

Summary
Objective
Benign epilepsy with centrotemporal spikes (BECTS) is a common, self‐limited epilepsy syndrome affecting school‐age children. Classic interictal epileptiform discharges (IEDs) confirm diagnosis, and BECTS is presumed to be pharmacoresponsive. As seizure risk decreases in time with this disease, we hypothesize that the impact of IEDs and anticonvulsive drug (ACD) treatment on the risk of subsequent seizure will differ based on disease duration.
Methods
We calculate subsequent seizure risk following diagnosis in a large retrospective cohort of children with BECTS (n = 130), evaluating the impact of IEDs and ACD treatment in the first, second, third, and fourth years of disease. We use a Kaplan–Meier survival analysis and logistic regression models. Patients were censored if they were lost to follow‐up or if they changed group status.
Results
Two‐thirds of children had a subsequent seizure within 2 years of diagnosis. The majority of children had a subsequent seizure within 3 years despite treatment. The presence of IEDs on electroencephalography (EEG) did not impact subsequent seizure risk early in the disease. By the fourth year of disease, all children without IEDs remained seizure free, whereas one‐third of children with IEDs at this stage had a subsequent seizure. Conversely, ACD treatment corresponded with lower risk of seizure early in the disease but did not impact seizure risk in later years.
Significance
In this cohort, the majority of children with BECTS had a subsequent seizure despite treatment. In addition, ACD treatment and IEDs predicted seizure risk at specific points of disease duration. Future prospective studies are needed to validate these exploratory findings.
Keywords: Benign epilepsy with centrotemporal spikes, Interictal epileptiform discharges, Treatment, Biomarker, Seizure risk
Key Points.
The impact of medication and IEDs on seizure risk depends on the duration of disease in BECTS
Medication lowers the risk of seizure in the first year, but does not affect seizure risk in subsequent years
Although presumed pharmacoresponsive, more than half of children treated with medication from presentation had a subsequent seizure
IEDs do not predict seizure risk in the first 3 years, but the absence of IEDs in the fourth year may indicate remission
Benign epilepsy with centrotemporal spikes (BECTS) is a common childhood epilepsy syndrome, accounting for 10–15% of all childhood epilepsies and characterized by a transient period of seizure susceptibility in school‐age children.1, 2, 3, 4, 5 Although nearly all children will enter remission spontaneously by age 18, there are no reliable clinical predictors for when remission will occur in an individual child.6 Fifteen percent of children with BECTS will have only a single seizure, two‐thirds will have 2–5 seizures, and still others have recurrent seizures over several years.6, 7
Interictal epileptiform discharges (IEDs) on electroencephalography (EEG) are a well‐characterized electrical feature for diagnosis in BECTS8, 9 and the most clinically useful biomarker to identify the risk of a second seizure in idiopathic childhood epilepsy in general.10, 11 In population studies, IEDs are highly specific for epilepsy, present in only 0.5–2.4% of the general population.12, 13, 14 IEDs also help stratify risk of subsequent seizure after drug taper in the general epilepsy population.15 Whether the presence of IEDs at different stages of disease predicts seizure risk in a self‐limited epilepsy syndrome, such as BECTS, is not known.
Given the uncertainty in individual seizure risk, there is wide variability in treatment strategies for BECTS. Most practitioners favor treatment of most children with anticonvulsant drugs (ACDs) until at least 1–2 years after their last clinical seizure, although there is much debate regarding the optimal approach due to competing concerns regarding seizure risk and ACD side‐effects.16, 17, 18, 19, 20, 21 Given the diminishing risk of seizure over time in BECTS, it follows that the benefits of ongoing ACD exposure over the course of the disease could decrease in a similar fashion, although this has not been previously evaluated.
We hypothesized that the impact of IEDs and ACD treatment on the risk of subsequent seizure differs based on disease duration in BECTS. To study this, we evaluated the risk of subsequent seizure in a large retrospective population of children with BECTS following initial diagnosis. We evaluated whether IEDs and ACD treatment predict subsequent seizure risk in each of the first 4 years of the disease.
Methods
Subjects
All patients seen at Massachusetts General Hospital (MGH) between June 2001 and June 2017 with a clinical diagnosis of BECTS made by a child neurologist, an EEG report that captured sleep, and sufficient clinical data to determine seizure course were included. Patients were identified via search of our institution's EEG database of EEG reports containing the words “rolandic,” “sleep‐activated,” “sleep activated,” “benign,” “BECTS,” “ECTS,” “centrotemporal,” “centro‐temporal,” or “horizontal dipole,” Patients were required to have had at least one clinical seizure characterized by a focal motor seizure or secondarily generalized convulsive seizure and an EEG showing sleep‐activated centrotemporal spikes, consistent with the 1989 International League Against Epilepsy (ILAE) electroclinical diagnosis of BECTS used in previous prospective and large retrospective studies.1, 3, 6, 22 Although the 1989 ILAE diagnostic criteria for BECTS include seizure onset between 3 and 13 years,23 several prospective studies and a large retrospective meta‐analysis have found that children that otherwise meet criteria for BECTS can present during infancy and toddler years,3, 4, 6, 7, 24, 25, 26 and the inclusion cut‐off at 3 years may inaccurately describe this disease. Here, we have included children with age at onset younger than 3 years. Most of the screened subjects that were excluded did not meet electroclinical criteria for BECTS and were not diagnosed with BECTS. Rarely, subjects were excluded because (1) the subject had an EEG study that was consistent with BECTS and was diagnosed by a provider with BECTS but had no documented clinical seizure; (2) the subject had an EEG study that was consistent with BECTS but no documented clinic visit at our institution; or (3) the subject had a comorbid diagnosis of symptomatic abnormal brain magnetic resonance imaging (MRI), abnormal neurologic examination findings, or an unrelated genetic disease. Twenty‐five subjects who met electroclinical criteria for BECTS were excluded because they did not have an EEG that captured sleep. We note that although we required all subjects to have an EEG that included sleep to improve the sensitivity to capture spikes,27, 28 EEG reports did not reliably distinguish between centrotemporal spikes captured during wakefulness, sleep, or both. Patients with clinical features consistent with BECTS but initial EEG without classic findings were included if subsequent EEG showed centrotemporal spikes. Chart review was performed to collect the clinical variables of interest through the duration of documented follow‐up at our institution: age at EEG in months, the presence of IEDs on EEG, presence of atypical findings on EEG (generalized or focal background slowing, generalized spike‐and‐wave discharges, non‐centrotemporal focal IEDs, and other findings), ACD status and dose, date of the first known seizure, and date of the most recent seizure following the EEG. A total of 130 unique patients (ages 0.6–13.4 years at first known seizure, mean = 7.6 years, median = 8.0 years) met inclusion criteria. From the 130 patients, 216 EEG studies obtained from the first, second, third, and fourth years of disease were evaluated. For each year of analysis, only one EEG (the first EEG obtained during the defined year) for each individual was included. Each year of analysis was evaluated based on status during that time period of interest, independent of status in prior years (e.g., year 2 analysis followed patient treatment status and seizure risk from year 2 on, independent of prior treatment status and seizure history in year 1). When all time points were evaluated together, all EEG recordings were studied.
Analysis
Seizure risk was analyzed using Kaplan‐Meier survival analysis and significance was computed using a log‐rank test.29 Results are reported with 95% confidence intervals (CIs) and figures displayed as Kaplan‐Meier survival curves with the cumulative probability of seizure recurrence plotted as a function of time from the first EEG obtained in the first, second, third, and fourth years of disease.
We report the risk of subsequent seizure from the time at EEG and compare the impact of IEDs and ACD treatment on subsequent seizure risk. To assess seizure risk at different stages of this disease, we evaluate the impact of IEDs and ACD treatment on subsequent seizure risk following EEG recordings obtained in the first year of disease and following EEG recordings obtained each year through the fourth year after diagnosis. Because the delay between first seizure and time to EEG and duration of follow‐up could each introduce a temporal bias in this age‐specific syndrome, we evaluated the significance of this delay between groups using a Mann‐Whitney test. Unless noted in the text, for all group comparisons, there was no difference between the median time from first seizure to EEG or duration of follow‐up between groups (p > 0.05 for all comparisons, Table S1).
Patients were censored if they were lost to follow‐up or if they changed group status (e.g., switched from off ACDs to on ACDs). Time to event was measured by the number of months between the EEG and when the patient was censored. We defined a patient's medication status as on or off ACD medication, excluding changes that lasted for less than 1 month. A patient was categorized as lost to follow‐up if they did not have a seizure at the time of the last available clinic note or at the time of a medication status change.
To test for an association between clinical and EEG features and subsequent seizure risk, we also built a logistic regression model of seizure risk and tested the predictors: ACD treatment, IED, age at onset, and atypical EEG findings. To account for censoring, we included duration of follow‐up as a predictor in each model. For each analysis, we initially tested the predictor variable of interest, added subsequent variables, and evaluated model quality using Akaike information criterion (AIC).30
Results
Overall risk of seizure in BECTS
The risk of a subsequent seizure in children with BECTS was high following diagnosis (n = 107 subjects, Fig. 1). Approximately one‐third of subjects (31.7%, 95% CI 21.7–40.4%) had a subsequent seizure within 6 months; one‐half of subjects (46.4, 95% CI 34.9– 55.8%) had a subsequent seizure within 12 months; and two‐thirds of subjects (60.7%, 95% CI 48.2–70.2%) had a subsequent seizure within 2 years. After 2 years without seizure following initial diagnosis, the risk of subsequent seizure was very low; only 3.3% and 5.1% of children had a subsequent seizure within 12 months after 2 and 3 years without seizure, respectively. In this cohort, the median time to first EEG after first clinical seizure was 1.0 month (range 0–11 months). The median duration of follow‐up was 17.5 months (range 0–103 months). The risk for subsequent seizure was not affected by whether patients had more than one seizure prior to EEG (n = 58) compared to those who had only a single seizure prior to EEG (n = 49, p = 0.56).
Figure 1.
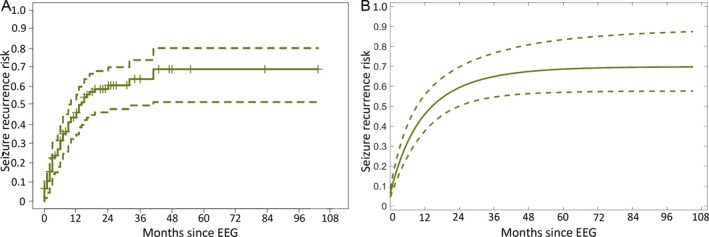
Seizure risk following electroclinical diagnosis. A, Results are displayed as a Kaplan‐Meier survival curve (±95% CI) with the cumulative probability of seizure recurrence plotted as a function of time from first EEG. Seizure risk follows an exponential curve, where approximately one‐third of children have a subsequent seizure within 6 months, one‐half within 12 months, and two‐thirds within 2 years. B, Risk of seizure recurrence computed from a logistic regression model following initial EEG (dashed lines indicate 95% CI computed from a bootstrap resampling procedure).
IEDs as a biomarker for seizure risk in BECTS
To determine whether interictal epileptiform spikes could serve as a biomarker for ongoing seizure risk in this disease, we evaluated the risk of subsequent seizures in children based on whether IEDs were present on their most recent EEG study. Among all subjects and all EEG data available, there was no difference in subsequent seizure risk based on the presence (n = 174) or absence of IEDs (n = 42) on the most recent EEG recording (p = 0.23). We note that, consistent with our hypothesis, the duration of time from first seizure to EEG was longer in the group without IEDs (p = 0.0002, Table S1). We subsequently evaluated the utility of this biomarker based on disease duration, below.
IEDs do not predict seizure risk in the first 3 years in BECTS
We found no difference in subsequent seizure risk based on the presence (n = 94) or absence of IEDs (n = 13) on the first EEG obtained after presentation (p = 0.94; Fig. 2A). Similarly, the presence (n = 38, n = 29) or absence (n = 9, n = 10) of IEDs on EEG obtained in the second or third year of disease did not predict subsequent seizure risk (p = 0.98, p = 0.64, respectively). We note that the second year cohort of children with IEDs on EEG had a longer duration of follow‐up compared to those without IEDs (p = 0.02, Table S1); because longer follow‐up would increase the likelihood of capturing a subsequent seizure, and this was not observed, we do not expect this difference to affect the interpretation of our results.
Figure 2.
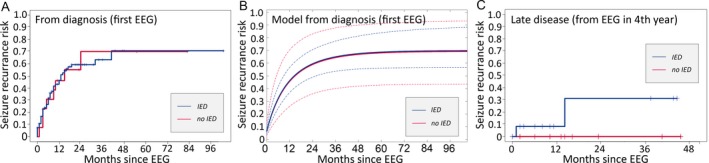
Impact of interictal epileptiform discharges (IEDs) on seizure risk. A, There is no difference in subsequent seizure risk based on the presence or absence of IEDs on the first EEG after initial presentation (p = 0.94). B, Risk of seizure recurrence computed from a logistic regression model following initial EEG based on the presence (red) or absence (blue) of IEDs (dashed lines indicate 95% CI computed from a bootstrap resampling procedure); IEDs do not correlate with seizure risk. C, Following EEG obtained in the fourth year of disease, all children without IEDs on EEG remained seizure‐free, whereas approximately one‐third of those with IEDs had a subsequent seizure.
Absence of IEDs may indicate remission in BECTS in later years of disease
During the fourth year of disease, no child without IEDs on EEG (n = 10) had a subsequent seizure. However, approximately one‐third of children for whom EEG captured IEDs (n = 13) at this stage of disease had a seizure over the subsequent 18 months of observation (31.3%, 95% CI 0–61.9%). Using a log‐rank test, the difference in seizure risk between the 2 groups was not statistically significant (p = 0.16) but was limited by small sample size. In the fourth year, the positive predictive value of IEDs to predict subsequent seizure was 0.31; however, the negative predictive value of IEDs to predict subsequent seizure was high at 1.0. The survival curve for this cohort, separated by whether IEDs were observed on the EEG, is shown in Figure 2C. The cumulative risk of subsequent seizure based on the presence or absence of IEDs for each year evaluated is summarized in Table 1.
Table 1.
Cumulative seizure risk over time by cohort
| Year | Feature | No. of children | 3‐month seizure risk (%) | 6‐month seizure risk (%) | 12‐month seizure risk (%) |
|---|---|---|---|---|---|
| 1 | ACD | 53 | 19.20 | 29.47 | 33.26 |
| No ACD | 54 | 26.30 | 33.67 | 56.11 | |
| IED | 94 | 22.6 | 31.77 | 46.26 | |
| No IED | 13 | 23.8 | 30.77 | 46.15 | |
| 2 | ACD | 32 | 31.25 | 34.52 | 41.80 |
| No ACD | 15 | 13.33 | 20.00 | 35.35 | |
| IED | 38 | 26.32 | 31.58 | 42.98 | |
| No IED | 9 | 22.22 | 22.22 | 22.22 | |
| 3 | ACD | 26 | 26.15 | 35.38 | 49.74 |
| No ACD | 13 | 35.83 | 35.83 | 35.83 | |
| IED | 29 | 34.05 | 34.05 | 43.10 | |
| No IED | 10 | 14.29 | 28.57 | 52.38 | |
| 4 | ACD | 9 | 12.50 | 12.50 | 12.50 |
| No ACD | 14 | 0 | 0 | 0 | |
| IED | 13 | 8.33 | 8.33 | 8.33 | |
| No IED | 10 | 0 | 0 | 0 |
ACD treatment modified seizure risk in BECTS early but not late in disease course
Among all subjects, at all time points, we found no difference in seizure risk between children treated with ACDs (n = 120) and children not treated with ACDs (n = 96, p = 0.80). We further evaluated the impact of ACD treatment on seizure risk throughout the disease course.
ACDs lower but do not eliminate seizure risk if started in the first year in BECTS
Children in whom ACD treatment was initiated early in the disease course (n = 53) had a lower risk of seizure compared to children not treated (n = 54; p = 0.03; Fig. 3A), and the majority of this benefit was achieved early. When initiated after the first EEG, ACD treatment was associated with a reduction in subsequent seizure risk by 17.9% and 26.2% at 12 months and 24 months, respectively. Although seizure risk was reduced with ACD treatment, it was not eliminated and the risk of seizure remained high in both groups; 38.3% of children (95% CI 23–50.5%) treated with ACD had a seizure within 12 months compared to 56.1% of children (95% CI 37.4–69.3%) who were not treated. Of those for whom treatment was initiated early and maintained, 55.8% (95% CI 33.1–70.8%) had a seizure within 36 months compared to 74.7% (95% CI 54.9–85.7%) of those never treated.
Figure 3.
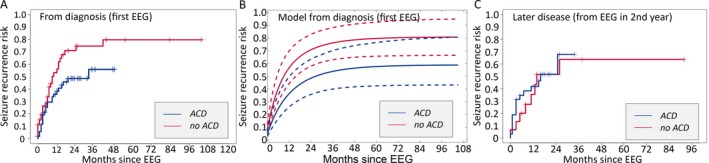
Impact of ACDs on seizure risk following all EEG studies. A, Children treated with ACDs at the time of the first EEG (blue) had a lower risk of subsequent seizure compared to those not treated (red, p = 0.03); however, more than half of treated children still had a subsequent seizure. B, Risk of subsequent seizure computed from a logistic regression model following initial EEG based on the absence (red) or presence (blue) of ACD treatment (dashed lines indicate 95% CI computed from a bootstrap resampling procedure); ACD treatment started in the first year is associated with a decreased risk of subsequent seizure (p = 0.03). (C) There was no difference in seizure risk between treated and untreated children in the second year of disease (shown here) or any subsequent year evaluated (not shown).
ACDs do not lower seizure risk after the first year in BECTS
Among children who underwent EEG evaluation in the second, third, and fourth years of disease, we found no difference in seizure risk among those treated with ACDs (n = 32, 26, 9) compared to those not treated (n = 15, 13, 14; p = 0.75, 0.34, 0.38, respectively). As expected with the natural history of this disease, there was a lower overall risk of subsequent seizure in later years of the disease. In the second year of disease, the risk of subsequent seizure in the untreated group was 35.4% at 12 months (95% CI 4.5–56.2%), which was comparable to the risk in those that received ACD treatment in the same time period 41.8% (95% CI 21.4–56.9%, Fig. 3C). By 24 months of follow‐up, more than half of treated and untreated children had a subsequent seizure (51.7% treated, 95% CI 28.1–67.5%; 51.5% untreated 95% CI 15.2–72.3%). In the third year, approximately one‐third of treated and untreated children (35.4% treated, 95% CI 12.1–52.5%; 35.8% untreated, 95% CI 0.35–58.7%) had a seizure within 6 months of follow‐up. Following an EEG in the fourth year of disease, 12.5% (95% CI 0.0–32.7%) of both treated and untreated children had a subsequent seizure within 24 months of follow‐up. We note that in the fourth year cohort, the duration of follow‐up was longer for children not treated with ACDs compared to those treated (p = 0.04, Table S1), but do not expect this difference to impact the interpretation of the findings, which were also evident in the second‐ and third‐year cohorts who had comparable durations of follow‐up. The cumulative risk of subsequent seizure for treated and untreated children for each year evaluated is summarized in Table 1.
Impact of atypical EEG findings on risk of seizure in BECTS
We explored the risk of subsequent seizure between children with at least one atypical finding on EEG (n = 25), only typical findings on EEG (e.g., centrotemporal spikes, n = 73), and normal EEG (n = 9) at diagnosis. Atypical EEG findings observed in our cohort included generalized background slowing (n = 3), focal background slowing (n = 9), generalized spike‐and‐wave complexes (n = 4), non‐centrotemporal spikes (n = 12), and other (n = 1, Table 2). There was no difference in subsequent seizure risk between groups (p = 0.87, Fig. 4).
Table 2.
Atypical EEG features in children with BECTS
| Atypical finding | No. of children |
|---|---|
| Extra‐rolandic spikes | 16 |
| Occipital | 3 |
| Frontal | 3 |
| Temporal | 2 |
| Parietal | 1 |
| Midline | 1 |
| Multifocal | 2 |
| Generalized | 4 |
| slowing | 12 |
| Hemispheric | 5 |
| Occipital | 3 |
| Frontal | 1 |
| Generalized | 4 |
| Sharp alpha | 1 |
| Any atypical finding | 25 |
Figure 4.
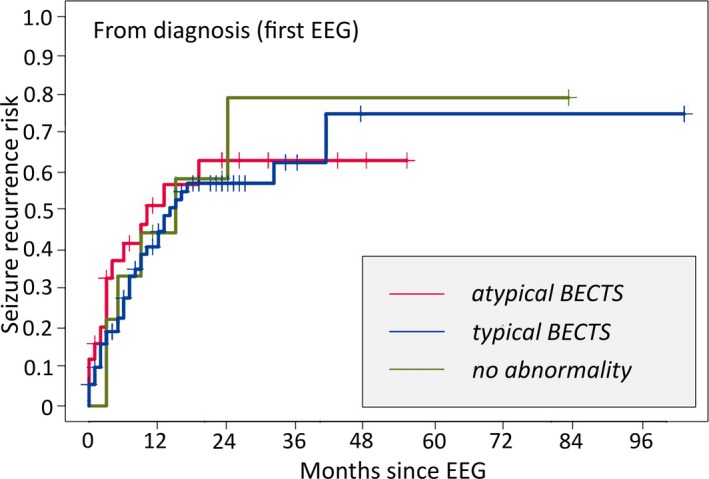
Impact of atypical EEG findings on subsequent seizure risk in BECTS. There was no difference in seizure risk between children who had normal EEG findings, typical centrotemporal spikes, or atypical findings on EEG at diagnosis.
Logistic regression models support empirical findings
To further examine the association between our features of interest and seizure risk, we built a logistic regression model of seizure risk and tested the predictors: time since EEG, ACD treatment, presence of IEDs, age at onset, and atypical EEG findings. As expected, following the first EEG, time since EEG was a significant predictor of seizure risk, where there was a decreased risk of seizure over time (p = 0.0006), such that there is a 5% reduction in the odds of seizure with each subsequent seizure‐free month.
Consistent with our empirical results, in a multivariate model using duration of follow‐up and the presence of IEDs as predictors, we found that the presence of IEDs did not correlate with seizure risk in the first, second, or third years of disease (p > 0.5 for all tests; Fig. 2B). Adding ACDs as a predictor did not impact these results (p > 0.5 for all tests). In the fourth year, because the absence of IEDs was a perfect predictor of no further seizures, maximum likelihood estimates for the effect of this predictor result in an estimated odds ratio of 0, but the significance of this effect cannot be computed by assuming approximate normality. However, when computing a Bayesian confidence interval we found that the probability that the absence of IEDs in the fourth year decreases seizure risk was 77%. Thus, the absence of spikes in the fourth year likely indicates decreased seizure risk.
Consistent with the empirical results, using a logistic regression model with ACD treatment status and duration of follow‐up as predictors, we found that ACD treatment started in the first year of disease is associated with a decreased risk of subsequent seizure (p = 0.03; Fig. 3B). The odds ratio of having a subsequent seizure is 0.53 when treated with ACDs started in the first year of disease. We found no effect of ACD treatment on seizure risk in the second, third, or fourth years of disease (p > 0.05 for all tests).
We further evaluated for an association between risk of seizure and age at first seizure, atypical EEG features, and number of ACDs beyond one. We found that for all data, there was no association between any of these predictors and seizure risk (p > 0.05 for all tests). In all of the above models tested, adding these predictors increased the AIC, thereby decreasing model quality.
Discussion
Although the electroclinical features for diagnosis in BECTS are stereotyped, the seizure course is highly variable across children, limiting the accuracy of the prognostic information available for any individual case. Here we evaluate a large retrospective cohort of children with BECTS to better characterize the seizure course and provide improved data to guide counseling discussions and treatment considerations. Because the risk of seizure decreases with time, we evaluated the impact of IEDs and ACD treatment on the risk of subsequent seizure at different stages in the disease course. We found that neither the presence of IEDs early in the disease nor atypical features on the EEG impact the risk of subsequent seizure; however, the absence of IEDs late in disease may indicate remission. We also found that treatment with ACDs early in the course of the disease reduces, but does not eliminate, the risk of seizure. After the first year of disease, ACD treatment did not significantly impact seizure risk.
These findings have several implications. First, BECTS is described as a pharmacoresponsive epilepsy syndrome, where children are expected to rapidly respond to anticonvulsant medication treatment.8, 9 However, this observation is confounded by the self‐limited nature of the disease, and no class I or class II evidence for medication efficacy currently exists.31, 32 In our cohort, over half of the children followed from diagnosis had a subsequent seizure despite receiving treatment with ACDs. The lack of complete seizure control in these children could indicate poor compliance or that this epilepsy is less pharmacoresponsive than previously thought. Because quality of life improves most dramatically in patients with epilepsy who achieve seizure freedom,33, 34, 35, 36 a better understanding of the true pharmacoresponsiveness of BECTS may influence the risk–benefit discussion when determining treatment plans with families.
Decisions on the duration of ACD treatment in BECTS rely on many variables, ranging from concerns of ACD medication side‐effects, to ongoing seizure risk, including recent attention to the low but present risk of sudden unexplained death in these children.18, 20, 37 We found that initiating ACD treatment at diagnosis corresponded with a decreased risk of subsequent seizure. However, after the first year, ACD treatment did not significantly lower seizure risk. Although selection bias may have contributed to these results, our findings are consistent with previous work that found that the duration of active disease is less than 3 years for most children with BECTS.6 In addition, a Cochrane review concluded that evidence supports only short‐term treatment for seizure control in BECTS.32 In our cohort, the risk of subsequent seizure in untreated children was much lower in the second year of disease compared to the first, and continued to decrease for each subsequent year. The lack of efficacy of ACDs in later years was thus likely influenced by spontaneous remission. These findings suggest that the diminishing returns of ACD treatment due to the expected rate of remission should be considered when weighing treatment options for a particular child.
In a large meta‐analysis, the decision to taper ACDs resulted in at least one seizure in 39% of children with BECTS.6 Biomarkers to help guide physicians in selecting who is ready for ACD discontinuation could limit unnecessary prolonged drug exposure and reduce discontinuation failures. A separate meta‐analysis evaluating the risk of ACD discontinuation among all epilepsies found that the presence of interictal spikes on EEG predicts increased seizure risk.15 Here, we found that this relationship may depend on disease duration in a self‐limited epilepsy syndrome. IEDs are poor biomarkers for ongoing seizure risk in children with BECTS early in disease. This is consistent with the observation that over 10% of BECTS children have been reported to still have IEDs on EEG after remission.6 The poor specificity of spikes to identify continued risk in BECTS has also been shown in kindred studies where classic centrotemporal IEDs were reported in one‐third of siblings of children with BECTS, most of whom did not have epilepsy.38, 39
Notably, we also found that all children without spikes on EEG during the fourth year of disease course remained seizure free (negative predictive value [NPV] 1.0), suggesting that that the absence of spikes late in the disease may provide a reliable, albeit delayed, marker of disease remission. Although the patient cohort to evaluate this feature was relatively small (n = 10), this result is consistent with data available from prior work showing that only 1 of 14 children with BECTS without spikes on EEG late in the disease (year 4 or after) had a subsequent seizure40; combined, these results suggest that the absence of spikes is a reliable indicator of disease remission. Although this exploratory finding requires validation in a larger, prospective study, this easy to obtain biomarker would enable confident identification of children for whom prolonged ACD therapy is no longer necessary, thereby curbing unnecessary drug exposure later in the disease course.
The primary limitation of this study is the retrospective collection of the data used. Because children require an EEG for diagnosis of this electroclinical syndrome, this limitation is unlikely to impact the selection of children evaluated early in the disease course. Moreover, the cumulative seizure risk in our cohort was similar to that reported from prospective datasets.1, 3 However, because only a subset of children underwent subsequent EEG evaluations in our study, the cohort of children evaluated later in the disease course diminished for each year of analysis and was likely enriched by both those being evaluated for medication taper due to seizure freedom and those being followed for more severe seizure course. Because the most common reason to obtain a late EEG study in children with BECTS is to guide a medication taper after a period seizure free, our later cohorts are most likely to be biased toward a cohort with lower seizure frequency. Thus, the finding that the presence of EEG spikes is not helpful to guide medication taper in years 2 and 3 may be particularly relevant to the population selected for repeat EEG. These findings, however, remain preliminary and should be validated in a prospective population‐based study. In addition, we were unable to control unmeasured factors, such as the presence of generalized seizures, seizures during wakefulness, ACD compliance, or other factors that may have contributed to a practitioner's decision to use ACDs. A prospective population‐based study with planned EEG and clinical follow‐up would be required to confirm the observations reported here.
We also note that we have included children with age at onset before 3 years of age in this study. To evaluate the impact of this decision, we evaluated age at onset as a predictor in our multivariate regression analysis. Consistent with a prior population‐based prospective study,1 we did not find that age at seizure onset predicted subsequent seizure risk.
Although BECTS is a common and well‐characterized electroclinical syndrome, reliable biomarkers for seizure course have not been identified and the impact and necessity of medication treatment remains controversial. This work helps to clarify the impact of IEDs and medication treatment on seizure risk in this electroclinical syndrome. In addition, this study highlights the importance of evaluating biomarkers and clinical risk factors with attention to the disease duration in age‐specific epilepsy syndromes.
Disclosure
The authors have no conflicts of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Table S1. Duration of follow‐up and EEG delay in all groups compared.
Acknowledgments
CJC is supported by National Institute of Neurological Disorders and Stroke K23‐NS092923. MAK is supported by National Science Foundation 1451384.
Biography
Wenting Xie is a post‐baccalaureate researcher in neurology at Massachusetts General Hospital.

References
- 1. Astradsson A, Olafsson E, Ludvigsson P, et al. Rolandic epilepsy: an incidence study in Iceland. Epilepsia 1998;39:884–886. [DOI] [PubMed] [Google Scholar]
- 2. Berg AT, Shinnar S, Levy SR, et al. Newly diagnosed epilepsy in children: presentation at diagnosis. Epilepsia 1999;40:445–452. [DOI] [PubMed] [Google Scholar]
- 3. Callenbach PMC, Bouma PAD, Geerts AT, et al. Long term outcome of benign childhood epilepsy with centrotemporal IEDs: Dutch study of epilepsy in childhood. Seizure 2010;19:501–506. [DOI] [PubMed] [Google Scholar]
- 4. Camfield CS, Camfield PR. Rolandic epilepsy has little effect on adult life 30 years later: a population‐based study. Neurology 2014;82:1162–1166. [DOI] [PubMed] [Google Scholar]
- 5. Larsson K, Eeg‐Olofsson O. A population based study of epilepsy in children from a Swedish county. Eur J Paediatr Neurol 2006;10:107–113. [DOI] [PubMed] [Google Scholar]
- 6. Bouma PAD, Bovenkerk AC, Westendorp RGJ, et al. The course of benign partial epilepsy of childhood with centrotemporal IEDs: a meta‐analysis. Neurology 1997;48:430–439. [DOI] [PubMed] [Google Scholar]
- 7. Loiseau P, Duche B, Cordova S, et al. Prognosis of benign childhood epilepsy with centrotemporal spikes: a follow‐up study of 168 patients. Epilepsia 1988;29:229–235. [DOI] [PubMed] [Google Scholar]
- 8. Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005‐2009. Epilepsia 2010;51:676–685. [DOI] [PubMed] [Google Scholar]
- 9. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58:512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shinnar S, Berg AT, Moshe SL, et al. The risk of seizure recurrence after a first unprovoked afebrile seizure in childhood: a prospective study. Pediatrics 1990;85:1076–1085. [PubMed] [Google Scholar]
- 11. Shinnar S, Berg AT, Moshe SL, et al. The risk of seizure recurrence after a first unprovoked afebrile seizure in childhood: an extended follow‐up. Pediatrics 1996;98:216–225. [PubMed] [Google Scholar]
- 12. Bennett DR. Spike‐wave complexes in “normal” flying personnel. Aerosp Med 1967;38:1276–1282. [PubMed] [Google Scholar]
- 13. Eeg Olofsson O, Peterson I, Sellden U. The development of the electroencephalogram in normal children from the age of 1 through 15 years. Proxysmal activity. Neuropediatrics 1971;2:375–404. [DOI] [PubMed] [Google Scholar]
- 14. Gregory RP, Oates T, Merry RT. Electroencephalogram epileptiform abnormalities in candidates for aircrew training. Electroencephalogr Clin Neurophysiol 1993;83:75–77. [DOI] [PubMed] [Google Scholar]
- 15. Berg AS, Shinnar S. Relapse following discontinuation of antiepileptic drugs: meta‐analysis. Neurology 1994;44:601–608. [DOI] [PubMed] [Google Scholar]
- 16. Hughes JR. Benign epilepsy of childhood with centrotemporal IEDs: to treat of not to treat, that is the question. Epilepsy Behav 2010;19:197–203. [DOI] [PubMed] [Google Scholar]
- 17. Mellish LC, Dunkley C, Ferrie CD, et al. Antiepileptic drug treatment of rolandic epilepsy and Panayiotopoulos syndrome: clinical practice survey and clinical trial feasibility. Arch Dis Child 2015;100:62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oguni H. Treatment of benign focal epilepsies in children: when and who should be treated. Brain Dev 2011;33:207–212. [DOI] [PubMed] [Google Scholar]
- 19. Perry S, Holt P, Benetar M. Levetiracetam versus carbamazepine monotherapy for partial epilepsy in children under 16 years of age. J Child Neurol 2008;23:515–519. [DOI] [PubMed] [Google Scholar]
- 20. Shields WD, Snead OC. Benign epilepsy with centrotemporal spikes. Epilepsia 2009;50:10–15. [DOI] [PubMed] [Google Scholar]
- 21. Wirrell E, Sherman EMS, Vanmastrigt R, et al. Deterioration in cognitive function in children with benign epilepsy of childhood with centro temporal IEDs treated with sulthiame. J Child Neurol 2008;23:14–21. [DOI] [PubMed] [Google Scholar]
- 22. Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia 2014;55:475–482. [DOI] [PubMed] [Google Scholar]
- 23. Commission on Classification and Terminology of the International League Against Epilepsy . Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30:389–399. [DOI] [PubMed] [Google Scholar]
- 24. Blom S, Heijbel J. Benign epilepsy of children with centrotemporal EEG foci: a follow‐up study in adulthood of patients initially studied as children. Epilepsia 1982;23:629–632. [DOI] [PubMed] [Google Scholar]
- 25. Arts WF, Tjiam AT, Tillema‐The P. Benign midtemporal epilepsy in childhood. Ned Tijdschr Geneeskd 1984;128:1704–1708. [PubMed] [Google Scholar]
- 26. Morikawa T, Seino M, Yagi K. Is rolandic discharge a hallmark of benign partial epilepsy of childhood? Epilepsy Res Suppl 1992;6:59–69. [PubMed] [Google Scholar]
- 27. Nicolai J, van der Linden I, Arends JB, et al. EEG characteristics related to educational impairments in children with benign childhood epilepsy with centrotemporal spikes. Epilepsia 2007;48:2093–2100. [DOI] [PubMed] [Google Scholar]
- 28. Tenney JR, Glauser T, Altaye M, et al. Longitudinal Stability of Interictal Spikes in Benign Epilepsy with Centrotemporal Spikes. Epilepsia 2016;57:805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bewick V, Cheeck L, Ball J. Statistics review 12: survival analysis. Crit Care 2000;8:389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Akaike H. A new look at the statistical model identification. IEEE Trans Autom Control 1974;19:716–723. [Google Scholar]
- 31. Glauser T, Ben‐Menachem E, Bourgeois B, et al. ILAE treatment guidelines: evidence‐based analysis of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia 2006;47:1094–1120. [DOI] [PubMed] [Google Scholar]
- 32. Tan HJ, Singh J, Gupta R, et al. Comparison of antiepileptic drugs, no treatment, or placebo, for children with benign epilepsy with centro temporal spikes. Cochrane Database Syst Rev 2014;9:CD006779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hermann BP, Wyler AR, Somes G. Preoperative psychological adjustment and surgical outcome are determinants of psychosocial status after anterior temporal lobectomy. J Neurol Neurosurg Psychiatry 1992;55:491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leidy NK, Elixhauser A, Vickrey B, et al. Seizure frequency and the health‐related quality of life of adults with epilepsy. Neurology 1999;53:162–166. [DOI] [PubMed] [Google Scholar]
- 35. Rausch R, Crandall PH. Psychological status related to surgical control of temporal lobe seizures. Epilepsia 1982;23:191–202. [DOI] [PubMed] [Google Scholar]
- 36. Stavem K, Loge JH, Kaasa S. Health status of people with epilepsy compared with a general reference population. Epilepsia 2000;41:85–90. [DOI] [PubMed] [Google Scholar]
- 37. Doumlele K, Friendman D, Buchhalter J, et al. Sudden unexpected death in epilepsy among patients with benign childhood epilepsy with centrotemporal spikes. JAMA Neurol 2017;74:645–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bray PF, Wiser WC. Hereditary characteristics of familial temporal‐central focal epilepsy. Pediatrics 1965;36:207–211. [PubMed] [Google Scholar]
- 39. Heijbel J, Blom S, Rasmussen M. Benign epilepsy of childhood with centrotemporal EEG foci: a genetic study. Epilepsia 1975;16:285–293. [DOI] [PubMed] [Google Scholar]
- 40. Kobayashi K, Yoshinaga H, Toda Y, et al. High‐frequency oscillations in idiopathic partial epilepsy of childhood. Epilepsia 2011;52:1812–1819. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Duration of follow‐up and EEG delay in all groups compared.