

Abstract
Schizophrenia is associated with core deficits in cognitive abilities and impaired functioning of the newly evolved prefrontal association cortex (PFC). In particular, neuropathological studies of schizophrenia have found selective atrophy of the pyramidal cell microcircuits in deep layer III of the dorsolateral PFC (dlPFC) and compensatory weakening of related GABAergic interneurons. Studies in monkeys have shown that recurrent excitation in these layer III microcircuits generates the precisely patterned, persistent firing needed for working memory and abstract thought. Importantly, excitatory synapses on layer III spines are uniquely regulated at the molecular level in ways that may render them particularly vulnerable to genetic and/or environmental insults. Glutamate actions are remarkably dependent on cholinergic stimulation, and there are inherent mechanisms to rapidly weaken connectivity, e.g. during stress. In particular, feedforward cyclic adenosine monophosphate (cAMP)-calcium signaling rapidly weakens network connectivity and neuronal firing by opening nearby potassium channels. Many mechanisms that regulate this process are altered in schizophrenia and/or associated with genetic insults. Current data suggest that there are “dual hits” to layer III dlPFC circuits: initial insults to connectivity during the perinatal period due to genetic errors and/or inflammatory insults that predispose the cortex to atrophy, followed by a second wave of cortical loss during adolescence, e.g. driven by stress, at the descent into illness. The unique molecular regulation of layer III circuits may provide a nexus where inflammation disinhibits the neuronal response to stress. Understanding these mechanisms may help to illuminate dlPFC susceptibility in schizophrenia and provide insights for novel therapeutic targets.
Keywords: Prefrontal cortex, pyramidal cells, schizophrenia, calcium, stress, inflammation
Graphical abstract
INTRODUCTION
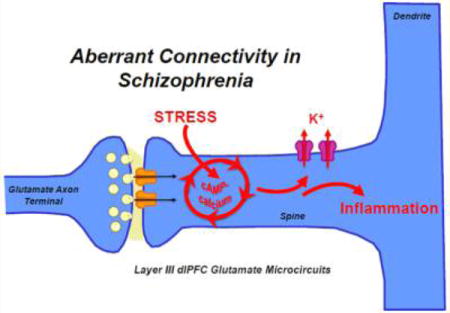
Schizophrenia was originally termed “dementia praecox” (premature dementia) by Pick and Kraepelin due to the marked cognitive deficits that are a core feature of this illness.1 Cognitive deficits in schizophrenia precede the onset of psychosis, continue throughout the lifetime of patients, and are the best predictor of long-term functional outcome.2,3 In particular, schizophrenia is characterized by profound alterations in the newly evolved prefrontal cortical (PFC) circuits that generate thought, manifesting in the clinical hallmarks of the disease.4 The PFC has extensive projections to provide top-down control of behavior, thought, and emotion (Figure 1A). Several lines of research indicate that the recurrent excitatory pyramidal cell microcircuits in deep layer III of the dorsolateral PFC (dlPFC) that generate thought are especially vulnerable in schizophrenia.5 These microcircuits are uniquely regulated at the molecular level in a manner that renders them susceptible to atrophy, as they contain the molecular machinery to magnify feedforward cyclic adenosine monophosphate (cAMP)-calcium signaling in spines, which may drive inflammation and weaken connections by opening nearby potassium channels. The molecular regulation of the dlPFC contrasts with “classical” sensory cortices (e.g., primary visual cortex), where cAMP–calcium signaling events strengthen connections,6 similar to those found in rodent sensory cortex. This review explores (1) how dlPFC circuits are regulated in a fundamentally different manner from classical circuits, (2) how genetic and environmental insults in schizophrenia may target these critical circuits, and (3) how strategies for protecting these synapses early in the course of illness may slow or lessen the progression of disease.
Figure 1.
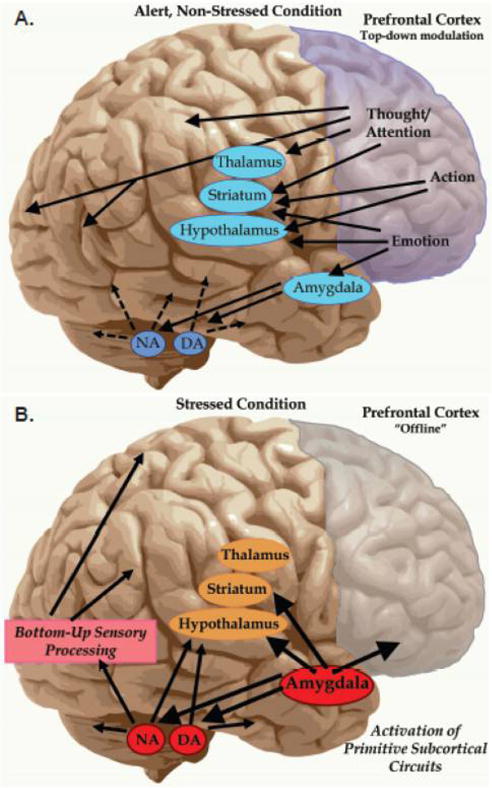
Differential sensitivity of neuronal circuits during stress. The newly evolved PFC subserves many higher cognitive functions, including abstract reasoning, executive functions such as planning and organization, and metacognitive operations, including insight about errors in thought and action. The PFC has extensive connections that allow top-down regulation of brain function. The PFC is also organized in a topographical manner with dorsal and lateral regions mediating thought and action, while ventral and medial regions mediate emotion. The PFC also has extensive interconnections with the arousal systems, including NE and DA cells in the brainstem. Panel A: Under alert, nonstressed conditions, there are optimal levels of catecholamine release in PFC, allowing top-down regulation of cortical and subcortical structures and strong cognitive function. Panel B: Under conditions of uncontrollable stress, high levels of catecholamine release take the dlPFC “offline”, while simultaneously strengthening subcortical and sensory cortical functions. Chronic stress exposure induces architectural changes, i.e. atrophy of layer III PFC connections. These actions may be relevant to dlPFC gray matter loss in schizophrenia.
SCHIZOPHRENIA: BURDEN, ETIOLOGY, AND SYMPTOMS
Schizophrenia is a neurodevelopmental psychiatric disease that afflicts a large population (0.5–1%) globally.7 The various symptoms of schizophrenia patients can be categorized into three categories: (A) positive symptoms such as hallucinations and delusions that alter perceptions of reality and falsify normative behaviors, (B) negative symptoms that reflect the absence of certain behaviors that are present in normal individuals such as flat affect, avolition, alogia, and anhedonia, and (C) cognitive symptoms such as thought disorder and working memory deficits.8 These symptoms usually manifest during late adolescence or early adulthood, with most patients experiencing a life-long course of the illness. Although these have been analyzed as distinct domains, the neurobiological underpinnings likely overlap, e.g. where symptoms such as alogia (poverty of speech), delusions, and hallucinations may all involve cognitive dysfunction of subsystems in the PFC.9,10 The disease is a highly heterogeneous syndrome, and the disease pathogenesis is multifactorial, involving several etiological factors that can aggravate the chance for developing the illness.11 In general, it is thought that gene–environment interactions are pivotal in driving the pathogenesis of schizophrenia, with genetic predispositions and various environmental insults during prenatal, perinatal, and postnatal development enhancing risk for schizophrenia.12,13
The “dual hit” hypothesis proposes that schizophrenia arises from initial insults during perinatal cortical formation, followed by a second wave of insults starting in adolescence that leads to onset of overt symptoms (Figure 2). Initial genetic and environmental insults would alter cortical formation in utero, afflicting cortical neuron migration and/or connectivity starting in the second trimester.14 These may include inherited or de novo genetic insults,15–19 including mosaic expression that may preferentially target the dlPFC and association cortices.20–22 A myriad of physiological stressors during pregnancy (e.g., maternal infection, immune dysregulation, maternal malnutrition, hypoxia) occurring at different stages of prenatal, perinatal, and postnatal development are associated with increased incidence of schizophrenia.12,23,24 The massively interconnected pyramidal cell circuits in layer III dlPFC may be especially vulnerable to insults that impact neuronal connectivity. The second age of vulnerability starts during adolescence (Figure 2). Recent neuroimaging data indicate an accelerated wave of cortical gray matter loss accompanies descent into illness,25,26 associated with increased signs of inflammation27,28 and often precipitated by stressful life events. Accelerated loss of gray matter may reflect exacerbations of the normal pruning process,29–32 although recent postmortem human data show the synaptic pruning in the PFC actually begins in childhood.32 Thus, additional, abnormal processes may be engaged during adolescence in schizophrenia. As stress exposure weakens PFC connectivity and function through increased catecholamine actions,33–35 and as there is increased dopamine (DA) innervation of layer III dlPFC in adolescence,36,37 stress exposure in adolescence may weaken already vulnerable dlPFC microcircuits.
Figure 2.
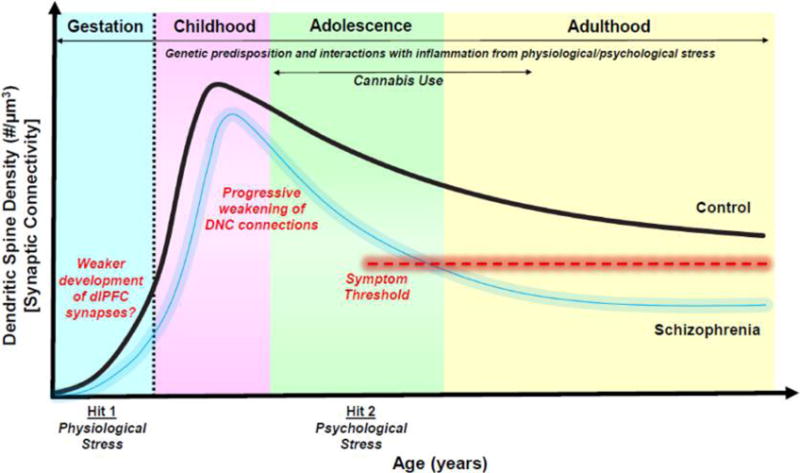
Cumulative impact of genetic predispositions and stress in the pathogenesis of schizophrenia. Schizophrenia is thought to arise from a dual hit, whereby perinatal, developmental insults set the stage for later neuropathological changes in adolescence. Healthy neuronal circuits (black line) undergo a stereotypical developmental pattern, whereby the number of excitatory connections via dendritic spines increase into childhood and then undergoes a normative pruning process. Increased risk of schizophrenia (blurred blue line) is associated with a myriad of environmental perturbations during gestation, including (1) environmental factors such as immune activation, infection, and malnutrition that may drive neuroinflammation and (2) genetic insults that impair synapse and circuit maturation. A second hit is seen in adolescence with an acceleration of gray matter loss at descent into illness. DNC regulation of deep layer III dlPFC connections may render these synapses particularly vulnerable to atrophy from physiological and psychological stress, especially given the increased DA innervation of layer III in adolescence. In healthy individuals, these trajectories are tightly regulated by molecular mechanisms (e.g., PDE4s), but in schizophrenia patients, a background of inflammation (genetic and/or environmental) would dysregulate stress signaling and drive circuit modifications, ultimately resulting in gray matter loss. Symptoms would emerge when synaptic connectivity decreases below a hypothetical threshold (illustrated by red dashed line) needed to support normal functioning.
COGNITIVE DEFICITS IN SCHIZOPHRENIA: LINKS TO THE DORSOLATERAL PFC
Cognitive deficits constitute a core feature of dysfunction in schizophrenia. They persist throughout the lifetime and are present in a majority of patients.1–3,38 Impairments in cognitive function can emerge prior to the onset of psychosis by almost a decade and are the best predictor of functional outcome.38–40 Impairments in cognitive function are present in medication-naïve, first-episode patients and also in a milder form in first-degree relatives of schizophrenia patients, suggesting that these deficits are not an artifact of neuroleptic treatment and are intrinsic to the disease process.41 The degree of cognitive impairments also predicts the ability of individuals with schizophrenia to integrate into society and future employment potential.42 Although an active area of debate, researchers have recently called for schizophrenia to be categorized not as a psychotic disorder but rather a “cognitive illness”,1 consistent with the original conceptualization of the illness as dementia praecox by Kraeplin in the Lehrbuch.
Imaging studies have a long history of identifying “hypofrontality” in the dlPFC of patients performing cognitive tasks that rely on the integrity of the PFC.43–45 These studies have also linked dlPFC cognitive dysfunction to symptoms of thought disorder and disorganization. For example, subjects with schizophrenia undergoing functional magnetic resonance imaging while performing the “n-back” sequential-letter working memory task, demonstrated hypofrontality of the dlPFC that strongly correlated with symptoms of thought disorder.46 These data suggest that both working memory dysfunction and thought disorder involve perturbations to the dlPFC in schizophrenia patients. Neuroimaging studies in healthy volunteers have revealed an “inverted U” shaped relationship between dlPFC activation and working memory, exhibiting marginal levels of activity at low and high loads but significantly elevated activity at intermediate loads.47 However, in schizophrenia, this inverted U appears to be left-shifted, indicating reduced activation at higher memory loads.48 In spite of the fact that cognitive deficits in patients are central to outcome, existing pharmacotherapies are unable to ameliorate these debilitating symptoms. Therefore, understanding the neurobiology of the dlPFC and working memory provides necessary clues to understanding the etiology of the illness and strategies for normalizing cognitive operations.
MICROCIRCUIT RESPONSIBLE FOR WORKING MEMORY: ROLE OF THE DORSOLATERAL PREFRONTAL CORTEX
The dlPFC is essential for working memory. Lesions to the dlPFC produce profound and permanent deficits in working memory abilities.49,50 The dlPFC subserves our highest order functions, generating the mental representations that are the foundation for abstract thought and higher reasoning. The dlPFC creates a “mental sketch pad” through neural networks that can maintain information in the absence of external stimulation.51 Similar to humans, macaque monkeys display profound age-dependent improvements in working memory performance from infancy through adolescence and into adulthood that are characterized by sophisticated refinements in dlPFC circuitry that contribute to working memory maturation.52–54
The primate PFC is topographically organized with extensive afferent and efferent projections with posterior cortex and subcortical regions55–57 (Figure 1A). The dlPFC has extensive reciprocal projections with the sensory and motor association cortices that are necessary for guiding thoughts, attention, and goal-directed actions.55 The dlPFC has direct connections with the posterior hippocampus and also has widespread subcortical projections down to caudate, thalamus (mediodorsal and reticular nuclei), and to the cerebellar cortex via pons.58,59 In contrast, the medial PFC (mPFC) and orbital PFC represent and regulate internal state and emotions by projecting to subcortical regions such as the amygdala, nucleus accumbens, and the hypothalamus.60–62 The PFC also can regulate our state of arousal through projections to the monoamine cell bodies located in the brain stem such as the locus coeruleus [source of norepinephrine (NE) projections], ventral tegmental area (VTA), and substantia nigra (SN) [the source of DA projections], and the dorsal raphe nucleus (DRN) to mediate serotonin/5-hydroxytryptamine (5-HT) cells.63–65 The orbital PFC also regulates cholinergic projections.66 Thus, the PFC has widespread projections for “top-down” control of arousal state and of thought, action, and emotion.
The physiological role of the dlPFC has been extensively interrogated in monkeys performing working memory tasks (Figure 3). Pioneering studies revealed neurons in dlPFC that can maintain firing and represent information in the absence of sensory stimulation.67–69 These so-called “delay cells” are able to maintain firing across the delay period when information must be held “in mind” without sensory stimulation. In visuospatial working memory tasks, delay cells are spatially tuned, firing to the memory of one location but not others, and are able to hold the representation of information in working memory over a period of several seconds to perform the behavioral task.51,67 During adolescence, the proportion of dlPFC pyramidal cells that demonstrate delay-period activity significantly increases, suggesting functional recruitment of this cell-type in mediating working memory, which improves during the same period.52
Figure 3.
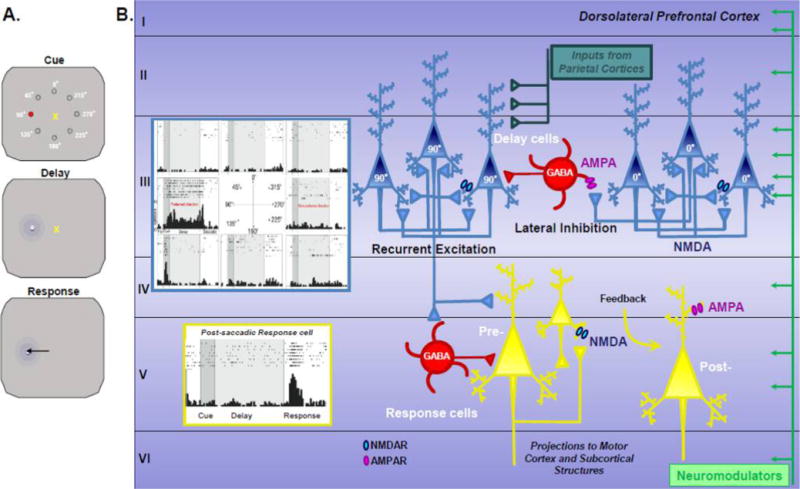
The dorsolateral prefrontal cortex and neuronal circuits mediating spatial working memory. Panel A: Schematic summarizing the oculomotor delayed response (ODR) task used to probe spatial working memory and the involvement of different cell-types in the dlPFC using electrophysiology measures. For each trial of the ODR task, the rhesus monkey must fixate on a central point and remember the spatial position of a cue in 1 out of 8 locations. Following a delay period of multiple seconds, the monkey must indicate the position with a saccade to the memorized spatial location. The cued position randomly changes over a plethora of trials. Panel B: Schematic illustration of the microcircuits within monkey dlPFC (Walker’s area 46) where neurons show persistent, spatially tuned firing during the delay period of the ODR task. Examples of two types of task-related neurons are shown: a delay cell with persistent firing for a preferred spatial location, and a postsaccadic response feedback cell. The dlPFC receives a large variety of inputs, including highly processed visuospatial information from the parietal association cortex and a variety of neuromodulators such as dopamine (green). Delay cells are thought to reside within microcircuits in deep layer III with extensive recurrent excitatory connections among neurons with shared preferred directions. This recurrent excitation is mediated by NMDAR (GluN2A and GluN2B) synapses on dendritic spines with marginal contribution from AMPARs. The spatial tuning activity of dlPFC layer III delay cells is sculpted by lateral inhibition from GABA interneurons, particularly involving parvalbumin-containing basket and chandelier cells. It is hypothesized that delay cells send their information to the motor systems via presaccadic response cells, and that postsaccadic response cells receive feedback (corollary discharge) regarding the motor response. Postsaccadic response cells depend on AMPAR as well as NMDAR stimulation and are powerfully modulated by DA D2R stimulation; as D2R expressing neurons are focused in layer V, it is likely that these cell types reside in dlPFC layer V.
It should be noted that the primate dlPFC also contains other types of task-related neurons, including cells that fire to the Cue, or in anticipation of the motor response67 (Figure 3). Most intriguingly, there are cells that fire during or just after the motor response (postsaccadic response “feedback” cells) that may be providing feedback about the response, i.e. corollary discharge from the motor system via mediodorsal thalamus.70 These cells likely reside in layer V and are greatly influenced by DA D2 receptor stimulation.71 Dysfunction of these neurons may be very relevant to symptoms such as delusions and hallucinations, where loss of a proper mental tag that a thought is self-generated may lead to voices or thoughts being attributed to an external agent. However, as much less is known about these postsaccadic response feedback neurons, this review will focus on delay cells and the neurobiology of mental representations.
The groundbreaking work of Goldman-Rakic and colleagues defined the microcircuit basis underlying delay cell firing.51 Pyramidal cells in dlPFC deep layer III with similar spatial tuning properties engage in recurrent excitation to maintain the persistent firing across the delay period.51 Anatomical tract tracing experiments in nonhuman primates revealed that pyramidal cells in deep layer III have wide-spreading, horizontal axon terminals that terminate in a stripe-like pattern in the supragranular layers.72–76 This highly recurrent excitatory connectivity allows neuronal firing to continue within deep layer III microcircuits in the absence of sensory stimulation.77 The spatial tuning properties are refined by parvalbumin-containing γ-aminobutyric acid (GABA) interneurons through lateral inhibition, thereby sculpting the pattern of activity within dlPFC layer III microcircuits78–83 (Figure 3).
These dlPFC layer III microcircuits expand during brain evolution, exhibiting striking differences in morphological, structural, and electrophysiological properties with the greatest expansion in human dlPFC.84–87 For example, there is an enormous expansion of the dendritic trees of layer III dlPFC pyramidal cells across evolution, and pyramidal cells of rhesus monkey layer III dlPFC have more than twice the number of spines as comparable neurons in primary visual cortex.84–87 In contrast, rodents have mPFC and orbital PFC but no dlPFC, with a large layer V and very small layer III.88 Indeed, rodent PFC neurons are less complex than those in primates.88 For example, comparative studies in mice have revealed that layer III pyramidal cells in frontal cortex and primary visual cortex (V1) have similar features in dendritic complexity, spine and excitatory synapse structure, and in electrophysiological metrics.89 These findings lend credence to the notion that layer III microcircuits in rodents are cytoarchitecturally conserved with comparable physiological profiles. In contrast, layer III pyramidal cells in primates are fundamentally distinctive across brain regions, with the greatest dendritic complexity and the richest spine density in deep layer III of dlPFC.84 The numerous spines on deep layer III dlPFC pyramidal cells are thought to subserve the extensive recurrent excitation that generate and sustain mental representations. In contrast, layer III pyramidal cells in primary sensory areas such as V1 are morphologically compact, have simple dendritic arbors, lower density of dendritic spines, and are characterized by higher input resistance, depolarized resting membrane potential, and higher action potential firing rates.90 In addition, spontaneous excitatory postsynaptic currents (sEPSCs) are diminished in amplitude and frequency and have kinetic profiles in V1 faster than those in dlPFC layer III pyramidal cells.87,90,91 The neuronal networks in nonhuman primate V1 have a constrained dynamic range, thereby enabling faithful synaptic transmission. Recent comparative ultrastructural analyses have revealed that nonhuman primate dlPFC axospinous synapses are characterized by larger presynaptic boutons and postsynaptic densities compared to those in V1,92 consistent with a greater preponderance of dendritic spines in dlPFC compared to V1. Consequently, the electrotonically complex dlPFC layer III pyramidal cells in nonhuman primates have a much larger dynamic range, permitting integration of synaptic inputs of various amplitudes over changing temporal scales, allowing mental representations of multimodal information streams needed for higher-order cognition.88,93
SPINE LOSS IN SCHIZOPHRENIA SELECTIVELY TARGETS DLPFC LAYER III MICROCIRCUITS
The microcircuits in dlPFC layer III that are necessary for mental representations are particularly susceptible in schizophrenia, and atrophy of these recurrent circuits likely contributes to a variety of symptoms, including cognitive deficits such as impaired working memory, thought disorder, and alogia. As described above, the onset of schizophrenia is accompanied by waves of dlPFC gray matter loss with cortical changes more pronounced in patients with shorter durations of prodromal symptoms26,94 and those associated with cannabis use.95,96 Studies conducted in postmortem tissue in subjects with schizophrenia have been particularly instrumental in elucidating the cellular, molecular, and circuit alterations in dlPFC layer III microcircuits (Figure 4). Neuropathological studies have identified significant atrophy in dlPFC deep layer III, including reduced neuropil.97 Morphological studies using Golgi impregnation methods have revealed decreased dendritic arborization and profound dendritic spine loss (∼25%) in dlPFC layer III pyramidal cells in schizophrenia,98 while dendritic spine density in deeper cortical layers such as layer V appear to be unaltered within the same subjects.99 Furthermore, somal volume of dlPFC layer III pyramidal cells are smaller compared to matched healthy subjects,100 consistent with dendritic atrophy. There is likely a loss of the presynaptic compartment as well, as reflected by a decrement in various proteins localized in axon terminals.101–104 Importantly, these morphological alterations are far more pronounced in dlPFC than V1, as alterations in dendritic spine density and dendritic complexity were not significantly reduced in the same schizophrenia subjects in the layer III pyramidal cells in V1.98 The loss of spine density in dlPFC was not attributable to antipsychotic medications or other potential confounds.
Figure 4.
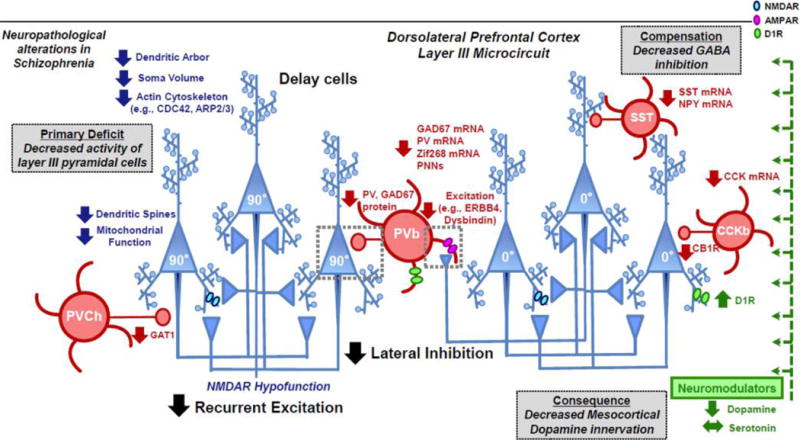
Alterations in dlPFC layer III microcircuits in schizophrenia. Schematic summarizing abnormalities observed from neuropathological postmortem studies in schizophrenia subjects.5,105–107 Inspired by the pioneering work from Lewis and colleagues and other groups, it has been surmised that the primary disturbance in dlPFC layer III microcircuits in schizophrenia lies in decreased recurrent excitation between pyramidal cells. Consistent with this notion, dlPFC layer III pyramidal cells (blue) in subjects with schizophrenia are characterized by numerous morphological changes, such as decreased density of dendritic spines, reduced dendritic arborization and decreased somal volume.98,100 Furthermore, dlPFC layer III pyramidal cells are characterized by molecular changes involving impairments in the actin cytoskeleton121,194 (e.g., CDC42, ARP2/3 signaling pathways) and mitochondrial function.108 To restore network activity, the upstream alterations in pyramidal cells are accompanied by compensatory changes in GABA interneurons (red) to reduce lateral inhibition. For example, the decreased activity of pyramidal cells is thought to reduce excitation of parvalbumin basket (PVb) cells through various activity-dependent mechanisms involving GAD67, PV, Zif268, and PNNs.216–219 Furthermore, dysregulated splicing of ERBB4 and lower expression of dysbindin may contribute to compromised excitatory drive to PVb cells.220–222 Additional changes to GABA interneurons include deficits in PV chandelier cells (PVCh), SST, and CCK cells.223–225 A downstream consequence of changes in excitation–inhibition in dlPFC layer III are changes in dopamine transmission, whereby dlPFC microcircuits receive reduced mesocortical dopamine innervation.189 These dopaminergic changes are associated with compensatory but insufficient upregulation of D1Rs.109 CDC42, cell division cycle 42; ARP2/3, actin-related protein 2/3; GAD67, glutamic acid decarboxylase 67; PV, parvalbumin; PNN, perineuronal nets; SST, somatostatin; NPY, neuropeptide Y; CCK, cholecystokinin; CB1, cannabinoid receptor 1; GAT1, GABA transporter 1; ERBB4, receptor tyrosine-protein kinase erbB-4; D1R, dopamine receptor D1.
Postmortem studies have also revealed that there are various pre- and postsynaptic alterations related to GABA neurotransmission in schizophrenia, which would weaken lateral inhibition in dlPFC layer III microcircuits. A summary of alterations in GABAergic interneurons in the dlPFC of patients with schizophrenia is shown in Figure 4 (for detailed review, please see refs 105–107). Although there was some consideration that insults to GABA interneurons may be a primary driver of disease, the more extensive evidence now suggests that impairments in dlPFC layer III pyramidal cells are a primary etiological disturbance in the disease and that GABA alterations are compensatory, by engaging in plasticity mechanisms to try to restore network activity.5,105 For example, the evidence now indicates that layer III dlPFC pyramidal cells are underactive in schizophrenia, not overactive, as would be predicted if GABA insults were the upstream cause.108
A plausible consequence of hypoactive dlPFC layer III microcircuits also might explain changes in the DA system in schizophrenia, ultimately leading to reduced DA actions in cortex but increased DA actions in caudate, producing the canonical positive symptoms associated with the illness (Figure 5). Human imaging data suggest heightened DA D1R actions in dlPFC at the onset of disease109 with a more complex picture at later stages with subcortical hyperdopaminergia and cortical hypodopaminergia.110 This supports previous studies suggesting that cognitive deficits in schizophrenia might be associated with reduced dopaminergic drive to the PFC.111,112 In fact, prior studies have shown that dlPFC activation is inversely related to the magnitude of striatal dopamine levels in schizophrenia patients.113
Figure 5.
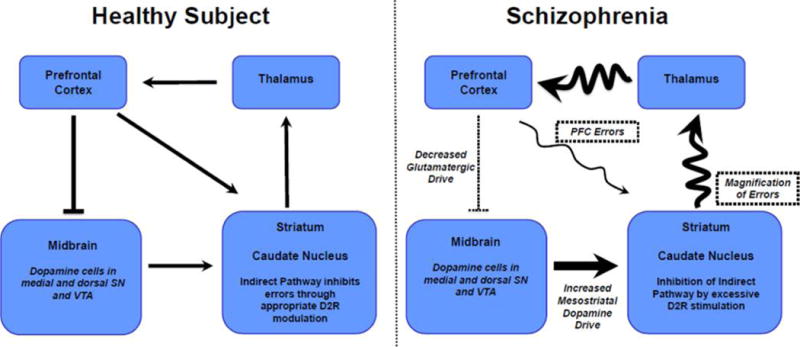
Cascade of changes in schizophrenia across cortical and subcortical systems. Healthy subject: Pyramidal cells in the dlPFC regulate DA neurons projecting to striatum via several mechanisms (see text) and convey information directly to the caudate nucleus as part of cortical-basal ganglia circuits mediating cognition. The indirect pathway emerging from the striatum normally serves to inhibit inappropriate thoughts and actions and performs this function under conditions of normal levels of DA release. Schizophrenia: Insults to dlPFC circuits cause errors in cortical processing. Hypoactive pyramidal cells in the PFC may also disinhibit DA release in caudate, inducing excessive DA stimulation of D2R, which inhibits the indirect pathway. The loss of indirect pathway regulation would magnify cortical errors, intensifying cognitive impairment and likely contributing to positive symptoms such as delusions and hallucinations. Antipsychotic medications with D2R-blocking actions would restore indirect pathway regulation and reduce symptoms but not correct the underlying errors.
Research in animals suggests that PFC can regulate DA neuronal activity. High-resolution tract-tracing studies in rat brain have shown that PFC pyramidal cells innervate DA cells in the VTA in the ventral mesencephalon, which project back to the PFC.114–117 In contrast, PFC projections innervate the GABA interneurons in the VTA that project to the nucleus accumbens.115 These circuit diagrams in rat suggest that reduced PFC activity would weaken DA projections back to PFC, but disinhibit DA projections to striatum. Parallel connections may occur in primates, although the connections from any one subregion of PFC to the midbrain are more subtle.118 Reduced PFC activity may also augment DA release in caudate via disinhibition of striasomal output to DA neurons.119,120 Thus, there are a number of mechanisms whereby reduced firing of dlPFC pyramidal cells may produce both diminished DA activity in the PFC, and especially elevated activity in the caudate.110 Recent investigations in transgenic mouse models, which mimic some aspects of PFC insults in schizophrenia, support this notion.121,122 Elevated DA stimulation of D2 receptors in caudate is well-established in schizophrenia123 and may magnify cortical errors by weakening the inhibitory, corrective actions of the indirect pathway (Figure 5). Thus, a major action of D2R antagonist antipsychotic medications would be to diminish magnification of cortical errors. However, this mechanism would not fix the cortical alterations that may be the primary cause of disease.
In summary, the dlPFC is a critical locus of pathology in schizophrenia with wide-reaching consequence to brain physiology and mental state. Thus, illuminating the molecular mechanisms that regulate these newly evolved circuits may provide clues as to why these neurons are especially vulnerable to atrophy and may suggest plausible novel protective targets.
DYNAMIC NETWORK CONNECTIVITY OF DLPFC LAYER III: UNIQUE NEUROTRANSMISSION AND NEUROMODULATION
Recent studies have shown that dlPFC delay cells have unique physiological properties, including unusual mechanisms for neurotransmission and neuromodulation. In contrast to classic neuronal circuits, delay cell synapses rely mostly on acetylcholine to permit glutamate transmission, and their connections are weakened rather than strengthened by cyclic adenosine monophosphate (cAMP) signaling, e.g. during exposure to uncontrollable stress. Thus, they are particularly vulnerable to changes in arousal state. The unique regulation of layer III dlPFC glutamatergic synapses is summarized in Figure 6.
Figure 6.
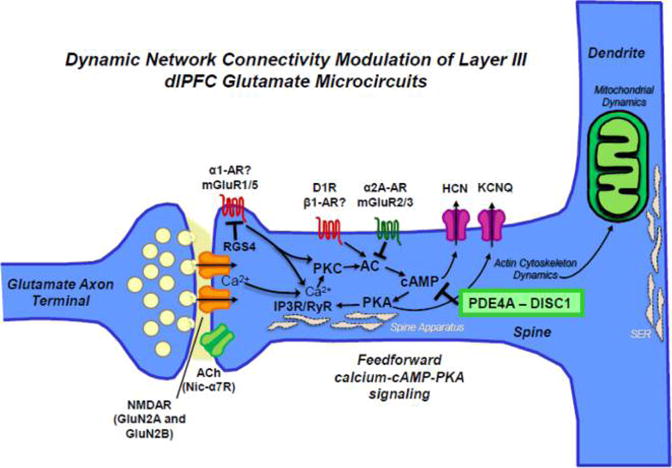
DNC in primate dlPFC layer III. Schematic summarizing the molecular regulators of DNC in dlPFC layer III NMDAR synapses on long, thin spines that permit biochemical/electrical compartmentalization of signaling. These synapses are mediated by NMDAR with either GluN2A or GluN2B subunits. In primate dlPFC, the permissive depolarization for NMDAR-containing synapses is provided by powerful influences of acetylcholine through nicotinic α7 receptors (nic-α7Rs). Negative feedback in these recurrent circuits is provided by feedforward calcium–cAMP– PKA signaling, which open HCN and KCNQ channels near the synapse to weaken connectivity. Internal calcium release from the spine apparatus (i.e., the SER in the spine) may be initiated by calcium influx through NMDAR by activation of type I metabotropic glutamate receptors (e.g., mGluR1 or mGluR5) activation of Gq signaling or by catecholamine activation of Gq or Gs-cAMP signaling, e.g. as occurs during stress exposure. RGS4 is positioned to regulate Gq signaling near the synapse. A variety of mechanisms regulate cAMP signaling. For example, moderate levels of norepinephrine release during nonstressed conditions engage high affinity α2A-ARs that inhibit cAMP opening of HCN and KCNQ channels to strengthen connections and enhance cognition. Recent data show that postsynaptic mGluR3 also performs this key strengthening action. cAMP signaling is also powerfully regulated by PDE4A, which is anchored to the spine apparatus by DISC1. As the spine apparatus interconnects with the SER in the dendrite, these mechanisms also allow communication between the synapse and the dendrite, e.g. with mitochondria residing in the nearby dendrite. Thus, regulation of feedforward cAMP–calcium signaling mechanisms is crucial to maintain normal mitochondrial function and the actin dynamics needed for the structural integrity of dendritic spines. Abbreviations: NMDAR, N-methyl-D-aspartate receptor; mGluR, metabotropic glutamate receptor; ACh, acetylcholine receptor; α2-AR/α1-AR, α2-A/α1 adrenoreceptors; D1R, dopamine receptor D1; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; PKC, protein kinase C; AC, adenylyl cyclase; RyR, ryanodine receptor; IP3R, inositol-1,4,5-triphosphate receptors; RGS4, regulator of G protein signaling 4; PDE4A, phosphodiesterase 4A; DISC1, Disrupted in Schizophrenia 1.
Neurotransmission at classic glutamate synapses, e.g. in V1, depends on AMPA receptor (AMPAR) stimulation,124 which depolarizes the membrane to relieve the Mg2+ block of N-methyl-D-aspartate receptors (NMDARs) to permit NMDAR actions. Classic synapses contain predominately NMDAR with GluN2A subunits, while those with slower, GluN2B subunits may reside outside the synapse. In contrast to these classic synapses, glutamate synapses in deep layer III of dlPFC are very different, with GluN2B subunit expressing NMDARs residing exclusively within the synapse and not at extrasynaptic sites.125 At the physiological level, delay cell firing depends on glutamate stimulation of NMDAR with both GluN2A and GluN2B playing key roles, while AMPAR blockade has surprisingly little effect on cell firing.125 GluN2B-containing NMDARs are particularly adapted to maintain DLPFC network firing in the absence of sensory stimulation due to the slower kinetics, where the faster kinetics of AMPARs can lead to dynamic instability and network collapse.125 These findings are supported by computational modeling of neural dynamics.77,126–128 In contrast, postsaccadic response feedback cells in layer V require AMPAR stimulation for permissive opening of NMDARs, similar to canonical circuits in sensory and subcortical systems.125 Consistent with this notion, transcript level expression of AMPARs is consistently higher in layer V pyramidal cells compared to that in layer III pyramidal cells in monkey dlPFC.129
If AMPAR plays such a small role in delay cell firing, what provides the membrane depolarization needed for removal of the Mg2+ block of NMDARs? Both electrophysiological and immunoEM data show that these key permissive actions are provided by cholinergic stimulation of nicotinic α7 receptors (nic-α7Rs), which reside within the synapse.130 These data are consistent with lesion data showing that cholinergic depletion from the dlPFC markedly impairs working memory performance in rhesus monkeys.131
In addition to these key cholinergic influences, dlPFC microcircuits are also powerfully modulated by the catecholamines, and depletion of catecholamines from the dlPFC is as harmful as lesioning the dlPFC itself.132 As schematized in Figure 7, both NE and DA have an inverted U influence on delay cell firing and cognitive performance, where moderate levels are essential to function, but excessive stimulation, e.g. during stress, suppresses neuronal firing and impairs cognitive abilities.33 Moderate levels of NE enhance delay cell firing by activating postsynaptic α2-A adrenoreceptors (α2A-AR), while high levels of NE (such as those occuring during stress exposure) suppress delay cell firing through α1 adrenoreceptor (α1-AR) stimulation.133,134 Similarly, either too little or too much DA D1R stimulation erodes delay cell firing.135,136 High levels of both NE and DA are released in the PFC during stress, rapidly taking PFC “off-line” to switch control of behavior to more primitive circuits that mediate habitual responses33 (Figure 1B). Thus, the stress-induced impairments in PFC neuronal firing and working memory performance can be mimicked by stimulating α1-AR or D1R in PFC and can be prevented by blocking these receptors.34 Similar effects have been seen in humans, where stress impairs working memory and reduces dlPFC activity137 and increases dopamine release in PFC.138,139 Human subjects with a weaker catechol-O-methyltransferase (COMT) Met-homozygote genotype were particularly vulnerable to stress-induced PFC dysfunction, consistent with a catecholamine dependent mechanism.140
Figure 7.
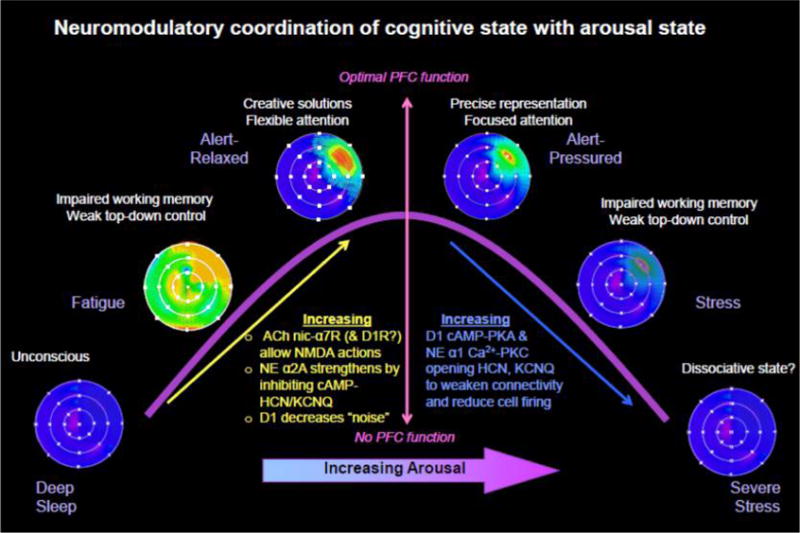
Impact of neuromodulators in dlPFC physiology and function. Schematic illustration of the inverted U-shaped influence of NE, ACh, and DA on the memory fields of dlPFC delay cells representing a position in visual space (blue = low firing, red = high firing). During deep sleep, no modulators are released, and dlPFC synapses are not able to communicate (unconscious). With drowsy waking, low levels of Ach and DA permit NMDAR in the postsynaptic density to respond to glutamate, but signaling is weak and noisy. Under optimal conditions, moderate levels of NE release engage α2A-ARs to inhibit cAMP–K+ channel signaling and strengthen persistent firing for the preferred direction. Moderate levels of DA D1R stimulation also reduce “noise”, and along with lateral inhibition, produce more refined representations, However, high levels of catecholamine release with uncontrollable stress engage α1-ARs and D1R to suppress all delay cell firing, eroding mental representations.
The molecular basis for these powerful catecholamine actions is now beginning to be understood, where these agents dynamically modulate feedforward cAMP–calcium signaling intracellular pathways in spines near NMDAR synapses to rapidly strengthen or weaken neural firing by altering the open state of nearby HCN and KCNQ potassium (K+) channels4 (Figure 6). This phenomenon has been termed dynamic network connectivity.141 These signaling pathways provide a mechanism to rapidly alter connectivity based on arousal state4 and provide important negative feedback in a microcircuit with extensive recurrent excitation and lateral (rather than feedforward) inhibition. Thus, in contrast to classical circuits where cAMP–PKA signaling strengthens connections by increasing glutamate release and driving long-term plastic changes;124,142 in layer III of dlPFC, cAMP–PKA signaling rapidly weakens connectivity and reduces delay cell firing by increasing the open state of HCN and KCNQ channels.34
ImmunoEM revealed a constellation of cAMP-related proteins in close proximity to the calcium-containing spine apparatus, the extension of the smooth endoplasmic reticulum (SER) into the spine, which are positioned to regulate feedforward cAMP–PKA-calcium signaling.141 Activation of cAMP–PKA can enhance internal calcium release through inositol-1,4,5-triphosphate receptors (IP3Rs)143 and ryanodine receptors, while calcium release can activate protein kinase C (PKC) to further drive the production of cAMP,133 thereby establishing a vicious, feedforward signaling process to activate K+ channels and weaken delay cell firing (Figure 6). These detrimental actions are induced by high levels of D1R and α1-AR stimulation during stress exposure.133,144
Conversely, a variety of mechanisms inhibit calcium– cAMP–K+ channel signaling in spines and strengthen connectivity to enhance cell firing (Figure 6). For example, both noradrenergic α2A-AR and glutamatergic mGluR3 are concentrated on spines, where they enhance delay cell firing by inhibiting cAMP–K+ channel signaling.134,145 In contrast, mGluR2 are localized presynaptically, where they inhibit glutamate release.146 Another critical regulator of feedforward cAMP–calcium signaling in newly evolved dlPFC layer III microcircuits is the phosphodiesterase, PDE4A, which catabolizes cAMP and is anchored to the spine apparatus by the scaffolding protein Disrupted in Schizophrenia (DISC1).147,148 Importantly, inflammation can reduce PDE4A actions and detach it from DISC1 via MAP kinase-activated protein kinase 2 (MK2) signaling.149 Normally, PKA signaling provides negative feedback to reign in cAMP signaling by activating PDE4s.149 However, this essential regulation is lost following MK2 activation,149 indicating a mechanism whereby inflammation can dysregulate cAMP–calcium signaling and weaken layer III dlPFC synapses. These mechanisms also provide a nexus whereby psychological stress may act on a background of inflammation to weaken neural connections and induce atrophy of layer III dlPFC spines.
Repeated stress exposure produces dendritic atrophy and loss of dendritic spines in layer II/III pyramidal cells.150,151 These morphological changes are correlated with deficits in working memory in the delayed alternation task152 and cognitive flexibility on perceptual set-shifting tasks153 and can be prevented blocking PKA154 or PKC152 signaling, consistent with an underlying role of dysregulated DNC signaling. Dendritic atrophy and loss of spines are brain region-specific; in contrast to the PFC, chronic stress actually increases dendrite morphogenesis in the amygdala.155 These findings lend credence to the notion that environmental stressors can detrimentally affect circuit and morphological properties of pyramidal cells, with the PFC being particularly vulnerable. Reduced PFC gray matter has also been seen in humans with repeated stress exposure.156 It is possible that these mechanisms contribute to the loss of PFC gray matter as patients descend from the prodrome into psychotic illness during late adolescence and early adulthood.26
RELEVANCE OF DYNAMIC NETWORK CONNECTIVITY TO SCHIZOPHRENIA
Perturbations to DNC mechanisms in dlPFC layer III microcircuits might play an important role in exacerbating susceptibility of these microcircuits in schizophrenia (Figure 8). Genome-wide association studies (GWAS) have identified a number of genes of relevance to layer III dlPFC synapses, including those associated with glutamate receptor transmission, HCN1 and calcium channels, and most recently, complement component 4A, which helps mediate synapse removal.18,157 Cognitive impairment in schizophrenia is particularly associated with genetic changes to signaling events associated with inflammation and oxidative stress.158 Although the consequences of genetic alterations to protein expression/function and neuronal physiology are still requiring elucidation for most genetic associations, we can begin to create speculative bridges between what we are learning about schizophrenia genetics, differences in transcript/protein expression postmortem, and the molecular regulation of layer III dlPFC synapses.
Figure 8.
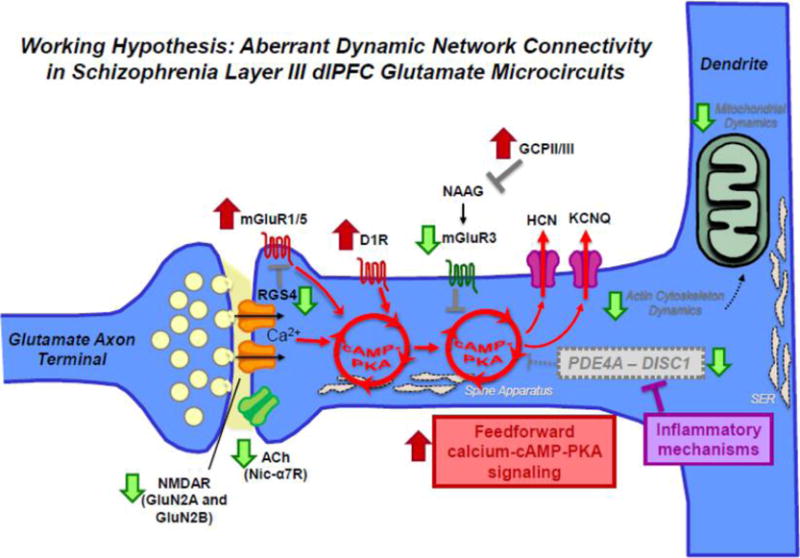
Aberrant DNC mechanisms in schizophrenia dlPFC layer III. Schematic summarizing changes in DNC mechanisms in schizophrenia dlPFC layer III due to genetic predispositions and neuropathological changes from postmortem studies. For example, both genetic and postmortem studies have revealed a decrement in acetylcholine nic-α7R and glutamate NMDAR signaling. Translocations in the Disc1 gene have been implicated in mental illness and in particular schizophrenia. DISC1 serves as a scaffold for the family of phosphodiesterases (e.g., PDE4A) that hydrolyze cAMP signaling, which have also been genetically linked to schizophrenia. ImmunoEM studies of monkey dlPFC layer III spines show that DISC1 anchors PDE4A near the spine apparatus, adjacent to HCN channels. In vitro studies show that PDE4 activity is inhibited by MK2 signaling during inflammation. If similar actions occur in human PFC in response to inflammation, e.g. in utero and/or during adolescence at the onset of illness, dysregulated cAMP–calcium signaling may lead to weaker connections and loss of spines. Schizophrenia is also associated with elevation in mGluR1α which drives internal calcium release. The activity of mGluR1α is inhibited by regulator of G protein signaling 4 (RGS4), positioned perisynaptically to gate signaling. Recent layer III pyramidal cell-type specific studies have also revealed downregulation of RGS4 mRNA in schizophrenia, which is predicted to result in dysregulated feedforward cAMP–calcium signaling. Finally, neuroimaging studies have suggested elevation in D1R in drug naïve patients which would further exaggerate a vicious cycle. These perturbations are thought to converge of impairments in actin cytoskeleton and mitochondria in dlPFC layer III pyramidal cells in schizophrenia, leading to enhanced vulnerability and weakening of network connectivity. In sum, various studies in schizophrenia suggest alterations that increase the generation of cAMP–calcium signaling and decrease mechanisms that moderate cAMP–calcium signaling.
Copy number variation (CNV) studies, GWAS, and exome sequencing studies have particularly identified enrichment in the NMDAR-signaling and PSD protein complexes, which are central elements to regulating synaptic strength at glutamatergic synapses and thought to be associated with increased risk for developing schizophrenia.17,159–161 These genetic studies are also corroborated by postmortem evaluations showing reductions in various NMDAR subunits.162–164 There is also genetic evidence implicating the cholinergic system and nic-α7Rs in the pathogenesis of schizophrenia.165–167
Various lines of genetic and postmortem data implicate dysfunctional feedforward cAMP–calcium signaling intracellular pathways in the pathogenesis of schizophrenia. In general, schizophrenia is characterized by weakening of mechanisms that control cAMP–calcium signaling and strengthening of mechanisms that generate cAMP–calcium signaling (Figure 8). For example, GWAS established links between schizophrenia and the voltage-gated calcium channel Cav1.2 coded by CACNA1C, where genetic insults lead to increased calcium influx.168 In cardiac muscle, calcium influx through this channel is increased by PKA phosphorylation169 and results in increased calcium levels in the SER.170 Similar actions in dlPFC neurons might exacerbate the stress response. Another gene linked to regulation of cAMP signaling is DISC1, which anchors PDE4s. Translocations in the DISC1 gene have been associated with mental illness in a large Scottish pedigree and are thought to be an important risk factor predisposing individuals to schizophrenia;171,172 they have been implicated in several other neuropsychiatric illnesses that involve PFC dysfunction. In general, DISC1 and its various protein interaction partners are thought to be a key hub for developmental pathways during neurogenesis and circuit maturation.173–175 Among the different proteins that DISC1 tethers, a crucial interaction exists between DISC1 and the phosphodiesterase PDE family of proteins.171 Our immunoEM analyses of layer III dlPFC spines have shown that DISC1 anchors PDE4A next to the spine apparatus, positioned to regulate cAMP drive of calcium release.148 Interestingly, genetic studies have also identified single-nucleotide polymorphisms (SNPs) in PDE proteins to be associated with schizophrenia that increase susceptibility.171,176 As described above, inflammation reduces PDE4 activity by preventing negative feedback by PKA and weakens PDE4 interactions with DISC1,149 which may cause PDE4 mislocalization. Therefore, genetic or inflammatory insults could weaken DISC1-PDE4A modulation of feedforward cAMP–calcium signaling and lead to reduced neuronal firing and susceptibility to atrophy. Studies in rodents indicate that mutations in DISC1 may only impact cognition under conditions of stress when PDE4 is needed to rein in elevated cAMP signaling.177 Thus, environmental factors as well as mosaic expression of DISC1 mutations in varying PFC subcircuits (e.g., dlPFC circuits, schizophrenia; medial PFC circuits, depression) may help to explain the heterogeneity in symptomology with DISC1 genetic alterations.
Another intracellular mediator of feedforward cAMP– calcium signaling is Regulator of G protein signaling 4 (RGS4) that normally inhibits G protein (especially Gq) signaling and regulates NMDAR function. RGS4 is concentrated next to excitatory synapses on spines in layer III primate dlPFC.178 RGS4 mRNA and protein levels are downregulated in the dlPFC in subjects with schizophrenia,179–181 and these changes are most prominent in dlPFC layer III pyramidal cells.182 Genetic association and linkage studies have also identified several SNPs for RGS4 at the genomic level.183 If RGS4 inhibits Gq signaling near the synapse in layer III dlPFC, the downregulation of RGS4 in schizophrenia may disinhibit calcium signaling and weaken layer III dlPFC synapses.
Finally, the group II metabotropic glutamate receptor, mGluR3, which couples to Gi/Go and inhibits cAMP–calcium signaling, has also been genetically linked to schizophrenia184,185 including GWAS confirmation.18 Postmortem studies have also shown evidence of reduced mGluR3 actions in the dlPFC of patients with schizophrenia, including increased expression of glutamate carboxypeptidase II (GCPII), the catabolic enzyme that destroys the endogenous mGluR3 ligand, N-acetylaspartylglutamate (NAAG).186 Recent studies in monkeys show that mGluR3 plays an expanded role in the primate dlPFC. In addition to its classic location in astrocytes, mGluR3 is localized postsynaptically on spines in monkey dlPFC layer III microcircuits, where its stimulation enhances dlPFC delay cell firing through inhibition of cAMP signaling.146 Conversely, maternal inflammation has been shown to increase microglial GCPII expression in neonate brain, which would reduce endogenous mGluR3 stimulation by NAAG.187 Taken together, these studies suggest that various DNC mechanisms that normally serve to inhibit feedforward cAMP–calcium signaling in dlPFC layer III microcircuits may be reduced in schizophrenia subjects.
Some DNC mechanisms that normally generate feedforward cAMP–calcium signaling are increased in schizophrenia (Figure 8). For example, schizophrenia subjects are associated with elevated expression levels of group I metabotropic glutamate receptors (i.e., mGluR1α), which elicits greater intracellular Ca2+ release.181 ImmunoEM experiments in monkey dlPFC layer III microcircuits have also revealed that mGluR1α are localized both presynaptically and on dendritic spines and are positioned to mobilize Ca2+ from intracellular stores.188 Similarly, in vivo imaging studies have revealed that drug-naïve and drug-free subjects show elevated levels of dopamine D1 receptor (D1R), which has been hypothesized to compensate for lower available dopamine in the dlPFC in subjects with schizophrenia109,189 but may lead to excessive D1R signaling during adolescence when DA levels may be especially high in layer III.36,37
Exacerbated feedforward cAMP–calcium signaling might also be associated with dysregulation of actin cytoskeleton pathways and mitochondrial function, e.g. due to toxic calcium dysregulation (Figure 4). In addition to its anchoring of PDE4 enzymes, DISC1 anchors Kalirin-7, a RAC guanine nucleotide exchange factor (GEF) that is highly concentrated in dendritic spines and regulates spine integrity through RAC1 and cell division cycle (CDC42).175,190 Previous postmortem studies in schizophrenia have revealed lower mRNA levels of Kalirin-7 and CDC42, and these changes were also correlated with spine density measures in dlPFC deep layer III pyramidal cells.191 The KALRN locus is associated with risk for schizophrenia192 and missense mutations have also been reported in a small cohort.193 Various actin cytoskeleton signaling pathways pertinent to CDC42 (e.g., CDC42-CDC42EP, CDC42-PAK-LIMK, and CDC42-NWASP-ARP2/3) are perturbed selectively in dlPFC layer III pyramidal cells in schizophrenia.121,191,194,195 In addition, de novo mutations in schizophrenia are overrepresented among loci encoding cytoskeleton-associated proteins that modulate actin filament dynamics, actin bundle assembly, interconnected with activity-regulated cytoskeleton-associated protein (ARC) and NMDAR signaling and could contribute to the pathogenesis of the illness.15 Impairments in actin cytoskeleton signaling pathways that are necessary to maintain mature dendritic spines, is predicted to destabilize dendritic spines, leading to spine loss.5 It is plausible that intrinsic genetic predisposition is modulated by mosaic expression and/or region and cell-type specific patterns of gene expression changes, to influence the selective vulnerability of dlPFC layer III microcircuits in schizophrenia.
In accordance with these findings, recent cell-type specific studies in dlPFC layer III pyramidal cells in schizophrenia have revealed downregulation of nuclear gene products regulating mitochondrial and ubiquitin–proteosomal signaling pathways.108 The molecular alterations germane to mitochondrial energy production and regulation of protein translation appear to be distinctive for schizophrenia as these transcriptome signatures are not observed in bipolar disorder or major depression.196 These neuropathological changes might be driven, at least in part, by aggravated feedforward cAMP– calcium signaling by inducing calcium leak from the smooth endoplasmic reticulum in a PKA dependent manner. This corroborates findings suggesting that dlPFC microcircuits in schizophrenia are hypoactive,45,197,198 with a greater propensity to affect layer III pyramidal cells. The vicious interaction between exaggerated feedforward cAMP–calcium signaling, actin cytoskeleton impairments and mitochondrial dysfunction might also shed light on attenuated γ-frequency neural oscillations (30–80 Hz) in patients with schizophrenia.199–202 Recordings from monkeys show that γ-frequency neural oscillations are associated with the persistent firing of dlPFC delay cells during working memory,203 possibly linking reduced γ-oscillations to weaker dlPFC layer III microcircuits. Consistent with this notion, the deficits in γ-frequency neural oscillations in schizophrenia are more pronounced in the dlPFC compared to those in posterior regions.204
Finally, we should add that genetic and/or environmental insults to the presumed layer V response feedback cells in primate dlPFC may also contribute to symptoms of schizophrenia. These cells appear to relay the “corollary discharge” that a response is being carried out by brain circuits. These cells are greatly influenced by AMPAR125 and dopamine D2 receptor (D2R) signaling.71 Alterations in these molecular events in response feedback cells may thus disrupt the mental tag that a neural event was self-generated, contributing to delusions and hallucinations. This is an important arena for future research.
HYPOTHETICAL CASCADE OF EVENTS IN THE PATHOGENESIS OF SCHIZOPHRENIA
The “dual-hit” working model of schizophrenia posits that insults to brain development in utero are compounded by a second wave of assaults during late adolescence to cause a descent into illness (Figure 2). A variety of genetic and environmental insults in utero and during early perinatal development could impair cortical circuit formation and maturation and set up an inflammatory framework that could exacerbate later responses to environmental insults (e.g., stress), causing atrophy of dlPFC circuits, profound dlPFC dysfunction, and the full emergence of schizophrenia symptoms. PFC circuits are particularly susceptible to environmental perturbations (e.g., stress) given the highly complex and protracted maturation, providing greater opportunities for insults to amplify alterations in developmental trajectories. For example, dysregulated feedforward cAMP–calcium signaling during stress could drive abnormal mitochondrial signaling, leading to actin cytoskeleton collapse and further activation of inflammatory pathways, establishing vicious cycles that result in atrophy of deep layer III spines. Increased vulnerability during adolescence might be compounded by genetic insults that impact synaptic pruning mechanisms and circuit reorganization. For example, variations in the major histocompatibility (MHC) locus and complex structural variations in complement C4 are strong contributors to schizophrenia risk.157 The elevated expression levels of C4 in schizophrenia subjects, in turn, might promote excessive pruning of spines and synapses during adolescence.157 Thus, gene–environment interactions appear to be a key factor in precipitating illness onset for patients with schizophrenia. Elucidating the neural underpinnings of dlPFC circuit alterations in nonhuman primates and in postmortem tissue in schizophrenia patients, might provide insight into pathophysiological impairments such as lower power of γ-frequency oscillations and working memory deficits.
RELEVANCE OF DNC MECHANISMS IN DLPFC LAYER III TO NOVEL THERAPEUTIC TREATMENTS FOR SCHIZOPHRENIA
Interrogation of DNC mechanisms in nonhuman primates and rodents, suggest that there are several plausible targets that might be pursued for the development of appropriate therapeutic targets to protect layer III dlPFC circuits during the prodrome. One key neuroprotective mechanism involves inhibition of cAMP–PKA signaling with α2A-AR agonist guanfacine. Although the neuroprotective mechanisms of guanfacine arise from several plausible routes, the primary mechanism revolves around stimulation of α2A-ARs on layer III spines, through inhibition of cAMP-HCN channel signaling.134 Electrophysiology studies in nonhuman primates show that α2A-AR inhibition of cAMP-HCN signaling enhances dlPFC firing,134 and either intra-PFC or systemic treatment with guanfacine strengthens PFC function.205 Particularly pertinent to schizophrenia, daily guanfacine treatment is also protective of PFC dendritic spines and cognitive functions in a chronic restrained stress paradigm,154 or from hypoxia,206 in rat medial PFC, encouraging this approach. As α2A-AR-agonists such as guanfacine are also anti-inflammatory, deactivating microglia,207 they may also be more generally protective of brain during the prodrome. As guanfacine is FDA approved for use in children 6–17 years, it would be feasible to test in teens at risk for schizophrenia.
Enhancing cholinergic actions in layer III dlPFC microcircuits might also strengthen network activity. For example, there has been a longstanding interest in the role of nic-α7Rs as a potential treatment strategy for schizophrenia.166 The nic-α7Rs are enriched in NMDAR glutamate network synapses and are permissive for NMDAR communication in dlPFC layer III pyramidal cell circuits.130 Administration of low-doses of nic-α7R agonists enhances delay cell firing and improves working memory, but higher doses produced excessive neuronal excitability and no longer improved working memory.130 Thus, it is essential to test very low doses in human studies. These findings might also shed light on why schizophrenia patients have a greater propensity for tobacco dependence, with over 85% of hospitalized schizophrenia patients engaging in smoking due to potential beneficial effects on cognitive function.208–210 Recent quantitative trait loci and gene expression analyses with high-resolution 3D chromatin contact maps during corticogenesis implicate acetylcholine as a plausible etiological mechanism in the pathogenesis of schizophrenia.167 In addition, previous studies suggest that muscarinic M1Rs might also be positioned in dlPFC layer III glutamate synapses to enhance spatial representation.211 Ongoing studies in nonhuman primates are delineating the relative contribution of M1Rs to persistent firing and if these receptors can be targeted by novel agonists and positive allosteric modulators (PAMs).
Finally, another possible candidate involves enhanced modulation of the metabotropic glutamate receptor mGluR3. As described earlier, mGluR3 are concentrated on layer III spines, while mGluR2 are mostly presynaptic in primate layer III dlPFC.146 Postsynaptically localized mGluR3 are activated by both glutamate and NAAG, which are both released from glutamate terminals.212 Physiological recordings from monkeys show that NAAG stimulation of mGluR3 in dlPFC greatly enhances delay cell firing by inhibiting cAMP–K+ channels signaling.145 NAAG activity is inhibited by the NAAG peptidase GCPII.213 A NAAG peptidase inhibitor markedly increased the firing of delay cells,146 suggesting these agents may have therapeutic potential. As postmortem studies have revealed increased protein levels of GCPII and reduced levels of mGluR3 in the dlPFC in subjects with schizophrenia,186 GCPII inhibitors might be an effective approach to increase endogenous NAAG levels to curb feedforward cAMP–calcium signaling. Low doses of mGluR2/3 agonists that preferentially engage postsynaptic actions also enhance delay cell firing and improve working memory in monkeys,145 and a meta-analysis of mGluR2/3 agonist effects in patients with schizophrenia has shown that low (but not high) doses of mGluR2/3 agonists can be beneficial in patients in early stages of the illness.214 Thus, data from patients with the illness encourage this strategy.
There are many fundamental challenges in drug discovery, including (1) differential drug effects in various brain regions, (2) refinement of drug dosage given inverted U-shaped drug response curves, and (3) a changing landscape of the disease process, where therapeutic targets may be lost (e.g., from spines) in later stages of disease. These putative pharmacological targets hold promise for protecting dlPFC circuits and preventing the emergence of symptoms of the illness. Because the dlPFC is a central locus of pathology in a range of neurological disorders, the putative therapeutic targets outlined in this review might also be suitable for treating age-related cognitive decline and neurodegenerative diseases (e.g., Alzheimer’s disease). Strategies for synergistic application of pharmacological intervention along with cognitive remediation and psychiatric rehabilitation215 might further enhance neuropsychological performance for subjects with schizophrenia.
CONCLUSIONS
The same molecular machinery that allows extraordinary flexibility in layer III dlPFC network connectivity may also confer vulnerability to atrophy when dysregulated by genetic perturbations and/or environmental insults. In contrast to the classical sensory circuits, the molecular mechanisms that govern network connectivity in the dlPFC may be particularly susceptible to stress exposure, as signaling events weaken connectivity. Signaling interactions in dlPFC layer III dendritic spines may provide a nexus whereby inflammation disinhibits the neuronal response to physiological and/or psychological stress, which may weaken connections at multiple stages of brain development. Therefore, understanding the unique regulation of these circuits may help to identify novel therapeutic strategies to protect circuits during the early stages of illness.
Acknowledgments
Funding
This research was supported by NIMH MH100064 and Pioneer DP1AG047744 to A.F.T.A.
Footnotes
ORCID
Dibyadeep Datta: 0000-0002-5415-3067
Author Contributions
D.D. and A.F.T.A had extensive discussions and cowrote the paper.
Notes
The authors declare no competing financial interest.
References
- 1.Kahn RS, Keefe RS. Schizophrenia is a cognitive illness: time for a change in focus. JAMA Psychiatry. 2013;70:1107–1112. doi: 10.1001/jamapsychiatry.2013.155. [DOI] [PubMed] [Google Scholar]
- 2.Elvevag B, Goldberg TE. Cognitive impairment in schizophrenia is the core of the disorder. Crit Rev Neurobiol. 2000;14:21. [PubMed] [Google Scholar]
- 3.Lesh TA, Niendam TA, Minzenberg MJ, Carter CS. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology. 2011;36:316–338. doi: 10.1038/npp.2010.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnsten AF, Wang MJ, Paspalas CD. Neuromodulation of thought: flexibilities and vulnerabilities in prefrontal cortical network synapses. Neuron. 2012;76:223–239. doi: 10.1016/j.neuron.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoftman GD, Datta D, Lewis DA. Layer 3 Excitatory and Inhibitory Circuitry in the Prefrontal Cortex: Developmental Trajectories and Alterations in Schizophrenia. Biol Psychiatry. 2017;81:862. doi: 10.1016/j.biopsych.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berardi N, Pizzorusso T, Ratto GM, Maffei L. Molecular basis of plasticity in the visual cortex. Trends Neurosci. 2003;26:369–378. doi: 10.1016/S0166-2236(03)00168-1. [DOI] [PubMed] [Google Scholar]
- 7.Lewis DA, Lieberman JA. Catching up on schizophrenia: natural history and neurobiology. Neuron. 2000;28:325–334. doi: 10.1016/s0896-6273(00)00111-2. [DOI] [PubMed] [Google Scholar]
- 8.Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th. American Psychiatric Press; Washington, DC: 1994. [Google Scholar]
- 9.Corlett PR, Murray GK, Honey GD, Aitken MR, Shanks DR, Robbins TW, Bullmore ET, Dickinson A, Fletcher PC. Disrupted prediction-error signal in psychosis: evidence for an associative account of delusions. Brain. 2007;130:2387–2400. doi: 10.1093/brain/awm173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marder SR, Galderisi S. The current conceptualization of negative symptoms in schizophrenia. World Psychiatry. 2017;16:14–24. doi: 10.1002/wps.20385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Insel TR, Scolnick EM. Cure therapeutics and strategic prevention: raising the bar for mental health research. Mol Psychiatry. 2006;11:11–17. doi: 10.1038/sj.mp.4001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Os J, Kenis G, Rutten BP. The environment and schizophrenia. Nature. 2010;468:203–212. doi: 10.1038/nature09563. [DOI] [PubMed] [Google Scholar]
- 13.van Os J, Rutten BP, Poulton R. Gene-environment interactions in schizophrenia: review of epidemiological findings and future directions. Schizophr Bull. 2008;34:1066–1082. doi: 10.1093/schbul/sbn117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Selemon LD, Zecevic N. Schizophrenia: a tale of two critical periods for prefrontal cortical development. Transl Psychiatry. 2015;5:e623. doi: 10.1038/tp.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, Carrera N, Humphreys I, Johnson JS, Roussos P, Barker DD, Banks E, Milanova V, Grant SG, Hannon E, Rose SA, Chambert K, Mahajan M, Scolnick EM, Moran JL, Kirov G, Palotie A, McCarroll SA, Holmans P, Sklar P, Owen MJ, Purcell SM, O’Donovan MC. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C, Chambert K, Bergen SE, Kahler A, Duncan L, Stahl E, Genovese G, Fernandez E, Collins MO, Komiyama NH, Choudhary JS, Magnusson PK, Banks E, Shakir K, Garimella K, Fennell T, DePristo M, Grant SG, Haggarty SJ, Gabriel S, Scolnick EM, Lander ES, Hultman CM, Sullivan PF, McCarroll SA, Sklar P. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, Stray SM, Rippey CF, Roccanova P, Makarov V, Lakshmi B, Findling RL, Sikich L, Stromberg T, Merriman B, Gogtay N, Butler P, Eckstrand K, Noory L, Gochman P, Long R, Chen Z, Davis S, Baker C, Eichler EE, Meltzer PS, Nelson SF, Singleton AB, Lee MK, Rapoport JL, King MC, Sebat J. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 20.Biesecker LG, Spinner NB. A genomic view of mosaicism and human disease. Nat Rev Genet. 2013;14:307–320. doi: 10.1038/nrg3424. [DOI] [PubMed] [Google Scholar]
- 21.Campbell IM, Shaw CA, Stankiewicz P, Lupski JR. Somatic mosaicism: implications for disease and transmission genetics. Trends Genet. 2015;31:382–392. doi: 10.1016/j.tig.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poduri A, Evrony GD, Cai X, Walsh CA. Somatic mutation, genomic variation, and neurological disease. Science. 2013;341:1237758. doi: 10.1126/science.1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoftman GD, Lewis DA. Postnatal developmental trajectories of neural circuits in the primate prefrontal cortex: identifying sensitive periods for vulnerability to schizophrenia. Schizophr Bull. 2011;37:493–503. doi: 10.1093/schbul/sbr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rapoport JL, Giedd JN, Gogtay N. Neurodevelopmental model of schizophrenia: update 2012. Mol Psychiatry. 2012;17:1228–1238. doi: 10.1038/mp.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cannon TD. How Schizophrenia Develops: Cognitive and Brain Mechanisms Underlying Onset of Psychosis. Trends Cognit Sci. 2015;19:744–756. doi: 10.1016/j.tics.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cannon TD, Chung Y, He G, Sun D, Jacobson A, van Erp TG, McEwen S, Addington J, Bearden CE, Cadenhead K, Cornblatt B, Mathalon DH, McGlashan T, Perkins D, Jeffries C, Seidman LJ, Tsuang M, Walker E, Woods SW, Heinssen R. Progressive reduction in cortical thickness as psychosis develops: a multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol Psychiatry. 2015;77:147–157. doi: 10.1016/j.biopsych.2014.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Howes OD, McCutcheon R. Inflammation and the neural diathesis-stress hypothesis of schizophrenia: a reconceptualization. Transl Psychiatry. 2017;7:e1024. doi: 10.1038/tp.2016.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muller N, Weidinger E, Leitner B, Schwarz MJ. The role of inflammation in schizophrenia. Front Neurosci. 2015;9:372. doi: 10.3389/fnins.2015.00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bourgeois JP, Goldman-Rakic PS, Rakic P. Synaptogenesis in the prefrontal cortex of rhesus monkeys. Cereb Cortex. 1994;4:78–96. doi: 10.1093/cercor/4.1.78. [DOI] [PubMed] [Google Scholar]
- 30.Feinberg I. Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J Psychiatr Res. 1982;17:319–334. doi: 10.1016/0022-3956(82)90038-3. [DOI] [PubMed] [Google Scholar]
- 31.Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol. 1997;387:167–178. doi: 10.1002/(sici)1096-9861(19971020)387:2<167::aid-cne1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 32.Petanjek Z, Judas M, Simic G, Rasin MR, Uylings HB, Rakic P, Kostovic I. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci U S A. 2011;108:13281–13286. doi: 10.1073/pnas.1105108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10:410–422. doi: 10.1038/nrn2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arnsten AF. Stress weakens prefrontal networks: molecular insults to higher cognition. Nat Neurosci. 2015;18:1376–1385. doi: 10.1038/nn.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79:16–29. doi: 10.1016/j.neuron.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenberg DR, Lewis DA. Changes in the dopaminergic innervation of monkey prefrontal cortex during late postnatal development: a tyrosine hydroxylase immunohistochemical study. Biol Psychiatry. 1994;36:272–277. doi: 10.1016/0006-3223(94)90610-6. [DOI] [PubMed] [Google Scholar]
- 37.Rosenberg DR, Lewis DA. Postnatal maturation of the dopaminergic innervation of monkey prefrontal and motor cortices: a tyrosine hydroxylase immunohistochemical analysis. J Comp Neurol. 1995;358:383–400. doi: 10.1002/cne.903580306. [DOI] [PubMed] [Google Scholar]
- 38.Reichenberg A, Caspi A, Harrington H, Houts R, Keefe RS, Murray RM, Poulton R, Moffitt TE. Static and dynamic cognitive deficits in childhood preceding adult schizophrenia: a 30-year study. Am J Psychiatry. 2010;167:160–169. doi: 10.1176/appi.ajp.2009.09040574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuller R, Nopoulos P, Arndt S, O’Leary D, Ho BC, Andreasen NC. Longitudinal assessment of premorbid cognitive functioning in patients with schizophrenia through examination of standardized scholastic test performance. Am J Psychiatry. 2002;159:1183–1189. doi: 10.1176/appi.ajp.159.7.1183. [DOI] [PubMed] [Google Scholar]
- 40.van Oel CJ, Sitskoorn MM, Cremer MP, Kahn RS. School performance as a premorbid marker for schizophrenia: a twin study. Schizophr Bull. 2002;28:401–414. doi: 10.1093/oxfordjournals.schbul.a006949. [DOI] [PubMed] [Google Scholar]
- 41.Sitskoorn MM, Aleman A, Ebisch SJ, Appels MC, Kahn RS. Cognitive deficits in relatives of patients with schizophrenia: a meta-analysis. Schizophr Res. 2004;71:285–295. doi: 10.1016/j.schres.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 42.Addington J, Addington D. Neurocognitive and social functioning in schizophrenia. Schizophr Bull. 1999;25:173–182. doi: 10.1093/oxfordjournals.schbul.a033363. [DOI] [PubMed] [Google Scholar]
- 43.Barch DM, Ceaser A. Cognition in schizophrenia: core psychological and neural mechanisms. Trends Cognit Sci. 2012;16:27–34. doi: 10.1016/j.tics.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berman KF, Weinberger DR, Shelton RC, Zec RF. A relationship between anatomical and physiological brain pathology in schizophrenia: lateral cerebral ventricular size predicts cortical blood flow. Am J Psychiatry. 1987;144:1277–1282. doi: 10.1176/ajp.144.10.1277. [DOI] [PubMed] [Google Scholar]
- 45.Andreasen NC, O’Leary DS, Flaum M, Nopoulos P, Watkins GL, Boles Ponto LL, Hichwa RD. Hypofrontality in schizophrenia: distributed dysfunctional circuits in neuroleptic-naive patients. Lancet. 1997;349:1730–1734. doi: 10.1016/s0140-6736(96)08258-x. [DOI] [PubMed] [Google Scholar]
- 46.Perlstein WM, Carter CS, Noll DC, Cohen JD. Relation of prefrontal cortex dysfunction to working memory and symptoms in schizophrenia. Am J Psychiatry. 2001;158:1105–1113. doi: 10.1176/appi.ajp.158.7.1105. [DOI] [PubMed] [Google Scholar]
- 47.Callicott JH, Mattay VS, Bertolino A, Finn K, Coppola R, Frank JA, Goldberg TE, Weinberger DR. Physiological characteristics of capacity constraints in working memory as revealed by functional MRI. Cereb Cortex. 1999;9:20–26. doi: 10.1093/cercor/9.1.20. [DOI] [PubMed] [Google Scholar]
- 48.Callicott JH, Mattay VS, Verchinski BA, Marenco S, Egan MF, Weinberger DR. Complexity of prefrontal cortical dysfunction in schizophrenia: more than up or down. Am J Psychiatry. 2003;160:2209–2215. doi: 10.1176/appi.ajp.160.12.2209. [DOI] [PubMed] [Google Scholar]
- 49.Goldman PS, Rosvold HE. Localization of function within the dorsolateral prefrontal cortex of the rhesus monkey. Exp Neurol. 1970;27:291–304. doi: 10.1016/0014-4886(70)90222-0. [DOI] [PubMed] [Google Scholar]
- 50.Jacobsen CF. The functions of the frontal association areas in monkeys. Comparative Psychology Monographs. 1936;13:1–60. [Google Scholar]
- 51.Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- 52.Alexander GE. Functional development of frontal association cortex in monkeys: behavioural and electrophysiological studies. Neurosci Res Program Bull. 1982;20:471–479. [PubMed] [Google Scholar]
- 53.Alexander GE, Goldman PS. Functional development of the dorsolateral prefrontal cortex: an analysis utlizing reversible cryogenic depression. Brain Res. 1978;143:233–249. doi: 10.1016/0006-8993(78)90566-8. [DOI] [PubMed] [Google Scholar]
- 54.Lewis DA. Development of the prefrontal cortex during adolescence: insights into vulnerable neural circuits in schizophrenia. Neuropsychopharmacology. 1997;16:385–398. doi: 10.1016/S0893-133X(96)00277-1. [DOI] [PubMed] [Google Scholar]
- 55.Goldman-Rakic PS. Development of cortical circuitry and cognitive function. Child Dev. 1987;58:601–622. [PubMed] [Google Scholar]
- 56.Goldman-Rakic PS. Motor control function of the prefrontal cortex. Ciba Found Symp. 1987;132:187–200. doi: 10.1002/9780470513545.ch12. [DOI] [PubMed] [Google Scholar]
- 57.Ongur D, Price JL. The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys and humans. Cereb Cortex. 2000;10:206–219. doi: 10.1093/cercor/10.3.206. [DOI] [PubMed] [Google Scholar]
- 58.Fuster JM. The prefrontal cortex–an update: time is of the essence. Neuron. 2001;30:319–333. doi: 10.1016/s0896-6273(01)00285-9. [DOI] [PubMed] [Google Scholar]
- 59.Goldman-Rakic PS. The prefrontal landscape: implications of functional architecture for understanding human mentation and the central executive. Philos Trans R Soc B. 1996;351:1445–1453. doi: 10.1098/rstb.1996.0129. [DOI] [PubMed] [Google Scholar]
- 60.Ghashghaei HT, Barbas H. Pathways for emotion: interactions of prefrontal and anterior temporal pathways in the amygdala of the rhesus monkey. Neuroscience. 2002;115:1261–1279. doi: 10.1016/s0306-4522(02)00446-3. [DOI] [PubMed] [Google Scholar]
- 61.Price JL, Amaral DG. An autoradiographic study of the projections of the central nucleus of the monkey amygdala. J Neurosci. 1981;1:1242–1259. doi: 10.1523/JNEUROSCI.01-11-01242.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Price JL, Carmichael ST, Drevets WC. Networks related to the orbital and medial prefrontal cortex; a substrate for emotional behavior? Prog Brain Res. 1996;107:523–536. doi: 10.1016/s0079-6123(08)61885-3. [DOI] [PubMed] [Google Scholar]
- 63.Barbas H, Saha S, Rempel-Clower N, Ghashghaei T. Serial pathways from primate prefrontal cortex to autonomic areas may influence emotional expression. BMC Neurosci. 2003;4:25. doi: 10.1186/1471-2202-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jankowski MP, Sesack SR. Prefrontal cortical projections to the rat dorsal raphe nucleus: ultrastructural features and associations with serotonin and gamma-aminobutyric acid neurons. J Comp Neurol. 2004;468:518–529. doi: 10.1002/cne.10976. [DOI] [PubMed] [Google Scholar]
- 65.Robbins TW. From arousal to cognition: the integrative position of the prefrontal cortex. Prog Brain Res. 2000;126:469–483. doi: 10.1016/S0079-6123(00)26030-5. [DOI] [PubMed] [Google Scholar]
- 66.Cavada C, Company T, Tejedor J, Cruz-Rizzolo RJ, Reinoso-Suarez F. The anatomical connections of the macaque monkey orbitofrontal cortex. A review. Cereb Cortex. 2000;10:220–242. doi: 10.1093/cercor/10.3.220. [DOI] [PubMed] [Google Scholar]
- 67.Funahashi S, Bruce CJ, Goldman-Rakic PS. Mnemonic coding of visual space in the monkey’s dorsolateral prefrontal cortex. J Neurophysiol. 1989;61:331–349. doi: 10.1152/jn.1989.61.2.331. [DOI] [PubMed] [Google Scholar]
- 68.Fuster JM, Alexander GE. Neuron activity related to short-term memory. Science. 1971;173:652–654. doi: 10.1126/science.173.3997.652. [DOI] [PubMed] [Google Scholar]
- 69.Kubota K, Niki H. Prefrontal cortical unit activity and delayed alternation performance in monkeys. J Neurophysiol. 1971;34:337–347. doi: 10.1152/jn.1971.34.3.337. [DOI] [PubMed] [Google Scholar]
- 70.Sommer MA, Wurtz RH. Brain circuits for the internal monitoring of movements. Annu Rev Neurosci. 2008;31:317–338. doi: 10.1146/annurev.neuro.31.060407.125627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang M, Vijayraghavan S, Goldman-Rakic PS. Selective D2 receptor actions on the functional circuitry of working memory. Science. 2004;303:853–856. doi: 10.1126/science.1091162. [DOI] [PubMed] [Google Scholar]
- 72.Cavada C, Goldman-Rakic PS. Posterior parietal cortex in rhesus monkey: I. Parcellation of areas based on distinctive limbic and sensory corticocortical connections. J Comp Neurol. 1989;287:393–421. doi: 10.1002/cne.902870402. [DOI] [PubMed] [Google Scholar]
- 73.Kritzer MF, Goldman-Rakic PS. Intrinsic circuit organization of the major layers and sublayers of the dorsolateral prefrontal cortex in the rhesus monkey. J Comp Neurol. 1995;359:131–143. doi: 10.1002/cne.903590109. [DOI] [PubMed] [Google Scholar]
- 74.Melchitzky DS, Gonzalez-Burgos G, Barrionuevo G, Lewis DA. Synaptic targets of the intrinsic axon collaterals of supragranular pyramidal neurons in monkey prefrontal cortex. J Comp Neurol. 2001;430:209–221. doi: 10.1002/1096-9861(20010205)430:2<209::aid-cne1026>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 75.Melchitzky DS, Sesack SR, Pucak ML, Lewis DA. Synaptic targets of pyramidal neurons providing intrinsic horizontal connections in monkey prefrontal cortex. J Comp Neurol. 1998;390:211–224. doi: 10.1002/(sici)1096-9861(19980112)390:2<211::aid-cne4>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 76.Schwartz ML, Goldman-Rakic PS. Callosal and intrahemispheric connectivity of the prefrontal association cortex in rhesus monkey: relation between intraparietal and principal sulcal cortex. J Comp Neurol. 1984;226:403–420. doi: 10.1002/cne.902260309. [DOI] [PubMed] [Google Scholar]
- 77.Wang XJ. Synaptic reverberation underlying mnemonic persistent activity. Trends Neurosci. 2001;24:455–463. doi: 10.1016/s0166-2236(00)01868-3. [DOI] [PubMed] [Google Scholar]
- 78.Constantinidis C, Goldman-Rakic PS. Correlated discharges among putative pyramidal neurons and interneurons in the primate prefrontal cortex. J Neurophysiol. 2002;88:3487–3497. doi: 10.1152/jn.00188.2002. [DOI] [PubMed] [Google Scholar]
- 79.Constantinidis C, Williams GV, Goldman-Rakic PS. A role for inhibition in shaping the temporal flow of information in prefrontal cortex. Nat Neurosci. 2002;5:175–180. doi: 10.1038/nn799. [DOI] [PubMed] [Google Scholar]
- 80.Melchitzky DS, Lewis DA. Pyramidal neuron local axon terminals in monkey prefrontal cortex: differential targeting of subclasses of GABA neurons. Cereb Cortex. 2003;13:452–460. doi: 10.1093/cercor/13.5.452. [DOI] [PubMed] [Google Scholar]
- 81.Rao SG, Williams GV, Goldman-Rakic PS. Isodirectional tuning of adjacent interneurons and pyramidal cells during working memory: evidence for microcolumnar organization in PFC. J Neurophysiol. 1999;81:1903–1916. doi: 10.1152/jn.1999.81.4.1903. [DOI] [PubMed] [Google Scholar]
- 82.Rao SG, Williams GV, Goldman-Rakic PS. Destruction and creation of spatial tuning by disinhibition: GABA(A) blockade of prefrontal cortical neurons engaged by working memory. J Neurosci. 2000;20:485–494. doi: 10.1523/JNEUROSCI.20-01-00485.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wilson FA, O’Scalaidhe SP, Goldman-Rakic PS. Functional synergism between putative gamma-aminobutyrate-containing neurons and pyramidal neurons in prefrontal cortex. Proc Natl Acad Sci U S A. 1994;91:4009–4013. doi: 10.1073/pnas.91.9.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Elston GN. Pyramidal cells of the frontal lobe: all the more spinous to think with. J Neurosci. 2000;20:RC95. doi: 10.1523/JNEUROSCI.20-18-j0002.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Elston GN. Cortex, cognition and the cell: new insights into the pyramidal neuron and prefrontal function. Cereb Cortex. 2003;13:1124–1138. doi: 10.1093/cercor/bhg093. [DOI] [PubMed] [Google Scholar]
- 86.Elston GN, Benavides-Piccione R, Elston A, Zietsch B, Defelipe J, Manger P, Casagrande V, Kaas JH. Specializations of the granular prefrontal cortex of primates: implications for cognitive processing. Anat Rec Part A. 2006;288:26–35. doi: 10.1002/ar.a.20278. [DOI] [PubMed] [Google Scholar]
- 87.Medalla M, Luebke JI. Diversity of glutamatergic synaptic strength in lateral prefrontal versus primary visual cortices in the rhesus monkey. J Neurosci. 2015;35:112–127. doi: 10.1523/JNEUROSCI.3426-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Defelipe J. The evolution of the brain, the human nature of cortical circuits, and intellectual creativity. Front Neuroanat. 2011;5:29. doi: 10.3389/fnana.2011.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gilman JP, Medalla M, Luebke JI. Area-Specific Features of Pyramidal Neurons-a Comparative Study in Mouse and Rhesus Monkey. Cereb Cortex. 2017;27:2078–2094. doi: 10.1093/cercor/bhw062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Amatrudo JM, Weaver CM, Crimins JL, Hof PR, Rosene DL, Luebke JI. Influence of highly distinctive structural properties on the excitability of pyramidal neurons in monkey visual and prefrontal cortices. J Neurosci. 2012;32:13644–13660. doi: 10.1523/JNEUROSCI.2581-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zaitsev AV, Povysheva NV, Gonzalez-Burgos G, Lewis DA. Electrophysiological classes of layer 2/3 pyramidal cells in monkey prefrontal cortex. J Neurophysiol. 2012;108:595–609. doi: 10.1152/jn.00859.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hsu A, Luebke JI, Medalla M. Comparative ultrastructural features of excitatory synapses in the visual and frontal cortices of the adult mouse and monkey. J Comp Neurol. 2017;525:2175–2191. doi: 10.1002/cne.24196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Luebke JI. Pyramidal Neurons Are Not Generalizable Building Blocks of Cortical Networks. Front Neuroanat. 2017;11:11. doi: 10.3389/fnana.2017.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vita A, De Peri L, Deste G, Sacchetti E. Progressive loss of cortical gray matter in schizophrenia: a meta-analysis and meta-regression of longitudinal MRI studies. Transl Psychiatry. 2012;2:e190. doi: 10.1038/tp.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rais M, Cahn W, Van Haren N, Schnack H, Caspers E, Hulshoff Pol H, Kahn R. Excessive brain volume loss over time in cannabis-using first-episode schizophrenia patients. Am J Psychiatry. 2008;165:490–496. doi: 10.1176/appi.ajp.2007.07071110. [DOI] [PubMed] [Google Scholar]
- 96.Van Haren NE, Cahn W, Hulshoff Pol HE, Kahn RS. Confounders of excessive brain volume loss in schizophrenia. Neurosci Biobehav Rev. 2013;37:2418–2423. doi: 10.1016/j.neubiorev.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 97.Selemon LD, Goldman-Rakic PS. The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biol Psychiatry. 1999;45:17–25. doi: 10.1016/s0006-3223(98)00281-9. [DOI] [PubMed] [Google Scholar]
- 98.Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- 99.Kolluri N, Sun Z, Sampson AR, Lewis DA. Lamina-specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia. Am J Psychiatry. 2005;162:1200–1202. doi: 10.1176/appi.ajp.162.6.1200. [DOI] [PubMed] [Google Scholar]
- 100.Pierri JN, Volk CL, Auh S, Sampson A, Lewis DA. Decreased somal size of deep layer 3 pyramidal neurons in the prefrontal cortex of subjects with schizophrenia. Arch Gen Psychiatry. 2001;58:466–473. doi: 10.1001/archpsyc.58.5.466. [DOI] [PubMed] [Google Scholar]
- 101.Castillo MA, Ghose S, Tamminga CA, Ulery-Reynolds PG. Deficits in syntaxin 1 phosphorylation in schizophrenia prefrontal cortex. Biol Psychiatry. 2010;67:208–216. doi: 10.1016/j.biopsych.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 102.Glantz LA, Lewis DA. Reduction of synaptophysin immunoreactivity in the prefrontal cortex of subjects with schizophrenia. Regional and diagnostic specificity. Arch Gen Psychiatry. 1997;54:660–669. doi: 10.1001/archpsyc.1997.01830190088009. [DOI] [PubMed] [Google Scholar]
- 103.Halim ND, Weickert CS, McClintock BW, Hyde TM, Weinberger DR, Kleinman JE, Lipska BK. Presynaptic proteins in the prefrontal cortex of patients with schizophrenia and rats with abnormal prefrontal development. Mol Psychiatry. 2003;8:797–810. doi: 10.1038/sj.mp.4001319. [DOI] [PubMed] [Google Scholar]
- 104.Karson CN, Mrak RE, Schluterman KO, Sturner WQ, Sheng JG, Griffin WS. Alterations in synaptic proteins and their encoding mRNAs in prefrontal cortex in schizophrenia: a possible neurochemical basis for ’hypofrontality’. Mol Psychiatry. 1999;4:39–45. doi: 10.1038/sj.mp.4000459. [DOI] [PubMed] [Google Scholar]
- 105.Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35:57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lewis DA, Gonzalez-Burgos G. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology. 2008;33:141–165. doi: 10.1038/sj.npp.1301563. [DOI] [PubMed] [Google Scholar]
- 107.Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- 108.Arion D, Corradi JP, Tang S, Datta D, Boothe F, He A, Cacace AM, Zaczek R, Albright CF, Tseng G, Lewis DA. Distinctive transcriptome alterations of prefrontal pyramidal neurons in schizophrenia and schizoaffective disorder. Mol Psychiatry. 2015;20:1397–1405. doi: 10.1038/mp.2014.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Abi-Dargham A, Mawlawi O, Lombardo I, Gil R, Martinez D, Huang Y, Hwang DR, Keilp J, Kochan L, Van Heertum R, Gorman JM, Laruelle M. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci. 2002;22:3708–3719. doi: 10.1523/JNEUROSCI.22-09-03708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lewis DA, Gonzalez-Burgos G. Pathophysiologically based treatment interventions in schizophrenia. Nat Med. 2006;12:1016–1022. doi: 10.1038/nm1478. [DOI] [PubMed] [Google Scholar]
- 111.Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. 1991;148:1474–1486. doi: 10.1176/ajp.148.11.1474. [DOI] [PubMed] [Google Scholar]
- 112.Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44:660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- 113.Meyer-Lindenberg A, Miletich RS, Kohn PD, Esposito G, Carson RE, Quarantelli M, Weinberger DR, Berman KF. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat Neurosci. 2002;5:267–271. doi: 10.1038/nn804. [DOI] [PubMed] [Google Scholar]
- 114.Carr DB, Sesack SR. Projections from the rat prefrontal cortex to the ventral tegmental area: target specificity in the synaptic associations with mesoaccumbens and mesocortical neurons. J Neurosci. 2000;20:3864–3873. doi: 10.1523/JNEUROSCI.20-10-03864.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Carr DB, Sesack SR. GABA-containing neurons in the rat ventral tegmental area project to the prefrontal cortex. Synapse. 2000;38:114–123. doi: 10.1002/1098-2396(200011)38:2<114::AID-SYN2>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 116.Sesack SR, Carr DB. Selective prefrontal cortex inputs to dopamine cells: implications for schizophrenia. Physiol Behav. 2002;77:513–517. doi: 10.1016/s0031-9384(02)00931-9. [DOI] [PubMed] [Google Scholar]
- 117.Sesack SR, Pickel VM. Prefrontal cortical efferents in the rat synapse on unlabeled neuronal targets of catecholamine terminals in the nucleus accumbens septi and on dopamine neurons in the ventral tegmental area. J Comp Neurol. 1992;320:145–160. doi: 10.1002/cne.903200202. [DOI] [PubMed] [Google Scholar]
- 118.Frankle WG, Laruelle M, Haber SN. Prefrontal cortical projections to the midbrain in primates: evidence for a sparse connection. Neuropsychopharmacology. 2006;31:1627–1636. doi: 10.1038/sj.npp.1300990. [DOI] [PubMed] [Google Scholar]
- 119.Crittenden JR, Graybiel AM. Basal Ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front Neuroanat. 2011;5:59. doi: 10.3389/fnana.2011.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Friedman A, Homma D, Bloem B, Gibb LG, Amemori KI, Hu D, Delcasso S, Truong TF, Yang J, Hood AS, Mikofalvy KA, Beck DW, Nguyen N, Nelson ED, Toro Arana SE, Vorder Bruegge RH, Goosens KA, Graybiel AM. Chronic Stress Alters Striosome-Circuit Dynamics, Leading to Aberrant Decision-Making. Cell. 2017;171:1191–1205. doi: 10.1016/j.cell.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Datta D, Arion D, Roman KM, Volk DW, Lewis DA. Altered Expression of ARP2/3 Complex Signaling Pathway Genes in Prefrontal Layer 3 Pyramidal Cells in Schizophrenia. Am J Psychiatry. 2017;174:163–171. doi: 10.1176/appi.ajp.2016.16020204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kim IH, Rossi MA, Aryal DK, Racz B, Kim N, Uezu A, Wang F, Wetsel WC, Weinberg RJ, Yin H, Soderling SH. Spine pruning drives antipsychotic-sensitive locomotion via circuit control of striatal dopamine. Nat Neurosci. 2015;18:883–891. doi: 10.1038/nn.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kegeles LS, Abi-Dargham A, Frankle WG, Gil R, Cooper TB, Slifstein M, Hwang DR, Huang Y, Haber SN, Laruelle M. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry. 2010;67:231–239. doi: 10.1001/archgenpsychiatry.2010.10. [DOI] [PubMed] [Google Scholar]
- 124.Yang S, Wang M, Paspalas CD, Crimins JL, Altman MT, Mazer JA, Arnsten AF. Core Differences in Synaptic Signaling Between Primary Visual and Dorsolateral Prefrontal Cortex. Cerebral Cortex. 2018;1 doi: 10.1093/cercor/bhx357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang M, Yang Y, Wang CJ, Gamo NJ, Jin LE, Mazer JA, Morrison JH, Wang XJ, Arnsten AF. NMDA receptors subserve persistent neuronal firing during working memory in dorsolateral prefrontal cortex. Neuron. 2013;77:736–749. doi: 10.1016/j.neuron.2012.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Compte A, Brunel N, Goldman-Rakic PS, Wang XJ. Synaptic mechanisms and network dynamics underlying spatial working memory in a cortical network model. Cereb Cortex. 2000;10:910–923. doi: 10.1093/cercor/10.9.910. [DOI] [PubMed] [Google Scholar]
- 127.Lisman JE, Fellous JM, Wang XJ. A role for NMDA-receptor channels in working memory. Nat Neurosci. 1998;1:273–275. doi: 10.1038/1086. [DOI] [PubMed] [Google Scholar]
- 128.Wang XJ. Synaptic basis of cortical persistent activity: the importance of NMDA receptors to working memory. J Neurosci. 1999;19:9587–9603. doi: 10.1523/JNEUROSCI.19-21-09587.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Datta D, Arion D, Lewis DA. Developmental Expression Patterns of GABAA Receptor Subunits in Layer 3 and 5 Pyramidal Cells of Monkey Prefrontal Cortex. Cereb Cortex. 2015;25:2295–2305. doi: 10.1093/cercor/bhu040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yang Y, Paspalas CD, Jin LE, Picciotto MR, Arnsten AF, Wang M. Nicotinic alpha7 receptors enhance NMDA cognitive circuits in dorsolateral prefrontal cortex. Proc Natl Acad Sci U S A. 2013;110:12078–12083. doi: 10.1073/pnas.1307849110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Croxson PL, Kyriazis DA, Baxter MG. Cholinergic modulation of a specific memory function of prefrontal cortex. Nat Neurosci. 2011;14:1510–1512. doi: 10.1038/nn.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brozoski TJ, Brown RM, Rosvold HE, Goldman PS. Cognitive deficit caused by regional depletion of dopamine in prefrontal cortex of rhesus monkey. Science. 1979;205:929–932. doi: 10.1126/science.112679. [DOI] [PubMed] [Google Scholar]
- 133.Birnbaum SG, Yuan PX, Wang M, Vijayraghavan S, Bloom AK, Davis DJ, Gobeske KT, Sweatt JD, Manji HK, Arnsten AF. Protein kinase C overactivity impairs prefrontal cortical regulation of working memory. Science. 2004;306:882–884. doi: 10.1126/science.1100021. [DOI] [PubMed] [Google Scholar]
- 134.Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley A, Nou E, Mazer JA, McCormick DA, Arnsten AF. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 135.Arnsten AF, Wang M, Paspalas CD. Dopamine’s Actions in Primate Prefrontal Cortex: Challenges for Treating Cognitive Disorders. Pharmacol Rev. 2015;67:681–696. doi: 10.1124/pr.115.010512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AF. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci. 2007;10:376–384. doi: 10.1038/nn1846. [DOI] [PubMed] [Google Scholar]
- 137.Qin S, Hermans EJ, van Marle HJ, Luo J, Fernandez G. Acute psychological stress reduces working memory-related activity in the dorsolateral prefrontal cortex. Biol Psychiatry. 2009;66:25–32. doi: 10.1016/j.biopsych.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 138.Lataster J, Collip D, Ceccarini J, Haas D, Booij L, van Os J, Pruessner J, Van Laere K, Myin-Germeys I. Psychosocial stress is associated with in vivo dopamine release in human ventromedial prefrontal cortex: a positron emission tomography study using [(1)(8)F]fallypride. NeuroImage. 2011;58:1081–1089. doi: 10.1016/j.neuroimage.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 139.Nagano-Saito A, Dagher A, Booij L, Gravel P, Welfeld K, Casey KF, Leyton M, Benkelfat C. Stress-induced dopamine release in human medial prefrontal cortex–18F-fallypride/PET study in healthy volunteers. Synapse. 2013;67:821–830. doi: 10.1002/syn.21700. [DOI] [PubMed] [Google Scholar]
- 140.Qin S, Cousijn H, Rijpkema M, Luo J, Franke B, Hermans EJ, Fernandez G. The effect of moderate acute psychological stress on working memory-related neural activity is modulated by a genetic variation in catecholaminergic function in humans. Front Integr Neurosci. 2012;6:16. doi: 10.3389/fnint.2012.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Arnsten AF, Paspalas CD, Gamo NJ, Yang Y, Wang M. Dynamic Network Connectivity: A new form of neuroplasticity. Trends Cognit Sci. 2010;14:365–375. doi: 10.1016/j.tics.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- 143.Brennan AR, Dolinsky B, Vu MA, Stanley M, Yeckel MF, Arnsten AF. Blockade of IP3-mediated SK channel signaling in the rat medial prefrontal cortex improves spatial working memory. Learn Mem. 2008;15:93–96. doi: 10.1101/lm.767408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gamo NJ, Lur G, Higley MJ, Wang M, Paspalas CD, Vijayraghavan S, Yang Y, Ramos BP, Peng K, Kata A, Boven L, Lin F, Roman L, Lee D, Arnsten AF. Stress Impairs Prefrontal Cortical Function via D1 Dopamine Receptor Interactions With Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels. Biol Psychiatry. 2015;78:860–870. doi: 10.1016/j.biopsych.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jin LE, Wang M, Yang ST, Yang Y, Galvin VC, Lightbourne TC, Ottenheimer D, Zhong Q, Stein J, Raja A, Paspalas CD, Arnsten AFT. mGluR2/3 mechanisms in primate dorsolateral prefrontal cortex: evidence for both presynaptic and postsynaptic actions. Mol Psychiatry. 2017;22:1615–1625. doi: 10.1038/mp.2016.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jin LE, Wang M, Galvin VC, Lightbourne TC, Conn PJ, Arnsten AF, Paspalas CD. mGluR2 versus mGluR3Metabotropic Glutamate Receptors in Primate Dorsolateral Prefrontal Cortex: Postsynaptic mGluR3 Strengthen Working Memory Networks. Cereb Cortex. 2017 doi: 10.1093/cercor/bhx005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Carlyle BC, Nairn AC, Wang M, Yang Y, Jin LE, Simen AA, Ramos BP, Bordner KA, Craft GE, Davies P, Pletikos M, Sestan N, Arnsten AF, Paspalas CD. cAMP–PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proc Natl Acad Sci U S A. 2014;111:5036–5041. doi: 10.1073/pnas.1322360111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Paspalas CD, Wang M, Arnsten AF. Constellation of HCN channels and cAMP regulating proteins in dendritic spines of the primate prefrontal cortex: potential substrate for working memory deficits in schizophrenia. Cereb Cortex. 2013;23:1643–1654. doi: 10.1093/cercor/bhs152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.MacKenzie KF, Wallace DA, Hill EV, Anthony DF, Henderson DJ, Houslay DM, Arthur JS, Baillie GS, Houslay MD. Phosphorylation of cAMP-specific PDE4A5 (phosphodiesterase-4A5) by MK2 (MAPKAPK2) attenuates its activation through protein kinase A phosphorylation. Biochem J. 2011;435:755–769. doi: 10.1042/BJ20101184. [DOI] [PubMed] [Google Scholar]
- 150.Radley JJ, Rocher AB, Janssen WG, Hof PR, McEwen BS, Morrison JH. Reversibility of apical dendritic retraction in the rat medial prefrontal cortex following repeated stress. Exp Neurol. 2005;196:199–203. doi: 10.1016/j.expneurol.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 151.Seib LM, Wellman CL. Daily injections alter spine density in rat medial prefrontal cortex. Neurosci Lett. 2003;337:29–32. doi: 10.1016/s0304-3940(02)01287-9. [DOI] [PubMed] [Google Scholar]
- 152.Hains AB, Vu MA, Maciejewski PK, van Dyck CH, Gottron M, Arnsten AF. Inhibition of protein kinase C signaling protects prefrontal cortex dendritic spines and cognition from the effects of chronic stress. Proc Natl Acad Sci U S A. 2009;106:17957–17962. doi: 10.1073/pnas.0908563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Liston C, Miller MM, Goldwater DS, Radley JJ, Rocher AB, Hof PR, Morrison JH, McEwen BS. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J Neurosci. 2006;26:7870–7874. doi: 10.1523/JNEUROSCI.1184-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hains AB, Yabe Y, Arnsten AF. Chronic Stimulation of Alpha-2A-Adrenoceptors With Guanfacine Protects Rodent Prefrontal Cortex Dendritic Spines and Cognition From the Effects of Chronic Stress. Neurobiol Stress. 2015;2:1–9. doi: 10.1016/j.ynstr.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci. 2002;22:6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ansell EB, Rando K, Tuit K, Guarnaccia J, Sinha R. Cumulative adversity and smaller gray matter volume in medial prefrontal, anterior cingulate, and insula regions. Biol Psychiatry. 2012;72:57–64. doi: 10.1016/j.biopsych.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van Doren V, Genovese G, Rose SA, Handsaker RE, Daly MJ, Carroll MC, Stevens B, McCarroll SA. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–183. doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fischer EK, Drago A. A molecular pathway analysis stresses the role of inflammation and oxidative stress towards cognition in schizophrenia. J Neural Transm (Vienna) 2017;124:765–774. doi: 10.1007/s00702-017-1730-y. [DOI] [PubMed] [Google Scholar]
- 159.Crowley JJ, Hilliard CE, Kim Y, Morgan MB, Lewis LR, Muzny DM, Hawes AC, Sabo A, Wheeler DA, Lieberman JA, Sullivan PF, Gibbs RA. Deep resequencing and association analysis of schizophrenia candidate genes. Mol Psychiatry. 2013;18:138–140. doi: 10.1038/mp.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, Moran J, Chambert K, Toncheva D, Georgieva L, Grozeva D, Fjodorova M, Wollerton R, Rees E, Nikolov I, van de Lagemaat LN, Bayes A, Fernandez E, Olason PI, Bottcher Y, Komiyama NH, Collins MO, Choudhary J, Stefansson K, Stefansson H, Grant SG, Purcell S, Sklar P, O’Donovan MC, Owen MJ. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2012;17:142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, Jonasdottir A, Jonasdottir A, Bjornsson A, Mattiasdottir S, Blondal T, Haraldsson M, Magnusdottir BB, Giegling I, M?ller HJ, Hartmann A, Shianna KV, Ge D, Need AC, Crombie C, Fraser G, Walker N, Lonnqvist J, Suvisaari J, Tuulio-Henriksson A, Paunio T, Toulopoulou T, Bramon E, Di Forti M, Murray R, Ruggeri M, Vassos E, Tosato S, Walshe M, Li T, Vasilescu C, Muhleisen TW, Wang AG, Ullum H, Djurovic S, Melle I, Olesen J, Kiemeney LA, Franke B, Sabatti C, Freimer NB, Gulcher JR, Thorsteinsdottir U, Kong A, Andreassen OA, Ophoff RA, Georgi A, Rietschel M, Werge T, Petursson H, Goldstein DB, Nothen MM, Peltonen L, Collier DA, St Clair D, Stefansson K. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Akbarian S, Sucher NJ, Bradley D, Tafazzoli A, Trinh D, Hetrick WP, Potkin SG, Sandman CA, Bunney WE, Jr, Jones EG. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J Neurosci. 1996;16:19–30. doi: 10.1523/JNEUROSCI.16-01-00019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology. 2008;33:2175–2186. doi: 10.1038/sj.npp.1301604. [DOI] [PubMed] [Google Scholar]
- 164.Weickert CS, Fung SJ, Catts VS, Schofield PR, Allen KM, Moore LT, Newell KA, Pellen D, Huang XF, Catts SV, Weickert TW. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol Psychiatry. 2013;18:1185–1192. doi: 10.1038/mp.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Freedman R, Leonard S, Gault JM, Hopkins J, Cloninger CR, Kaufmann CA, Tsuang MT, Farone SV, Malaspina D, Svrakic DM, Sanders A, Gejman P. Linkage disequilibrium for schizophrenia at the chromosome 15q13–14 locus of the alpha7-nicotinic acetylcholine receptor subunit gene (CHRNA7) Am J Med Genet. 2001;105:20–22. [PubMed] [Google Scholar]
- 166.Martin LF, Freedman R. Schizophrenia and the alpha7 nicotinic acetylcholine receptor. Int Rev Neurobiol. 2007;78:225–246. doi: 10.1016/S0074-7742(06)78008-4. [DOI] [PubMed] [Google Scholar]
- 167.Won H, de la Torre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, Gandal MJ, Sutton GJ, Hormozdiari F, Lu D, Lee C, Eskin E, Voineagu I, Ernst J, Geschwind DH. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. 2016;538:523–527. doi: 10.1038/nature19847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yoshimizu T, Pan JQ, Mungenast AE, Madison JM, Su S, Ketterman J, Ongur D, McPhie D, Cohen B, Perlis R, Tsai LH. Functional implications of a psychiatric risk variant within CACNA1C in induced human neurons. Mol Psychiatry. 2015;20:162–169. doi: 10.1038/mp.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Nystoriak MA, Nieves-Cintron M, Patriarchi T, Buonarati OR, Prada MP, Morotti S, Grandi E, Fernandes JD, Forbush K, Hofmann F, Sasse KC, Scott JD, Ward SM, Hell JW, Navedo MF. Ser1928 phosphorylation by PKA stimulates the L-type Ca2+ channel CaV1.2 and vasoconstriction during acute hyperglycemia and diabetes. Sci Signaling. 2017;10:eaaf9647. doi: 10.1126/scisignal.aaf9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Shaw RM, Colecraft HM. L-type calcium channel targeting and local signalling in cardiac myocytes. Cardiovasc Res. 2013;98:177–186. doi: 10.1093/cvr/cvt021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Millar JK, Pickard BS, Mackie S, James R, Christie S, Buchanan SR, Malloy MP, Chubb JE, Huston E, Baillie GS, Thomson PA, Hill EV, Brandon NJ, Rain JC, Camargo LM, Whiting PJ, Houslay MD, Blackwood DH, Muir WJ, Porteous DJ. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science. 2005;310:1187–1191. doi: 10.1126/science.1112915. [DOI] [PubMed] [Google Scholar]
- 172.St Clair D, Blackwood D, Muir W, Carothers A, Walker M, Spowart G, Gosden C, Evans HJ. Association within a family of a balanced autosomal translocation with major mental illness. Lancet. 1990;336:13–16. doi: 10.1016/0140-6736(90)91520-k. [DOI] [PubMed] [Google Scholar]
- 173.Bradshaw NJ, Porteous DJ. DISC1-binding proteins in neural development, signalling and schizophrenia. Neuropharmacology. 2012;62:1230–1241. doi: 10.1016/j.neuropharm.2010.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Brandon NJ, Sawa A. Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat Rev Neurosci. 2011;12:707–722. doi: 10.1038/nrn3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hayashi-Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H, Dunlop AJ, Makino Y, Seshadri AJ, Ishizuka K, Srivastava DP, Xie Z, Baraban JM, Houslay MD, Tomoda T, Brandon NJ, Kamiya A, Yan Z, Penzes P, Sawa A. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci. 2010;13:327–332. doi: 10.1038/nn.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tomppo L, Hennah W, Lahermo P, Loukola A, Tuulio-Henriksson A, Suvisaari J, Partonen T, Ekelund J, Lonnqvist J, Peltonen L. Association between genes of Disrupted in schizophrenia 1 (DISC1) interactors and schizophrenia supports the role of the DISC1 pathway in the etiology of major mental illnesses. Biol Psychiatry. 2009;65:1055–1062. doi: 10.1016/j.biopsych.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gamo NJ, Duque A, Paspalas CD, Kata A, Fine R, Boven L, Bryan C, Lo T, Anighoro K, Bermudez L, Peng K, Annor A, Raja A, Mansson E, Taylor SR, Patel K, Simen AA, Arnsten AF. Role of disrupted in schizophrenia 1 (DISC1) in stress-induced prefrontal cognitive dysfunction. Transl Psychiatry. 2013;3:e328. doi: 10.1038/tp.2013.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Paspalas CD, Selemon LD, Arnsten AF. Mapping the regulator of G protein signaling 4 (RGS4): presynaptic and postsynaptic substrates for neuroregulation in prefrontal cortex. Cereb Cortex. 2009;19:2145–2155. doi: 10.1093/cercor/bhn235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Erdely HA, Tamminga CA, Roberts RC, Vogel MW. Regional alterations in RGS4 protein in schizophrenia. Synapse. 2006;59:472–479. doi: 10.1002/syn.20265. [DOI] [PubMed] [Google Scholar]
- 180.Mirnics K, Middleton FA, Stanwood GD, Lewis DA, Levitt P. Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol Psychiatry. 2001;6:293–301. doi: 10.1038/sj.mp.4000866. [DOI] [PubMed] [Google Scholar]
- 181.Volk DW, Eggan SM, Lewis DA. Alterations in metabotropic glutamate receptor 1alpha and regulator of G protein signaling 4 in the prefrontal cortex in schizophrenia. Am J Psychiatry. 2010;167:1489–1498. doi: 10.1176/appi.ajp.2010.10030318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kimoto S, Glausier JR, Fish KN, Volk DW, Bazmi HH, Arion D, Datta D, Lewis DA. Reciprocal Alterations in Regulator of G Protein Signaling 4 and microRNA16 in Schizophrenia. Schizophr Bull. 2016;42:396–405. doi: 10.1093/schbul/sbv139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chowdari KV, Mirnics K, Semwal P, Wood J, Lawrence E, Bhatia T, Deshpande SN, B KT, Ferrell RE, Middleton FA, Devlin B, Levitt P, Lewis DA, Nimgaonkar VL. Association and linkage analyses of RGS4 polymorphisms in schizophrenia. Hum Mol Genet. 2002;11:1373–1380. doi: 10.1093/hmg/11.12.1373. [DOI] [PubMed] [Google Scholar]
- 184.Harrison PJ, Lyon L, Sartorius LJ, Burnet PW, Lane TA. The group II metabotropic glutamate receptor 3 (mGluR3, mGlu3, GRM3): expression, function and involvement in schizophrenia. J Psychopharmacol. 2008;22:308–322. doi: 10.1177/0269881108089818. [DOI] [PubMed] [Google Scholar]
- 185.Sartorius LJ, Weinberger DR, Hyde TM, Harrison PJ, Kleinman JE, Lipska BK. Expression of a GRM3 splice variant is increased in the dorsolateral prefrontal cortex of individuals carrying a schizophrenia risk SNP. Neuropsychopharmacology. 2008;33:2626–2634. doi: 10.1038/sj.npp.1301669. [DOI] [PubMed] [Google Scholar]
- 186.Ghose S, Gleason KA, Potts BW, Lewis-Amezcua K, Tamminga CA. Differential expression of metabotropic glutamate receptors 2 and 3 in schizophrenia: a mechanism for antipsychotic drug action? Am J Psychiatry. 2009;166:812–820. doi: 10.1176/appi.ajp.2009.08091445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zhang Z, Bassam B, Thomas AG, Williams M, Liu J, Nance E, Rojas C, Slusher BS, Kannan S. Maternal inflammation leads to impaired glutamate homeostasis and up-regulation of glutamate carboxypeptidase II in activated microglia in the fetal/newborn rabbit brain. Neurobiol Dis. 2016;94:116–128. doi: 10.1016/j.nbd.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Muly EC, Maddox M, Smith Y. Distribution of mGluR1alpha and mGluR5 immunolabeling in primate prefrontal cortex. J Comp Neurol. 2003;467:521–535. doi: 10.1002/cne.10937. [DOI] [PubMed] [Google Scholar]
- 189.Akil M, Pierri JN, Whitehead RE, Edgar CL, Mohila C, Sampson AR, Lewis DA. Lamina-specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am J Psychiatry. 1999;156:1580–1589. doi: 10.1176/ajp.156.10.1580. [DOI] [PubMed] [Google Scholar]
- 190.Cahill ME, Xie Z, Day M, Photowala H, Barbolina MV, Miller CA, Weiss C, Radulovic J, Sweatt JD, Disterhoft JF, Surmeier DJ, Penzes P. Kalirin regulates cortical spine morphogenesis and disease-related behavioral phenotypes. Proc Natl Acad Sci U S A. 2009;106:13058–13063. doi: 10.1073/pnas.0904636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hill JJ, Hashimoto T, Lewis DA. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2006;11:557–566. doi: 10.1038/sj.mp.4001792. [DOI] [PubMed] [Google Scholar]
- 192.Ikeda M, Aleksic B, Kinoshita Y, Okochi T, Kawashima K, Kushima I, Ito Y, Nakamura Y, Kishi T, Okumura T, Fukuo Y, Williams HJ, Hamshere ML, Ivanov D, Inada T, Suzuki M, Hashimoto R, Ujike H, Takeda M, Craddock N, Kaibuchi K, Owen MJ, Ozaki N, O’Donovan MC, Iwata N. Genome-wide association study of schizophrenia in a Japanese population. Biol Psychiatry. 2011;69:472–478. doi: 10.1016/j.biopsych.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 193.Kushima I, Nakamura Y, Aleksic B, Ikeda M, Ito Y, Shiino T, Okochi T, Fukuo Y, Ujike H, Suzuki M, Inada T, Hashimoto R, Takeda M, Kaibuchi K, Iwata N, Ozaki N. Resequencing and association analysis of the KALRN and EPHB1 genes and their contribution to schizophrenia susceptibility. Schizophr Bull. 2012;38:552–560. doi: 10.1093/schbul/sbq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Datta D, Arion D, Corradi JP, Lewis DA. Altered expression of CDC42 signaling pathway components in cortical layer 3 pyramidal cells in schizophrenia. Biol Psychiatry. 2015;78:775–785. doi: 10.1016/j.biopsych.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ide M, Lewis DA. Altered cortical CDC42 signaling pathways in schizophrenia: implications for dendritic spine deficits. Biol Psychiatry. 2010;68:25–32. doi: 10.1016/j.biopsych.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Arion D, Huo Z, Enwright JF, Corradi JP, Tseng G, Lewis DA. Transcriptome Alterations in Prefrontal Pyramidal Cells Distinguish Schizophrenia From Bipolar and Major Depressive Disorders. Biol Psychiatry. 2017;82:594–600. doi: 10.1016/j.biopsych.2017.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Glahn DC, Laird AR, Ellison-Wright I, Thelen SM, Robinson JL, Lancaster JL, Bullmore E, Fox PT. Meta-analysis of gray matter anomalies in schizophrenia: application of anatomic likelihood estimation and network analysis. Biol Psychiatry. 2008;64:774–781. doi: 10.1016/j.biopsych.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P. Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J Neurosci. 2002;22:2718–2729. doi: 10.1523/JNEUROSCI.22-07-02718.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Cho RY, Konecky RO, Carter CS. Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proc Natl Acad Sci U S A. 2006;103:19878–19883. doi: 10.1073/pnas.0609440103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Minzenberg MJ, Firl AJ, Yoon JH, Gomes GC, Reinking C, Carter CS. Gamma oscillatory power is impaired during cognitive control independent of medication status in first-episode schizophrenia. Neuropsychopharmacology. 2010;35:2590–2599. doi: 10.1038/npp.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci. 2010;11:100–113. doi: 10.1038/nrn2774. [DOI] [PubMed] [Google Scholar]
- 202.Gonzalez-Burgos G, Cho RY, Lewis DA. Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol Psychiatry. 2015;77:1031–1040. doi: 10.1016/j.biopsych.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lundqvist M, Rose J, Herman P, Brincat SL, Buschman TJ, Miller EK. Gamma and Beta Bursts Underlie Working Memory. Neuron. 2016;90:152–164. doi: 10.1016/j.neuron.2016.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ferrarelli F, Sarasso S, Guller Y, Riedner BA, Peterson MJ, Bellesi M, Massimini M, Postle BR, Tononi G. Reduced natural oscillatory frequency of frontal thalamocortical circuits in schizophrenia. Arch Gen Psychiatry. 2012;69:766–774. doi: 10.1001/archgenpsychiatry.2012.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Arnsten AF, Wang M. Targeting Prefrontal Cortical Systems for Drug Development: Potential Therapies for Cognitive Disorders. Annu Rev Pharmacol Toxicol. 2016;56:339–360. doi: 10.1146/annurev-pharmtox-010715-103617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kauser H, Sahu S, Kumar S, Panjwani U. Guanfacine is an effective countermeasure for hypobaric hypoxia-induced cognitive decline. Neuroscience. 2013;254:110–119. doi: 10.1016/j.neuroscience.2013.09.023. [DOI] [PubMed] [Google Scholar]
- 207.Gyoneva S, Traynelis SF. Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J Biol Chem. 2013;288:15291–15302. doi: 10.1074/jbc.M113.458901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ahlers E, Hahn E, Ta TM, Goudarzi E, Dettling M, Neuhaus AH. Smoking improves divided attention in schizophrenia. Psychopharmacology (Berl) 2014;231:3871–3877. doi: 10.1007/s00213-014-3525-2. [DOI] [PubMed] [Google Scholar]
- 209.Brunzell DH, McIntosh JM. Alpha7 nicotinic acetylcholine receptors modulate motivation to self-administer nicotine: implications for smoking and schizophrenia. Neuropsychopharmacology. 2012;37:1134–1143. doi: 10.1038/npp.2011.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hajos M, Rogers BN. Targeting alpha7 nicotinic acetylcholine receptors in the treatment of schizophrenia. Curr Pharm Des. 2010;16:538–554. doi: 10.2174/138161210790361434. [DOI] [PubMed] [Google Scholar]
- 211.Mrzljak L, Levey AI, Goldman-Rakic PS. Association of m1 and m2 muscarinic receptor proteins with asymmetric synapses in the primate cerebral cortex: morphological evidence for cholinergic modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A. 1993;90:5194–5198. doi: 10.1073/pnas.90.11.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Neale JH, Olszewski RT, Zuo D, Janczura KJ, Profaci CP, Lavin KM, Madore JC, Bzdega T. Advances in understanding the peptide neurotransmitter NAAG and appearance of a new member of the NAAG neuropeptide family. J Neurochem. 2011;118:490–498. doi: 10.1111/j.1471-4159.2011.07338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Zhou J, Neale JH, Pomper MG, Kozikowski AP. NAAG peptidase inhibitors and their potential for diagnosis and therapy. Nat Rev Drug Discovery. 2005;4:1015–1026. doi: 10.1038/nrd1903. [DOI] [PubMed] [Google Scholar]
- 214.Kinon BJ, Millen BA, Zhang L, McKinzie DL. Exploratory analysis for a targeted patient population responsive to the metabotropic glutamate 2/3 receptor agonist pomaglumetad methionil in schizophrenia. Biol Psychiatry. 2015;78:754–762. doi: 10.1016/j.biopsych.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 215.Wykes T, Huddy V, Cellard C, McGurk SR, Czobor P. A meta-analysis of cognitive remediation for schizophrenia: methodology and effect sizes. Am J Psychiatry. 2011;168:472–485. doi: 10.1176/appi.ajp.2010.10060855. [DOI] [PubMed] [Google Scholar]
- 216.Enwright JF, Sanapala S, Foglio A, Berry R, Fish KN, Lewis DA. Reduced Labeling of Parvalbumin Neurons and Perineuronal Nets in the Dorsolateral Prefrontal Cortex of Subjects with Schizophrenia. Neuropsychopharmacology. 2016;41:2206–2214. doi: 10.1038/npp.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci. 2003;23:6315–6326. doi: 10.1523/JNEUROSCI.23-15-06315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kimoto S, Bazmi HH, Lewis DA. Lower expression of glutamic acid decarboxylase 67 in the prefrontal cortex in schizophrenia: contribution of altered regulation by Zif268. Am J Psychiatry. 2014;171:969–978. doi: 10.1176/appi.ajp.2014.14010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA. Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry. 2000;57:237–245. doi: 10.1001/archpsyc.57.3.237. [DOI] [PubMed] [Google Scholar]
- 220.Chung DW, Fish KN, Lewis DA. Pathological Basis for Deficient Excitatory Drive to Cortical Parvalbumin Interneurons in Schizophrenia. Am J Psychiatry. 2016;173:1131–1139. doi: 10.1176/appi.ajp.2016.16010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Chung DW, Volk DW, Arion D, Zhang Y, Sampson AR, Lewis DA. Dysregulated ErbB4 Splicing in Schizophrenia: Selective Effects on Parvalbumin Expression. Am J Psychiatry. 2015;173:60. doi: 10.1176/appi.ajp.2015.15020150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Weickert CS, Straub RE, McClintock BW, Matsumoto M, Hashimoto R, Hyde TM, Herman MM, Weinberger DR, Kleinman JE. Human dysbindin (DTNBP1) gene expression in normal brain and in schizophrenic prefrontal cortex and midbrain. Arch Gen Psychiatry. 2004;61:544–555. doi: 10.1001/archpsyc.61.6.544. [DOI] [PubMed] [Google Scholar]
- 223.Eggan SM, Hashimoto T, Lewis DA. Reduced cortical cannabinoid 1 receptor messenger RNA and protein expression in schizophrenia. Arch Gen Psychiatry. 2008;65:772–784. doi: 10.1001/archpsyc.65.7.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Morris HM, Hashimoto T, Lewis DA. Alterations in somatostatin mRNA expression in the dorsolateral prefrontal cortex of subjects with schizophrenia or schizoaffective disorder. Cereb Cortex. 2008;18:1575–1587. doi: 10.1093/cercor/bhm186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Volk DW, Pierri JN, Fritschy JM, Auh S, Sampson AR, Lewis DA. Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb Cortex. 2002;12:1063–1070. doi: 10.1093/cercor/12.10.1063. [DOI] [PubMed] [Google Scholar]