is a 10 KDa, single pass transmembrane protein with aspartate rich C-terminus. EMRE was shown to be essential for MCU-mediated mCa2+ uptake [23]. Although MCU protein was present in EMRE KO cells, IMCU was significantly reduced, suggesting that EMRE was required for the open configuration of the channel. Bioinformatics sequence analysis revealed that EMRE is absent in lower organisms including yeast and is only specific to metazoans. Although reconstitution of MCU alone was sufficient to conduct Ca2+ in lipid bi-layers, EMRE along with MCU was required in human mitoplasts, suggesting that Ca2+ entry into the metazoan mitochondria depended on EMRE. Using the yeast model system that lack mitochondrial Ca2+ uptake machinery, it was demonstrated that DdMCU can be reconstituted using a single genetic component, while the minimum metazoan uniplex components are MCU along with EMRE [16]. EMRE has been proposed to be the matrix sensor regulating MCU activity by sensing Ca2+ through the acidic patch at its carboxy terminal and mediating interaction of MICU1 and MICU2 with MCU [41]. However, deletion of the acidic amino acids did not affect mCa2+ uptake [42]. Furthermore, co-immunoprecipitation and bimolecular fluorescence complementation (BiFC) analysis revealed a direct interaction of MICU1 with MCU without EMRE bridging [39].
Transcriptional Regulation of MCU
Regulation of cCa2+ is critical for cell survival. The ability of mitochondria to shape cytosolic Ca2+ transients is well documented and hence one might predict that cytosolic Ca2+ may stimulate the genes responsible for mitochondrial Ca2+ uptake [43–45]. In most non-excitable cells cytosolic Ca2+ is released from (inositol 1,4,5-trisphosphate) IP3-induced ER store release and store-operated Ca2+ entry (SOCE) [46–53]. Ca2+ released from ER and SOCE is rapidly cleared by the plasma membrane components and the mitochondria thus underscoring the interdependence of the upstream cytosolic Ca2+ signaling with the downstream mitochondrial Ca2+ transport mechanisms. Any perturbation in the anterograde Ca2+ signaling including loss of IP3Rs or STIM1 or Orai1 has been shown to alter mitochondrial Ca2+ uptake, reducing basal matrix Ca2+ and current through the channel by a reduction in MCU protein. This decrease in protein levels of MCU in the absence of proximal Ca2+ signals was shown to be mediated by Ca2+-regulated transcription factor CREB (cyclic adenosine monophosphate response element). Chromatin immune-precipitation (Chip) assay revealed the binding of CREB onto the MCU promoter. Lymphocytes lacking IP3R, or SOCE had undetectable levels of phosphorylated CREB, thus decreased MCU abundance and mCa2+ uptake [54]. This study was the first to reveal a mechanistic link of regulation of mitochondrial transport by proximal Ca2+ signaling components.
Multiple layers of regulation exist in cells, in the event that one fails, other mechanisms in place take over. One such mode of regulation is by microRNAs (miRNAs). miRNAs are small, non-coding nucleotides that regulates gene expression by sequence complementarity based binding of the target mRNA. Binding to the target mRNA results in either degradation of the target mRNA or inhibition of translation [55]. Target prediction algorithms identified five miRNAs (miR-15, miR-17, miR-21, miR-25 and miR-137) that have sequence complementarity to the 3′UTR of MCU. Of these, only miR-25 shows a 100% seed sequence match to the 3′UTR of MCU. In vitro luciferase assays reveal a down-regulation of MCU RNA and protein by miR-25 but not that of other MCU complex components. Immuno-histochemistry confirmed the inverse regulation of MCU by miR-25 in mucosal tissues of colon cancer. The increased levels of miR-25 in colon cancer down-regulated MCU levels thus protecting the tumor cells from cell death [56]. This study was the first to propose MCU regulation by miRNAs and progression of cancer due to protection from MCU-mediated tumor cell death but it lacked in vivo data to show tumor cell protection by miR-25. Later studies show that in pulmonary arterial hypertension (PAH) patients where the pulmonary smooth muscle cells (PSMCs) proliferate uncontrollably (cancer-like phenotype), migrate and are resistant to apoptosis, MCU expression is down-regulated without any changes on the MCU complex components. This study demonstrates the post-transcriptional regulation of MCU by miR-25 and miR-138 in PSMCs from PAH patients. The authors validate their findings by using MCU over-expression and anti-miR25 and anti-miR138 that restores mCa2+ homeostasis and reverses PAH [57]. To further corroborate the regulation of MCU by miRNA in vivo, a lung metastasis model of animals was generated. Nude mice were injected via tail vein with estrogen receptor-negative breast cancer cells MDA-MB-231, MDA-MB-231 cells with down regulated MCU expression (MDA-MB-231 with shRNA MCU) or cells over expressing miR-340. Hemoxylin and eosin staining of the lung tissue from each animal revealed significantly fewer metastatic lung nodules in mice that had been injected with MCU-downregulated or miR-340–overexpressing MDA-MB-231 cells than in the control group, indicating that miR-340 silencing of MCU reduces cell migration and metastasis [58]. An interplay of mCa2+ and miRNAs has been implicated in other pathological conditions. An example of this is the down-regulation of MCU by miR-1 in cardiac hypertrophy by interfering with protein translation. miR-1 is well characterized miRNA that plays a key role in cardiac development and stress conditions. Analysis of mice subjected to exercise induced-cardiac hypertrophy or transaortic constriction (TAC), a condition that corresponds to the initial, compensatory response to a hypertrophying stimulus with cardiomyocyte growth accompanied by no contractile failure demonstrated decreased miR-1 with concomitant increase in MCU levels. Thus, the initial cardiomyocyte adaptation to increased heart work is characterized by increased MCU levels that are specifically regulated by miR-1 [59]. Collectively, these findings suggest new modes of regulation of MCU under various physiological and pathological conditions.
Regulation of MCU by Anions and Nucleotides
The primary driving force for mitochondrial Ca2+ uptake is the highly negative proton gradient generated by the pumping of the H+ ions into the IMS. Ca2+ uptake into the mitochondria requires active anions like Pi, acetate, β-hydroxybutyrate, glutamate, bicarbonate that can provide an H+ source. Transport of active anions makes the matrix more negative, thus generating a pulling force for the uptake of cations. Early studies on isolated mitochondria postulated two modes of Ca2+ uptake: i) limited Ca2+ uptake in the absence of Pi and ii) massive uptake in the presence of Pi [60]. The phosphate facilitated Ca2+ uptake was explained by the formation of calcium-phosphate precipitates in the matrix to maintain the electrochemical gradient for Ca2+ uptake and also to balance the charge [61]. The major carrier of Pi into the matrix is through PiC encoded by the nuclear gene SLC25A3 with two isoforms PiC A and PiC B. PiC B is ubiquitously expressed while PiC A is specific to the skeletal muscle [5, 62–65]. PiC belongs to the super family of mitochondrial carriers characterized by six transmembrane domains with both the C and N-terminal domains in the IMS. Inhibitor stop assay revealed the flux of an estimate 50,000 Pi ions per second at 25°C with H+ symport. The first functional studies on PiC were performed in yeast where genetic deletion of MIR1, yeast homologues of PiC1 failed to grow on non-fermentable carbon source [66]. Patients harboring frame-shift mutation (c.158–9A→G and c.215G→A) in the gene have been identified and present with hypertrophic cardiomyopathy, muscular hypotonia and lactic acidosis [66–68]. Assessment of skin fibroblasts from control and PiC mutation revealed a decrease in oxygen consumption capacity in the mutants compared to the control [69] Although the fundamental requirement of Pi for calcium uptake and ox-phos was long established, a direct involvement of Pi in Ca2+ uptake in vivo has not been shown. An attempt to answer this question was made in cardiac specific knock-out of slc25A3. 2 weeks post tamoxifen administration resulted in significant reduction in Pi with ~ 45% decrease in cardiac mitochondrial ATP while total cardiac tissue ATP levels remained unaltered with minimal cardiac phenotype. Additionally, MCU-mediated Ca2+ overload was reduced in cardiomyocytes deleted from PiC [70, 71]. Possible explanations for the limited phenotype are either decreased mitochondrial Ca2+ uptake due to the absence of Pi transport or compensation of Pi transport through other mitochondrial solute carriers.
mCa2+ is known to generate ATP and vice-versa ATP is essential for mCa2+ uptake. It is known that ATP is required to maintain mitochondrial ion homeostasis which is key for MCU-mediated Ca2+ uptake [72]. The ATP-Mg solute carriers (SLC25A23, SLC25A24 and SLC25A25) transport adenine nucleotides to the matrix of the mitochondria in response to cytosolic Ca2+ [73–77]. Silencing of SLC25A23 exhibited a significant decrease in mCa2+ uptake and rate of mCa2+ with sustained cCa2+. In complement, SLC25A23 KD cells displayed reduced IMCU. This decrease in mCa2+ uptake was not a result of decreased ΔΨm and SLC25A23 KD cells showed no changes in ΔΨm. SLC25A23 was shown to facilitate mCa2+ uptake and mitochondrial reactive oxygen species (mROS) production by directly interacting with MCU possibly through hydrophobic interactions [24]. One key factor by which SLC25A23 contributes to mCa2+ uptake could be by Ca2+-activated of Pi flow that balances the net matrix charge.
Oxidative Regulation of MCU
Post translational modifications are key mechanisms utilized by cells to fine-tune and expand a proteins function. Two predominant forms of PTM are oxidation and phosphorylation and MCU has been shown to undergo both [78, 79]. MCU has three conserved cysteines containing free thiols in the NTD that get oxidized. Of the three conserved cysteines (C67, C97 and C191), Cys97 was susceptible to nucleophilic reaction. Oxidative stress causes S-glutathionylation of C97 and addition of glutathione to MCU remodels the NTD conformation promoting persistent MCU activity and increased rate of mCa2+ entry. Size exclusion chromatographic analysis of MCUC97A or S-glutathionated MCU revealed higher order oligomers or MCU super complex formation in the mutant. To further study the detailed assembly and regulation mechanisms upon oxidative stress photo-activated localization microscopy (PALM) was utilized. PALM imaging revealed well organized, clustered distribution of the channel in MCUC97A, in contrast with the randomly distributed MCU along the IMM under un-oxidative conditions without altering its interactions with the regulatory proteins. Thus, both biochemical and super resolution microscopy reveal the increased rate of mCa2+ uptake to be due to MCU super complex formation. It is intriguing that though MICU1, MCUR1, MCUb contain several conserved cysteines, the comprehensive biochemical gel shift assay revealed only MCU as the luminal mROS sensor. Thus, conditions of inflammation or I/R injury that promote mROS production in the matrix of the mitochondria tipping the GSH/GSSG balance resulting in mROS-mediated modification of MCU. S-glutathione conjugated MCU exhibits sustained channel activity leading to mCa2+overload-mediated death [18].
A second PTM that MCU has been proposed to undergo is phosphorylation by Calmodulin kinase II (CaMKII) at S57 and S92 of the NTD of MCU [80]. It has been demonstrated that mCa2+ overload during IR can be prevented by inhibiting phosphorylation of S92 in MCU by CaMKII. Electrophysiological studies on mitoplasts from WT and CaMKIIN (transgenic mice expressing CaMKII inhibitor) mice reveal reduced IMCU from CaMKII heart mitochondria compared to the WT mitochondria. Furthermore, IMCU reversed when constitutively active form of CaMKII was added to the pipette solution only in the presence of ATP. Absence of ATP or addition of catalytically incompetent CaMKII (K43M) failed to increase IMCU, indicating that catalytically active CaMKII was required to increase IMCU. Structural analysis of MCU revealed that S92 is a conserved residue in the L2 loop of NTD that stabilizes by hydrogen bond formation with D119. Either phosphorylation of S92 by CaMKII or mutating S92 to alanine abrogates mCa2+ uptake but does not destabilize the MCU complex and does not inhibit interaction with the regulatory molecules. S92A modulates channel activity by changing the conformation of L2-L4 loops such that S92 no longer forms hydrogen bond with D119 [17]. Although mutation analysis demonstrates the significant of S92, a direct phosphorylation assay by CaMKII needs to be investigated.
MCU in Ca2+ Overload and PTP Opening
Having established that Ca2+ that enters through MCU channel shapes cytosolic Ca2+ transients, a major question arises. What may be the implications of impaired cCa2+ signaling? Sudden elevation of cCa2+ due to sustained release from the stores as well as acidic endo-lysosomal compartments through two-pore channels (TPC1 and TPC2) upon nicotinic acid adenine dinucleotide phosphate (NAADP) agonist stimulation under conditions of ischemia-reperfusion (IR) injury results in persistent mCa2+ uptake [81]. This sustained elevation of mCa2+ disrupts the physiological Ca2+ cycling of influx and efflux leading to the activation of permeability transition pore (PTP) [45, 82, 83]. PTP is a voltage dependent, high conductance pore comprising of proteins spanning the OMM, IMS, IMM and matrix. Two known triggers of the pore are permissive amounts of matrix Ca2+ and ROS. Opening of the pore results in dissipation of the proton gradient, inhibition of the ETC and ATP hydrolysis, rapid influx of water and solutes leading to swelling of the mitochondria and eventually cell death by necrosis [84–86]. Hence, it was thought that preventing Ca2+ uptake by the mitochondria under conditions of Ca2+ overload would offer protection from PTP-mediated cell death. To test this hypothesis, mice genetically deleted for Mcu were generated. Mice were born in Mendelian ratio and developed without any gross phenotypic changes. A loss of mCa2+ uptake was observed in various cell types thus confirming the global KO. Surprisingly, WT and MCU−/− MEFs subjected to various stressors that trigger apoptotic and necrotic pathways including ceramide, H2O2, doxorubicin, thapsigargin and tunicamycin failed to offer any protection from Ca2+-overload induced death. In complement, MCU−/− animals subjected to IR injury exhibited unchanged post ischemic recovery as that of the control animals, although the in vitro indices of PTP opening were absent. Similar infarct size, magnitude of ischemic contracture and cell death was observed between the WT and MCU−/− mice following global IR injury [87]. In support, acute conditional deletion of MCU (MCUfl/fl x αMHC-MCM) in the heart exhibited no difference in basal NADH production and oxygen consumption capacity but a metabolic failure under acute stress. In contrast to the previous finding, MCUfl/fl x αMHC-MCM animals demonstrated protection from IR injury and PTP-mediated cell death [88]. The discrepancy in the studies has been attributed to acute vs chronic deletion of the channel where global germ line deletion of MCU animals do not display phenotypic changes possibly due to compensation by other mitochondrial solute carriers. Additionally, differences in the genetic backgrounds of the animals may be a contributing factor to the discrepancy in the two studies observed. Although both the findings report the absence of mCa2+ uptake, upstream cytosolic components were not assessed.
Role of MCU in Mitochondrial shape transition (MiST)
There is long standing evidence supporting the interdependence of mitochondrial function and its shape. Tubular long filamentous network of mitochondria are known to be actively respiring and channel ATP to different part of the cell [89–92]. On the other hand, short circular fragmented mitochondria are often associated with various pathological conditions [93]. The canonical opinion in the field is that mitochondria undergo changes in shape in response to insults such as fragmentation or Ca2+ overload mediated mitochondrial swelling. However, recent findings from two independent groups show a novel mechanism by which mitochondria transition from long filamentous to short circular [94, 95]. The authors term this phenomenon of mitochondrial transition from long spaghetti-like to short donut-like mitochondrial shape transition (MiST). They demonstrate that MiST is triggered by sustained elevation of cCa2+ but not mCa2+. MiST occurs in hepatocytes isolated from MCU liver specific knock out animal (MCUΔHEP) at the same rate as WT hepatocytes even though MCU-mediated Ca2+ overload was absent. MiST was shown to be temporally separate from PTP-mediated mitochondrial swelling, where HeLa cells exhibited a change in mitochondrial shape ~200 s before mitochondrial swelling observed as release of calcein from mitochondria. These data were further validated in genetic deletion of components of OMM (Bax−/−Bak−/− MEFs) and PTP (SPG7−/− and VDAC1/3−/− MEFs). Ca2+-induced MiST was then demonstrated to be independent of mitochondrial fragmentation. MiST was observed in multiple cells that were genetically, RNAi and pharmacologically inhibited of the mitochondrial fission machinery (Drp1−/−, MFF−/−/Fis1−/−, Dyn2 KD, Mid49 KD and Mid51 KD MEFs). The molecular factor mediating MiST in response to cCa2+ changes was identified to be an OMM tethered EF hand containing atypical Rho GTPase, Miro1. Mutations in EF hand 1 of Miro1 that limit Ca2+ binding drastically limit Ca2+-induced MiST. Miro1 senses rise in cCa2+ with its EF hands and depending on the cCa2+ transients (sustained elevation by Ca2+ stressor or transient rise by GPCR agonists) relays either an irreversible MiST or reversible MiST. The Kd for MiST was determined to be > 1 μM cCa2+. Miro1EF1-mediated MiST was revealed to be a pre-requisite for mitochondrial quality control. Long filamentous mitochondria need to transition to sterically small entities to be engulfed by the autophagosomes and inhibiting this change in shape halts autophagosome formation. This was confirmed in Miro1EF1 mutants that do not undergo MiST and thereby do not get engulfed by the autophagosome or degraded by the lysosome [95]. A second group report the same mitochondrial transition independent of mitochondrial fission and swelling to be necessary for mitochondrial DNA release during an inflammatory response. In an attempt to elucidate how mtDNA located in the matrix of the mitochondria releases into the cytosol and activates the cGAS STING pathway that causes cells to secrete type 1 interferon, the authors observed a dynamic change in mitochondrial phenotype from filamentous to globular upon treatment with Bcl2 inhibitor. Herniation of IMM releases mtDNA through OMM pores formed by Bax Bak oligomers [94]. It is possible that other triggers of MiST exist to function under varied pathological conditions and remain to be explored.
Perspectives
A large body of evidence on the molecular identity, stoichiometry, its regulatory proteins has accumulated since the discovery of the mitochondrial calcium uniporter. The generation of germline and tissue specific MCU KO animals permitted understanding the physiological significance of the channel. A lot remains to be explored on the role of MCU under pathological conditions.
Binding of Mg2+ to the MRAP region in the NTD of MCU has been shown to close MCU channel activity. Studies on supplementing Mg2+ to limit MCU channel activity under conditions of Ca2+ overload may be warranted. Studies for the first time have shown like most ion channels, MCU also undergoes oxidative modifications, based on this studying if MCU can serve as matrix ROS set point and understanding the molecular mechanisms of MCU oxidation in inflammatory diseases may open therapeutic avenues. Finally, genetic ablation of MCU did not offer protection from mCa2+ overload-mediated death. A prospective reason is that MCU KO cells triggered MiST due to elevated Ca2+ leading to MiST-mediated cell death. Future studies on MiST mediated pathways need to be explored.
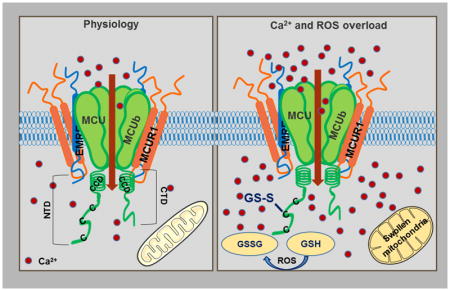
Figure 1.

MCU super complex in physiology and pathology.
Ca2+ enters the mitochondria through MCU channel. MCU, the pore-forming subunit is a pentamer with N and C terminal domains in the matrix. MCUR1 and EMRE are transmembrane proteins that interact with MCU and regulate mCa2+ uptake by the channel. Under conditions of oxidative stress, C97 at the NTD of MCU gets S-glutathionylated promoting MCU oligomerization and increasing mCa2+ uptake leading to Ca2+ overload and swelling of the mitochondria.
Figure 2.

Loss of MCU elicits elevated [Ca2+]c – induced MiST.
Actively respiring filamentous mitochondria that are tethered to microtubules through interactions of Miro and kinesin, take up cCa2+ in response to agonist stimulation through the MCU channel for bioenergetic output. Loss of mCa2+ uptake in the absence of MCU causes elevated cCa2+. Sustained elevated cCa2+ binds the EF hand of Mior1 and releases the mitochondria from microtubules thus causing a change in mitochondrial shape.
Highlights.
The discovery of the molecular identity and structure of the N- terminal domain of mitochondrial calcium uniporter (MCU) has identified MCU to be the pore forming subunit.
MCU is regulated on many levels from protein-protein interaction with its regulatory components (MCUb, MICUs, MCUR1, EMRE and SLC25A23), transcriptional regulation by CREB and micro RNAs, post translational modifications of oxidation and phosphorylation to divalent cations of Ca2+ and Mg2+.
Loss of mitochondrial Ca2+ uptake in the absence of MCU promotes elevated cytosolic Ca2+-induced mitochondrial shape change (MiST)
Acknowledgments
This research was funded by the NIH (R01GM109882, R01HL086699, R01HL119306, and 1S10RR027327 to M.M.). N.N. is supported by the AHA fellowship (17PRE33660720). S.S. is supported by a NIH K99/R00 grant (1K99HL138268-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nature Reviews Molecular Cell Biology. 2000;1:11. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 2.Berridge MJ, Bootman MD, Roderick HL. Calcium: Calcium signalling: dynamics, homeostasis and remodelling. Nature Reviews Molecular Cell Biology. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 3.Penner R, Fasolato C, Hoth M. Calcium influx and its control by calcium release. Curr Opin Neurobiol. 1993;3(3):368–74. doi: 10.1016/0959-4388(93)90130-q. [DOI] [PubMed] [Google Scholar]
- 4.Putney JW, McKay RR. Capacitative calcium entry channels. BioEssays. 1999;21(1):38–46. doi: 10.1002/(SICI)1521-1878(199901)21:1<38::AID-BIES5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 5.Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258(5 Pt 1):C755–86. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- 6.Glancy B, Balaban RS. Role of Mitochondrial Ca2+in the Regulation of Cellular Energetics. Biochemistry. 2012;51:2959–2973. doi: 10.1021/bi2018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J Cell Biol. 1997;137(3):633–48. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilabert JA, Parekh AB. Respiring mitochondria determine the pattern of activation and inactivation of the store-operated Ca(2+) current I(CRAC) Embo j. 2000;19(23):6401–7. doi: 10.1093/emboj/19.23.6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lemasters JJ, et al. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta. 1998;1366(1–2):177–96. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- 10.Nakagawa T, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434(7033):652–8. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 11.Baughman JM, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Stefani D, et al. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476(7360):336–40. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luft JH. Ruthenium red and violet. I. Chemistry, purification, methods of use for electron microscopy and mechanism of action. Anat Rec. 1971;171(3):347–68. doi: 10.1002/ar.1091710302. [DOI] [PubMed] [Google Scholar]
- 14.Reed KC, Bygrave FL. The inhibition of mitochondrial calcium transport by lanthanides and Ruthenium Red. Biochem J. 1974;140(2):143–55. doi: 10.1042/bj1400143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 16.Kovacs-Bogdan E, et al. Reconstitution of the mitochondrial calcium uniporter in yeast. Proc Natl Acad Sci U S A. 2014;111(24):8985–90. doi: 10.1073/pnas.1400514111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee Y, et al. Structure and function of the N-terminal domain of the human mitochondrial calcium uniporter. EMBO Rep. 2015;16(10):1318–33. doi: 10.15252/embr.201540436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee SK, et al. Structural Insights into Mitochondrial Calcium Uniporter Regulation by Divalent Cations. Cell chemical biology. 2016;23:1157–1169. doi: 10.1016/j.chembiol.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oxenoid K, et al. Architecture of the mitochondrial calcium uniporter. Nature. 2016;533(7602):269–73. doi: 10.1038/nature17656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mallilankaraman K, et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. 2012;14(12):1336–43. doi: 10.1038/ncb2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perocchi F, GV, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010 doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plovanich M, et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One. 2013;8(2):e55785. doi: 10.1371/journal.pone.0055785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yasemin Sancak ALM, Kitami Toshimori, Kovács-Bogdán Erika, Kamer Kimberli J, Udeshi Namrata D, Carr Steven A, Chaudhuri Dipayan, Clapham David E, Li Andrew A, Calvo Sarah E, Goldberger Olga, Mootha Vamsi K. EMRE Is an Essential Component of the Mitochondrial Calcium Uniporter Complex. Science. 2013:342. doi: 10.1126/science.1242993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffman NE, et al. SLC25A23 augments mitochondrial Ca(2)(+) uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Mol Biol Cell. 2014;25(6):936–47. doi: 10.1091/mbc.E13-08-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raffaello A, et al. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013;32(17):2362–76. doi: 10.1038/emboj.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mallilankaraman K, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. 2012;151(3):630–44. doi: 10.1016/j.cell.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Csordás GGT, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Fuente Perez S, Bogorad R, Koteliansky V, Adijanto J, Mootha VK, Hajnóczky G. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metabolism. 2013 doi: 10.1016/j.cmet.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoffman NE, et al. MICU1 motifs define mitochondrial calcium uniporter binding and activity. Cell Rep. 2013;5(6):1576–1588. doi: 10.1016/j.celrep.2013.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L, et al. Structural and mechanistic insights into MICU1 regulation of mitochondrial calcium uptake. Embo j. 2014;33(6):594–604. doi: 10.1002/embj.201386523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu JC, et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep. 2016;16(6):1561–1573. doi: 10.1016/j.celrep.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Logan CV, et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet. 2014;46(2):188–93. doi: 10.1038/ng.2851. [DOI] [PubMed] [Google Scholar]
- 32.Bhosale G, et al. Pathological consequences of MICU1 mutations on mitochondrial calcium signalling and bioenergetics. Biochim Biophys Acta. 2017;1864(6):1009–1017. doi: 10.1016/j.bbamcr.2017.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antony AN, et al. MICU1 regulation of mitochondrial Ca(2+) uptake dictates survival and tissue regeneration. Nat Commun. 2016;7:10955. doi: 10.1038/ncomms10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamer KJ, V, Mootha K. MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. 2014;15(3):299–307. doi: 10.1002/embr.201337946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bick AG, et al. Cardiovascular homeostasis dependence on MICU2, a regulatory subunit of the mitochondrial calcium uniporter. Proc Natl Acad Sci U S A. 2017;114(43):E9096–E9104. doi: 10.1073/pnas.1711303114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamer KJ, Grabarek Z, Mootha VK. High-affinity cooperative Ca(2+) binding by MICU1-MICU2 serves as an on-off switch for the uniporter. EMBO Rep. 2017;18(8):1397–1411. doi: 10.15252/embr.201643748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vais H, et al. MCUR1, CCDC90A, Is a Regulator of the Mitochondrial Calcium Uniporter. Cell Metab. 2015;22(4):533–5. doi: 10.1016/j.cmet.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paupe V, et al. CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab. 2015;21(1):109–16. doi: 10.1016/j.cmet.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 39.Tomar D, et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016;15(8):1673–85. doi: 10.1016/j.celrep.2016.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chaudhuri D, et al. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc Natl Acad Sci U S A. 2016;113(13):E1872–80. doi: 10.1073/pnas.1602264113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vais H, et al. EMRE Is a Matrix Ca(2+) Sensor that Governs Gatekeeping of the Mitochondrial Ca(2+) Uniporter. Cell Rep. 2016;14(3):403–410. doi: 10.1016/j.celrep.2015.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamamoto T, et al. Analysis of the structure and function of EMRE in a yeast expression system. Biochim Biophys Acta. 2016;1857(6):831–9. doi: 10.1016/j.bbabio.2016.03.019. [DOI] [PubMed] [Google Scholar]
- 43.Hajnóczky G, et al. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82(3):415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- 44.Robb-Gaspers LD, et al. Integrating cytosolic calcium signals into mitochondrial metabolic responses. Embo j. 1998;17(17):4987–5000. doi: 10.1093/emboj/17.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529(Pt 1):57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4(7):517–29. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 47.Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feske S, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441(7090):179–85. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 49.Roos J, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169(3):435–45. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang SL, et al. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci U S A. 2006;103(24):9357–62. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liou J, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15(13):1235–41. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luik RM, et al. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454(7203):538–42. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McNally BA, et al. Gated regulation of CRAC channel ion selectivity by STIM1. Nature. 2012;482(7384):241–5. doi: 10.1038/nature10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shanmughapriya S, et al. Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci Signal. 2015;8(366):ra23. doi: 10.1126/scisignal.2005673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9(2):102–14. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 56.Marchi S, et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol. 2013;23(1):58–63. doi: 10.1016/j.cub.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hong Z, et al. MicroRNA-138 and MicroRNA-25 Down-regulate Mitochondrial Calcium Uniporter, Causing the Pulmonary Arterial Hypertension Cancer Phenotype. Am J Respir Crit Care Med. 2017;195(4):515–529. doi: 10.1164/rccm.201604-0814OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu C, et al. Mitochondrial calcium uniporter as a target of microRNA-340 and promoter of metastasis via enhancing the Warburg effect. Oncotarget. 2017;8(48):83831–83844. doi: 10.18632/oncotarget.19747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zaglia T, et al. Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc Natl Acad Sci U S A. 2017;114(43):E9006–E9015. doi: 10.1073/pnas.1708772114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carafoli E. The fateful encounter of mitochondria with calcium: how did it happen? Biochim Biophys Acta. 2010;1797(6–7):595–606. doi: 10.1016/j.bbabio.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 61.Brierley GP, Murer E, Bachmann E. STUDIES ON ION TRANSPORT. III. THE ACCUMULATION OF CALCIUM AND INORGANIC PHOSPHATE BY HEART MITOCHONDRIA. Arch Biochem Biophys. 1964;105:89–102. doi: 10.1016/0003-9861(64)90239-5. [DOI] [PubMed] [Google Scholar]
- 62.Fiermonte G, Dolce V, Palmieri F. Expression in Escherichia coli, functional characterization, and tissue distribution of isoforms A and B of the phosphate carrier from bovine mitochondria. J Biol Chem. 1998;273(35):22782–7. doi: 10.1074/jbc.273.35.22782. [DOI] [PubMed] [Google Scholar]
- 63.Deluca HF, Engstrom GW. Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci U S A. 1961;47:1744–50. doi: 10.1073/pnas.47.11.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rossi CS, Lehninger AL. STOICHIOMETRY OF RESPIRATORY STIMULATION, ACCUMULATION OF CA++ AND PHOSPHATE, AND OXIDATIVE PHOSPHORYLATION IN RAT LIVER MITOCHONDRIA. J Biol Chem. 1964;239:3971–80. [PubMed] [Google Scholar]
- 65.Zoeteweij JP, et al. Calcium-induced cytotoxicity in hepatocytes after exposure to extracellular ATP is dependent on inorganic phosphate. Effects on mitochondrial calcium. J Biol Chem. 1993;268(5):3384–8. [PubMed] [Google Scholar]
- 66.Mayr JA, et al. Mitochondrial phosphate-carrier deficiency: a novel disorder of oxidative phosphorylation. Am J Hum Genet. 2007;80(3):478–84. doi: 10.1086/511788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mayr JA, et al. Deficiency of the mitochondrial phosphate carrier presenting as myopathy and cardiomyopathy in a family with three affected children. Neuromuscul Disord. 2011;21(11):803–8. doi: 10.1016/j.nmd.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 68.Bhoj EJ, et al. Pathologic Variants of the Mitochondrial Phosphate Carrier SLC25A3: Two New Patients and Expansion of the Cardiomyopathy/Skeletal Myopathy Phenotype With and Without Lactic Acidosis. JIMD Rep. 2015;19:59–66. doi: 10.1007/8904_2014_364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seifert EL, et al. Natural and Induced Mitochondrial Phosphate Carrier Loss: DIFFERENTIAL DEPENDENCE OF MITOCHONDRIAL METABOLISM AND DYNAMICS AND CELL SURVIVAL ON THE EXTENT OF DEPLETION. J Biol Chem. 2016;291(50):26126–37. doi: 10.1074/jbc.M116.744714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kwong JQ, et al. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 2014;21(8):1209–17. doi: 10.1038/cdd.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gutiérrez-Aguilar M, et al. Genetic Manipulation of The Cardiac Mitochondrial Phosphate Carrier does not affect Permeability Transition. J Mol Cell Cardiol. 2014;72:316–25. doi: 10.1016/j.yjmcc.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Buttgereit F, Brand MD. A hierarchy of ATP-consuming processes in mammalian cells. Biochem J. 1995;312(Pt 1):163–7. doi: 10.1042/bj3120163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fiermonte G, et al. Identification of the mitochondrial ATP-Mg/Pi transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution. J Biol Chem. 2004;279(29):30722–30. doi: 10.1074/jbc.M400445200. [DOI] [PubMed] [Google Scholar]
- 74.Aprille JR. Regulation of the mitochondrial adenine nucleotide pool size in liver: mechanism and metabolic role. Faseb j. 1988;2(10):2547–56. doi: 10.1096/fasebj.2.10.3290024. [DOI] [PubMed] [Google Scholar]
- 75.Palmieri F. The mitochondrial transporter family SLC25: identification, properties and physiopathology. Mol Aspects Med. 2013;34(2–3):465–84. doi: 10.1016/j.mam.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 76.Tewari SG, et al. A biophysical model of the mitochondrial ATP-Mg/P(i) carrier. Biophys J. 2012;103(7):1616–25. doi: 10.1016/j.bpj.2012.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Amigo I, et al. Glucagon regulation of oxidative phosphorylation requires an increase in matrix adenine nucleotide content through Ca2+ activation of the mitochondrial ATP-Mg/Pi carrier SCaMC-3. J Biol Chem. 2013;288(11):7791–802. doi: 10.1074/jbc.M112.409144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Theillet FX, et al. Cell signaling, post-translational protein modifications and NMR spectroscopy. J Biomol NMR. 2012;54(3):217–36. doi: 10.1007/s10858-012-9674-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brewer TF, et al. Chemical approaches to discovery and study of sources and targets of hydrogen peroxide redox signaling through NADPH oxidase proteins. Annu Rev Biochem. 2015;84:765–90. doi: 10.1146/annurev-biochem-060614-034018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Joiner ML, et al. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491(7423):269–73. doi: 10.1038/nature11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Davidson SM, et al. Inhibition of NAADP signalling on reperfusion protects the heart by preventing lethal calcium oscillations via two-pore channel 1 and opening of the mitochondrial permeability transition pore. Cardiovasc Res. 2015;108(3):357–66. doi: 10.1093/cvr/cvv226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79(4):1127–55. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 83.Halestrap AP, Griffiths EJ, Connern CP. Mitochondrial calcium handling and oxidative stress. Biochem Soc Trans. 1993;21(2):353–8. doi: 10.1042/bst0210353. [DOI] [PubMed] [Google Scholar]
- 84.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87(1):99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 85.Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112(4):481–90. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- 86.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4(7):552–65. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 87.Pan X, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15(12):1464–72. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Luongo TS, et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015;12(1):23–34. doi: 10.1016/j.celrep.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.VOTH JB-HAM. Dynamics of Mitochondria in Living Cells: Shape Changes, Dislocations, Fusion, and Fission of Mitochondria. MICROSCOPY RESEARCH AND TECHNIQUE. 1994 doi: 10.1002/jemt.1070270303. [DOI] [PubMed] [Google Scholar]
- 90.Youle MKaR. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003 doi: 10.1038/sj.cdd.4401260. [DOI] [PubMed] [Google Scholar]
- 91.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337(6098):1062–5. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yaffe MP. The machinery of mitochondrial inheritance and behavior. Science. 1999;283(5407):1493–7. doi: 10.1126/science.283.5407.1493. [DOI] [PubMed] [Google Scholar]
- 93.Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. 2014;15(10):634–46. doi: 10.1038/nrm3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McArthur KW, Heddleston LW, Li JM, Padman L, Oorschot BS, Geoghegan V, Chappaz Davidson ND, San Chin S, Lane H, Dramicanin RM, Saunders M, Sugiana TL, Lessene C, Osellame R, Chew LD, Dewson TL, Lazarou G, Ramm M, Lessene G, Ryan G, Rogers MT, van Delft KL, Kile MFBT. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. 2018;359(6378) doi: 10.1126/science.aao6047. [DOI] [PubMed] [Google Scholar]
- 95.Nemani N, et al. MIRO-1 Determines Mitochondrial Shape Transition upon GPCR Activation and Ca(2+) Stress. Cell Rep. 2018;23(4):1005–1019. doi: 10.1016/j.celrep.2018.03.098. [DOI] [PMC free article] [PubMed] [Google Scholar]