

Abstract
Neoangiogenesis is a fundamental process which helps to meet energy requirements, tissue growth, and wound healing. Although previous studies showed that Peroxisome proliferator‐activated receptor (PPAR‐γ) regulates neoangiogenesis via upregulation of vascular endothelial growth factor (VEGF), and both VEGF and PPAR‐γ expressions were inhibited during hyperhomocysteinemic (HHcy), whether these two processes could trigger pathological effects in skeletal muscle via compromising neoangiogenesis has not been studied yet. Unfortunately, there are no treatment options available to date for ameliorating HHcy‐mediated neoangiogenic defects. Hydrogen sulfide (H2S) is a novel gasotransmitter that can induce PPAR‐γ levels. However, patients with cystathionine‐β‐synthase (CBS) mutation(s) cannot produce a sufficient amount of H2S. We hypothesized that exogenous supplementation of H2S might improve HHcy‐mediated poor neoangiogenesis via the PPAR‐γ/VEGF axis. To examine this, we created a hind limb femoral artery ligation (FAL) in CBS +/− mouse model and treated them with GYY4137 (a long‐acting H2S donor compound) for 21 days. To evaluate neoangiogenesis, we used barium sulfate angiography and laser Doppler blood flow measurements in the ischemic hind limbs of experimental mice post‐FAL to assess blood flow. Proteins and mRNAs levels were studied by Western blots and qPCR analyses. HIF1‐α, VEGF, PPAR‐γ and p‐eNOS expressions were attenuated in skeletal muscle of CBS +/− mice after 21 days of FAL in comparison to wild‐type (WT) mice, that were improved via GYY4137 treatment. We also found that the collateral vessel density and blood flow were significantly reduced in post‐FAL CBS +/− mice compared to WT mice and these effects were ameliorated by GYY4137. Moreover, we found that plasma nitrite levels were decreased in post‐FAL CBS +/− mice compared to WT mice, which were mitigated by GYY4137 supplementation. These results suggest that HHcy can inhibit neoangiogenesis via antagonizing the angiogenic signal pathways encompassing PPAR‐γ/VEGF axis and that GYY4137 could serve as a potential therapeutic to alleviate the harmful metabolic effects of HHcy conditions.
Keywords: Angiogenesis, hydrogen sulfide, stress response
Introduction
Homocysteine (Hcy) has been studied extensively for over 30 years for its unique involvement in an increasing number of human diseases (Hankey and Eikelboom 1999; Narayanan et al. 2014; Stipanuk and Ueki 2011). The Hcy level is controlled by two major processes: around 50% of Hcy enters the transsulfuration pathway to produce cysteine, and the other half is remethylated back to methionine (Met) via the folate 1‐carbon cycle (Cascella et al. 2015; Veeranki and Tyagi 2013). Hcy is generally present in blood in four different forms: approximately 1% as free thiol, 70–80% as a disulfide‐bound to plasma proteins and the remaining 20–30% as a homodimer or heterodimer with other thiols (Hankey and Eikelboom 1999). Normal total plasma Hcy concentration in our body ranges from 5 to 10 μmol/L; however, in a diseased condition, such as in hyperhomocysteinemia (HHcy), plasma total Hcy levels are increased (>15 μmol/L) (Lehotsky et al. 2016). HHcy can be classified as moderate (15–30 μmol/L), intermediate (30–100 μmol/L), and severe (>100 μmol/L) (Lehotsky et al. 2016). Notable, there are four ways people can develop HHcy: (1) consumption of a Met‐rich protein diet; (2) vitamin B12/folate deficiency; (3) presence of heterozygous/homozygous status for cystathionine‐β synthase (CBS+/−/CBS−/−); and (4) obstruction of renal clearance (Sen et al. 2010). Apart from these factors, genetic variants in Hcy metabolism enzymes such as 677C>T and 1298A>C in the MTHFR gene can also lead to HHcy (Brustolin et al. 2010; Iqbal et al. 2009; Kharb et al. 2016; Verhoef et al. 2005). There are also several other drivers like age, sex, physical activity, alcohol intake, certain medications, and different disease conditions (such as type 2‐diabetes) that can also modulate the functioning Met cycle leading to an increased total Hcy concentration in the blood (Diakoumopoulou et al. 2005). Hyperhomocysteinemia (HHcy) has been associated with severe skeletal muscle dysfunction (Brustolin et al. 2010; Kanwar et al. 1976; Kolling et al. 2013; Majumder et al. 2017; Miller et al. 2000; Valentino et al. 2010; Veeranki et al. 2015; Voskoboeva et al. 2018; Zoccolella et al. 2008), but the precise mechanism(s) is still unknown. Previous studies found that HHcy causes endothelial cell (EC) injury (Wang et al. 1997), inhibition of EC proliferation (Zou et al. 2011), reduction of bioavailability of vasoregulatory mediators (nitric oxide and endothelin) (Upchurch et al. 1997), and induction of oxidative/ER‐stress (Singh et al. 2018; Tyagi et al. 2005; Werstuck et al. 2001). How HHcy reduces neovascularization in the skeletal muscle is not precisely known.
The growth of new blood vessels from the preexisting vascular network is known as neoangiogenesis. When oxygen supply is low in a tissue or organ, it activates hypoxia‐inducible factor 1 (HIF1α), which sends a signal to nearest blood vessel, activating endothelial nitric oxide synthase (eNOS) and producing NO needed for vasodilation. Vascular endothelial growth factor (VEGF) increases permeability and separates pericytes leading to the degradation of the basement membrane, thereby activating metalloproteases such as MMP‐2 and 9 (Carmeliet and Jain 2011). These changes lead to EC proliferation and a concomitant migration in order to form new blood vessels (Duran et al. 2017). In recent years, it has been shown that Peroxisome proliferator‐activated receptor (PPAR‐γ) might be involved in neoangiogenesis via growth factors and cytokines that in turn stimulate migration, proliferation, and survival of these ECs (Biscetti et al. 2008).
PPAR‐γ belongs to the nuclear hormone receptor superfamily, and when specific ligands bind to the ligand‐binding domain of PPAR‐γ, a conformational change releases the bound corepressors (Chawla et al. 1994; Tontonoz et al. 1995). This allows coactivators like PGC‐1α and other coactivators to be recruited to the PPAR‐γ responsive genes’ promoters, thereby promoting the PPAR‐γ‐mediated transcription (Costa et al. 2010; Laha et al. 2018; Murphy and Holder 2000). HHcy reduces PPAR‐γ expression in ECs (Mishra et al. 2010). Studies have shown that PPAR‐γ could regulate neoangiogenesis via upregulating VEGF (Biscetti et al. 2008) and that can further activate eNOS (Kroll and Waltenberger 1998). Although, previous works showed that HHcy impaired neoangiogenic growth in muscle via reduction of HIF1α and VEGF levels, whether PPAR‐γ plays any role in this process had not been studied (Veeranki et al. 2014).
Hydrogen sulfide (H2S) is increasingly being recognized as an important signaling molecule in cardiovascular and nervous systems via its ability to neutralize a variety of reactive oxygen species (ROS) (Kimura et al. 2010; Mironov et al. 2017; Yang et al. 2015), as well as via increased cellular glutathione levels through activation of gamma‐glutamylcysteine synthetase, and reduction of the disulfide bonds (Calvert et al. 2010; Elsey et al. 2010; Fiorucci et al. 2006; Gadalla and Snyder 2010; George et al. 2018; Kimura 2010; Predmore and Lefer 2010; Szabo 2007; Wang 2003). Cystathionine γ‐lyase (CSE) and CBS are the main H2S‐generating enzymes, producing H2S from Hcy in the transsulfuration pathway. Patients with CBS deficiency tend to produce a lesser amount of H2S (Beard and Bearden 2011; Kozich et al. 2016); suggesting that these patients are likely more prone to oxidative stress‐mediated damages due to excessive production of Hcy (Szabo 2007). A study revealed that endogenous H2S could induce mRNA and protein expression of PPAR‐γ (Cai et al. 2016), indicating that exogenous H2S supplementation could be employed as a beneficial strategy to improve neoangiogenesis defect in HHcy patients. Hence, the purpose of our study was to answer the following questions: (i) Does HHcy inhibit neoangiogenesis via downregulation of angiogenic signals like HIF1α and VEGF in the postfemoral artery ligation (FAL) hind limb of CBS+/− mice? (ii) Does HHcy inhibit PPAR‐γ expression which can further downregulate VEGF/eNOS signaling in the post‐FAL hind limb of CBS+/− mice? And finally, (iii) does GYY4137 treatment improve neoangiogenesis via PPAR‐γ/VEGF axis after 21 days of FAL in the hind limb of experimental CBS+/− mice?
CBS is one of the key enzymes in the transsulfuration pathway, and heterozygous CBS deficiency (CBS+/−) has proved to be a useful model for analyzing the effects of mild to a severe endogenous elevation in the levels of Hcy (Familtseva et al. 2014; Nandi and Mishra 2017; Narayanan et al. 2013; Tyagi et al. 2011, 2012; Watanabe et al. 1995; Winchester et al. 2018; Yang et al. 2018). Hence, in this study, we used CBS+/− mouse model to dissect the effect(s) of HHcy on neoangiogenesis in the skeletal muscle and evaluate whether exogenous administration of GYY4137 (an H2S donor) could improve this effect(s). Our results indicate that H2S could be developed as a potential therapeutic agent to treat the neoangiogenic defects in skeletal muscle wherein HHcy is linked with a barrage of metabolic dysfunctions.
Materials and Methods
Animal maintenance, genotyping, and diet protocol
Male WT (C57BL/6J) and CBS+/− (B6.129P2‐Cbstm1Unc/J 002853) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All animals were ∼8–10 weeks‐old and maintained in 12:12 h light–dark cycle with regular mouse chow diet in the animal facility of the University of Louisville. All animal protocols and care were carried out according to the guidelines of National Institute of Health (NIH Pub. No. 86–23, revised 1985) and were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Louisville, KY.
After purchasing mice were cross‐bred, yielding around 10% CBS−/−, 60% CBS+/−, and 25% CBS+/+. For genotyping, tail samples were collected, and DNA was isolated using DNeasy Blood & Tissue Kits (Qiagen, Germantown, MD). Genotypic analysis was performed using PCR by targeted disruption of the CBS gene at loci (representative images from each group of post‐FAL mice are shown in Fig. 1A and genotyping in Fig. 1B). CBS+/− heterozygote mice produced two bands (450 and 308 bp), while wild‐type (CBS+/+) mice represented only one band (308 bp).
Figure 1.
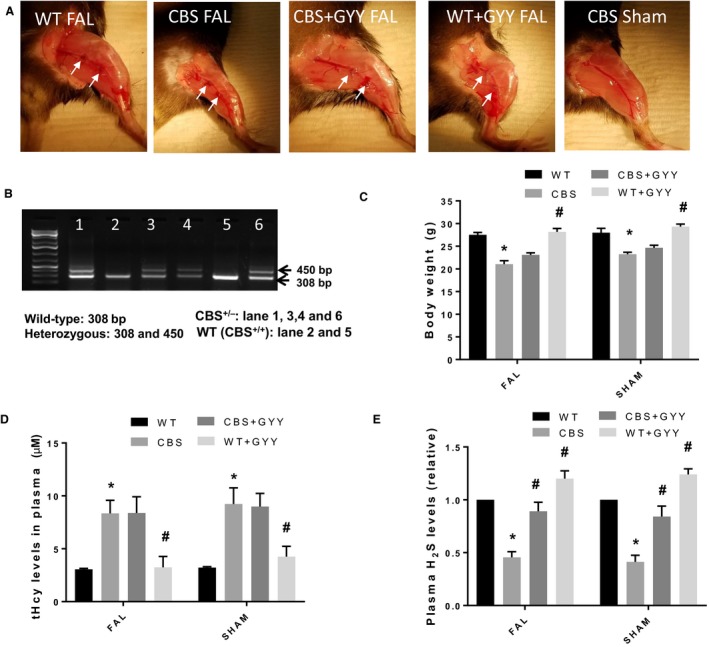
Phenotypic and genotypic correlations between cystathionine‐β‐synthase (CBS +/−) and wild‐type (WT) mice groups. (A) Hind limb images after 21 days of GYY4137 treatments. (B) Genotyping for CBS +/− and WT mice. (C) Body weight measurements of experimental mice. (D) tHcy measurements from the plasma of experimental mice. (E) H2S measurements from the plasma of experimental mice. Data are shown as Mean ± SEM and mice number (n) = 4, statistical difference *P < 0.05 versus WT and # P < 0.05 versus CBS. (tHcy = total homocysteine, FAL = femoral artery ligation).
Animals were divided into four experimental groups: (1) Wild‐type C57BJ/L6 mice (WT), (2) CBS+/−heterozygous mice (CBS), (3) GYY4137‐supplemented CBS+/− (CBS+GYY), and (4) GYY4137‐supplemented wild‐type mice (WT+GYY). A dose of 0.25 mg intraperitoneal injection was administered for mouse of GYY4137/kg body weight every day for a total 21 days after FAL surgery and while the WT mice were given 0.9% normal saline (vehicle control) (John et al. 2017).
Femoral artery ligation
To create hypoxic condition, FAL (unilateral) was performed under intraperitoneal pentobarbital sodium (50 mg/kg) anesthesia as previously described (Beard and Bearden 2011; Kozich et al. 2016). Briefly, after separation of the femoral artery from the vein and nerve, it was ligated using 6‐0 silk suture at proximal and distal places (keeping same distance in all animals). We used separate mice groups [WT, CBS, CBS+GYY, and WT+GYY] as sham control where we passed the suture underneath the femoral artery but was not ligated. The skin was sutured using 6‐0 silk thread. After the skin closure, betadine was applied. After recovery, laser Doppler blood perfusion was carried out to confirm the induction of ischemia.
Laser Doppler tissue perfusion imaging and flowmetry
MoorLDI (Moor Instruments) was used to measure tissue perfusion intensity and blood flow rates as described (Bhargava et al. 2014).
Barium angiograms
To determine neoangiogenesis barium sulfate angiography was performed in mice as described (Givvimani et al. 2011). In brief, after pentobarbital anesthesia mice were infused with barium sulfate (0.1 g/mL) in 50 mmol/L Tris‐buffer (pH 5.0) at a constant flow (∼1 mL/min) and pressure with a syringe pump through the common carotid artery. Heparin (20 U/mL) was used along with barium sulfate to visualize the nascent neoangiogenesis. Angiograms were captured using the Carestream whole animal X‐ray imaging system (Carestream Molecular Imaging, Woodbridge, CT) as a previously described method (Machens et al. 2006) and the vessel density was quantified using VesSeg tool (Institute for Signal Processing, University of Luebeck, Lübeck, Germany).
Reagents and antibodies
All reagents and chemicals were ordered from Sigma–Aldrich or available elsewhere but with highest grade.
The antibodies for HIF1α (ab51608), VEGF (ab51745), and eNOS (ab66127) were ordered from Abcam (Cambridge, USA). Whereas PPAR‐γ (sc‐7273), p‐eNOS Ser 1177 (sc‐12972), rabbit anti‐mouse (sc‐358914), mouse anti‐rabbit (sc‐2357), and mouse anti‐goat (sc‐2354) were from Santa Cruz Biotechnology (Dallas, TX). The antibody for GAPDH (MAB374) was from EMD Millipore (Burlington, MA), and used for Western blots analyses as per the manufacturers’ protocols.
Western Blotting
Protein expressions were assessed by Western blots as described (John et al. 2017). Briefly, at the time of sacrifice, gastrocnemius muscle from the ischemic leg of each mouse was quickly removed, snap‐frozen, and stored at –80°C until further use. Protein from samples was extracted by homogenizing in the ice‐cold RIPA buffer (Boston BioProducts, Worcester, MA) containing 1 mmol/L Phenylmethylsulfonyl fluoride (Sigma, Saint Louis, MO), and 1% protease inhibitors cocktail (Sigma) and sonicated employing the Sonifier 450 (Branson Ultrasonics, Danbury, CT). The homogenates were centrifuged 17,400g for 20 min at 4°C, and the supernatants were quickly stored at –80°C until further use. The protein contents were estimated by the Bradford assay. Equal amounts of proteins (50 μg) were resolved on SDS‐PAGE (8%, 10%, 12%) and then transferred to polyvinylidene difluoride (PVDF) membranes. The respective blots were incubated with primary and secondary antibodies before visualizing them using the ECL Luminata Forte (Millipore, Temecula, CA) in a Bio‐Rad ChemiDoc system. The intensities of the bands were normalized to the housekeeping GAPDH for all the proteins examined. The quantification was performed using Image Lab™ Software (Bio‐Rad, Hercules, CA).
Reverse transcription and real‐time quantitative PCR
Total RNA was extracted from muscle samples using a Trizol method as described (Rio et al. 2010). Then, RNA quality was determined by NanoDrop ND‐1000, and RNA with high purity (260/280~2.00 and 260/230~2.00) was used for q‐PCR analysis. Reverse transcription was performed according to manufacturer's protocol using high‐capacity cDNA RT kit from Applied Biosystems (Foster City, USA) for the primer sequences listed in Table 1. For RT‐qPCR, SYBR green‐based kit was used to measure the relative expression of each mRNA specific primers. Briefly, three steps cycling protocol was performed using 20 ng of cDNA template in a 20 μL reaction volume under the following conditions: denaturation at 95°C for 15 min followed by 40 cycles of 94°C for 15 s, 55°C for 30 s, and 70°C for 34 s in which fluorescence was acquired and detected by Roche LightCycler® 96 Real‐Time PCR System (Roche Diagnostics, IN). Following RT‐qPCR, analysis of melt curve was performed to validate the specific generation of the expected PCR product. GAPDH was used as an endogenous control (Quanta Biosciences, Beverly, MA).
Table 1.
List of primers used for RT‐qPCR experiments
| Genes | Forward primers | Reverse primers |
|---|---|---|
| HIF1α | 5′‐TCAAGTCAGCAACGTGGAAG‐3′ | 5′‐TATCGAGGCTGTGTCGACTG‐3′ |
| VEGF | 5′‐CAGGCTGCTGTAACGATGAA‐3′ | 5′‐CAATTTGGCTCCTCCTACCA‐3′ |
| PPAR‐γ | 5′‐TTTTCAAGGGTGCCAGTTTC‐3′ | 5′‐AATCCTTGGCCCTCTGAGAT‐3′ |
| NOS3 | 5′‐GACCCTCACCGCTACAACAT‐3′ | 5′‐TCTGGCCTTCTGCTCATTTT‐3′ |
Total plasma Homocysteine, H2S, and nitrite measurement
Blood samples were collected in tubes containing a 1/10 volume of 3.8% sodium citrate from each mouse by cardiac puncture after euthanasia. Then, plasma was isolated by centrifugation at 2500g for 15 min at 4°C. Total plasma Hcy concentrations were measured in samples using the homocysteine assay kit (Crystal Chem, USA) as per manufacturer's instructions.
Plasma H2S was measured as a previously described method from our laboratory (Kundu et al. 2015).
Nitrite from the plasma was measured by a Griess reagent [0.1% N‐(1‐naphthyl) ethylenediamine dihydrochloride, 1% sulfanilamide, and 2.5% phosphoric acid] and using sodium nitrite (0.01–100 μg) as a standard as described previously (Kalani et al. 2014).
Statistics
All values are expressed as mean ± SEM. The interaction between groups was determined by one‐way or two‐way ANOVA, including a Tukey's post hoc analysis when significant interactions were observed. The threshold for significance was set at P < 0.05, and total number of mice (n) = 4–5 was subjected to experimentation from each group. For statistical analyses, GraphPad Prism (Ver 7, GraphPad Software) was used.
Results
The phenotypic feature and genotype of WT and CBS+/− mice are depicted in Figure 1A and B, respectively. In this study, we noticed that CBS mice had significantly lower body weights in comparison to WT mice; however, we did not see any difference in body weights between CBS versus WT after GYY4137 treatment for 21 days (Fig. 1C). We observed CBS mice had significantly higher levels of plasma tHcy compared to WT mice, and GYY4137‐supplemented CBS mice also had similarly higher plasma tHcy levels as that of the CBS mice (Fig. 1D). After 21 days of GYY4137 treatment, we wanted to examine the plasma H2S concentrations in the experimental mice. Results showed that plasma H2S levels were significantly lower in the untreated CBS mice compared to that of the untreated WT mice as expected; however, after administration of GYY4137 for 21 days we did notice that plasma H2S levels were significantly elevated in both the CBS and WT mice (Fig. 1E).
As a marker of a hypoxia induction in post‐FAL hind limb, we measured the HIF1α levels by Western blotting. We found that HIF1α expression was higher in FAL mice compared to sham mice (Fig. 2A–D). However, when we examined the HIF1α levels between each group of FAL mice, we noticed that HIF1α levels were downregulated in post‐FAL CBS mice in comparison to WT mice, and GYY4137 treatment was found to mitigate this effect (Fig. 2A). To confirm mRNA expression of HIF1α, we did qPCR analysis, and it did not show any significant decrease in mRNA levels of HIF1α in post‐FAL CBS mice compared to post‐FAL WT (P = 0.4824), and GYY4137 treatment could not improve the mRNA levels in post‐FAL CBS mice (P = 0.9781) (Fig. 2B). We found that the protein expressions of VEGF and PPAR‐γ were reduced in post‐FAL CBS mice as compared to post‐FAL WT mice, whereas this effect was improved after GYY4137 administration (Fig. 2A). Additionally, in the qPCR analysis, we found mRNA expression of VEGF was significantly reduced in the post‐FAL CBS mice compared to post‐FAL WT mice (P = 0.0365); however, this effect was not improved upon GYY4137 administration (P = 0.2139). We did not notice any significant change in mRNA expression of PPAR‐γ among the four experimental groups (Fig. 2B). Besides, we did not find any difference in proteins and mRNA levels for HIF1α and VEGF among individual groups of sham mice as shown in Figure 2C–D. However, we did notice that the expression of PPAR‐γ was reduced in sham CBS mice compared to sham WT mice and that GYY4137 supplementation could not mitigate this effect. The observed reduction of PPAR‐γ mRNA level in sham CBS mice, compared to sham WT mice, was not statistically significant (P = 0.7423). Similarly, we did not observe, any significant improvement in mRNA expression for PPAR‐γ in sham CBS mice after GYY4237 treatment (P = 0.3549) (Fig. 2D).
Figure 2.
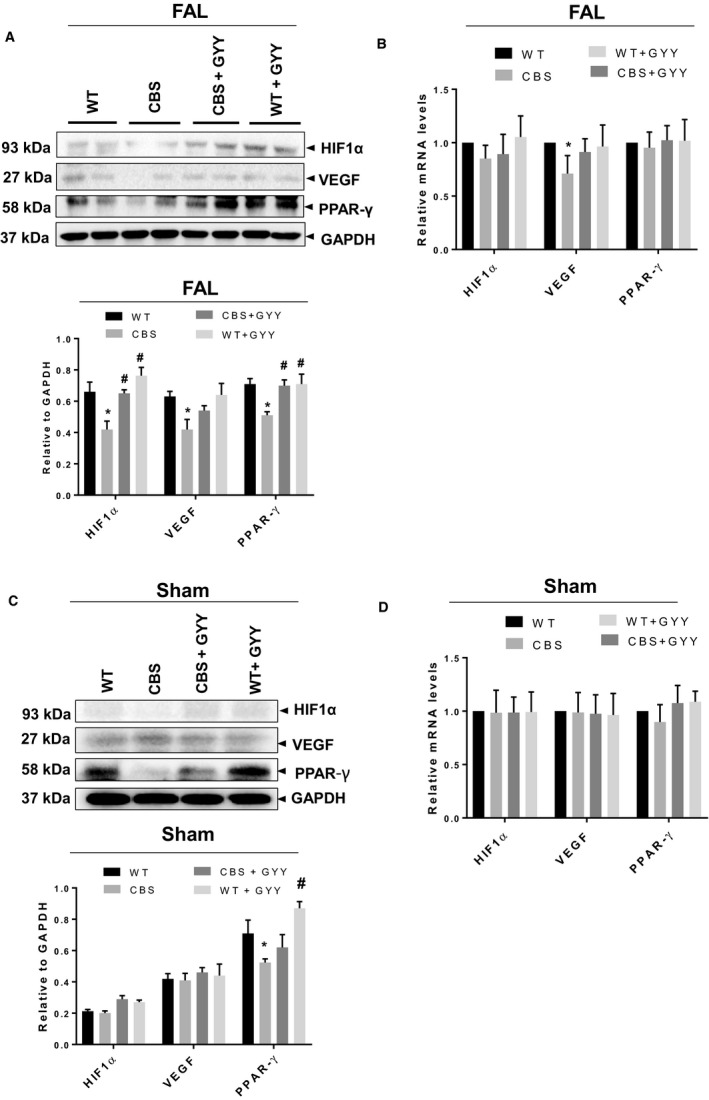
(A and B) Effect of GYY4137 on the improvement of neoangiogenic signals in skeletal muscle of post‐FAL cystathionine‐β‐synthase (CBS +/−) mice. (A) Western blots analysis showing protein expressions: HIF1‐α, VEGF, and PPAR‐γ in the top panel and densitometric analysis of Western blots images are shown in the bottom panel. (B) mRNA expression for HIF1‐α, VEGF, and PPAR‐γ (log transformed). Data are shown as Mean ± SEM and mice number (n) = 4, statistical difference *P < 0.05 versus wild‐type (WT) and # P < 0.05 versus CBS (FAL = femoral artery ligation). (C and D) Effect of GYY4137 on angiogenic signals in skeletal muscle in sham CBS +/− and WT mice. (C) Western blots analysis showing protein expressions: HIF1‐α, VEGF, and PPAR‐γ in the top panel and densitometric analysis of Western blots images are shown in the bottom panel. (D) mRNA expression for HIF1‐α, VEGF, and PPAR‐γ (log transformed). Data are shown as Mean ± SEM and mice number (n) = 4, statistical difference *P < 0.05 versus WT and # P < 0.05 versus CBS.
Finally, we measured the vessel density employing barium sulfate angiography after 21 days of FAL surgery. We found that total collateral vessels’ number was significantly less in the post‐FAL CBS mice in comparison to post‐FAL WT mice, and this effect was further significantly improved upon GYY4137 treatment (Fig. 3A–B). Besides, we found blood flow in the hind limb after 21 days of FAL was reduced considerably in CBS mice compared to WT mice. It was improved by GYY4137 treatment as could be seen in Figure 3C–D. We did not notice any difference in the mRNA expression levels of NOS3; however, we observed that the p‐eNOS levels were reduced in the post‐FAL CBS mice as compared to post‐FAL WT mice, and interestingly, this effect was improved via GYY4137 treatment (Fig. 3E). The changes in the plasma nitrite levels in experimental mice were also monitored as the marker of nitric oxide (NO) production. Although not statistically significant, our findings revealed a reduction of plasma nitrite levels in post‐FAL CBS mice as compared to post‐FAL WT mice (P = 0.1050) with the GYY4137 administration improving this effect (P = 0.2923) (Fig. 3F). Based upon above findings, we have proposed a model that we firmly believe in application of H2S as a potential therapeutic intervention in treating neoangiogenic defects in skeletal muscle due HHcy (Fig. 3F).
Figure 3.
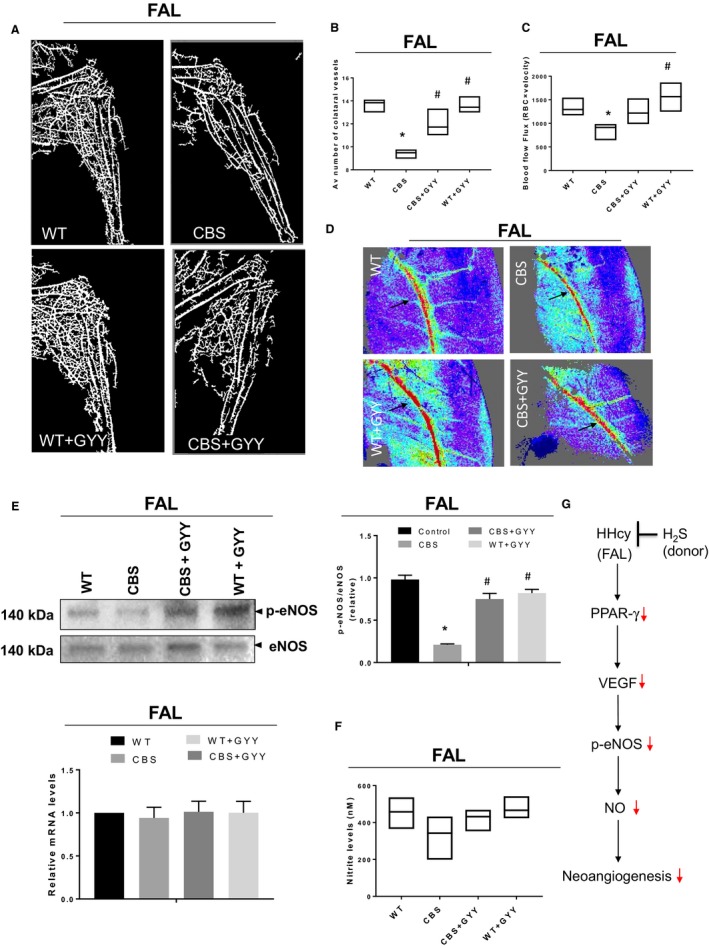
(A–D) GYY4137 supplementation improves neoangiogenic profile in the hind limb muscle of mice after 21 days of FAL in cystathionine‐β‐synthase (CBS +/−) mice. (A) Barium angiogram images showing vascular density in the hind limb of skeletal muscle. (B) Quantitation of collateral vessel numbers in hind limb post‐FAL. (C) Blood flow rate measurements in hind limb post‐FAL. (D) Laser Doppler perfusion imaging showing the intensity of limb perfusion on 21 days after FAL. Data are shown as Mean ± SEM and mice number (n) = 4, statistical difference *P < 0.05 versus wild‐type (WT) and # P < 0.05 versus CBS (FAL = femoral artery ligation). (E–G) GYY4137 supplementation activates eNOS‐phosphorylation and plasma nitrite levels in CBS +/− mice. (E) Western blots analysis of protein expressions: p‐eNOS and eNOS in the top panel and densitometric analysis of p‐eNOS/eNOS ratio from Western blots images are shown in the right panel, and mRNA expression of NOS3 is shown in bottom panel. (F) Nitrite levels in plasma of post‐FAL mice measured by Griess assay. (G) Proposed model of this study is outlined. Data are shown as Mean ± SEM and mice number (n) = 4, statistical difference *P < 0.05 versus WT and # P < 0.05 versus CBS and # P < 0.05 versus CBS (FAL = femoral artery ligation).
Discussion
Previous reports suggested several mechanisms for detrimental outcomes during the HHcy condition in different tissue types, including oxidative stress (Racek et al. 2005; Tyagi et al. 2005), protein homocysteinylation (Jakubowski 1999; Jakubowski et al. 2000), hypo/hypermethylation (Jiang et al. 2007; Narayanan et al. 2014; Pushpakumar et al. 2015; Yi et al. 2000), and endoplasmic reticulum stress (Perna et al. 2003). However, it is important to note that most of these observations are from in vitro studies using a supra‐physiological concentration of Hcy (~1 mmol/L) unlike as seen in HHcy patients. There are many in vivo models to study neoangiogenesis, either by passing a flexible wire or by applying a laser or an electrical current; however, none of these are relevant to clinical settings (Carmeliet et al. 1998; Lindner et al. 1993; Rosen et al. 2001). In this study, we used the FAL model by employing a genetically engineered mouse model (CBS+/−) mimicking HHcy conditions as seen in HHcy patients for the purpose of studying the postischemic neoangiogenesis phenomenon during HHcy. Our results add to the growing body of evidence that HHcy is associated with defective neoangiogenesis as shown in other in vivo experiments (Tan et al. 2006; Tawfik et al. 2013). Earlier, studies also showed that high Hcy (HHcy) has a profound inhibitory effect on EC proliferation and their migration (Cai et al. 2007; Li et al. 2006; Ozsvari et al. 2010; Papapetropoulos et al. 2009; Tan et al. 2006).
Neoangiogenesis is a natural process during chronic regional ischemia, which requires EC proliferation, migration, differentiation, and survival to form new blood vessels in order to compensate the hypoxic environment (Norton and Popel 2016). VEGF is a prototypical angiogenic cytokine that plays a vital role in this process and has been widely studied (Carmeliet 2000; Isner and Asahara 1999; Liu et al. 2000). A previous report involving hind limb ischemia in CBS+/− mice showed no difference in VEGF levels after seven days of ischemia (Bosch‐Marce et al. 2005). However, they did notice a significant reduction in capillary density in the CBS mice compared to WT mice. Interestingly, our results demonstrate a significant decrease in HIF1α and VEGF expression after 21 days of FAL, suggesting that lower expressions of postischemic HIF‐1α may be responsible for delayed induction of VEGF in CBS mice compared to that of WT mice. The present study also explored mechanistic role of HHcy for the reduction of VEGF through PPAR‐γ‐dependent pathway. A study demonstrated that NaHS (H2S donor) treatment significantly improved capillary density and angiographic scores resulting in enhancement of blood flow in the ischemic hind limb (Wang et al. 2010). Similarly, we also noticed that exogenous administration of GYY4137 (H2S donor) could successfully mitigate the HHcy‐mediated neoangiogenic defects in skeletal muscle of CBS+/− mice (Fig. 3).
H2S has been studied extensively for its salubrious effects in the cardiovascular system demonstrating profound vasodilatation, vascular protection, homeostatic regulation of blood pressure, and many others (Calvert et al. 2010; Elsey et al. 2010; Gadalla and Snyder 2010; Predmore and Lefer 2010; Szabo 2007). A previous study using chicken chorioallantoic membrane model revealed that H2S increased the length and complexity of the vascular network (Papapetropoulos et al. 2009). Similarly, in this study, we also noticed that exogenous supplementation of GYY4137 could improve collateral vessels’ density after 21 days of FAL in the CBS mice. In agreement with our study, Moore and colleagues were able to show that intraperitoneal administration of NaHS (an H2S donor) induced neovascularization in an in vivo mouse model using a Matrigel plug assay (Cai et al. 2007). A previous report showed that genetic deletion/silencing of CSE (another H2S‐producing enzyme) in the endothelium, reduced migration and sprouting of ECs in vitro, where in VEGF played a critical mediator (Papapetropoulos et al. 2009). In this present study, we observed that PPAR‐γ and VEGF expressions were significantly downregulated in CBS mice compared to WT mice. We also demonstrated that these effects were mitigated via GYY4137 administration. This suggests that most likely VEGF is regulated via the PPAR‐γ‐dependent pathway further corroborating Biscetti et al. (2008) findings wherein they clearly showed that activation of PPAR‐γ led to endothelial tube formation and induction of VEGF in ECs. Similarly, other investigators revealed that inhibiting PDE activity by H2S induces PPAR‐γ protein and mRNA expressions (Cai et al. 2016).
NO is also an endogenous gasotransmitter that, like H2S, is involved in vasorelaxation and stimulation of angiogenesis (Mustafa et al. 2009; Szabo 2010). HHcy was also found to quench NO (a vasodilator) by the formation of peroxynitrite anion (ONOO−) and uncoupling of eNOS, further reducing the bioavailability of NO (Dimmeler et al. 1999; Morbidelli et al. 2003; Topal et al. 2004). Similarly, we noticed nitrite levels and phosphorylation of eNOS were found to be reduced in CBS mice in comparison to WT mice. eNOS is known to produce NO during neoangiogenesis via VEGF (Kroll and Waltenberger 1998), and thus it appears that impaired angiogenesis in HHcy could be due to the reduction of NO availability. In this work, we demonstrated that nitrite levels and eNOS activation were reduced in the CBS mice compared to WT mice, and their levels could be mitigated via GYY4137 treatment. These findings are also highly consistent with previous reports where H2S was shown to stimulate Akt in ECs leading to the induction of eNOS through phosphorylation of Ser1177 (activation site) and a parallel dephosphorylation Thr495 (inhibitory site) (Coletta et al. 2012; Osipov et al. 2009).
In conclusion, our work embodies the proangiogenic role of H2S molecule. Pertinent findings from this study have been elaborated in a flowchart/model (Fig. 3G) highlighting the plausible intracellular signaling pathway of how H2S could mitigate the neoangiogenic defects during HHcy. We opine that additional pathways might be at work during neoangiogenesis (Majumder et al. 2018); however, further investigation needs to be undertaken involving similar but not identical scenarios wherein muscle dysfunction is the outcome of metabolic derangement. In brief, H2S does hold potential ramifications toward developing it as a clinically relevant therapeutic option for chronic conditions that are implicated in a host of inflammatory and cellular stress injury including the apparent defect in the neoangiogenesis (Majumder et al. 2018).
Conflicts of Interest
No conflicts of interest, financial or otherwise, are declared by the authors.
Majumder A., Singh M., George A. K., Behera J., Tyagi N., Tyagi S. C.. Hydrogen sulfide improves postischemic neoangiogenesis in the hind limb of cystathionine‐β‐synthase mutant mice via PPAR‐γ/VEGF axis. Physiol Rep, 6 (17), 2018, e13858, https://doi.org/10.14814/phy2.13858
Funding Information
The work was supported by grants from the National Institute of Health (Heart, Lung, and Blood Institute; No. HL‐74815, HL‐107640) and the Institute of Neurological Disorders and Stroke (No. NS‐084823).
Contributor Information
Avisek Majumder, Email: avisek.majumder@louisville.edu.
Mahavir Singh, Email: mahavir.singh@louisville.edu.
References
- Beard Jr, R. S. , and Bearden S. E.. 2011. Vascular complications of cystathionine beta‐synthase deficiency: future directions for homocysteine‐to‐hydrogen sulfide research. Am. J. Physiol. Heart Circ. Physiol. 300:H13–H26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava, S. , Pushpakumar S., Metreveli N., Givvimani S., and Tyagi S. C.. 2014. MMP‐9 gene ablation mitigates hyperhomocystenemia‐induced cognition and hearing dysfunction. Mol. Biol. Rep. 41:4889–4898. [DOI] [PubMed] [Google Scholar]
- Biscetti, F. , Gaetani E., Flex A., Aprahamian T., Hopkins T., Straface G., et al. 2008. Selective activation of peroxisome proliferator‐activated receptor (PPAR)alpha and PPAR gamma induces neoangiogenesis through a vascular endothelial growth factor‐dependent mechanism. Diabetes 57:1394–1404. [DOI] [PubMed] [Google Scholar]
- Bosch‐Marce, M. , Pola R., Wecker A. B., Silver M., Weber A., Luedemann C., et al. 2005. Hyperhomocyst(e)inemia impairs angiogenesis in a murine model of limb ischemia. Vasc. Med. 10:15–22. [DOI] [PubMed] [Google Scholar]
- Brustolin, S. , Giugliani R., and Felix T. M.. 2010. Genetics of homocysteine metabolism and associated disorders. Braz. J. Med. Biol. Res. 43:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, W. J. , Wang M. J., Moore P. K., Jin H. M., Yao T., and Zhu Y. C.. 2007. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 76:29–40. [DOI] [PubMed] [Google Scholar]
- Cai, J. , Shi X., Wang H., Fan J., Feng Y., Lin X., et al. 2016. Cystathionine gamma lyase‐hydrogen sulfide increases peroxisome proliferator‐activated receptor gamma activity by sulfhydration at C139 site thereby promoting glucose uptake and lipid storage in adipocytes. Biochem. Biophys. Acta. 1861:419–429. [DOI] [PubMed] [Google Scholar]
- Calvert, J. W. , Coetzee W. A., and Lefer D. J.. 2010. Novel insights into hydrogen sulfide–mediated cytoprotection. Antioxid. Redox Signal. 12:1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet, P. 2000. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 6:389–395. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P. , and Jain R. K.. 2011. Molecular mechanisms and clinical applications of angiogenesis. Nature 473:298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet, P. , Moons L., and Collen D.. 1998. Mouse models of angiogenesis, arterial stenosis, atherosclerosis and hemostasis. Cardiovasc. Res. 39:8–33. [DOI] [PubMed] [Google Scholar]
- Cascella, M. , Arcamone M., Morelli E., Viscardi D., Russo V., De Franciscis S., et al. 2015. Erratum to: multidisciplinary approach and anesthetic management of a surgical cancer patient with methylene tetrahydrofolate reductase deficiency: a case report and review of the literature. J. Med. Case Rep. 9:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla, A. , Schwarz E. J., Dimaculangan D. D., and Lazar M. A.. 1994. Peroxisome proliferator‐activated receptor (PPAR) gamma: adipose‐predominant expression and induction early in adipocyte differentiation. Endocrinology 135:798–800. [DOI] [PubMed] [Google Scholar]
- Coletta, C. , Papapetropoulos A., Erdelyi K., Olah G., Modis K., Panopoulos P., et al. 2012. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium‐dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 109:9161–9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa, V. , Gallo M. A., Letizia F., Aprile M., Casamassimi A., and Ciccodicola A.. 2010. PPARG: gene expression regulation and next‐generation sequencing for unsolved issues. PPAR Res, pii: 409168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diakoumopoulou, E. , Tentolouris N., Kirlaki E., Perrea D., Kitsou E., Psallas M., et al. 2005. Plasma homocysteine levels in patients with type 2 diabetes in a mediterranean population: relation with nutritional and other factors. Nutr. Metab. Cardiovasc. Dis. 15:109–117. [DOI] [PubMed] [Google Scholar]
- Dimmeler, S. , Fleming I., Fisslthaler B., Hermann C., Busse R., and Zeiher A. M.. 1999. Activation of nitric oxide synthase in endothelial cells by Akt‐dependent phosphorylation. Nature 399:601–605. [DOI] [PubMed] [Google Scholar]
- Duran, C. L. , Howell D. W., Dave J. M., Smith R. L., Torrie M. E., Essner J. J., et al. 2017. Molecular regulation of sprouting angiogenesis. Compr. Physiol. 8:153–235. [DOI] [PubMed] [Google Scholar]
- Elsey, D. J. , Fowkes R. C., and Baxter G. F.. 2010. Regulation of cardiovascular cell function by hydrogen sulfide (H(2)S). Cell Biochem. Funct. 28:95–106. [DOI] [PubMed] [Google Scholar]
- Familtseva, A. , Kalani A., Chaturvedi P., Tyagi N., Metreveli N., and Tyagi S. C.. 2014. Mitochondrial mitophagy in mesenteric artery remodeling in hyperhomocysteinemia. Physiol. Rep. 2:e00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorucci, S. , Distrutti E., Cirino G., and Wallace J. L.. 2006. The emerging roles of hydrogen sulfide in the gastrointestinal tract and liver. Gastroenterology 131:259–271. [DOI] [PubMed] [Google Scholar]
- Gadalla, M. M. , and Snyder S. H.. 2010. Hydrogen sulfide as a gasotransmitter. J. Neurochem. 113:14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George, A. K. , Singh M., Homme R. P., Majumder A., Sandhu H. S., and Tyagi S. C.. 2018. A hypothesis for treating inflammation and oxidative stress with hydrogen sulfide during age‐related macular degeneration. Int. J. Ophthalmol. 11:881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givvimani, S. , Sen U., Tyagi N., Munjal C., and Tyagi S. C.. 2011. X‐ray imaging of differential vascular density in MMP‐9‐/‐, PAR‐1‐/+, hyperhomocysteinemic (CBS‐/+) and diabetic (Ins2‐/+) mice. Arch. Physiol. Biochem. 117:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankey, G. J. , and Eikelboom J. W.. 1999. Homocysteine and vascular disease. Lancet 354:407–413. [DOI] [PubMed] [Google Scholar]
- Iqbal, M. P. , Lindblad B. S., Mehboobali N., Yusuf F. A., Khan A. H., and Iqbal S. P.. 2009. Folic acid and vitamin B6 deficiencies related hyperhomocysteinemia in apparently healthy Pakistani adults; is mass micronutrient supplementation indicated in this population? J. Coll. Physicians Surg. Pak. 19:308–312. [PubMed] [Google Scholar]
- Isner, J. M. , and Asahara T.. 1999. Angiogenesis and vasculogenesis as therapeutic strategies for postnatal neovascularization. J. Clin. Investig. 103:1231–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubowski, H. 1999. Protein homocysteinylation: possible mechanism underlying pathological consequences of elevated homocysteine levels. FASEB J. 13:2277–2283. [PubMed] [Google Scholar]
- Jakubowski, H. , Zhang L., Bardeguez A., and Aviv A.. 2000. Homocysteine thiolactone and protein homocysteinylation in human endothelial cells: implications for atherosclerosis. Circ. Res. 87:45–51. [DOI] [PubMed] [Google Scholar]
- Jiang, Y. , Sun T., Xiong J., Cao J., Li G., and Wang S.. 2007. Hyperhomocysteinemia‐mediated DNA hypomethylation and its potential epigenetic role in rats. Acta Biochim. Biophys. Sin. 39:657–667. [DOI] [PubMed] [Google Scholar]
- John, A. , Kundu S., Pushpakumar S., Fordham M., Weber G., Mukhopadhyay M., et al. 2017. GYY4137, a hydrogen sulfide donor modulates miR194‐dependent collagen realignment in diabetic kidney. Sci. Rep. 7:10924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalani, A. , Kamat P. K., Givvimani S., Brown K., Metreveli N., Tyagi S. C., et al. 2014. Nutri‐epigenetics ameliorates blood‐brain barrier damage and neurodegeneration in hyperhomocysteinemia: role of folic acid. J. Mol. Neurosci. 52:202–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanwar, Y. S. , Manaligod J. R., and Wong P. W.. 1976. Morphologic studies in a patient with homocystinuria due to 5, 10‐methylenetetrahydrofolate reductase deficiency. Pediatr. Res. 10:598–609. [DOI] [PubMed] [Google Scholar]
- Kharb, S. , Aggarwal D., Bala J., and Nanda S.. 2016. Evaluation of homocysteine, vitamin B12 and folic acid levels during all the trimesters in pregnant and preeclamptic women. Curr. Hypertens. Rev. 12:234–238. [DOI] [PubMed] [Google Scholar]
- Kimura, H. 2010. Hydrogen sulfide: from brain to gut. Antioxid. Redox Signal. 12:1111–1123. [DOI] [PubMed] [Google Scholar]
- Kimura, Y. , Goto Y., and Kimura H.. 2010. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal. 12:1–13. [DOI] [PubMed] [Google Scholar]
- Kolling, J. , Scherer E. B., Siebert C., Hansen F., Torres F. V., Scaini G., et al. 2013. Homocysteine induces energy imbalance in rat skeletal muscle: is creatine a protector? Cell Biochem. Func. 31:575–584. [DOI] [PubMed] [Google Scholar]
- Kozich, V. , Krijt J., Sokolova J., Melenovska P., Jesina P., Vozdek R., et al. 2016. Thioethers as markers of hydrogen sulfide production in homocystinurias. Biochimie 126:14–20. [DOI] [PubMed] [Google Scholar]
- Kroll, J. , and Waltenberger J.. 1998. VEGF‐A induces expression of eNOS and iNOS in endothelial cells via VEGF receptor‐2 (KDR). Biochem. Biophys. Rese. Commun. 252:743–746. [DOI] [PubMed] [Google Scholar]
- Kundu, S. , Pushpakumar S., and Sen U.. 2015. MMP‐9‐ and NMDA receptor‐mediated mechanism of diabetic renovascular remodeling and kidney dysfunction: hydrogen sulfide is a key modulator. Nitric Oxide 46:172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laha, A. , Majumdar A., Singh M., and Tyagi S. C.. 2018. Connecting homocysteine and obesity through pyroptosis, gut microbiome, epigenetics, peroxisome proliferator‐activated receptor gamma, and zinc finger protein 407. Can. J. Physiol. Pharmacol. 2018:1–6. [DOI] [PubMed] [Google Scholar]
- Lehotsky, J. , Tothova B., Kovalska M., Dobrota D., Benova A., Kalenska D., et al. 2016. Role of homocysteine in the ischemic stroke and development of ischemic tolerance. Front. Neurosci. 10:538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. B. , Ge Y. K., Zhang L., and Zheng X. X.. 2006. Astragaloside IV improved barrier dysfunction induced by acute high glucose in human umbilical vein endothelial cells. Life Sci. 79:1186–1193. [DOI] [PubMed] [Google Scholar]
- Lindner, V. , Fingerle J., and Reidy M. A.. 1993. Mouse model of arterial injury. Circ. Res. 73:792–796. [DOI] [PubMed] [Google Scholar]
- Liu, W. , Ahmad S. A., Reinmuth N., Shaheen R. M., Jung Y. D., Fan F., et al. 2000. Endothelial cell survival and apoptosis in the tumor vasculature. Apoptosis 5:323–328. [DOI] [PubMed] [Google Scholar]
- Machens, H. G. , Grzybowski S., Bucsky B., Spanholtz T., Niedworok C., Maichle A., et al. 2006. A technique to detect and to quantify fasciocutaneous blood vessels in small laboratory animals ex vivo. J. Surg. Res. 131:91–96. [DOI] [PubMed] [Google Scholar]
- Majumder, A. , Behera J., Jeremic N., and Tyagi S. C.. 2017. Hypermethylation: causes and consequences in skeletal muscle myopathy. J. Cell. Biochem. 118:2108–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder, A. , Singh M., Behera J., Theilen N. T., George A. K., Tyagi N., et al. 2018. Hydrogen sulfide alleviates hyperhomocysteinemia‐1 mediated skeletal muscle atrophy via mitigation of oxidative and endoplasmic reticulum stress injury. Am. J. Physiol. Cell Physiol. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, A. , Mujumdar V., Shek E., Guillot J., Angelo M., Palmer L., et al. 2000. Hyperhomocyst(e)inemia induces multiorgan damage. Heart Vessels 15:135–143. [DOI] [PubMed] [Google Scholar]
- Mironov, A. , Seregina T., Nagornykh M., Luhachack L. G., Korolkova N., Lopes L. E., et al. 2017. Mechanism of H2S‐mediated protection against oxidative stress in Escherichia coli. Proc. Natl. Acad. Sci. USA 114:6022–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra, P. K. , Tyagi N., Sen U., Joshua I. G., and Tyagi S. C.. 2010. Synergism in hyperhomocysteinemia and diabetes: role of PPAR gamma and tempol. Cardiovasc. Diabetol. 9:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morbidelli, L. , Donnini S., and Ziche M.. 2003. Role of nitric oxide in the modulation of angiogenesis. Curr. Pharm. Des. 9:521–530. [DOI] [PubMed] [Google Scholar]
- Murphy, G. J. , and Holder J. C.. 2000. PPAR‐gamma agonists: therapeutic role in diabetes, inflammation and cancer. Trends Pharmacol. Sci. 21:469–474. [DOI] [PubMed] [Google Scholar]
- Mustafa, A. K. , Gadalla M. M., and Snyder S. H.. 2009. Signaling by gasotransmitters. Sci. Signal. 2:re2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi, S. S. , and Mishra P. K.. 2017. H2S and homocysteine control a novel feedback regulation of cystathionine beta synthase and cystathionine gamma lyase in cardiomyocytes. Sci. Rep. 7:3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan, N. , Tyagi N., Shah A., Pagni S., and Tyagi S. C.. 2013. Hyperhomocysteinemia during aortic aneurysm, a plausible role of epigenetics. Int. J. Physiol. Pathophysiol. Pharmacol. 5:32–42. [PMC free article] [PubMed] [Google Scholar]
- Narayanan, N. , Pushpakumar S. B., Givvimani S., Kundu S., Metreveli N., James D., et al. 2014. Epigenetic regulation of aortic remodeling in hyperhomocysteinemia. FASEB J. 28:3411–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton, K. A. , and Popel A. S.. 2016. Effects of endothelial cell proliferation and migration rates in a computational model of sprouting angiogenesis. Sci. Rep. 6:36992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipov, R. M. , Robich M. P., Feng J., Liu Y., Clements R. T., Glazer H. P., et al. 2009. Effect of hydrogen sulfide in a porcine model of myocardial ischemia‐reperfusion: comparison of different administration regimens and characterization of the cellular mechanisms of protection. J. Cardiovasc. Pharmacol. 54:287–297. [DOI] [PubMed] [Google Scholar]
- Ozsvari, B. , Puskas L. G., Nagy L. I., Kanizsai I., Gyuris M., Madacsi R., et al. 2010. A cell‐microelectronic sensing technique for the screening of cytoprotective compounds. Int. J. Mol. Med. 25:525–530. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos, A. , Pyriochou A., Altaany Z., Yang G., Marazioti A., Zhou Z., et al. 2009. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 106:21972–21977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perna, A. F. , Ingrosso D., and De Santo N. G.. 2003. Homocysteine and oxidative stress. Amino Acids 25:409–417. [DOI] [PubMed] [Google Scholar]
- Predmore, B. L. , and Lefer D. J.. 2010. Development of hydrogen sulfide‐based therapeutics for cardiovascular disease. Cardiovasc. Transl. Res. 3:487–498. [DOI] [PubMed] [Google Scholar]
- Pushpakumar, S. , Kundu S., Narayanan N., and Sen U.. 2015. DNA hypermethylation in hyperhomocysteinemia contributes to abnormal extracellular matrix metabolism in the kidney. FASEB J. 29:4713–4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racek, J. , Rusnakova H., Trefil L., and Siala K. K.. 2005. The influence of folate and antioxidants on homocysteine levels and oxidative stress in patients with hyperlipidemia and hyperhomocysteinemia. Physiol. Res. 54:87–95. [DOI] [PubMed] [Google Scholar]
- Rio, D. C. , Ares M. Jr, Hannon G. J., and Nilsen T. W.. 2010. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010:pdb.prot5439. [DOI] [PubMed] [Google Scholar]
- Rosen, E. D. , Raymond S., Zollman A., Noria F., Sandoval‐Cooper M., Shulman A., et al. 2001. Laser‐induced noninvasive vascular injury models in mice generate platelet‐ and coagulation‐dependent thrombi. Am. J. Pathol. 158:1613–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen, U. , Mishra P. K., Tyagi N., and Tyagi S. C.. 2010. Homocysteine to hydrogen sulfide or hypertension. Cell Biochem. Biophys. 57:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, M. , George A. K., Homme R. P., Majumder A., Laha A., Sandhu H. S., et al. 2018. Circular RNAs profiling in the cystathionine‐beta‐synthase mutant mouse reveals novel gene targets for hyperhomocysteinemia induced ocular disorders. Exp. Eye Res. 174:80–92. [DOI] [PubMed] [Google Scholar]
- Stipanuk, M. H. , and Ueki I.. 2011. Dealing with methionine/homocysteine sulfur: cysteine metabolism to taurine and inorganic sulfur. J. Inherit. Metab. Dis. 34:17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo, C. 2007. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discovery 6:917–935. [DOI] [PubMed] [Google Scholar]
- Szabo, C . 2010. Gaseotransmitters: new frontiers for translational science. Sci. Transl. Med. 2: 59ps54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, H. , Jiang X., Yang F., Li Z., Liao D., Trial J., et al. 2006. Hyperhomocysteinemia inhibits post‐injury reendothelialization in mice. Cardiovasc. Res. 69:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfik, A. , Al‐Shabrawey M., Roon P., Sonne S., Covar J. A., Matragoon S., et al. 2013. Alterations of retinal vasculature in cystathionine‐Beta‐synthase mutant mice, a model of hyperhomocysteinemia. Invest. Ophthalmol. Vis. Sci. 54:939–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz, P. , Hu E., and Spiegelman B. M.. 1995. Regulation of adipocyte gene expression and differentiation by peroxisome proliferator activated receptor gamma. Curr. Opin. Genet. Dev. 5:571–576. [DOI] [PubMed] [Google Scholar]
- Topal, G. , Brunet A., Millanvoye E., Boucher J. L., Rendu F., Devynck M. A., et al. 2004. Homocysteine induces oxidative stress by uncoupling of NO synthase activity through reduction of tetrahydrobiopterin. Free Radic. Biol. Med. 36:1532–1541. [DOI] [PubMed] [Google Scholar]
- Tyagi, N. , Sedoris K. C., Steed M., Ovechkin A. V., Moshal K. S., and Tyagi S. C.. 2005. Mechanisms of homocysteine‐induced oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 289:H2649–H2656. [DOI] [PubMed] [Google Scholar]
- Tyagi, N. , Qipshidze N., Sen U., Rodriguez W., Ovechkin A., and Tyagi S. C.. 2011. Cystathionine beta synthase gene dose dependent vascular remodeling in murine model of hyperhomocysteinemia. Int. J. Physiol. Pathophysiol. Pharmacol. 3:210–222. [PMC free article] [PubMed] [Google Scholar]
- Tyagi, N. , Qipshidze N., Munjal C., Vacek J. C., Metreveli N., Givvimani S., et al. 2012. Tetrahydrocurcumin ameliorates homocysteinylated cytochrome‐c mediated autophagy in hyperhomocysteinemia mice after cerebral ischemia. J. Molecul. Neurosci. 47:128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upchurch Jr, G. R. , Welch G. N., Fabian A. J., Pigazzi A., Keaney J. F. Jr, and Loscalzo J.. 1997. Stimulation of endothelial nitric oxide production by homocyst(e)ine. Atherosclerosis 132:177–185. [DOI] [PubMed] [Google Scholar]
- Valentino, F. , Bivona G., Butera D., Paladino P., Fazzari M., Piccoli T., et al. 2010. Elevated cerebrospinal fluid and plasma homocysteine levels in ALS. Eur. J. Neurol. 17:84–89. [DOI] [PubMed] [Google Scholar]
- Veeranki, S. , and Tyagi S. C.. 2013. Defective homocysteine metabolism: potential implications for skeletal muscle malfunction. Int. J. Mol. Sci. 14:15074–15091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeranki, S. , Givvimani S., Pushpakumar S., and Tyagi S. C.. 2014. Hyperhomocysteinemia attenuates angiogenesis through reduction of HIF‐1alpha and PGC‐1alpha levels in muscle fibers during hindlimb ischemia. Am. J. Physiol. Heart Circ. Physiol. 306:H1116–H1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeranki, S. , Winchester L. J., and Tyagi S. C.. 2015. Hyperhomocysteinemia associated skeletal muscle weakness involves mitochondrial dysfunction and epigenetic modifications. Biochim. Biophys. Acta 1852:732–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoef, P. , van Vliet T., Olthof M. R., and Katan M. B.. 2005. A high‐protein diet increases postprandial but not fasting plasma total homocysteine concentrations: a dietary controlled, crossover trial in healthy volunteers. Am. J. Clin. Nutri. 82:553–558. [DOI] [PubMed] [Google Scholar]
- Voskoboeva, E. , Semyachkina A., Yablonskaya M., and Nikolaeva E.. 2018. Homocystinuria due to cystathionine beta‐synthase (CBS) deficiency in Russia: molecular and clinical characterization. Mol. Genet. Metab. Rep. 14:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R. 2003. The gasotransmitter role of hydrogen sulfide. Antioxid. Redox Signal. 5:493–501. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Yoshizumi M., Lai K., Tsai J. C., Perrella M. A., Haber E., et al. 1997. Inhibition of growth and p21ras methylation in vascular endothelial cells by homocysteine but not cysteine. J. Biol. Chem. 272:25380–25385. [DOI] [PubMed] [Google Scholar]
- Wang, M. J. , Cai W. J., Li N., Ding Y. J., Chen Y., and Zhu Y. C.. 2010. The hydrogen sulfide donor NaHS promotes angiogenesis in a rat model of hind limb ischemia. Antioxid. Redox Signal. 12:1065–1077. [DOI] [PubMed] [Google Scholar]
- Watanabe, M. , Osada J., Aratani Y., Kluckman K., Reddick R., Malinow M. R., et al. 1995. Mice deficient in cystathionine beta‐synthase: animal models for mild and severe homocyst(e)inemia. Proc. Natl. Acad. Sci. USA 92:1585–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werstuck, G. H. , Lentz S. R., Dayal S., Hossain G. S., Sood S. K., Shi Y. Y., et al. 2001. Homocysteine‐induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Invest. 107:1263–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winchester, L. J. , Veeranki S., Pushpakumar S., and Tyagi S. C.. 2018. Exercise mitigates the effects of hyperhomocysteinemia on adverse muscle remodeling. Physiol. Rep. 6:e13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, G. , An S. S., Ji Y., Zhang W., and Pei Y.. 2015. Hydrogen sulfide signaling in oxidative stress and aging development. Oxid. Med. Cell. Longev. 2015:357–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, A. , Jiao Y., Yang S., Deng M., Yang X., Mao C., et al. 2018. Homocysteine activates autophagy by inhibition of CFTR expression via interaction between DNA methylation and H3K27me3 in mouse liver. Cell Death Dis. 9:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, P. , Melnyk S., Pogribna M., Pogribny I. P., Hine R. J., and James S. J.. 2000. Increase in plasma homocysteine associated with parallel increases in plasma S‐adenosylhomocysteine and lymphocyte DNA hypomethylation. J. Biol. Chem. 275:29318–29323. [DOI] [PubMed] [Google Scholar]
- Zoccolella, S. , Simone I. L., Lamberti P., Samarelli V., Tortelli R., Serlenga L., et al. 2008. Elevated plasma homocysteine levels in patients with amyotrophic lateral sclerosis. Neurology 70:222–225. [DOI] [PubMed] [Google Scholar]
- Zou, T. , Liu W. J., Li S. D., Zhou W., Yang J. F., and Zou C. G.. 2011. TRB3 mediates homocysteine‐induced inhibition of endothelial cell proliferation. J. Cell. Physiol. 226:2782–2789. [DOI] [PubMed] [Google Scholar]