

Abstract
Objective
Primary progressive multiple sclerosis (PPMS) causes accumulation of neurological disability from disease onset without clinical attacks typical of relapsing multiple sclerosis (RMS). However, whether genetic variation influences the disease course remains unclear. We aimed to determine whether mutations causative of neurological disorders that share features with multiple sclerosis (MS) contribute to risk for developing PPMS.
Methods
We examined whole‐genome sequencing (WGS) data from 38 PPMS and 81 healthy subjects of European ancestry. We selected pathogenic variants exclusively found in PPMS patients that cause monogenic neurological disorders and performed two rounds of replication genotyping in 746 PPMS, 3,049 RMS, and 1,000 healthy subjects. To refine our findings, we examined the burden of rare, potentially pathogenic mutations in 41 genes that cause hereditary spastic paraplegias (HSPs) in PPMS (n = 314), secondary progressive multiple sclerosis (SPMS; n = 587), RMS (n = 2,248), and healthy subjects (n = 987) genotyped using the MS replication chip.
Results
WGS and replication studies identified three pathogenic variants in PPMS patients that cause neurological disorders sharing features with MS: KIF5A p.Ala361Val in spastic paraplegia 10; MLC1 p.Pro92Ser in megalencephalic leukodystrophy with subcortical cysts, and REEP1 c.606 + 43G>T in Spastic Paraplegia 31. Moreover, we detected a significant enrichment of HSP‐related mutations in PPMS patients compared to controls (risk ratio [RR] = 1.95; 95% confidence interval [CI], 1.27–2.98; p = 0.002), as well as in SPMS patients compared to controls (RR = 1.57; 95% CI, 1.18–2.10; p = 0.002). Importantly, this enrichment was not detected in RMS.
Interpretation
This study provides evidence to support the hypothesis that rare Mendelian genetic variants contribute to the risk for developing progressive forms of MS. Ann Neurol 2018;83:51–63
Primary progressive multiple sclerosis (PPMS) is a rare form of multiple sclerosis (MS) characterized by progressive accumulation of disability from disease onset without the attacks typically observed in the relapsing form of the disease (RMS).1 PPMS represents an unmet need in the care of neurological patients because of its poor response to MS disease‐modifying therapies and its relentless clinical course that resembles neurodegenerative disorders.2 Compared to RMS, PPMS patients are older at onset, men and women are equally affected, and the most common clinical presentation is a progressive spastic paraparesis.3, 4 Moreover, some family studies demonstrate a higher concordance in MS disease course (PPMS versus RMS) within affected siblings than expected by chance.5, 6, 7 These observations suggest that unique genetic and environmental susceptibility factors may, in part, influence risk for PPMS.
To date, genetic studies have not shown a difference between PPMS and RMS susceptibility,8 either because of a strong shared genetic susceptibility or because of a lack of power given the lower prevalence of primary progressive disease and consequent under‐representation in genome‐wide association studies (GWAS) and other screens. Furthermore, the role of Mendelian genes has not been systematically studied in MS, despite the observation that there is widespread comorbidity among Mendelian and complex diseases.9 PPMS shares clinical features with specific Mendelian neurological disorders (ie, potential MS phenocopies) that cause progressive neurological disability attributed to injury to the central nervous system (CNS). Examples of genetic disorders that resemble PPMS include hereditary spastic paraplegias (HSPs), inherited leukodystrophies, and mitochondrial disorders.3, 10, 11
This study aims to determine whether mutations causative of genetic disorders that share features with MS contribute to disability in PPMS. Furthermore, we hypothesized that genomes from subjects with PPMS are enriched for mutations in genes involved in monogenic disorders that share clinicopathological features of MS. To this end, we performed whole‐genome sequencing (WGS) in a well‐characterized PPMS cohort and validated identified variants in multiple independent PPMS cohorts. We next examined whether PPMS patients carried mutations in specific classes of MS phenocopy disorders. Specifically, we hypothesized that PPMS patients were enriched for mutations in genes that caused hereditary spastic paraplegias, a rare group of conditions that cause progressive leg weakness and spasticity resembling PPMS.12 Last, we performed similar analyses in RMS and secondary progressive multiple sclerosis (SPMS) patients to determine whether MS phenocopy mutations identified in this study were unique to PPMS.
Patients and Methods
Cohorts
All human studies were approved by each respective institutional ethics review committee, and all participants provided written informed consent. To investigate the role of MS phenocopy mutations in PPMS pathogenesis, we examined WGS data in a discovery cohort of 38 PPMS patients of European descent and 81 ethnicity‐matched controls. PPMS patients in this group were recruited at or referred to the University of California San Francisco (UCSF) between 1996 and 2013 and satisfied 2010 International Panel Criteria13 (Table S1). WGS data for controls were obtained from the 1000 Genomes Project,14 Complete Genomics Inc. (CGI) Public Genomes,15 and healthy individuals recruited at UCSF. We subsequently performed replication genotyping in 142 PPMS patients from Germany and in 269 PPMS and 460 RMS patients recruited at UCSF (Phase 1 replication). We performed a second round of replication in 335 PPMS and 340 RMS patients from Italy and in 2,249 RMS patients and 1,000 healthy controls recruited at UCSF (Phase 2 replication). In total, the discovery and replication cohorts included 784 PPMS and 3,049 RMS patients and 1,081 controls (Table 1; Fig 1A).
Table 1.
Demographic Data in Study Cohorts Used to Identify MS Phenocopy Mutations
| Discovery Phase | Phase 1 Replication | Phase 2 Replication | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Platform | Complete Genomics Inc. | Illumina OpenArray | Targeted Genotyping | ||||||
| Variants | 15.2 million | 15 | 4 | ||||||
| Cohort source | UCSF | 1KGa (64), CGIb (11), UCSF (6) | Germany | UCSF | Italy | UCSF | |||
| Phenotype | PPMSc | Controls | PPMS | PPMS | RMS | PPMS | RMS | RMS | Controls |
| Sample size | 38 | 81 | 142 | 269 | 460 | 335 | 340 | 2,249 | 1,000 |
| Age at onset | |||||||||
| Mean ± SD | 42.9 ± 9.9 | NA | — | 40.1 ± 11.1 | 31.2 ± 9.2 | 40.7 ± 9.5 | 29.5 ± 9.4 | 31.9 ± 9.5 | NA |
| Median (range) | 47 (25–54) | NA | — | 40 (5–66) | 31 (4–61) | 40 (18–66) | 28 (10–64) | 31 (5–69) | NA |
| Disease duration | |||||||||
| Mean ± SD | 11.3 ± 8.1 | NA | — | 19.2 ± 11.3 | 22.6 ± 9.9 | 11.1 ± 7.3 | 9.0 ± 5.3 | 13.8 ± 10.6 | NA |
| Median (range) | 10 (0–37) | NA | — | 18 (1–56) | 20 (10–62) | 10 (1–45) | 8 (1–39) | 12 (0–66) | NA |
| Female sex (%) | 22 (58) | 39 (48) | 93 (65) | 155 (58) | 475 (60) | 171 (50.6) | 179 (52.6) | 1,886 (84) | 516 (52) |
1000 Genomes Project, Complete Genomics Data.
Complete Genomics Inc. Public Genomes.
Patients meet 2010 International Panel Criteria for PPMS.
MS = multiple sclerosis; SD = standard deviation; UCSF = University of California San Francisco; PPMS = primary progressive multiple sclerosis; NA = not applicable; HSP = hereditary spastic paraplegia; PPMS = primary progressive multiple sclerosis; RMS = relapsing multiple sclerosis.
Figure 1.
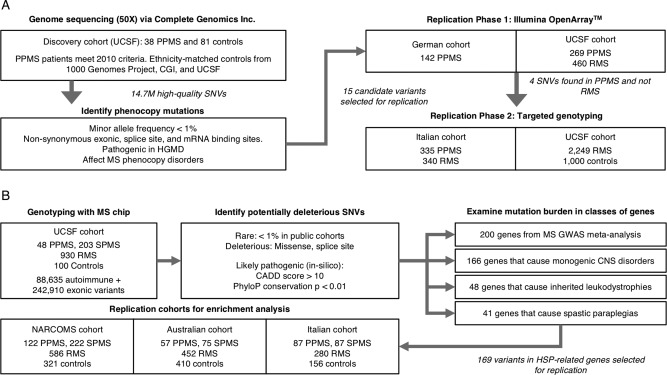
Summary of study cohorts, genotyping platforms, and variant selection. (A) Schematic of study design used for identifying MS phenocopy variants. The WGS discovery cohort included 38 PPMS patients (who met 2010 International Panel Criteria) and 81 ethnicity‐matched controls sequenced using the Complete Genomics Inc. (CGI) platform. Fifteen candidate variants were selected for Phase 1 replication genotyping in 411 PPMS and 460 RMS patients using OpenArray. Four top candidate variants exclusively found in PPMS and not RMS patients were selected for Phase 2 replication in 335 PPMS and 340 RMS patients from an Italian cohort and in 2,249 RMS and 1,000 controls from UCSF. (B) Schematic of study design used for determining the burden of HSP‐related mutations in PPMS. The discovery cohort comprised of 48 PPMS patients (who met 2010 International Panel Criteria) and 100 controls genotyped on the MS replication chip. Replication patients included an additional 266 PPMS, 1,702 RMS, and 887 control subjects from three additional cohorts (NARCOMS, Australian, and Italian) genotyped on the same platform. All subjects examined were of European ancestry. CADD = Combined Annotation Dependent Depletion; CNS = central nervous system; GWAS = genome‐wide association studies; HGMD = Human Gene Mutation Database; HSP = hereditary spastic paraplegia; MS = multiple sclerosis; NARCOMS = North America Research Committee on Multiple Sclerosis; PhyloP = phylogenetic conservation p value; PPMS = primary progressive multiple sclerosis; RMS = relapsing multiple sclerosis; SNVs = single‐nucleotide variants; SPMS = secondary progressive multiple sclerosis; UCSF = University of California San Francisco; WGS = whole‐genome sequencing.
To test the hypothesis that PPMS patients may be enriched for mutations in genes that cause spastic paraplegias, we examined a cohort of 48 PPMS patients of European descent recruited at UCSF and 100 ethnicity‐matched controls who were genotyped using the MS replication chip.16 These PPMS patients met 2010 International Panel Criteria and included 25 patients from our WGS cohort and 23 patients who were exclusively genotyped on the MS chip (Table S1). For replication, we examined three additional cohorts of European ancestry genotyped on the same platform. These included the North America Research Committee on Multiple Sclerosis (NARCOMS; 122 PPMS and 321 controls), an Australian cohort (57 PPMS and 410 controls), and an Italian cohort (87 PPMS and 156 controls). Last, we examined RMS and SPMS patients from these cohorts to investigate whether MS phenocopy mutations are unique to patients with PPMS. In total, we performed enrichment analysis in 314 PPMS, 587 SPMS, 2,248 RMS, and 987 healthy subjects (Table 2; Fig 1B). A description of all cohorts and genotyping methods is provided in Tables 1 and 2 and Figure 1.
Table 2.
Discovery and Replication Cohorts Used to Examine Burden of Spastic Paraplegia Mutations
| Discovery Cohort | Replication Cohort 1 | Replication Cohort 2 | Replication Cohort 3 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Platform | MS Replication Chip | MS Replication Chip | MS Replication Chip | MS Replication Chip | ||||||||||||
| Variants | 331,536 | 169a | 169a | 169a | ||||||||||||
| Cohort | UCSF | NARCOMSb | Australia | Italy | ||||||||||||
| Diagnosisd | CTL | RMS | SPMS | PPMS | CTL | RMS | SPMS | PPMS | CTL | RMS | SPMS | PPMS | CTL | RMS | SPMS | PPMS |
| Samples | 100 | 930 | 203 | 48c | 321 | 586 | 222 | 122 | 410 | 452 | 75 | 57c | 156 | 280 | 87 | 87 |
| Age at onset | ||||||||||||||||
| Mean ± SD | NA | 32.8 ± 9.4 | 31.3 ± 9.3 | 42.3 ± 11.2 | NA | 32.6 ± 9.8 | 31.0 ± 9.2 | 40.7 ± 11.1 | NA | 36 ± 9.8 | 35 ± 9.4 | 42 ± 10.8 | NA | 28.2 ± 8.9 | 29.1 ± 9.4 | 39.9 ± 10.1 |
| Median, range | NA | 32 (5–64) | 31 (11–58) | 42 (22–66) | NA | 33 (4–61) | 30 (10–65) | 41 (12–66) | NA | 36 (14–64) | 33 (15–59) | 41 (20–72) | NA | 27 (7–52) | 27 (15–59) | 40 (19–60) |
| Disease duration | ||||||||||||||||
| Mean ± SD | NA | 12.6 ± 9.5 | 23.0 ± 10.0 | 11.2 ± 7.4 | NA | 21.9 ± 11.3 | 28 ± 10.9 | 22.6 ± 10.4 | NA | 8 ± 6.5 | 21 ± 10.2 | 13 ± 9.9 | NA | 8.1 ± 7.0 | 17.3 ± 7.9 | 10.8 ± 8.2 |
| Median range | NA | 12 (0–54) | 22 (3–48) | 10 (0–37) | NA | 20 (1–66) | 28 (0–58) | 20 (2–56) | NA | 6 (0–41) | 22 (3–44) | 10 (1–43) | NA | 6 (0–37) | 17 (3–41) | 9 (1–45) |
| Female sex | 22% | 74% | 68% | 40% | 47% | 81% | 81% | 61% | 77% | 77% | 76% | 61% | 38% | 66% | 70% | 57% |
One hundred sixty‐nine HSP‐related variants were examined in these replication cohorts.
North American Research Committee on Multiple Sclerosis.
Patients met 2010 International Panel Criteria for PPMS.
CTL = control subjects; RMS = relapsing MS patients excluding those with known secondary progression; SPMS = secondary progressive MS; PPMS = primary progressive MS.
SD = standard deviation; UCSF = University of California San Francisco; NA = not applicable; HSP = hereditary spastic paraplegia.
WGS and Replication Genotyping
For each sample selected for genome sequencing, we derived lymphoblastoid cell lines (LCLs) from whole blood samples17 and extracted 15 μg of DNA from each of these cell lines for sequencing using the CGI platform.15 The LCL is a convenient research tool for obtaining virtually unlimited amounts of biological material from an individual, and there is high concordance for single‐nucleotide variant (SNV) calls obtained from WGS using LCL and whole blood.18 CGI performed DNA read mapping to the human genome (reference hg19) and provided variant calls using CGI proprietary software.19 We performed additional quality control by removing low‐quality calls (heterozygous calls with VarScoreVAF <40 and homozygous calls with VarScoreVAF <20) and variants with less than 95% call rate. We confirmed European ancestry using Identity‐By‐Descent analysis of WGS variants.20 We annotated WGS variants with curated data from the Human Gene Mutation Database21, 22 (HGMD) and selected SNVs that were functionally deleterious (nonsynonymous exonic, splice site, or mRNA binding site), rare (<1% in public cohorts), classified as pathogenic, affected monogenic neurological disorders, and were exclusively found in PPMS patients. We used an allele frequency cutoff of 1% to identify potentially pathogenic mutations, which, in Mendelian disorders, have allele frequencies below 0.1%.23 Candidate MS phenocopy variants were then selected for replication genotyping using the Illumina OpenArray system (Illumina, San Diego, CA) in Phase 1 replication and using targeted individual genotyping in Phase 2 replication.
Mutation Enrichment Analysis
To determine whether PPMS patients are enriched for mutations in genes that cause spastic paraplegias, we examined cases and controls who were genotyped using the custom MS replication chip, which includes 88,635 autoimmune markers and 242,910 exonic variants from the Illumina HumanExome BeadChip v1.1. Quality control included the following SNV‐level exclusion criteria: (1) missingness > 0.05; (2) Hardy‐Weinberg equilibrium p value < 10−6; and (3) differential missingness in cases and controls p value < 0.001. We annotated variants using Ingenuity Variant Analysis24 and extracted all rare (minor allele frequency [MAF] < 1% in public data sets), functionally deleterious (missense and splice site), and potentially pathogenic (Combined Annotation Dependent Depletion [CADD] score > 10; Phylogenetic conservation p value [PhyloP] < 0.01) variants within 41 genes that cause spastic paraplegias (Table S4).12, 25, 26, 27, 28, 29 We performed logistic regression on the number of potentially pathogenic variants per individual adjusted for subject sex and used p = 0.05 as the threshold for significance. To understand the impact of variant selection on enrichment results, we performed sensitivity analysis using different in silico predicted pathogenicity scores including CADD, PhyloP, Sorting Intolerant From Tolerant (SIFT), and Polymorphism Genotyping v2 [PolyPhen‐2]; Table S5). To evaluate the likelihood of finding an enrichment of spastic paraplegia variants in PPMS patients by chance, we randomly permuted PPMS and healthy control status in our discovery cohort 10,000 times and determined the permutation p value as the likelihood of observing an enrichment in HSP‐related genes greater than or equal to that found in our discovery cohort.
To replicate our findings from mutation enrichment analysis, we examined the same spastic paraplegia variants in three additional PPMS cohorts genotyped on the MS replication chip. We performed meta‐analysis across these four (discovery plus three replication) cohorts using a random‐effects model examining the mean number of mutations per individual in PPMS compared to controls. To determine whether the enrichment of HSP‐related mutations is unique to PPMS, we also examined RMS and SPMS patients genotyped on the MS replication chip from these four cohorts. We calculated the average number of spastic paraplegia variants per individual in each phenotype and used a t test to determine whether the average burden of HSP‐related variants differed between MS disease‐course subtypes.
To investigate the relationship between spastic paraplegia variants and the burden of common MS susceptibility variants, we calculated the MS Genetic Burden (MSGB) using MS replication chip genotypes. The MSGB is obtained by summing the number of independently associated MS risk alleles weighted by their beta coefficients, obtained from a large GWAS meta‐analysis, at 177 (of 200) non‐MHC (major histocompatibility complex) loci and 18 (of 32) MHC variants, which includes the HLA‐DRB1*15:01‐tagging single‐nucleotide polymorphism (SNP) rs3135388.16 Subsequently, we examined whether the average MSGB differed between PPMS (n = 170), SPMS (n = 425), RMS (n = 1516), and healthy subjects (n = 421) in the UCSF and NARCOMS cohorts using pair‐wise t tests between groups. Finally, we examined whether the mean MSGB differed in PPMS and SPMS patients who carried a HSP‐related variant compared to those who did not carry any such variants.
Results
Genome sequencing in a well‐characterized cohort of 38 PPMS patients and 81 ethnicity‐matched controls using CGI yielded on average greater than 50 × depth of coverage and identified more than 3 million SNVs, 5,000 insertion deletions, 1,500 structural variants, and 250 copy number variants (CNVs) per sample. After performing quality control, we found 14,709,637 high‐quality SNV calls in the autosomal and sex chromosomes across these 119 genomes that comprised our discovery cohort (Fig 1).
We searched for candidate MS phenocopy variants, by annotating the 14.7 million SNVs identified in the 119 genomes, and found 1,287 pathogenic variants directly involved in 714 Mendelian disorders. Of these, 691 variants involved in 474 disorders were identified in PPMS patients, and 1,029 variants involved in 609 disorders were identified in controls. This included 52 variants on the sex chromosomes; however, none of these affected neurological disorders that share features with MS. Fifteen of the 691 variants found in PPMS patients were rare, functionally deleterious, affected neurological disorders and were absent in controls. We attempted an independent replication to validate the WGS calls in the discovery cohort by genotyping these 15 variants in an additional 411 PPMS and 460 RMS patients using the Illumina OpenArray platform (Phase 1 replication). Twelve of the 15 candidate variants were confirmed, and four were exclusively found in PPMS (and not in RMS) patients in the combined discovery and initial replication cohort. To further assess whether these four pathogenic variants were PPMS specific, we selectively genotyped them in an additional set of PPMS (n = 335) and RMS (n = 2,589) patients, in addition to healthy subjects (n = 1,000; Phase 2 replication). The variants selected for replication genotyping are summarized in Table 3.
Table 3.
MS Phenocopy Variants Selected for Phase 1 and Phase 2 Replication
| Genetic Variant | Number of Individuals Carrying a Heterozygous Mutation | Minor Allele Frequency (%)a | Risk vs ExAC EUR | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Discovery Cohort (WGS) | Phase 1 Replication (OpenArray) | Phase 2 Replication (targeted typing) | ||||||||||||
| Gene | Variantb | Disorder (inheritance)c | 38 PPMS | 81 CTRLS | 411PPMS | 460 RMS | 335 PPMS | 2589 RMS | 1000 CTRLS | All PPMS | All RMS | All CTRLS | ExAC EUR | RR for PPMS |
| KIF5A | p.A361V | SPG10 (AD) | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0.064 | 0.016 | 0.000 | 0.003 | 23.3 |
| TSC2 | p.E75K | TSd (AD) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0.064 | 0.000 | 0.000 | 0.019 | 3.3 |
| MLC1 | p.P92S | MLC (AR) | 1 | 0 | 0 | 0 | 0 | 4 | 0 | 0.064 | 0.066 | 0.000 | 0.034 | 1.9 |
| REEP1 | c.606 +43G>T | SPG31 (AD) | 1 | 0 | 2 | 0 | 0 | 2 | 2 | 0.191 | 0.033 | 0.093 | 0.115 | 1.7 |
| SCN9A | p.W1538R | PE (AD) | 1 | 0 | 6 | 3 | Not selected for Phase 2: | 0.67 | 0.33 | 0.00 | 0.173 | 3.9 | ||
| HPD | p.I335M | TYRSN3 (AR) | 1 | 0 | 3 | 3 | Variant was found in at least | 0.33 | 0.33 | 0.00 | 0.158 | 2.1 | ||
| CACNA1A | p.P897R | EA2 (AD) | 1 | 0 | 1 | 2 | one RMS patient in Phase 1. | 0.22 | 0.22 | 0.00 | 0.141 | 1.6 | ||
| DCTN1 | p.T1249I | ALS (AD) | 1 | 0 | 3 | 4 | 0.22 | 0.43 | 0.00 | 0.416 | 0.5 | |||
| D2HGDH | p.A426T | D‐2‐HGA (AR) | 3 | 0 | 7 | 3 | 0.89 | 0.33 | 0.00 | 1.101 | 0.8 | |||
| ADAR | p.P193A | AGS (AR, AD) | 1 | 0 | 1 | 1 | 0.22 | 0.11 | 0.00 | 0.281 | 0.8 | |||
| NOTCH3 | p.S497L | CADASIL (AD) | 2 | 0 | 7 | 4 | 0.67 | 0.43 | 0.00 | 1.395 | 0.5 | |||
| SLC6A5 | p.T690T | HKPX3d (AD) | 1 | 0 | 0 | 4 | 0.11 | 0.43 | 0.00 | 0.271 | 0.4 | |||
| NF1 | p.D176E | NF1 (AD) | 1c | 0 | 2 | 5 | Not selected for Phase 2: | 0.24 | 0.54 | 0.00 | 0.512 | NA | ||
| TSC2 | p.L1423L | TSd AD) | 1c | 0 | 0 | 0 | CGI genotypes not | 0.00 | 0.00 | 0.00 | 0.423 | |||
| LRRK2 | p.E334K | PD (AD) | 1c | 0 | 0 | 0 | validated on OA | 0.00 | 0.00 | 0.00 | 0.484 | |||
Minor allele frequencies (MAFs) are calculated for 784 PPMS, 3,049 RMS, and 1,081 control subjects for the top four candidate variants. For all other variants, MAF is calculated for 449 PPMS, 460 RMS, and 81 control subjects. The ExAC European (including Finnish) cohort was used as the reference to calculate relative risk for candidate variants.
“c” denotes coding DNA sequence position; “p” denotes protein amino acid position. Only the variant corresponding to the primary transcript according to ExAC is provided.
Phenocopy disorder abbreviations: SPG = spastic paraplegia; TS = tuberous sclerosis; MLC = megalencephalic leukodystrophy with subcortical cysts; PE = primary erythromelalgia; TYRSN3 = tyrosinemia type 3; EA2 = episodic ataxia 2; ALS = amyotrophic lateral sclerosis; D‐2‐HGA = D‐2‐hydroxyglutaric aciduria; AGS = Aicardi‐Goutières syndrome; CADASIL = cerebral autosomal‐dominant arteriopathy with subcortical infarcts and leukoencephalopathy; HKPX3 = hyperekplexia 3; NF1 = neurofibromatosis type 1; PD = Parkinson's disease.
These variants were not validated during Phase 1 replication genotyping.
These variants have not yet been reported to be pathogenic in this disorder.
The four variants selected for Phase 2 replication were observed at a higher frequency in PPMS patients (n = 784) compared to over 36,000 European individuals from the Exome Aggregation Consortium (ExAC‐EUR).30 Of note, three of these variants had been previously reported in disorders that potentially mimic MS: KIF5A p.Ala361Val (risk ratio [RR] = 23), a dominant variant for spastic paraplegia 10 (SPG10 [MIM: 604187])27; MLC1 p.Pro92Ser (RR = 1.8), a recessive variant for megalencephalic leukodystrophy with subcortical cysts (MLC [MIM: 604004])31; and REEP1 c.606 + 43G>T (RR = 1.6), a dominant variant affecting mRNA binding at the 3′ untranslated region (UTR) of REEP1 causing spastic paraplegia 31 (SPG31 [MIM: 610250]).25 The last variant, TSC2 p.Glu75Lys (RR = 3.3), is a variant of unknown significance for tuberous sclerosis (TSC2 [MIM: 613254]).
REEP1 c.606 + 43G>T was found in a PPMS patient (65‐0008) with spastic paraparesis from our WGS discovery cohort and in 2 additional PPMS patients (52‐0139 and 52‐1859) with a progressive spinal cord syndrome in our Phase 1 replication genotyping. This variant was also found in 2 RMS patients (21‐0003, MSGENE02‐528) and 2 controls (9961‐50050, 9961‐51000901) in our Phase 2 replication genotyping at a frequency comparable to the ExAC European cohort. KIF5A c.C1082T p.Ala361Val was found in a PPMS patient (02‐0069) with spastic paraparesis from our discovery cohort and was also found in 1 RMS patient (MSGENE02‐539) in our Phase 2 replication genotyping at a lower frequency compared to that observed in PPMS patients. MLC1 c.274C>T p.Pro92Ser was found in a PPMS patient (04‐1225) with brain‐predominant disease from our discovery cohort. Of note, the MLC1 variant was also found in 4 RMS patients (52‐1463, 05‐0032, 60‐0354, and 60‐0362) with the same frequency as that observed in PPMS patients, and in 0 controls, in our Phase 2 replication genotyping. Last, TSC2 c.G223A p.Glu75Lys was found in a PPMS patient (60‐0385) with a mild disease course from our discovery cohort. These results are summarized in Table 3, and detailed clinical information is provided in Table 4 and Tables S2 and S3.
Table 4.
PPMS Patients Who Carry a Reported Phenocopy Variant
| Clinical characteristics | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2010 International Panel Criteriaa | Genetic Variant | ||||||||||||
| ID | Sex | Age at Onset | Progressive course | Positive CSF (elevated IgG index, OCBs) | Brain MRI | Spine MRI | Gad + | Meets criteria | Gene | Disorder (inheritance) | Reported Variantb | Typec | Platformd |
| 020069 | F | 37 | + | ND | + | + | ‐ | + | KIF5A | SPG10 (AD) | p.A361V | mis | CGI |
| 041225 | F | 51 | + | + ( + , + ) | + | ‐ | ‐ | + | MLC1 | MLC (AR) | p.P92S | mis | CGI |
| 650008 | F | 50 | + | + ( + , + ) | + | + | ‐ | + | REEP1 | SPG31 (AD) | c.606 + 43G>T | 3′ | CGI |
| 650084 | F | 48 | + | + (‐, + ) | + | + | ‐ | + | SPG7 | SPG7 (AR,AD) | c.1552 + 1G>T | ss | MS chip |
| 700019 | F | 61 | + | + ( + , + ) | + | + | + | + | SPG7 | SPG7 (AR,AD) | p.A510V | mis | MS chip |
| 520139e | F | 26 | + | + | ‐ | + | ‐ | + | REEP1 | SPG31 (AD) | c.606 + 43G>T | 3′ | OA |
| 521859e | M | 57 | + | ND | + | ND | ‐ | ‐ | REEP1 | SPG31 (AD) | c.606 + 43G>T | 3′ | OA |
2010 International Panel Criteria includes (1) progression since onset without relapses and (2) two of the following three criteria: positive CSF (elevated IgG index or oligoclonal bands), brain lesions consistent with MS, and spinal cord lesions consistent with MS. “+” denotes satisfies criteria, “–” denotes does not satisfy criteria, “ND” denotes test was not done. GAD denotes gadolinium enhancement on at least one magnetic resonance imaging brain or spinal cord.
“c” denotes coding DNA sequence position; “p” denotes protein amino acid position. Only the variant corresponding to the primary transcript according to ExAC is provided.
Variant types: mis = missense; ss = splice site; 3′ = 3′UTR.
Genotyping platforms: CGI = WGS via Complete Genomics Inc.; MS chip = MS replication chip; OA = OpenArray.
PPMS patients from Phase 1 replication cohort. CSF for patient 520139, performed at the NIH in 1977, was reported to be consistent with MS; however, results were not available.
Given that two of the top four genes identified by WGS (REEP1 and KIF5A) are associated with progressive spinal cord injury, we hypothesized that PPMS patients may be generally enriched for deleterious mutations in genes that associate with the hereditary spastic paraplegias. Spastic paraplegias are a rare group of conditions that cause degeneration of motor axons in the corticospinal tract resulting in progressive leg weakness and spasticity,12 providing a plausible basis for phenotypic mimicry (ie, phenocopy) with MS. To test this hypothesis, we examined 48 PPMS patients of European ancestry who satisfied 2010 International Panel Criteria and 100 matched controls genotyped using the custom MS (ie, replication) chip, which includes more than 240,000 exonic variants. We extracted 169 rare, functionally deleterious, and potentially pathogenic variants within the 41 genes known to cause spastic paraplegias12, 25, 26, 27, 28, 29 (Table S4). Interestingly, PPMS patients harbored, on average, significantly more variants (0.29 per individual) in these genes than did controls (0.11), and the risk for PPMS increased with the number of potentially pathogenic variants (RR = 2.65; 95% confidence interval [CI], 1.18–5.96; likelihood ratio [LR], p = 0.028).
To evaluate the likelihood of finding an enrichment of spastic paraplegia variants in PPMS patients by chance, we randomly permuted PPMS and healthy control status in our discovery cohort 10,000 times and calculated the enrichment of HSP‐related variants determined using the same criteria. Strikingly, the 2.7‐fold enrichment of spastic paraplegia variants in PPMS was greater than in 98.5% of case‐control permutations (p = 0.015), suggesting that the observed enrichment was unlikely attributed to chance (Fig 2). Sensitivity analysis demonstrated a persistent enrichment of HSP variants in PPMS after applying various in silico predicted pathogenicity criteria (Table S5). No significant enrichment was detected in 48 genes affecting inherited leukodystrophies,11 166 genes affecting other Mendelian disorders that involve the CNS,32 or in the 200 genes associated with MS in a recent GWAS meta‐analysis.16 Whereas we observed a trend toward enrichment of variants in 48 genes affecting inherited leukodystrophies, this was not significant (RR = 1.9; 95% CI, 0.9–4.2; LR, p = 0.091). A summary of discovery cohort PPMS patients who carry a reported MS phenocopy variant is shown in Table 4.
Figure 2.
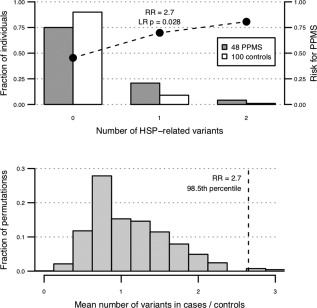
Burden of spastic paraplegia mutations in PPMS. (A) Forty‐eight PPMS patients genotyped on the MS replication chip are enriched for rare (MAF < 1% in public data sets), functionally deleterious (missense and splice site), and potentially pathogenic (CADD score, > 10; PhyloP conservation, p < 0.01) variants in 41 genes known to cause spastic paraplegias, and the relative risk for PPMS increases with the number of HSP‐related variants carried by an individual (cases mean = 0.29; controls mean = 0.11; RR = 2.7; logistic regression, p = 0.028). (B) Random permutation of PPMS case and control status shows that the 2.7‐fold enrichment of pathogenic variants in 41 HSP‐related genes is greater than in 98.5% of 10,000 permutations (p = 0.015). HSP = hereditary spastic paraplegia; LR = likelihood ratio; MAF = minor allele frequency; MS = multiple sclerosis; PhyloP = phylogenetic conservation p value; PPMS = primary progressive multiple sclerosis; RR = risk ratio.
To replicate our finding that PPMS patients are enriched for mutations in genes that cause spastic paraplegias, we examined three additional cohorts of European ancestry genotyped on the MS replication chip. Using the same variant selection criteria as described previously, we detected an enrichment of HSP‐related mutations in 122 PPMS patients (0.3 variants per individual) recruited through NARCOMS compared to 321 controls (0.12; RR = 2.56; 95% CI, 1.64–4.01; LR, p = 6.0 × 10−4), as well as in an Australian cohort of 57 PPMS patients (0.26) compared to 410 controls (0.14; RR = 1.83; 95% CI, 1.00–3.35; LR, p = 0.049). No enrichment was observed in an Italian cohort of 87 PPMS patients (0.08) compared to 156 controls (0.1; RR = 0.84; 95% CI, 0.35–2.02; p = 0.8). Meta‐analysis of the discovery and three replication cohorts confirmed a significant enrichment of potentially pathogenic HSP mutations in 315 PPMS patients compared to 987 controls (RR = 1.95; 95% CI, 1.27–2.98; random‐effects model, p = 0.002; Fig 3).
Figure 3.
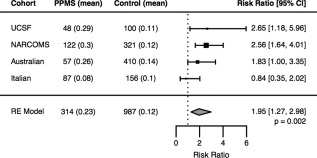
Meta‐analysis of HSP‐related mutations across multiple cohorts. Examination of 314 PPMS patients and 987 controls genotyped on the MS Replication Chip across four cohorts (UCSF, NARCOMS, Australian, and Italian) confirmed the observation that PPMS patients harbor significantly more potentially pathogenic HSP mutations compared to controls (RR = 1.95; random effects model, p = 0.002). HSP = hereditary spastic paraplegia; MS = multiple sclerosis; NARCOMS = North America Research Committee on Multiple Sclerosis; PPMS = primary progressive multiple sclerosis; RR = risk ratio; UCSF = University of California San Francisco; RE = random effects.
To determine whether the enrichment of variants observed in spastic paraplegia genes was unique to PPMS patients or whether this also contributes to the risk for SPMS, we examined genotypes from SPMS (n = 587) and RMS (n = 2,248) patients of European ancestry from the four cohorts in our meta‐analysis. On average, PPMS (n = 315) patients harbored a significantly higher number of spastic paraplegia variants (0.23 per individual) compared to RMS patients (0.14; n = 2,248; mean difference [MD] = 0.10; 95% CI, 0.06–0.14; t test, p = 9.6 × 10−4) and compared to controls (0.12; n = 987; MD = 0.11; 95% CI, 0.05–0.17; p = 3.8 × 10−4). Interestingly, SPMS patients (n = 587) also harbored a higher number of spastic paraplegia variants (0.17 per individual) compared to RMS patients (0.14; n = 2,248; MD = 0.04; 95% CI, 0.00–0.07; p = 0.048) and compared to controls (0.12; n = 987; MD = 0.05; 95% CI, 0.01–0.09; p = 0.018). By contrast, no significant enrichment was found in RMS patients compared to controls (MD = 0.01; 95% CI, –0.02 to 0.04; p = 0.4; Fig 4). Whereas we observed a trend toward enrichment of HSP‐related variants in PPMS patients (0.23) compared to SPMS patients (0.17), this was not significant (MD = 0.06; 95% CI, –0.01 to 0.12; p = 0.07). These results suggest that the enrichment of spastic paraplegia variants is unique to patients with a progressive disease course and is not present in all forms of MS.
Figure 4.
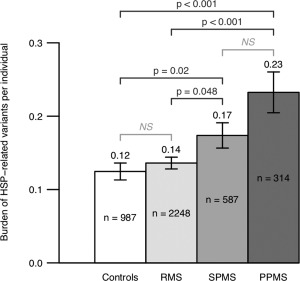
HSP‐related mutation burden in PPMS, SPMS, RMS, and controls. Examination of 314 PPMS, 2,248 RMS, 587 SPMS, and 987 control subjects from four cohorts (UCSF, NARCOMS, Australian, and Italian) showed that PPMS patients (0.23 variants per individual) on average harbored a significantly higher number of potentially pathogenic HSP‐related mutations compared to RMS (0.14) and controls (0.12; t test, p = 3.8 × 10−4 for PPMS vs controls; p = 9.6 × 10−4 for PPMS vs RMS). Moreover, SPMS patients (0.17), on average, also harbored a higher number of HSP‐related mutations compared to RMS (0.14) and controls (0.12; p = 0.018 for SPMS vs controls, p = 0.048 for SPMS vs RMS). Importantly, no significant enrichment was detected in RMS patients compared to healthy controls (p = 0.4). HSP = hereditary spastic paraplegia; NS = not significant; PPMS = primary progressive multiple sclerosis; RMS = relapsing multiple sclerosis; SPMS = secondary progressive multiple sclerosis.
We subsequently hypothesized that the risk for developing a progressive form of MS is related to the accumulation of rare deleterious variants that directly affect degenerative neurological disorders, and this risk is independent of the genetic burden that confers susceptibility for MS. To test this hypothesis, we calculated the MS Genetic Burden (MSGB) in PPMS (n = 170), SPMS (n = 425), RMS (n = 1516), and healthy subjects (n = 421) from our UCSF and NARCOMS MS replication chip cohorts. We found that, on average, MS patients (n = 2,111) have a higher MSGB (mean = 22.7) compared to healthy controls (21.7; (MD = 1.0; 95% CI, 0.86–1.13; p = 2.2 × 10−42). We did not detect a significant difference in MSGB between PPMS (22.9), SPMS (22.8), and RMS (22.7) patients (PPMS versus RMS, MD = 0.15; 95% CI, –0.06 to 0.37; p = 0.17; SPMS versus RMS, MD = 0.07; 95% CI, –0.07 to 0.21; p = 0.34; PPMS versus SPMS, MD = 0.8; 95% CI, –0.16 to 0.33; p = 0.49). Importantly, we observed no significant difference between PPMS patients who carry a HSP variant (22.9; n = 44) and those who do not (22.9; n = 126; MD = 0.02; 95% CI, –0.55 to 0.48; p = 0.95). Likewise, we detected no difference between SPMS patients who carry a HSP variant (23.0; n = 62) and those who do not (22.7; n = 363; MD = 0.22; 95% CI, –0.14 to 0.58; p = 0.23). These results suggest that rare HSP‐related variants modulate the risk for developing a progressive disease course independent of the overall genetic burden that confers risk for developing MS.
Discussion
Although many disorders resemble MS clinically and radiographically, we are unaware of previous reports associating Mendelian disorder genes in PPMS. Systematic review of clinical records indicates that patients who carried a MS phenocopy‐related mutation were not misdiagnosed with PPMS, but rather carry clinical characteristics of both PPMS and the phenocopy disorder. Specifically, the PPMS patients with pathogenic mutations in REEP1 and KIF5A have demyelinating‐appearing lesions on magnetic resonance imaging. Cerebrospinal fluid (CSF) was obtained in 2 of 3 REEP1 carriers and both had elevated intrathecal gammaglobulin synthesis. CSF was not obtained in the KIF55A pA361V carrier. Carriers of these phenocopy mutations experienced progressive spastic paraparesis typical of both PPMS and HSP. Additional studies are needed to better understand the impact of these MS phenocopy mutations on disease severity in PPMS.
Furthermore, to our knowledge, this is the first study to report that PPMS patients are enriched for mutations in genes that cause spastic paraplegias. We show that the enrichment of HSP‐related variants in PPMS is significantly higher than expected by chance, is validated in meta‐analysis across multiple cohorts, and is not present in RMS patients. Importantly, SPMS patients also harbor a detectable enrichment of spastic paraplegia mutations, suggesting that there might be a shared genetic susceptibility to progressive forms of MS, and that carrying such variants might increase the risk for developing secondary progression after an earlier relapsing‐onset course. Last, we observe no significant difference in the burden of common MS susceptibility variants in PPMS and SPMS patients who carry a spastic paraplegia variant compared to those who do not. These findings suggest that rare mutations in genes that cause degenerative neurological disorders contribute to a progressive disease course, and this effect is independent of the burden of common MS susceptibility variants that influences the risk for developing MS.
We acknowledge a number of limitations. First, although we comprehensively examined pathogenic SNVs, which are better documented in literature, we did not analyze CNVs, structural variants, or intergenic regulatory mutations that might also be pathogenic. Second, the size of the discovery WGS cohort was limited by the high cost of genome sequencing. After performing WGS in the pilot PPMS cohort, we subsequently devoted resources to independent replication rather than additional sequencing. Third, we did not identify pathogenic SNVs on the sex chromosomes, and thus our results do not explain the difference in gender distribution between PPMS (equally affects males and females) and RMS (female predominant). Fourth, although we used an expert‐curated and experimentally validated list of spastic paraplegia genes, a more comprehensive gene list will emerge as additional HSP‐related loci are discovered. Therefore, our analysis might underestimate the prevalence of such mutations in progressive forms of MS. Fifth, the enrichment of spastic paraplegia mutations in PPMS patients was not detected in the Italian cohort. The genetic variation of Italian Europeans can be distinguished from that of other European populations,8 and known common pathogenic spastic paraplegia mutations in this population33, 34, 35 were not captured on the MS replication chip. Last, whereas clinical genetic testing may be useful in refining a diagnosis when it is in question, the utility of genetic testing in PPMS patients is currently limited given the lack of disease‐modifying treatments for the Mendelian genetic variants that might contribute to a progressive disease course.
Proposed mechanisms for PPMS pathogenesis include compartmentalized leptomeningeal inflammation behind a relatively intact blood–brain barrier, oxidative stress driving mitochondrial injury, chronic microglial activation causing oligodendrocyte dysfunction and axonal injury, and age‐related iron accumulation.36, 37, 38, 39, 40 By examining MS phenocopy mutations, this study identified genes encoding neuroaxonal proteins (KIF5A), mitochondrial function (REEP1, SPG7), and astrocyte osmoregulation (MLC1), supporting the hypothesis that genetic variation contributes to progressive neuronal and glial dysfunction in PPMS. However, our approach does not comprehensively assess all proposed mechanisms for PPMS, including complex local autoimmune and glial‐mediated pathways that do not manifest as monogenic disorders.
KIF5A c.C1082T p.Ala361Val (a reported dominant mutation for SPG1027) was found in a PPMS patient (02‐0069) from our WGS discovery cohort. We considered the presence of this variant to be responsible for a progressive myelopathy characteristic of SPG10, highlighting a potential MS phenocopy. This variant was previously reported in a SPG10 patient with adult‐onset (age 35) spastic paraparesis from an affected family spanning 4 generations.27 KIF5A encodes an axonal motor protein responsible for anterograde transport. Reduced expression of KIF5A has been observed in MS white matter lesions,41 and some SPG10 patients have demyelinating‐appearing lesions in the spinal cord.26 Whereas KIF5A is located in the MS susceptibility locus CYP28B1‐OS9, the top SNP in this region (rs701006) does not influence KIF5A expression,42 and there is no clear linkage between this common MS susceptibility variant and the rare KIF5A variant (rs121434444).14 We hypothesize that disruption to axonal transport may be, in part, responsible for neurodegeneration and spinal cord injury in progressive forms of MS; however, additional studies are needed to confirm this association and to understand its functional impact.
REEP1 c.606 + 43G>T (a reported dominant variant for SPG3125) was found in a PPMS patient (65‐0008) from our discovery cohort and 2 PPMS patients (52‐0139, 52‐1859) from our replication cohorts. Reported SPG31 patients who carried this variant had heterogeneous clinical characteristics, ranging from mild paraparesis to severe tetraparesis with bulbar dysfunction. Functional studies show that REEP1 facilitates mitochondrial‐endoplasmic reticulum (ER) interactions, and altered ER‐mitochondrial contacts cause intracellular Ca2+ overload resulting in axonal injury. We found additional evidence for the role of mitochondrial dysfunction in PPMS by identifying reported pathogenic mutations for spastic paraplegia 7 (SPG7 [MIM: 607259])12, 43, 44, 45, 46 in two PPMS patients (70‐0019, 65‐0084) from our MS chip discovery cohort (Table 4). SPG7 encodes the mitochondrial protein paraplegin, and mutations in this gene cause complex spastic paraplegia attributed to complex I deficiency and increased sensitivity to oxidative stress.47 These findings provide evidence for a pathogenic role of REEP1 and SPG7 in PPMS, and supports the hypothesis that mitochondrial dysfunction and diminished tolerance to oxidative stress may contribute to progressive myelopathy in PPMS.
The MLC1 c.274C>T p.Pro92Ser variant (a reported recessive and isolated heterozygous mutation in MLC,31 a leukodystrophy characterized by myelin swelling and cystic changes arising from dysfunction of MLC1 cell junction proteins on astrocytic foot processes) was found in a PPMS patient (04‐1225) from our WGS discovery cohort. Of note, this variant was also found in 4 RMS patients (52‐1463, 05‐0032, 60‐0354, and 60‐0362) with the same frequency as that observed in PPMS patients, and in 0 controls, in our Phase 2 replication genotyping. This variant is rare (MAF 0.03%) and, to our knowledge, has not been examined through GWAS. Neuropathology studies show that active MS lesions have reduced staining for perivascular astrocytic MLC1, whereas chronic lesions demonstrate upregulation of MLC1 attributed to astrogliosis.48, 49 Astrocytes are hypothesized to play a role in MS disease progression through participation in the innate immune system, production of cytotoxic factors, and inhibition of remyelination by forming glial scar.50 Despite these observations, the exact role of MLC1 in MS pathogenesis remains unclear. Given that MLC1 p.Pro92Ser has been reported in leukodystrophy patients and is well characterized in functional studies, we hypothesize that this MLC1 variant affects white matter injury through astrocyte‐mediated osmoregulatory dysfunction in both PPMS and RMS patients, albeit with incomplete penetrance and variable expressivity.
TSC2 p.Glu75Lys was found in a PPMS patient (60‐0385) from our WGS discovery cohort. White matter lesions in tuberous sclerosis are often caused by abnormal cortical development or neuronal migration rather than demyelination.51 Because of the lack of reported pathogenic cases, we consider TSC2 p.Glu75Lys a variant of uncertain significance without a clear role in PPMS pathogenesis.
Understanding the role of phenocopies in PPMS has important clinical implications. The finding that PPMS patients are enriched for HSP‐related mutations that cause progressive axonal injury is consistent with the observation that the most common clinical presentation in PPMS is a progressive spastic paraparesis3, 52, 53 and might help explain why these patients respond poorly to immunomodulatory therapies. Moreover, carrying a pathogenic mutation for a MS phenocopy disorder does not cause MS, but rather modulates the disease course through mechanisms independent of immune‐mediated pathways implicated by reported MS susceptibility loci. Larger studies are needed to identify additional phenocopy disorders that might contribute to a progressive disease course in MS. Longitudinal studies are needed to examine the predictive value of rare phenocopy variants on transition to secondary progression in patients with relapsing‐onset disease. Last, translational studies are required to develop disease‐modifying therapies for spastic paraplegias and other Mendelian disorders that share clinical features with MS. These efforts are instrumental for developing effective treatments that slow and prevent disability in patients with progressive forms of MS.
Author Contributions
S.E.B., J.R.O., B.A.C.C., X.J., and S.L.H. were responsible for conception and design of the study. All authors were responsible for data acquisition and analysis. X.J., L.M. and S.E.B. were responsible for drafting the manuscript. All authors reviewed and accepted the final draft of the manuscript.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Additional supporting information can be found in the online version of this article.
Supporting Information Table S1
Supporting Information Table S2
Supporting Information Table S3
Supporting Information Table S4
Supporting Information Table S5
Supporting Information Table S6
Supporting Information Table S7
Acknowledgment
This work was supported by NIH/NINDS grants R01NS026799 (S.L.H. and J.R.O.), R01NS049477 (S.L.H.), and R01NS088155, (S.E.B.).
The authors thank the multiple sclerosis patients and healthy controls who participated in this study. The authors acknowledge the contributions of Rosa Guerrero and Hourieh Mousavi for sample processing and management.
References
- 1. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014;83:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Louapre C, Lubetzki C, Matute C, Seze J, Giovannoni G, Hemmer B. Neurodegeneration in multiple sclerosis is a process separate from inflammation: yes. Mult Scler J 2015;21:1626–1628. [DOI] [PubMed] [Google Scholar]
- 3. Cree BA. Genetics of primary progressive multiple sclerosis. Handb Clin Neurol 2014;122:211–230. [DOI] [PubMed] [Google Scholar]
- 4. Jenkins TM, Khaleeli Z, Thompson AJ. Diagnosis and management of primary progressive multiple sclerosis. Minerva Med 2008;99:141–155. [PubMed] [Google Scholar]
- 5. Chataway J, Mander A, Robertson N, et al. Multiple sclerosis in sibling pairs: an analysis of 250 families. J Neurol Neurosurg Psychiatry 2001;71:757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oturai AB, Ryder LP, Fredrikson S, et al. Concordance for disease course and age of onset in Scandinavian multiple sclerosis coaffected sib pairs. Mult Scler 2004;10:5–8. [DOI] [PubMed] [Google Scholar]
- 7. Robertson NP, Clayton D, Fraser M, Deans J, Compston DA. Clinical concordance in sibling pairs with multiple sclerosis. Neurology 1996;47:347–352. [DOI] [PubMed] [Google Scholar]
- 8. International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium , Sawcer S, et al. Genetic risk and a primary role for cell‐mediated immune mechanisms in multiple sclerosis. Nature 2011;476:214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blair DR, Lyttle CS, Mortensen JM, et al. A nondegenerate code of deleterious variants in mendelian loci contributes to complex disease risk. Cell 2013;155:70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ars E, Kruyer H, Morell M, et al. Recurrent mutations in the NF1 gene are common among neurofibromatosis type 1 patients. J Med Genet 2003;40:e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vanderver A, Tonduti D, Schiffmann R, Schmidt J, van der Knaap MS. Leukodystrophy overview In: Adam MP, Ardinger HH, Pagon RA, et al, eds. SourceGeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993‐2018. 2014 Feb 6. [Google Scholar]
- 12. Fink JK. Hereditary spastic paraplegia: clinico‐pathologic features and emerging molecular mechanisms. Acta Neuropathol 2013;126:307–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 Revisions to the McDonald criteria. Ann Neurol 2011;69:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. A global reference for human genetic variation. The 1000 Genomes Project Consortium. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed]
- 15. Drmanac R, Sparks AB, Callow MJ, et al. Human genome sequencing using unchained base reads on self‐assembling DNA nanoarrays. Science 2009;327:78–81. [DOI] [PubMed] [Google Scholar]
- 16. International Multiple Sclerosis Genetics Consortium , Patsopoulos N, Baranzini SE, et al. The Multiple Sclerosis Genomic Map: Role of peripheral immune cells and resident microglia in susceptibility. bioRxiv 2017:143933. doi: 10.1101/143933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ling PD, Huls HM. Isolation and immortalization of lymphocytes. Curr Protoc Mol Biol 2005;Chapter 28:Unit 28.2. [DOI] [PubMed] [Google Scholar]
- 18. Nickles D, Madireddy L, Yang S, et al. In depth comparison of an individual's DNA and its lymphoblastoid cell line using whole genome sequencing. BMC Genomics 2012;13:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carnevali P, Baccash J, Halpern AL, et al. Computational techniques for human genome resequencing using mated gapped reads. J Comput Biol 2012;19:279–292. [DOI] [PubMed] [Google Scholar]
- 20. Browning BL, Browning SR. Improving the accuracy and efficiency of identity by descent detection in population data. Genetics 2013;194:459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014. 133(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. QIAGEN . Genome Trax—QIAGEN Bioinformatics. [Web Resource] 2017 [cited 2017]. Available at: https://www.qiagenbioinformatics.com/products/genome-trax/. Accessed on July 1, 2014.
- 23. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. QIAGEN . Ingenuity Variant Analysis. 2017. [cited 2017]. Available at: https://www.qiagenbioinformatics.com/products/ingenuity-variant-analysis/. Accessed on June 27, 2017.
- 25. Beetz C, Schüle R, Deconinck T, et al. REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain 2008;131:1078–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Crimella C, Baschirotto C, Arnoldi A, et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot‐Marie‐Tooth type 2. Clin Genet 2012;82:157–164. [DOI] [PubMed] [Google Scholar]
- 27. Lo Giudice M, Neri M, Falco M, et al. A missense mutation in the coiled‐coil domain of the KIF5A gene and late‐onset hereditary spastic paraplegia. Arch Neurol 2006;63:284–287. [DOI] [PubMed] [Google Scholar]
- 28. Mead SH, Proukakis C, Wood N, et al. A large family with hereditary spastic paraparesis due to a frame shift mutation of the spastin (SPG4) gene: association with multiple sclerosis in two affected siblings and epilepsy in other affected family members. J Neurol Neurosurg Psychiatry 2001;71:788–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Romagnolo A, Masera S, Mattioda A, et al. Atypical hereditary spastic paraplegia mimicking multiple sclerosis associated with a novel SPG11 mutation. Eur J Neurol 2014;21:e14–e15. [DOI] [PubMed] [Google Scholar]
- 30. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ben‐Zeev B, Levy‐Nissenbaum E, Lahat H, et al. Megalencephalic leukoencephalopathy with subcortical cysts; a founder effect in Israeli patients and a higher than expected carrier rate among Libyan Jews. Hum Genet 2002;111:214–218. [DOI] [PubMed] [Google Scholar]
- 32. Kibbe WA, Arze C, Felix V, et al. Disease Ontology 2015 update: an expanded and updated database of human diseases for linking biomedical knowledge through disease data. Nucleic Acids Res 2015;43(Database issue):D1071–D1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Racis L, Tessa A, Di Fabio R, et al. The high prevalence of hereditary spastic paraplegia in Sardinia, insular Italy. J Neurol 2014;261:52–59. [DOI] [PubMed] [Google Scholar]
- 34. Magariello A, Muglia M, Patitucci A, et al. Mutation analysis of the SPG4 gene in Italian patients with pure and complicated forms of spastic paraplegia. J Neurol Sci 2010;288:96–100. [DOI] [PubMed] [Google Scholar]
- 35. Magariello A, Muglia M, Patitucci A, et al. Novel spastin (SPG4) mutations in Italian patients with hereditary spastic paraplegia. Neuromuscul Disord 2006;16:387–390. [DOI] [PubMed] [Google Scholar]
- 36. Correale J, Gaitan MI, Ysrraelit MC, Fiol MP. Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain 2017;140:527–546. [DOI] [PubMed] [Google Scholar]
- 37. Abdelhak A, Weber MS, Tumani H. Primary progressive multiple sclerosis: putting together the puzzle. Front Neurol 2017;8:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fitzner D, Simons M. Chronic progressive multiple sclerosis—pathogenesis of neurodegeneration and therapeutic strategies. Curr Neuropharmacol 2010;8:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 2012;8:647–656. [DOI] [PubMed] [Google Scholar]
- 40. Greenfield AL, Hauser SL. B cell therapy for multiple sclerosis: entering an era. Ann Neurol 2018;83:13‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hares K, Redondo J, Kemp K, et al. Axonal motor protein KIF5A and associated cargo deficits in multiple sclerosis lesional and normal‐appearing white matter. Neuropathol Appl Neurobiol 2017;43:227–241. [DOI] [PubMed] [Google Scholar]
- 42. Carithers LJ, Ardlie K, Barcus M, et al. A novel approach to high‐quality postmortem tissue procurement: The GTEx Project. Biopreserv Biobank 2015;13:311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roxburgh RH, Marquis‐Nicholson R, Ashton F, et al. The p.Ala510Val mutation in the SPG7 (paraplegin) gene is the most common mutation causing adult onset neurogenetic disease in patients of British ancestry. J Neurol 2013;260:1286–1294. [DOI] [PubMed] [Google Scholar]
- 44. Sánchez‐Ferrero E, Coto E, Beetz C, et al. SPG7. mutational screening in spastic paraplegia patients supports a dominant effect for some mutations and a pathogenic role for p.A510V. Clin Genet 2013;83:257–262. [DOI] [PubMed] [Google Scholar]
- 45. Klebe S, Depienne C, Gerber S, et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain 2012;135:2980–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Warnecke T, Duning T, Schirmacher A, et al. A novel splice site mutation in the SPG7 gene causing widespread fiber damage in homozygous and heterozygous subjects. Mov Disord 2010;25:413–420. [DOI] [PubMed] [Google Scholar]
- 47. Atorino L, Silvestri L, Koppen M, et al. Loss of m‐AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J Cell Biol 2003;163:777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kovacs GG, Wagner U, Dumont B, et al. An antibody with high reactivity for disease‐associated alpha‐synuclein reveals extensive brain pathology. Acta Neuropathol 2012;124:37–50. [DOI] [PubMed] [Google Scholar]
- 49. Masaki K, Masaki K, et al. Acta Neuropathol. 2012. (https://www.ncbi.nlm.nih.gov/m/pubmed/22438105/) [Google Scholar]
- 50. Correale J, Farez MF. The role of astrocytes in multiple sclerosis progression. Front Neurol 2015;6:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Umeoka S, Koyama T, Miki Y, et al. Pictorial review of tuberous sclerosis in various organs. Radiographics 2008;28:e32. [DOI] [PubMed] [Google Scholar]
- 52. Andersson PB, Waubant E, Gee L, Goodkin DE. Multiple sclerosis that is progressive from the time of onset. Arch Neurol 1999;56:1138–1142. [DOI] [PubMed] [Google Scholar]
- 53. Salter A, Thomas NP, Tyry T, Cutter GR, Marrie RA. A contemporary profile of primary progressive multiple sclerosis participants from the NARCOMS Registry. Mult Scler J 2017:135245851771127. doi: 10.1177/1352458517711274. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information can be found in the online version of this article.
Supporting Information Table S1
Supporting Information Table S2
Supporting Information Table S3
Supporting Information Table S4
Supporting Information Table S5
Supporting Information Table S6
Supporting Information Table S7