

Abstract
HIV splicing involves five splice donor and eight splice acceptor sequences which, together with cryptic splice sites, generate over 100 mRNA species. Ninety percent of both partially spliced and fully spliced transcripts utilize the intrinsically weak A4/A5 3′ splice site cluster. We show that DDX17, but not its close paralog DDX5, specifically controls the usage of this splice acceptor group. In its absence, production of the viral envelope protein and other regulatory and accessory proteins is grossly reduced, while Vif, which uses the A1 splice acceptor, is unaffected. This is associated with a profound decrease in viral export from the cell. Loss of Vpu expression causing upregulation of cellular Tetherin compounds the phenotype. DDX17 utilizes distinct RNA binding motifs for its role in efficient HIV replication, and we identify RNA binding motifs essential for its role, while the Walker A, Walker B (DEAD), Q motif and the glycine doublet motif are all dispensable. We show that DDX17 interacts with SRSF1/SF2 and the heterodimeric auxiliary factor U2AF65/35, which are essential splicing factors in the generation of Rev and Env/Vpu transcripts.
Keywords: DDX17/HIV-1, splicing factors, A4/A5 3′ splice site cluster
Abbreviations: ESEs, exonic splicing enhancers; DAPI, 4′,6-diamidino-2-phenlindole
Graphical Abstract

Highlights
-
•
Regulation of HIV splicing is significantly dependent on host cellular factors.
-
•
DDX17 interacts with splicing factors and is essential for efficient HIV-1 splicing.
-
•
DDX17 utilizes distinct RNA/ATPase motifs and acts independently of DDX5.
-
•
DDX17 C-terminal domain is critical for the role of DDX17 in HIV-1 replication.
Introduction
HIV differs from a number of other viruses that encode their own RNA helicases (such as the NS3 protein of hepatitis C) [1], [2], instead depending entirely on cellular helicases as essential cofactors for several stages of its replication cycle. The first of these to be identified was DDX3, which facilitates Rev-mediated export of unspliced and partially spliced viral transcripts [3]. Subsequently, DDX1 was shown to be a cofactor of Rev [4], while RNA helicase A (RHA) promotes HIV-1 reverse transcription, transcription and translation [4], [5], [6], [7], [8]. The possible roles of helicases in HIV-1 replication and latency have been reviewed extensively [9], [10], [11], [12], [13], [14].
We previously published a comprehensive siRNA knockdown screen of a library of 59 human cellular helicases including the ubiquitous DEAD box helicase family that are involved in all stages of RNA processing [15], [16], [17]. Among those identified were two closely related DEAD box helicases, DDX5 and DDX17 [18].
DDX5 and DDX17 share 90% homology in their core region and 60% and 30% homology in their N and C termini, respectively [19]. DDX5 and DDX17 can exist as monomers, homodimers or heterodimers [20]. Because of their functional overlap and structural homology, they are often considered together [21]; however, they have distinct cellular roles [22]. DDX17 exists as two isoforms (p72 and p82) due to an alternative upstream start codon (AUG) [23], although there is no documented functional difference between the two isoforms. DDX17 has been implicated in alternative splicing [24] and found to interact with known splicing factors in in vitro experiments [25], [26]. In addition, DDX17 has been suggested to modulate HIV-1 RNA stability, associating with Rev to promote nuclear export, genomic RNA packaging and Gag-Pol frameshifting [27], [28], [29]. Silencing DDX17 in TZM-bl cells resulted in a reduction in unspliced and spliced HIV transcripts [29].
HIV-1 gene regulation has been extensively reviewed [30] and HIV-1 splicing has been specifically reviewed in Refs. [31], [32], [33]. From a polycistronic transcript, HIV-1 generates more than 100 different mRNA species by alternative splicing [34]. The major structural polyproteins, Gag and Gag/Pol, are encoded by unspliced RNA; Env, Vpu, Vif and Vpr are encoded by partially spliced transcripts and the regulatory proteins, Rev, Tat and Nef derive from fully spliced species. Early gene expression is characterized by the production of 2-kb fully spliced transcripts leading to accumulation of the Rev protein which acts as a switch from early to late gene expression by mediating nuclear export of unspliced and partially spliced transcripts.
Selection of different 3′ splice site (ss) acceptors is regulated by positively acting exonic splicing enhancers (ESEs), intronic splicing enhancers (ISEs), negative exonic splicing silencers (ESSs) and intronic splicing silencers (ISSs) [35]. Cellular splicing factors facilitate or repress splicing by binding to cis-acting elements in the RNA. Recognition and usage of suboptimal viral splice sites depends heavily on cellular proteins [36].
Ninety-two percent of Env/Vpu coding transcripts derive from 3′ss A5 and 90% of fully spliced transcripts encoding Rev and Nef are generated from the A4/A5 3′ splice site cluster. Splice site A3 is used for the vast majority of Tat transcripts. Vif mRNA is expressed in low abundance (approx. 1% of the incompletely spliced transcripts) and is formed by splicing 5′ss D1 to 3′ss A1 [33].
Usage of the 3′ss A4/A5 cluster is potentiated by ESE3 located in exon 5. ESE3 is a unique bidirectional GA-rich enhancer, highly conserved in most viral subtypes with three SRSF1/ASF binding sites and one SRSF5/SRp40 binding site. ESE3 regulates expression of env, vpu, rev and nef mRNAs [37]. SRSF1/SF2 and SRSF5/SRp40 activate ESE3 and are essential for efficient 3′ss A4/A5 cluster usage and binding of the U1 snRNP to the downstream D4 5′ss. The binding of U1snRNP to D4 is required for the efficient expression of rev and nef mRNAs and to increase expression of the partially spliced env mRNA [31], [37], [38]. The bidirectional enhancer interacts upstream with the heterodimeric auxillary factor U2AF65/35 and downstream with U1snRNP. This not only potentiates the usage of the A4/A5 cluster but is also thought to stabilize partially spliced transcripts [38].
We investigated the role of DDX17 as an HIV dependency factor. Using siRNA specific for knockdown of DDX17 and rescue with an siDDX17-resistant construct,we have confirmed its essential nature for HIV replication in the physiologically relevant Jurkat cell line, knockdown having no detectable adverse effect on cellular viability. Knockdown of DDX17 strikingly reduces partially and fully spliced transcripts arising from the A4/A5 splice site cluster, with a parallel increase in unspliced viral mRNA. These findings were confirmed at the protein level where Env/Vpu/Rev and Nef were significantly reduced while Gag accumulated intracellularly. Knockdown of DDX17 profoundly reduced production of infectious viral particles. This was due to a combination of reduced levels of viral envelope and regulatory and structural proteins compounded by an increase in cellular Tetherin. By mutagenesis, we identified the domains of DDX17 critical for this activity. We show that, despite its ability to substitute for DDX17 for many cellular functions, DDX5 cannot perform the critical controlling role of DDX17 in regulating the production of HIV spliced mRNA and that this unique function may make this a vulnerable therapeutic target.
Results
DDX17 is essential for release of infectious HIV-1
We knocked down DDX17 by siRNA in cells transfected with a replication-competent proviral clone of HIV-1 pLAI and monitored the effects of DDX17 reduction upon viral infectivity. HeLa cells were sequentially transfected with siDDX17 that targets both isoforms of DDX17 and then a second round of siRNA together with pLAI, with or without increasing concentrations of the siDDX17 rescue plasmid (Figs. 1a and S1a). Up to 90% of endogenous DDX17 was depleted in siDDX17 transfected cells (Fig. 1a). Supernatants from these transfected cells were used to infect the TZM-bl indicator cell line, which, in the presence of the HIV-1 Tat protein, expresses luciferase. Knockdown of endogenous DDX17 significantly reduced both viral supernatant CA-p24 and detectable infectious virus by 4- to 5-fold (Fig. 1b and c). Both parameters were successfully restored upon co-expression of the siDDX17-resistant DDX17 expressor, confirming that the phenotypes were specifically due to DDX17 depletion. There was no detectable cellular toxicity following treatment with siDDX17 with or without increasing concentrations of rescue plasmid (Fig. S1b). DDX17 knockdown does not reduce intracellular CA p24, instead it increases (Fig. 1d). Expression of the rescue plasmid restored virus production and supernatant CA-p24; however, overexpression of DDX17 appeared to augment supernatant and intracellular levels above that seen in siControl-treated cells (Fig. 1b–d). To further evaluate this, we performed a DDX17 dose–response experiment using a plasmid expressing the wild-type DDX17 sequence. HeLa cells were co-transfected with increasing amounts of this plasmid and a constant amount of pLAI. Overexpression of wild-type DDX17 resulted in a detectable increase in HIV-1 virus production (Fig. 1e and f), indicating that overexpression of DDX17 can modestly increase production of infectious virions.
Fig. 1.

DDX17 is essential for release of infectious HIV-1. (a) Western blot analysis of DDX17 knockdown and rescue. HIV-1 proviral clone pLAI, and siDDX17 with or without increasing concentrations of siDDX17 rescue expressor were transfected into HeLa cells. (b) HIV-1 relative virus production following DDX17 knockdown and rescue. Cells were transfected with siDDX17 and then a second round of siRNA together with pLAI, with or without increasing concentrations of siDDX17 rescue construct. Supernatant was used to infect CD4 + TZM-bl cells. (c) Supernatants from panel b were harvested and CA-p24 quantified by ELISA. (d) Cell lysates from virus producing cells in panel b were harvested and CA-p24 quantified by ELISA. Each graph is a representative of three independent experiments done in triplicate. (e) Supernatant was harvested 48 h after co-transfection of a constant amount of pLAI with increasing concentrations of pCDNA4-DDX17, and virus quantitated by infection of CD4 + TZM-bl cells. The graph shown is a representative of two independent experiments done in duplicate. Data are represented as mean of duplicate samples ± SEM. (f) Cell lysates from producer cells in panel e were harvested and subjected to Western blotting. Statistical significance: *P < 0.05, **P < 0.01. See also Fig. S1.
DDX17 is essential for release of infectious HIV-1. (a) Western blot analysis of DDX17 knockdown and rescue. HIV-1 proviral clone pLAI, and siDDX17 with or without increasing concentrations of siDDX17 rescue expressor were transfected into HeLa cells. (b) HIV-1 relative virus production following DDX17 knockdown and rescue. Cells were transfected with siDDX17 and then a second round of siRNA together with pLAI, with or without increasing concentrations of siDDX17 rescue construct. Supernatant was used to infect CD4 + TZM-bl cells. (c) Supernatants from panel b were harvested and CA-p24 quantified by ELISA. (d) Cell lysates from virus producing cells in panel b were harvested and CA-p24 quantified by ELISA. Each graph is a representative of three independent experiments done in triplicate. (e) Supernatant was harvested 48 h after co-transfection of a constant amount of pLAI with increasing concentrations of pCDNA4-DDX17, and virus quantitated by infection of CD4 + TZM-bl cells. The graph shown is a representative of two independent experiments done in duplicate. Data are represented as mean of duplicate samples ± SEM. (f) Cell lysates from producer cells in panel e were harvested and subjected to Western blotting. Statistical significance: *P < 0.05, **P < 0.01. See also Fig. S1.
DDX17 and HIV-1 replication in Jurkat cells
To ensure our findings were applicable to cell lines physiologically relevant to HIV-1 infection, we carried out nucleofection of Jurkat cells using the HIV-1 proviral clone pLAI together with siDDX17 with or without increasing concentrations of siDDX17 rescue plasmid. Up to 60% of endogenous DDX17 was depleted upon treatment with siDDX17 (Fig. 2). The same phenotype that we observed in HeLa cells was recapitulated. siRNA knockdown of DDX17 in Jurkat cells reduces relative virus production by 4- to 5-fold compared to siControl-treated cells, and this effect was successfully restored by the expression of siDDX17-resistant DDX17 construct (Fig. 2).
Fig. 2.
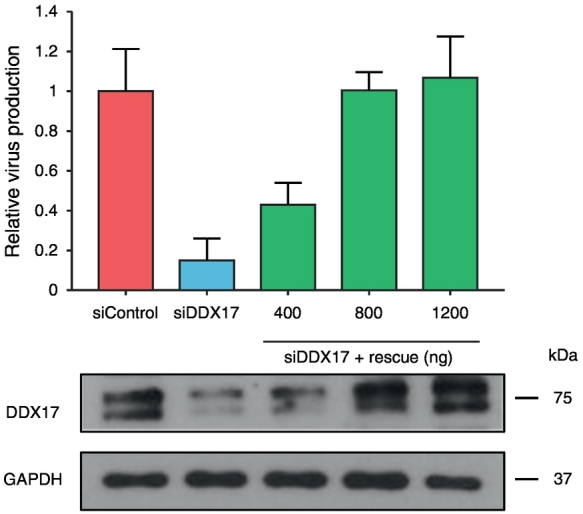
DDX17 and HIV-1 replication in Jurkat cells. HIV-1 relative virus production following DDX17 knockdown and rescue in Jurkat cells. Supernatant was harvested 96 h post-nucleofection and used to infect TZM-bl cells. The graph is a representative of two independent experiments done in duplicate. Bars represent mean of duplicate samples ± SEM. Lower panel shows the corresponding Western blot.
Knockdown of DDX17 differentially affects viral splice variants
Since DDX17 has been implicated in alternative splicing in other models, we focused on the effect of DDX17 knockdown on the different HIV splice variants. DDX17 knockdown does not inhibit HIV-1 transcription, indeed there is a slight increase in the amount of full-length HIV RNA (Fig. 3a) and a decrease in the partially spliced HIV-1 mRNAs (Fig. 3b). There is, however, a clear reduction in fully spliced HIV-1 mRNAs compared to siControl-treated cells (Fig. 3c). Endogenous DDX17 depletion thus differentially affects the different types of viral transcript. The rise in unspliced mRNA likely reflects a population of transcripts that would otherwise have been spliced. Gag production rises as quantified by ELISA (Fig. 1d), immunofluorescence (Fig. 3d) and cellular CA-p24 Western blot (Fig. 3e). There is an overall accumulation of intracellular Gag correlating with the rise in Gag mRNA and a failure of virus export (Fig. 3d). DDX17 knockdown results in an 80%–90% reduction of Rev and Nef compared to the siControl-treated cells, quantified by ImageJ (Fig. 3f). The partially spliced mRNA encoding Vif originates from the use of a different splice acceptor from Rev and Nef and although there is possibly a very minor decrease in Vif expression in siDDX17-treated cells, this is trivial compared to the effects on Rev, Nef and Vpu (Fig. 3f).
Fig. 3.

Effect of DDX17 knockdown on HIV-1 splicing and Gag. (a) RT-qPCR analysis of unspliced, (b) partially and (c) fully spliced HIV-1 mRNA respectively. Cells were sequentially transfected with siControl or siDDX17 and then a second round of siRNA together with pLAI. Data shown are representative of three independent repeats, with triplicate samples for each siRNA. Data have been normalized to siControl. Error bars represent ± SEM. Values are scored as a fold-change relative to that of siControl-treated cells. Statistical significance *P < 0.05. (d) Confocal microscopy results showing intracellular HIV-1 Gag distribution following siRNA treatment and rescue in HeLa M cells. From left to right, DAPI, Gag and merged columns. Cells were sequentially transfected with siControl or siDDX17 and then a second round of siRNA together with pLAI, with or without increasing concentrations of siDDX17 rescue expressor construct. Cells were harvested 48 h post-co-transfection and dual stained for DAPI and Gag. (e) Western blot showing the effect of DDX17 knockdown on cellular CA-p24. Cells were sequentially transfected with siControl or siDDX17 and then a second round of siRNA together with pLAI, with or without increasing concentrations of siDDX17 rescue expressor construct. (f) Western blot showing the effect of DDX17 knockdown on Rev, Nef and Vif levels. Cells were sequentially transfected with siControl or siDDX17 and then a second round of siRNA together with pLAI, with or without increasing concentrations of siDDX17 rescue expressor construct. (g) Western blot showing the effect of DDX17 knockdown and rescue on Env and Vpu levels. (h) Western blot showing the effect of DDX17 knockdown on Tetherin levels.
HIV-1 env/vpu mRNA is bicistronic, with the Vpu start codon upstream of the Env AUG codon. Knockdown of endogenous DDX17 resulted in 80%–90% reduction of expression of both viral proteins, as quantified using ImageJ (Fig. 3g).
Since Vpu downregulates Tetherin [39], [40], [41], [42], the reduced Vpu expression upon knockdown of DDX17 would be predicted to result in upregulation of Tetherin and indeed we observed enhanced Tetherin expression (Fig. 3h) that was reversed on DDX17 rescue. This increased Tetherin undoubtedly reduces virion release contributing to the decrease in detectable supernatant CA-p24. Hence, the intracellular Gag accumulation noted on immunofluorescence, ELISA and cellular CA-p24 Western blot and the decrease in extracellular p24 are a result of both reduced splicing and reduced viral release. Tetherin downregulation, which occurs readily on restoration of Vpu rescue, then assists depletion of the intracellular Gag; however, the modest upregulation of HIV production with increasing DDX17 means that the intracellular level does not return to baseline.
DDX17 interacts with splicing factors associated with the 3′ss A4/A5 cluster
A schematic diagram of the HIV-1 genome with relevant splice site positions is shown in Fig. 4a. DDX17 has been implicated in alternative splicing [24]. Given the observation that depletion of DDX17 reduces levels of spliced RNA, we sought evidence of functional links with splicing factors. We found that DDX17 co-immunoprecipitates with the upstream heterodimer (U2AF65/35) and the ESE binding factor SRSF1/SF2 raising the possibility that it acts as a bridge between the two. DDX17 also interacts with U1C (Fig. 4b). The interaction with SRSF1/SF2 and U1C is RNA dependent as there were no detectable bands on subjecting the co-immunoprecipitated samples to RNase (Fig. 4b). The interaction between DDX17 and U2AF65 was unaffected by RNase, but this interaction might be mediated via double-stranded RNA since the RNase we use digests only single-stranded RNA (Fig. 4b). DDX17 knockdown does not affect the level of U2AF65 or that of the trans-acting ESEs, SR protein SRSF1/SF2 (Fig. 4c). Similarly, knockdown of DDX17 does not alter the expression of U1C, an essential component of the U1snRNP complex required for the selection and stabilization of the downstream 5′ss donor (Fig. 4c).
Fig. 4.

DDX17 interacts with essential splicing factors. (a) Schematic representation of HIV-1 genome organization and mapping of the splice sites described in this manuscript. Other known sites have been omitted for simplicity. (b) Exogenously expressed Myc-DDX17 co-immunoprecipitates with endogenous U2AF65, SRSF1/SF2 and U1-C. IgG lane, immunoprecipitation of proteins treated with isotype control matched antibody. In RNase-treated lane, samples were subjected to digestion with RNase A. (c) HeLa M cells were sequentially transfected with siControl or siDDX17 and then a second round of siRNA together with pLAI, with or without increasing concentrations of siDDX17 rescue expressor construct. Expression of U2AF65, SRSF1/SF2 and U1-C, following knockdown and rescue of DDX17 was detected by Western blotting, using their respective antibodies. GAPDH was detected to monitor sample loading. (d) Effect of overexpression of DDX5 as attempted rescue of HIV-1 virus production following endogenous DDX17 depletion. Cells were sequentially transfected with siControl or siDDX17 and then a second round of siRNA together with pLAI, with or without increasing concentrations of DDX5 expressor construct. (e) Western blot for DDX17 and DDX5 expression. The graph is a representative of three independent experiments done in triplicate. Bars represent mean of triplicate samples ± SEM. Statistical significance: **P < 0.01.
DDX17 acts independently of DDX5
DDX5 and DDX17 have many functions in common and are often treated as functional analogs. Previous studies have established that the two helicases exist as monomers, homodimers or heterodimers [20]. Thus, we next queried whether the splicing role of DDX17 in HIV-1 was dependent or independent of DDX5. We attempted to rescue DDX17 knockdown by overexpression of DDX5, but despite successful overexpression (Fig. 4d), this failed to restore virus production (Fig. 4e). These findings underscore that the role of DDX17 is critical for HIV-1 replication and that it cannot be substituted by DDX5.
DDX17 utilizes distinct RNA binding motifs for efficient HIV replication
DDX17 has many functional domains common to DEAD box proteins (Fig. 5a). We introduced individual point mutations into known RNA and ATPase binding motifs and cloned each mutant into the siDDX17 rescue backbone. There was no detectable cellular toxicity following treatment with siDDX17 with or without increasing concentrations of any of the various rescue plasmids (Fig. S1c).
Fig. 5.

DDX17 utilizes distinct RNA binding motifs. (a) Schematic representation of DDX17 and the annotated individual point mutations that are defective for the glycine doublet function and RNA/ATPase binding activities. HeLa M cells were sequentially transfected with siControl or siRNA targeting DDX17 (siDDX17 with or without increasing concentrations of siDDX17-resistant DDX17–T253R + E255V + L256A; siDDX17-resistant DDX17–S356L; siDDX17-resistant DDX17–R508Q or siDDX17-resistant G280D). (b) Effect on HIV-1 relative virus production following endogenous DDX17 depletion and rescue with DDX17–T253R + E255V + L256A. (c) Western blot showing DDX17 knockdown and rescue with DDX17–T253R + E255V + L256A. (d) Effect on HIV-1 relative virus production following DDX17 knockdown and rescue with DDX17–T301D. (e) Western blot showing DDX17 knockdown and rescue with DDX17–T301D. (f) Effect on HIV-1 relative virus production following DDX17 knockdown and rescue with DDX17–S356L. (g) Effect on HIV-1 relative virus production following DDX17 knockdown and rescue with DDX17–R508Q. (h) Western blot showing DDX17 knockdown and rescue with DDX17–S356L. (i) Western blot showing DDX17 knockdown and rescue with DDX17–R508Q. (j) Effect on HIV-1 relative virus production following DDX17 knockdown and rescue with DDX17–G280D. (k) Western blot showing DDX17 knockdown and rescue with DDX17–G280D. Each graph is a representative of two independent experiments done in triplicate. Bars represent mean of triplicate samples ± SEM. Statistical significance *P < 0.05, **P < 0.01.
We mapped the motifs required for control of HIV replication by DDX17. Motif 1a (PTRELA) and 1b (TPGR) are known to be necessary for RNA binding in conjunction with motifs IV and V [43]. Individual point mutations were introduced into motif 1a (PTRELA to PRRVAA) or motif 1b (TPGR to DPGR) to abrogate ATP/helicase and RNA binding activities, respectively [44]. The latter mutant also has a detrimental effect on ATPase, RNA crosslinking and helicase activity [44]. In the context of rescue experiments, despite successful expression, the motif 1a mutation failed to restore relative virus production (Fig. 5b and c) indicating its importance for splicing control. By contrast, the point mutation in motif 1b (DDX17–T301D) successfully restored relative virus production (Fig. 5d and e) confirming that it is dispensable. DDX17 RNA binding was also disrupted by introducing individual point mutations in known RNA binding motifs, DDX17–S356L and DDX17–R508Q, respectively (Fig. 5a). Both mutants failed to restore virus production in DDX17-depleted cells (Fig. 5f and g), despite successful expression (Fig. 5h and i), indicating that both are required for function. Between these two motifs is a highly conserved glycine doublet that facilitates the formation of a sharp turn within the loop between them. It is important for protein–protein interactions [45] and it is needed for motifs 1a and 1b to retain RNA binding activity. Substituting the second glycine residue with aspartic acid disrupts the formation of the turn. This mutant has also been shown to lack ATPase and RNA helicase activity but retains ATP binding activity [44]. We generated DDX17–G280D (Fig. 5a) to interrogate the functionality of the glycine doublet in HIV rescue experiment. DDX17–G280D successfully restored virus production (Fig. 5j and k). These findings show that neither the conformational effect of the GG doublet sharp turn between motif 1a and 1b nor its protein–protein interaction function is required in HIV-1 replication. This confirms the finding that the two motifs act independently in affecting HIV (Fig. 5b and d).
At its N terminus, DDX17 has three RGG boxes, which feature in other well-characterized cellular splicing factors [46]. RGG boxes facilitate protein–protein interactions and are involved in RNA binding activities. Substitution of arginine for glutamine (DDX17–R100Q) in the second RGG box (Fig. S2a) prevented rescue (Fig. S2b) despite successful expression (Fig. S2c). Thus, the second RGG box is essential for HIV replication.
DDX17 Q motif, Walker A and DEAD box signature motifs (Walker B) are dispensable for HIV-1 replication
The Q motif is necessary for binding of ssRNA, and through its interaction with motif 1a, it is important for ATP binding, acting as a regulator of ATPase activity. Substituting the highly conserved glutamine residue (aa198) by alanine abrogates ATP/RNA binding [47], [48]. The siDDX17-resistant DDX17–Q198A construct successfully restored virus production (Fig. 6a–c).
Fig. 6.

DDX17 Q, Walker A and Walker B motifs are dispensable in HIV-1 replication. (a) Schematic representation of DDX17 and the annotated individual point mutations in the Q motif that abrogates ATP binding and hydrolysis, and in the Walker A and Walker B (DEAD) motifs that are defective for ATPase and helicase activity, respectively. HeLa M cells were sequentially transfected with siControl or siRNA targeting DDX17 (siDDX17 with or without increasing concentrations of siDDX17-resistant DDX17–Q198A or siDDX17-resistant DDX17–K221A or siDDX17-resistant DDX17–E326Q). (b) Effect on HIV-1 relative virus production following endogenous DDX17 knockdown and rescue with DDX17–Q198A. (c) Western blot showing DDX17 knockdown and rescue with DDX17–Q198A. (d) Effect on HIV-1 relative virus production following endogenous DDX17 knockdown and rescue with silent Walker A (DDX17–K221A). (e) Western blot showing DDX17 knockdown and rescue with DDX17–K221A. (f) Effect on HIV-1 relative virus production following endogenous DDX17 knockdown and rescue with silent Walker B (DDX17–E326Q). (g) Western blot showing DDX17 knockdown and rescue with DDX17–E326Q. Each graph is a representative of three independent experiments done in triplicate ± SEM. Statistical significance: *P < 0.05, **P < 0.01.
Both ATP and helicase unwinding activities are required for most of the known functions of DEAD box helicases. We introduced siRNA-resistant DDX17 constructs with either abrogated Walker A or Walker B activity to DDX17-depleted cells (Fig. 6a). Mutating the conserved lysine to an alanine residue in Walker A abolishes its ATPase activity by reducing the affinity for and the rate of hydrolysis of ATP [49]. However, DDX17–K221A successfully restores virus production (Fig. 6d and e).
The Walker B motif (DEAD) is well characterized for its role in helicase activity [45]. A point mutation (DEAD to DQAD) that abolishes helicase activity (siDDX17-resistant DDX17–E326Q) (Fig. 6a) successfully restored virus production (Fig. 6f and g). Thus, none of the three motifs—Q, Walker A or Walker B—are required for facilitating HIV replication.
The C terminus of DDX17 is essential for HIV-1 replication
The DDX17 C terminus has a serine–glycine motif [19] characteristically found in RNA binding proteins [50], [51] where it facilitates homo- and heterodimeric interactions that lead to higher order RNP complex formation and allow co-operative RNA binding [52], [53]. Co-operative RNA binding increases the efficiency of RNA splicing. The DDX17 C terminus also contains a poly-proline tract and a transactivation domain believed to be involved in protein–protein interactions [51], [54], [55]. Having shown interactions between DDX17 and certain splicing factors, we constructed a series of DDX17 C terminal truncation mutants to seek interaction regions responsible for this.
Expressing C-terminal truncations of DDX17 did not cause cytotoxicity (Fig. S3a). DDX17–T516, an siDDX17-resistant expressor with a stop codon at amino acid 516, failed to restore virus production despite successful rescue of DDX17 protein production (Fig. 7a and b), indicating that the DDX17 C terminus is essential for facilitating efficient HIV-1 splicing. DDX17–T690 failed to restore virus production (Fig. 7c and d). However, DDX17–T710 successfully did (Fig. 7e and f). These data suggest that the 20-amino-acid region between position 690 and 710 in the C terminus region is critical for DDX17 to exert its role in rescuing HIV.
Fig. 7.

DDX17 C terminus is critical for HIV-1 replication. HeLa M cells were sequentially transfected with siControl or siRNA targeting DDX17 (siDDX17 with increasing concentrations of siDDX17-resistant DDX17–T516 or siDDX17-resistant DDX17–T690 or siDDX17-resistant DDX17–T710). Supernatant was used to infect CD4 + TZM-bl indicator cells for infectivity assay, 48 h after second dose transfection. (a) Effect on HIV-1 relative virus production following endogenous DDX17 depletion and rescue with DDX17–T516. (b) Western blot showing DDX17 knockdown and rescue with DDX17–T516. (c) Effect on HIV-1 relative virus production following endogenous DDX17 depletion and rescue with DDX17–T690. (d) Western blot showing DDX17 knockdown and rescue with DDX17–T690. (e) Effect on HIV-1 relative virus production following endogenous DDX17 depletion and rescue with DDX17–T710. (f) Western blot showing DDX17 knockdown and rescue with DDX17–T710. Each graph is a representative of at least two independent experiments done in triplicate. Bars represent mean of triplicate samples ± SEM. Statistical significance *P < 0.05.
We investigated whether a mutant just proximal to DDX17–T690 or distal to DDX17–T710 would exhibit the same phenotype. siDDX17-resistant expressor DDX17–T680 failed to restore virus production (Fig. S3b), despite being sufficiently expressed (Fig. S3c). However, siDDX17 rescue expressor DDX17–T717 successfully restored virus production (Fig. S3d and e). It is likely that the failure of DDX17–T690 to restore virus production is a result of loss of interaction with U2AF65 and SRSF1/SF2 (Fig. S3f). Therefore, the region between 690 and 710 at the C terminus of DDX17 is also essential.
Discussion
Splicing is the major post-transcriptional mechanism used by HIV to control its lifecycle and splicing control is a potentially vulnerable therapeutic target. DDX17 has been suggested to be involved in several aspects of HIV replication including interaction with HIV Rev [29] and modulation of Gag processing [27]; however, detailed functional information about its targets of activity and mechanism of action has been absent until now. Previous studies have sought evidence for the role of DDX17 in HIV and there is consensus that it has a significant effect on viral replication. Naji et al. investigated DDX17 as a Rev interacting protein. Their study involved prior knockdown of DDX17 followed by HIV infection of knocked down cells, and like us, they detected reduced HIV production. Their studies demonstrated reduction in Env protein but also Gag levels unlike ours. This may reflect a more profound generalized effect of DDX17 depletion on processes other than splicing caused by the prior knockdown, and unlike us, they did not investigate the effects of subsequent rescue. We showed that DDX17 depletion was accompanied by a slight increase in unspliced HIV transcripts unlike Lorgeoux et al. [27]. The difference in our observations might be attributable to different experimental techniques used. We depleted DDX17 using sequential siRNA knockdown conditions, achieving high-efficiency reduction in levels of the helicase, and in our studies, the timing of assays differed. Lorgeoux et al. used shRNA knockdown and harvested their cells at 48 h, whereas our studies were done at 72 h post-knockdown when possibly further reduction of residual DDX17 had been achieved [27]. Both Lorgeoux and ourselves agree that DDX17 affects HIV splicing. In our hands, the selective effects on splicing are highly reproducible and confirm our previous siRNA knockdown screen, indicating that DDX17 is essential for HIV replication. We agree that our studies do not exclude additional functional roles for this helicase in HIV replication as suggested by Naji et al. especially given its versatility in many aspects of RNA processing and work to define whether aspects such as viral RNA stability are influenced by DDX17 knockdown, among other possibilities, would be of value. From our data, however, the clear conclusion is that DDX17 is a pivotal component of splicing regulation in HIV. We detected no evidence that DDX17 had a significant effect on transcription since knockdown resulted in reduced viral export without reducing unspliced viral RNA or total Gag protein production, consistent with a lack of effect on production of Tat which predominantly utilizes the A3 splice site. In the context of HIV, DDX17 interacts with U2AF65, SRSF1/SF2 and U1C splicing factors likely acting as an essential co-factor or stabilizing the RNP complex (see graphical abstract). We suggest that DDX17 may act as a bridge between U2AF65 and SRSF1/SF2; however, this does not exclude that these proteins may interact directly without DDX17. Hence, the graphical abstract provided is only one of several possible models of how DDX17 could be working. Alternatives include the alteration by DDX17 of RNA folding to expose binding sites for host factors that modulate splicing. Such effects may only require the RNA binding capacity of DDX17 and not its helicase function as determined by the mutational analysis. Our findings suggest that the helicase RNA function of the Walker B motif (Fig. 6a and f) is dispensable for the role of DDX17 in potentiating HIV replication. Interestingly, Lorgeoux et al. [27] observed that Walker B motif maybe required for Gag processing. In this case, the differences are likely due to experimental design as Lorgeoux et al. used the Walker B motif (DQAD) mutant in overexpression experiments, whereas we used these mutants in the context of knockdown and rescue. The DQAD mutant has a dominant negative effect when overexpressed, possibly due to recruitment of different interacting factors.
Other differences between our findings and those of previous studies may be attributable to the complex interplay between DDX5 and DDX17 as they are capable of forming heterodimers, DDX5 also exerts non-reciprocal control over DDX17 production, and DDX5 and DDX17 do have some interchangeable cellular roles [21], [22]. Our study used siRNA knockdown that was proven to specifically affect either DDX17 or DDX5 without reciprocal effects, and our work is the first to demonstrate a comprehensive analysis by specific rescue of expression using wild-type and mutated DDX17 expressors. We clearly showed that we could not rescue DDX17 knockdown-mediated inhibition of HIV production by overexpression of DDX5. We did attempt to construct rescue mutants with mutations in regions involved in DDX5/DDX17 heterodimer formation, but these were not successful as the interacting regions are also required in HIV replication.
By selectively inhibiting usage of the A4/A5 ss DDX17 knockdown results in an 80%–90% reduction in Rev, Nef and Env/Vpu protein expression. Despite significant depletion of Rev, there was still enough of this regulatory protein to affect export of unspliced and partially spliced transcripts, indicating that a low abundance of Rev molecules is sufficient for export of HIV-1 transcripts presumably since Rev can shuttle and it is known to have a long half-life of 16 h [56]. Reduction in Env expression is likely a major component of the impaired infectivity of virions that are produced under DDX17 knockdown particularly given the paucity (~ 10 copies) of gp120/gp41 heterotrimeric spikes on normal virions [57]. This is compounded by the raised Tetherin levels secondary to Vpu depletion.
Our analysis of DDX17 functional regions revealed a surprising selectivity in the domains necessary for carrying out splicing control, with distinct RNA binding and ATPase motifs, namely, Ia, III and VI motifs and the C terminus being essential for efficient HIV replication. The Q motif and the Walker A, Walker B (DEAD), glycine doublet and Ib regions were all dispensable in contrast to their known functions in other DEAD box proteins and in keeping with the multifunctional nature of the helicase family. Perhaps the most unexpected was the independence of motifs 1a (necessary) and motif 1b (dispensable) as these regions are suggested to act cooperatively as was shown for eIF4A [43]. However, this was consistent with finding that the glycine doublet was dispensable since its major role is as a structural link between them facilitating their combined activity.
HIV replication does not require the Walker B helicase in DDX17. Mutations in motif III, which is essential for splicing at A4/A5 ss, have been shown to cause minor effects on ATP binding, hydrolysis and RNA binding but cause a significant loss of helicase activity [24], [58]. Motif III is situated in a region essential for conformational changes that facilitate efficient RNA binding. Hence, it is likely that it is the RNA binding activity of motif III that is important and plausible that HIV splicing is independent of any specific helicase-mediated unwinding activity.
Superimposing the mutagenic data on a reconstructed core domain of DDX17 shows that regions dispensable for splicing control are clustered around the AMP binding site, suggesting that either AMP is not functionally required or there are no dynamic processes dependent on the structural effects of bound AMP (Fig. 8b). Motif 3 is critical for helicase activity, and its enzymatic function is necessary for the role of DDX17 (Fig. 8b). Motif 1a is surface exposed and its ssRNA binding function appears crucial, whereas the second ssRNA binding domain in motif 1b does not appear to be necessary (Fig. 8b).
Fig. 8.

DDX17 rescue mutants and core DDX17 structure/function model. Domains marked in blue are essential for DDX17 splicing function in HIV-1 while those marked in green are dispensable.
In addition to the previously published roles of DDX17 in modulating Gag processing [27] and interactions with Rev and Gag [29], [59], we now propose that DDX17 is essential for modulating HIV splicing. We postulate based on our findings that DDX17 modulates HIV splicing by interacting with splicing factors that are critical for the selection of the A4/5 splice site cluster. DDX17 may exert its effect by acting as a bridge or co-factor between the U2AF65/35 heterodimeric factor and the downstream U1C in complex with the U1C snRNP. The resultant complex likely adds to the stability and potentiation of the otherwise intrinsically weak A4/5 cluster. Our identification of a critical and unique role for DDX17 and the functional mapping of motifs required in potentiating HIV-1 splicing has revealed a specific and potentially vulnerable therapeutic target to control HIV replication. In none of our studies did manipulation of DDX17 levels or expression of mutants lead to detectable adverse effects on the cells, suggesting that the level of functional redundancy for cellular processes would leave the cell unharmed by antiviral strategies targeting this protein.
Materials and Methods
Cell lines and plasmid DNA
HeLa M, a derivative of HeLa cells [60], was obtained from ATCC. TZM-bl, carrying two HIV-1 LTR-driven reporter genes, firefly luciferase and Escherichia coli β-galactosidase [61], [62], is a HeLa cell clone that stably expresses high levels of CD4, CXCR4 and CCR5 receptors, and was obtained from NIH AIDS Research and Reference Reagent Program. HeLa M and TZM-bl cells were grown in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum. Jurkat cells were grown in RPMI supplemented with 10% fetal bovine serum. Generation of the siDDX17-resistant constructs was performed using a site-directed mutagenesis kit (Agilent). See Table S1 for primer sequences. Myc DX17 was a kind gift from Dr Frances-Fuller Pace (Dundee). An empty cloning vector pBluescript (Stratagene) was used to maintain a constant amount of DNA in transfection experiments. pLAI is a full-length molecular clone of HIV-1 strain LAI for the expression of wild-type virus [63].
Cell viability
CellTiter-Glo Luminescent Cell Viability Assay (Promega) was adapted for 96-well half-area plates and assay performed as previously described [64].
siRNA and plasmid DNA transfections
siDDX17, siDDX3 and siControl were purchased from Life Technologies. See Table S2 for siRNA sequences. Sequential transfections were performed at 24 h (1 μl of 20 pmol siRNA alone) and 48 h (siRNA + DNA plasmids) post-seeding in 24-well plates. A constant amount, 200 ng, of pLAI was used. In the rescue experiments, in addition to siDDX17 and pLAI, HeLa cells were co-transfected with increasing amounts of various siDDX17-resistant constructs. All transfections were carried out using jetPRIME(Polyplus) transfection reagent per the manufacturer’s protocol. Cells and supernatant were harvested 48 h post-co-transfection and the supernatant was used to infect CD4 + TZM-bl cells to assay for viral infectivity. To assess siRNA-mediated knockdown and rescue, cell lysates were subjected to Western blotting.
Nucleofection
Amaxa Nucleofector II Device and Amaxa cell line Nucleofector Kit V from Lonza were used for nucleofection of Jurkat cells per the manufacturer’s protocol. Cells were sub-cultured in 12-well plates for 48 h and 1 × 106 cells per condition were used for nucleofection with 2 μg DNA plasmid of either test sample or control plasmid (pmaxGFP Vector) together with 30 pmol/sample siRNA (either siControl or siDDX17). Forty-eight hours post-nucleofection, the medium was replaced with fresh RPMI 1640. Cell culture supernatant and cell lysates were harvested 96 h post-nucleofection.
Virus production assay
Virus infectivity in culture supernatants was determined by infecting TZM-bl indicator cells, based on Tat-dependent upregulation of LTR-driven luciferase expression. Cell lysates were harvested 48 h post-infection. Five microliters of cell lysate was transferred to a 96-well half-area plate, to which 25 μl of Luciferase Assay Reagent was added. Firefly luciferase activity was quantified using the Luciferase Assay System with the Glomax 96 Microplate Luminometer (Promega).
Nuclear extracts preparation and co-immunoprecipitation
HeLa cells were co-transfected with the appropriate Myc-tagged constructs and pLAI. Cells were harvested 48 h post-transfection and nuclear extracts prepared as previously described [65], [66] with the following buffers and modifications: Lysis buffer containing 10 mM Hepes (pH 7.9), 1.5 mM MgCl2, 10 mM KCl and 0.5 mM DTT supplemented with protease inhibitors (Roche). This was followed by 5 μl per 500-μl reaction mix RNase A (Sigma) treatment for 10 min at 4 °C. Samples were incubated overnight with 2 μg of either Myc or isotype control antibody in CO-IP buffer [20 mM Hepes (pH 7.9), 150 mM NaCl, 0.5 mM DTT, 20% (v/v) glycerol, 10 mM NaF and protease inhibitors]. Samples were then incubated with A/G ultralink Sepharose beads (Thermoscientific) for 3 h at 4 °C. After washing with CO-IP buffer, samples were eluted in 2 × Laemmli sample buffer. Proteins were separated by SDS-PAGE followed by Western blotting using standard protocol.
Western blotting
Proteins were separated by SDS-PAGE before being subjected to Western blot. The following antibodies were from Santa Cruz: p24 IF antibody (sc69728), Myc (9E10), DDX17 N Terminus (sc-398168) and DDX17 (sc-271112); Abcam: SRSF1/SF2 (ab129108), Tetherin/BST2 (ab14694), Vpu (ab134061), U2AF65 (ab37530), rabbit isotype-matched control IgG (ab27478), rabbit c-Myc (ab39688), Nef (ab42355), Rev (ab855299) and GAPDH (ab9485). Vif antibody was from GeneTex (GTX80393). U1C antibody was obtained from Bethyl Laboratories (A303-947A). DDX5 antibody PAb-204 was a kind gift from Dr. Frances Fuller-Pace (Dundee). HIV-1 p55/p24 (ARP, NIBSC) [67]. Envelope/gp120 Mouse Monoclonal was obtained from CFAR. Mouse IgG isotype control (401402) was obtained from Biolegend. The following secondary antibodies were from Cell Signalling: horseradish peroxidase-conjugated anti-mouse (#7076, 1:2000), and from Santa Cruz: horseradish peroxidase-conjugated anti-rabbit (#2123, 1:2000). Detection was carried out using ECL prime (Amersham) per the manufacturer’s instructions.
Immunofluorescence assay
HeLa cells were seeded in 8-well chamber slides (Millipore). Cells were sequentially transfected with siControl or siDDX17 and then a second round of siRNA together with pLAI, with or without increasing concentrations of siDDX17 rescue expressor construct. Cells were harvested 48 h post-co-transfection and fixed with 4% paraformaldehyde followed by washing with PBS and permeabilizing with 0.1% Triton X-100 for 10 min at room temperature. Cells were then treated with 5% BSA blocking solution for 45 min, followed by incubation with the relevant primary antibodies (Gag, Isotype matched control or DDX17) for 1 h. Cells were washed with PBST prior to incubation with the relevant secondary antibodies. Secondary antibodies used were as follows: Alexa-Fluor 647 donkey anti-mouse conjugated secondary antibody, Alexa Fluor 555 goat anti-rabbit, or isotype-matched control antibodies (Santa Cruz). Vectashield with 4′,6-diamidino-2-phenlindole (DAPI) (Vector) was applied before coverslip was mounted. Cells were visualized using Leica Sp5 confocal laser microscope. All images were digitally recorded and merged using the Leica software.
Enzyme-linked immunosorbent assay
Extracellular and intracellular/supernatant CA-p24 levels were quantified by ELISA (Alto) with slight modification [68].
RT-qPCR
HeLa cells were sequentially transfected with siRNA and DNA plasmids as previously described in 24-well plates. Total RNA was extracted using RNeasy kit (Qiagen) per the manufacturer’s instructions. Five micrograms of total RNA was used for complementary DNA synthesis with random hexamers, using reverse-transcription kit (Applied Biosystems). Applied Biosystems 7500 HT Fast Real Time PCR System (Life Technologies) and SYBR green qPCR reagent (Applied Biosystems) were used for qPCR. Primer sequences and thermo-cycling conditions have been previously described [69], [70] and are shown in Tables S1 and S3, respectively. The specificity of qPCR products was examined by a dissociation curve and relative abundance of transcripts was calculated using the 2−∆∆CT method [71]. Results were normalized to actin, and mean results of triplicates were converted to ratios relative to siControl samples.
Statistical analyses
Statistical analyses were performed in Excel and GraphPad Prism version 4. The data for viral infectivity and CA-p24 production were analyzed by unpaired two-tailed Student’s t-test, with Welch’s correction.
Modeling methods
The sequence of the N-terminal and C-terminal domains was submitted individually to the PHYRE modeling server [72]. PDB helicase structure 3fe2 was the template for the N-terminal domain subsequently extended using 4a4d. PDB helicase structure 4pxa was used for the C-terminal domain. Finally, PDB helicase structure 2i4i was used to guide the relative positions of the two domains and add the AMP at the active site. The overall domain arrangements and geometry of the model were manually improved by rebuilding in COOT [73]. The model was renumbered to the DD17_HUMAN Isoform 2 UniProt residue numbering and covered UniProt residues 49-473. Molecular graphic display used ICM browser (http://www.molsoft.com/).
The following are the supplementary data related to this article.
Cell viability. (a) DDX17 plasmid DNA was mutated in the seed sequence of both siRNAs which target DDX17 knockdown. Red arrows and letters indicate the base-pair change in the rescue plasmid. All base changes are silent mutations and do not alter the amino acid sequence of DDX17. (b and c) HeLa cells were seeded in a 96-well at 4 × 103 cells per well and sequentially transfected with 10 pmol of siControl or siDDX17 and then a second round of siRNA together with or without increasing concentrations of various siDDX17 rescue constructs. The number of viable cells was determined by measuring the amount of ATP present as an indicator of metabolically active cells. This was then calculated as a percentage relative to untransfected cells. Puromycin-treated cells were used as positive controls. Bars represent mean of duplicate samples ± SEM.
DDX17 second RGG box is indispensable. (a) Schematic representation of DDX17 and the annotated individual point mutation that is defective for RNA binding activity. HeLa M cells were sequentially transfected with siControl or siRNA targeting DDX17 (siDDX17 with or without increasing concentrations of siDDX17-resistant DDX17–R100Q). (b) Effect on HIV-1 relative virus production following endogenous DDX17 knockdown and rescue with DDX17–R100Q. (c) Western blot showing DDX17 knockdown and rescue with DDX17–R100Q. Each graph is a representative of at least two independent experiments done in triplicate. Bars represent mean of triplicate samples ± SEM. Statistical significance **P < 0.01.
DDX17 C terminus is essential for HIV infectivity. (a) HeLa cells were seeded in a 96-well at 4 × 103 cells per well and sequentially transfected with 10 pmol of siControl or siDDX17 and then a second round of siRNA together with or without increasing concentrations of various DDX17 C terminal truncated siDDX17 rescue constructs. The number of viable cells was determined by measuring the amount of ATP present as an indicator of metabolically active cells. This was then calculated as a percentage relative to untransfected cells. Puromycin-treated cells were used as positive controls. Bars represent mean of duplicate samples ± SEM. (b) Effect on HIV-1 relative virus production following endogenous DDX17 depletion and rescue with DDX17–T680. (c) Western blot showing DDX17 knockdown and rescue with DDX17–T680. (d) Effect on HIV-1 relative virus production following endogenous DDX17 depletion and rescue with DDX17–T717. (e) Western blot showing DDX17 knockdown and rescue with DDX17–T717. (f) Exogenously expressed Myc-DDX17 and Myc-DDX17–T717 co-immunoprecipitates with endogenous U2AF65 and SRSF1/SF2. IgG lane, immunoprecipitation of proteins treated with isotype control matched antibody. Myc-DD17-T690 fails to co-immunoprecipitate with both U2AF65 and SRSF1/SF2. Each graph is a representative of three independent experiments done in triplicate ± SEM. Statistical significance: *P < 0.05.
Supplementary tables
Acknowledgments
We thank Frances Fuller-Pace for the kind gifts of plasmids (pMyc DDX5 and pMyc DDX17) and PAb-204 antibody. We would like to thank Dr Martyn Symmons for his helpful contributions in assembling the DDX17 molecular model and the images obtained. The work was supported by the Wellcome Trust and the Cambridge Biomedical Research Centre. A.M.L.L. is supported through Clinical Academic Reserve funding. Past collaborations with the late K.-T. Jeang underpinning these studies is warmly acknowledged. The authors declare that they have no competing interests.
Author Contributions: Conceptualization, N.S. and A.M.L.L.; Methodology, N.S., C.A.W., A.M.V., J.C.K. and A.M.L.L.; Investigation, N.S., A.M.V. and C.A.W.; Writing – Original Draft, N.S., and A.M.L.L.; Writing – Review and Editing, N.S., C.A.W., A.M.V., J.C.K., and A.M.L.L.; Funding Acquisition, N.S., and A.M.L.L.; Resources, N.S., C.A.W., A.M.V., and A.M.L.L.; Supervision, J.C.K., and A.M.L.L.
Declarations/Conflict of Interests: The authors declare that no competing interests exist.
Edited by Eric O. Freed
References
- 1.Kim D.W., Gwack Y., Han J.H., Choe J. C-terminal domain of the hepatitis C virus NS3 protein contains an RNA helicase activity. Biochem. Biophys. Res. Commun. 1995;215:160–166. doi: 10.1006/bbrc.1995.2447. [DOI] [PubMed] [Google Scholar]
- 2.Kadare G., Haenni A.L. Virus-encoded RNA helicases. J. Virol. 1997;71:2583–2590. doi: 10.1128/jvi.71.4.2583-2590.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yedavalli V.S., Neuveut C., Chi Y.H., Kleiman L., Jeang K.T. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell. 2004;119:381–392. doi: 10.1016/j.cell.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 4.Fang J., Kubota S., Yang B., Zhou N., Zhang H., Godbout R., Pomerantz R.J. A DEAD box protein facilitates HIV-1 replication as a cellular co-factor of Rev. Virology. 2004;330:471–480. doi: 10.1016/j.virol.2004.09.039. [DOI] [PubMed] [Google Scholar]
- 5.Brass A.L., Dykxhoorn D.M., Benita Y., Yan N., Engelman A., Xavier R.J., Lieberman J., Elledge S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 6.Roy B.B., Hu J., Guo X., Russell R.S., Guo F., Kleiman L., Liang C. Association of RNA helicase a with human immunodeficiency virus type 1 particles. J. Biol. Chem. 2006;281:12625–12635. doi: 10.1074/jbc.M510596200. [DOI] [PubMed] [Google Scholar]
- 7.Bolinger C., Sharma A., Singh D., Yu L., Boris-Lawrie K. RNA helicase A modulates translation of HIV-1 and infectivity of progeny virions. Nucleic Acids Res. 2010;38:1686–1696. doi: 10.1093/nar/gkp1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xing L., Liang C., Kleiman L. Coordinate roles of Gag and RNA helicase A in promoting the annealing of formula to HIV-1 RNA. J. Virol. 2011;85:1847–1860. doi: 10.1128/JVI.02010-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lever A.M., Jeang K.T. Insights into cellular factors that regulate HIV-1 replication in human cells. Biochemistry. 2011;50:920–931. doi: 10.1021/bi101805f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeang K.-T., Yedavalli V. Role of RNA helicases in HIV-1 replication. Nucleic Acids Res. 2010;34:4198–4205. doi: 10.1093/nar/gkl398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ranji A., Boris-Lawrie K. RNA helicases: emerging roles in viral replication and the host innate response. RNA Biol. 2010;7:775–787. doi: 10.4161/rna.7.6.14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ajamian L., Mouland A.J. Implications of RNA helicases in HIV-1 replication: possible roles in latency. Curr. HIV Res. 2011;9:588–594. doi: 10.2174/157016211798998835. [DOI] [PubMed] [Google Scholar]
- 13.Lorgeoux R.P., Guo F., Liang C. From promoting to inhibiting: diverse roles of helicases in HIV-1 Replication. Retrovirology. 2012;9:79. doi: 10.1186/1742-4690-9-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen C.Y., Liu X., Boris-Lawrie K., Sharma A., Jeang K.T. Cellular RNA helicases and HIV-1: insights from genome-wide, proteomic, and molecular studies. Virus Res. 2013;171:357–365. doi: 10.1016/j.virusres.2012.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jarmoskaite I., Russell R. DEAD-box proteins as RNA helicases and chaperones. Wiley Interdiscip. Rev. RNA. 2011;2:135–152. doi: 10.1002/wrna.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woodson S.A. Taming free energy landscapes with RNA chaperones. RNA Biol. 2010;7:677–686. doi: 10.4161/rna.7.6.13615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parsyan A., Svitkin Y., Shahbazian D., Gkogkas C., Lasko P., Merrick W.C., Sonenberg N. mRNA helicases: the tacticians of translational control. Nat. Rev. Mol. Cell Biol. 2011;12:235–245. doi: 10.1038/nrm3083. [DOI] [PubMed] [Google Scholar]
- 18.Williams C.A., Abbink T.E., Jeang K.T., Lever A.M. Identification of RNA helicases in human immunodeficiency virus 1 (HIV-1) replication—a targeted small interfering RNA library screen using pseudotyped and WT HIV-1. J. Gen. Virol. 2015;96:1484–1489. doi: 10.1099/vir.0.000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamm G.M., Nicol S.M., Fuller-Pace F.V., Lamond A.I. p72: a human nuclear DEAD box protein highly related to p68. Nucleic Acids Res. 1996;24:3739–3747. doi: 10.1093/nar/24.19.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogilvie V.C., Wilson B.J., Nicol S.M., Morrice N.A., Saunders L.R., Barber G.N., Fuller-Pace F.V. The highly related DEAD box RNA helicases p68 and p72 exist as heterodimers in cells. Nucleic Acids Res. 2003;31:1470–1480. doi: 10.1093/nar/gkg236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dardenne E., Polay Espinoza M., Fattet L., Germann S., Lambert M.P., Neil H., Zonta E., Mortada H., Gratadou L., Deygas M., Chakrama F.Z., Samaan S., Desmet F.O., Tranchevent L.C., Dutertre M., Rimokh R., Bourgeois C.F., Auboeuf D. RNA helicases DDX5 and DDX17 dynamically orchestrate transcription, miRNA, and splicing programs in cell differentiation. Cell Rep. 2014;7:1900–1913. doi: 10.1016/j.celrep.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 22.Fuller-Pace F.V. The DEAD box proteins DDX5 (p68) and DDX17 (p72): multi-tasking transcriptional regulators. Biochim. Biophys. Acta. 2013;1829:756–763. doi: 10.1016/j.bbagrm.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 23.Uhlmann-Schiffler H., Rossler O.G., Stahl H. The mRNA of DEAD box protein p72 is alternatively translated into an 82-kDa RNA helicase. J. Biol. Chem. 2002;277:1066–1075. doi: 10.1074/jbc.M107535200. [DOI] [PubMed] [Google Scholar]
- 24.Honig A., Auboeuf D., Parker M.M., O'Malley B.W., Berget S.M. Regulation of alternative splicing by the ATP-dependent DEAD-box RNA helicase p72. Mol. Cell. Biol. 2002;22:5698–5707. doi: 10.1128/MCB.22.16.5698-5707.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Havugimana P.C., Hart G.T., Nepusz T., Yang H., Turinsky A.L., Li Z., Wang P.I., Boutz D.R., Fong V., Phanse S., Babu M., Craig S.A., Hu P., Wan C., Vlasblom J., Dar V.U., Bezginov A., Clark G.W., Wu G.C., Wodak S.J., Tillier E.R., Paccanaro A., Marcotte E.M., Emili A. A census of human soluble protein complexes. Cell. 2012;150:1068–1081. doi: 10.1016/j.cell.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hegele A., Kamburov A., Grossmann A., Sourlis C., Wowro S., Weimann M., Will C.L., Pena V., Luhrmann R., Stelzl U. Dynamic protein–protein interaction wiring of the human spliceosome. Mol. Cell. 2012;45:567–580. doi: 10.1016/j.molcel.2011.12.034. [DOI] [PubMed] [Google Scholar]
- 27.Lorgeoux R.P., Pan Q., Le Duff Y., Liang C. DDX17 promotes the production of infectious HIV-1 particles through modulating viral RNA packaging and translation frameshift. Virology. 2013;443:384–392. doi: 10.1016/j.virol.2013.05.026. [DOI] [PubMed] [Google Scholar]
- 28.Zhu Y., Chen G., Lv F., Wang X., Ji X., Xu Y., Sun J., Wu L., Zheng Y.T., Gao G. Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proc. Natl. Acad. Sci. U. S. A. 2011;108:15834–15839. doi: 10.1073/pnas.1101676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naji S., Ambrus G., Cimermancic P., Reyes J.R., Johnson J.R., Filbrandt R., Huber M.D., Vesely P., Krogan N.J., Yates J.R., III, Saphire A.C., Gerace L. Host cell interactome of HIV-1 Rev includes RNA helicases involved in multiple facets of virus production. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.M111.015313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karn J., Stoltzfus C.M. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb. Perspect. Med. 2012;2 doi: 10.1101/cshperspect.a006916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stoltzfus C.M., Madsen J.M. Role of viral splicing elements and cellular RNA binding proteins in regulation of HIV-1 alternative RNA splicing. Curr. HIV Res. 2006;4:43–55. doi: 10.2174/157016206775197655. [DOI] [PubMed] [Google Scholar]
- 32.Tazi J., Bakkour N., Marchand V., Ayadi L., Aboufirassi A., Branlant C. Alternative splicing: regulation of HIV-1 multiplication as a target for therapeutic action. FEBS J. 2010;277:867–876. doi: 10.1111/j.1742-4658.2009.07522.x. [DOI] [PubMed] [Google Scholar]
- 33.Stoltzfus C.M., A. J. S. Regulation of HIV-1 alternative RNA splicing and its role in virus replication. In: Maramorosch K., Murphy F.A., editors. vol. 74. Elsevier, Academic Press; USA: 2009. pp. 1–33. (Advances in Virus Research). [DOI] [PubMed] [Google Scholar]
- 34.Ocwieja K.E., Sherrill-Mix S., Mukherjee R., Custers-Allen R., David P., Brown M., Wang S., Link D.R., Olson J., Travers K., Schadt E., Bushman F.D. Dynamic regulation of HIV-1 mRNA populations analyzed by single-molecule enrichment and long-read sequencing. Nucleic Acids Res. 2012;40:10345–10355. doi: 10.1093/nar/gks753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naftelberg S., Schor I.E., Ast G., Kornblihtt A.R. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu. Rev. Biochem. 2015;84:165–198. doi: 10.1146/annurev-biochem-060614-034242. [DOI] [PubMed] [Google Scholar]
- 36.Staknis D., Reed R. SR proteins promote the first specific recognition of pre-mRNA and are present together with the U1 small nuclear ribonucleoprotein particle in a general splicing enhancer complex. Mol. Cell. Biol. 1994;14:7670–7682. doi: 10.1128/mcb.14.11.7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caputi M., Freund M., Kammler S., Asang C., Schaal H. A bidirectional SF2/ASF- and SRp40-dependent splicing enhancer regulates human immunodeficiency virus type 1 rev, env, vpu, and nef gene expression. J. Virol. 2004;78:6517–6526. doi: 10.1128/JVI.78.12.6517-6526.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Asang C., Hauber I., Schaal H. Insights into the selective activation of alternatively used splice acceptors by the human immunodeficiency virus type-1 bidirectional splicing enhancer. Nucleic Acids Res. 2008;36:1450–1463. doi: 10.1093/nar/gkm1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tokarev A., Guatelli J. Misdirection of membrane trafficking by HIV-1 Vpu and Nef: keys to viral virulence and persistence. Cell Logist. 2011;1:90–102. doi: 10.4161/cl.1.3.16708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neil S.J., Eastman S.W., Jouvenet N., Bieniasz P.D. HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2006;2:e39. doi: 10.1371/journal.ppat.0020039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Damme N., Goff D., Katsura C., Jorgenson R.L., Mitchell R., Johnson M.C., Stephens E.B., Guatelli J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. 2008;3:245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neil S.J., Zang T., Bieniasz P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 43.Rogers G.W., Jr., Komar A.A., Merrick W.C. eIF4A: the godfather of the DEAD box helicases. Prog. Nucleic Acid Res. Mol. Biol. 2002;72:307–331. doi: 10.1016/s0079-6603(02)72073-4. [DOI] [PubMed] [Google Scholar]
- 44.Svitkin Y.V., Pause A., Haghighat A., Pyronnet S., Witherell G., Belsham G.J., Sonenberg N. The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA. 2001;7:382–394. doi: 10.1017/s135583820100108x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cordin O., Banroques J., Tanner N.K., Linder P. The DEAD-box protein family of RNA helicases. Gene. 2006;367:17–37. doi: 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 46.Kiledjian M., Dreyfuss G. Primary structure and binding activity of the hnRNP U protein: binding RNA through RGG box. EMBO J. 1992;11:2655–2664. doi: 10.1002/j.1460-2075.1992.tb05331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cordin O., Tanner N.K., Doere M., Linder P., Banroques J. The newly discovered Q motif of DEAD-box RNA helicases regulates RNA-binding and helicase activity. EMBO J. 2004;23:2478–2487. doi: 10.1038/sj.emboj.7600272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tanner N.K., Cordin O., Banroques J., Doere M., Linder P. The Q motif: a newly identified motif in DEAD box helicases may regulate ATP binding and hydrolysis. Mol. Cell. 2003;11:127–138. doi: 10.1016/s1097-2765(03)00006-6. [DOI] [PubMed] [Google Scholar]
- 49.Rozen F., Pelletier J., Trachsel H., Sonenberg N. A lysine substitution in the ATP-binding site of eucaryotic initiation factor 4A abrogates nucleotide-binding activity. Mol. Cell. Biol. 1989;9:4061–4063. doi: 10.1128/mcb.9.9.4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vance C., Rogelj B., Hortobagyi T., De Vos K.J., Nishimura A.L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P., Ganesalingam J., Williams K.L., Tripathi V., Al-Saraj S., Al-Chalabi A., Leigh P.N., Blair I.P., Nicholson G., de Belleroche J., Gallo J.M., Miller C.C., Shaw C.E. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sachs A.B., Bond M.W., Kornberg R.D. A single gene from yeast for both nuclear and cytoplasmic polyadenylate-binding proteins: domain structure and expression. Cell. 1986;45:827–835. doi: 10.1016/0092-8674(86)90557-x. [DOI] [PubMed] [Google Scholar]
- 52.Steinert P.M., Rice R.H., Roop D.R., Trus B.L., Steven A.C. Complete amino acid sequence of a mouse epidermal keratin subunit and implications for the structure of intermediate filaments. Nature. 1983;302:794–800. doi: 10.1038/302794a0. [DOI] [PubMed] [Google Scholar]
- 53.Godin K.S., Varani G. How arginine-rich domains coordinate mRNA maturation events. RNA Biol. 2007;4:69–75. doi: 10.4161/rna.4.2.4869. [DOI] [PubMed] [Google Scholar]
- 54.Pinol-Roma S., Swanson M.S., Gall J.G., Dreyfuss G. A novel heterogeneous nuclear RNP protein with a unique distribution on nascent transcripts. J. Cell Biol. 1989;109:2575–2587. doi: 10.1083/jcb.109.6.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heinrichs V., Bach M., Winkelmann G., Luhrmann R. U1-specific protein C needed for efficient complex formation of U1 snRNP with a 5′ splice site. Science. 1990;247:69–72. doi: 10.1126/science.2136774. [DOI] [PubMed] [Google Scholar]
- 56.Kubota S., Duan L., Furuta R.A., Hatanaka M., Pomerantz R.J. Nuclear preservation and cytoplasmic degradation of human immunodeficiency virus type 1 Rev protein. J. Virol. 1996;70:1282–1287. doi: 10.1128/jvi.70.2.1282-1287.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu P., Chertova E., Bess J., Jr., Lifson J.D., Arthur L.O., Liu J., Taylor K.A., Roux K.H. Electron tomography analysis of envelope glycoprotein trimers on HIV and simian immunodeficiency virus virions. Proc. Natl. Acad. Sci. U. S. A. 2003;100:15812–15817. doi: 10.1073/pnas.2634931100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pause A., Sonenberg N. Mutational analysis of a DEAD box RNA helicase: the mammalian translation initiation factor eIF-4A. EMBO J. 1992;11:2643–2654. doi: 10.1002/j.1460-2075.1992.tb05330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Le Sage V., Cinti A., Valiente-Echeverría F., Mouland A.J. Proteomic analysis of HIV-1 Gag interacting partners using proximity-dependent biotinylation. Virol. J. 2015;12:138. doi: 10.1186/s12985-015-0365-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tiwari R.K., Kusari J., Sen G.C. Functional equivalents of interferon-mediated signals needed for induction of an mRNA can be generated by double-stranded RNA and growth factors. EMBO J. 1987;6:3373–3378. doi: 10.1002/j.1460-2075.1987.tb02659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Platt E.J., Wehrly K., Kuhmann S.E., Chesebro B., Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 1998;72:2855–2864. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wei X., Decker J.M., Liu H., Zhang Z., Arani R.B., Kilby J.M., Saag M.S., Wu X., Shaw G.M., Kappes J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002;46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peden K., Emerman M., Montagnier L. Changes in growth properties on passage in tissue culture of viruses derived from infectious molecular clones of HIV-1LAI, HIV-1MAL, and HIV-1ELI. Virology. 1991;185:661–672. doi: 10.1016/0042-6822(91)90537-l. [DOI] [PubMed] [Google Scholar]
- 64.Meng B., Ip N.C., Prestwood L.J., Abbink T.E., Lever A.M. Evidence that the endosomal sorting complex required for transport-II (ESCRT-II) is required for efficient human immunodeficiency virus-1 (HIV-1) production. Retrovirology. 2015;12:72. doi: 10.1186/s12977-015-0197-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dignam J.D., Lebovitz R.M., Roeder R.G. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fuller-Pace F.V., Nicol S.M. DEAD-box RNA helicases as transcription cofactors. Methods Enzymol. 2012;511:347–367. doi: 10.1016/B978-0-12-396546-2.00016-4. [DOI] [PubMed] [Google Scholar]
- 67.Ferns R.B., Partridge J.C., Spence R.P., Hunt N., Tedder R.S. Epitope location of 13 anti-gag HIV-1 monoclonal antibodies using oligopeptides and their cross reactivity with HIV-2. AIDS. 1989;3:829–834. doi: 10.1097/00002030-198912000-00008. [DOI] [PubMed] [Google Scholar]
- 68.Abbink T.E., Berkhout B. RNA structure modulates splicing efficiency at the human immunodeficiency virus type 1 major splice donor. J. Virol. 2008;82:3090–3098. doi: 10.1128/JVI.01479-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Duffy S., Cochrane A. Alternative Pre-mRNA Splicing. Wiley-VCH Verlag GmbH & Co. KGaA; 2012. Analysis of HIV-1 RNA Splicing; pp. 438–448. [Google Scholar]
- 70.Jablonski J.A., Buratti E., Stuani C., Caputi M. The secondary structure of the human immunodeficiency virus type 1 transcript modulates viral splicing and infectivity. J. Virol. 2008;82:8038–8050. doi: 10.1128/JVI.00721-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(− Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 72.Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cell viability. (a) DDX17 plasmid DNA was mutated in the seed sequence of both siRNAs which target DDX17 knockdown. Red arrows and letters indicate the base-pair change in the rescue plasmid. All base changes are silent mutations and do not alter the amino acid sequence of DDX17. (b and c) HeLa cells were seeded in a 96-well at 4 × 103 cells per well and sequentially transfected with 10 pmol of siControl or siDDX17 and then a second round of siRNA together with or without increasing concentrations of various siDDX17 rescue constructs. The number of viable cells was determined by measuring the amount of ATP present as an indicator of metabolically active cells. This was then calculated as a percentage relative to untransfected cells. Puromycin-treated cells were used as positive controls. Bars represent mean of duplicate samples ± SEM.
DDX17 second RGG box is indispensable. (a) Schematic representation of DDX17 and the annotated individual point mutation that is defective for RNA binding activity. HeLa M cells were sequentially transfected with siControl or siRNA targeting DDX17 (siDDX17 with or without increasing concentrations of siDDX17-resistant DDX17–R100Q). (b) Effect on HIV-1 relative virus production following endogenous DDX17 knockdown and rescue with DDX17–R100Q. (c) Western blot showing DDX17 knockdown and rescue with DDX17–R100Q. Each graph is a representative of at least two independent experiments done in triplicate. Bars represent mean of triplicate samples ± SEM. Statistical significance **P < 0.01.
DDX17 C terminus is essential for HIV infectivity. (a) HeLa cells were seeded in a 96-well at 4 × 103 cells per well and sequentially transfected with 10 pmol of siControl or siDDX17 and then a second round of siRNA together with or without increasing concentrations of various DDX17 C terminal truncated siDDX17 rescue constructs. The number of viable cells was determined by measuring the amount of ATP present as an indicator of metabolically active cells. This was then calculated as a percentage relative to untransfected cells. Puromycin-treated cells were used as positive controls. Bars represent mean of duplicate samples ± SEM. (b) Effect on HIV-1 relative virus production following endogenous DDX17 depletion and rescue with DDX17–T680. (c) Western blot showing DDX17 knockdown and rescue with DDX17–T680. (d) Effect on HIV-1 relative virus production following endogenous DDX17 depletion and rescue with DDX17–T717. (e) Western blot showing DDX17 knockdown and rescue with DDX17–T717. (f) Exogenously expressed Myc-DDX17 and Myc-DDX17–T717 co-immunoprecipitates with endogenous U2AF65 and SRSF1/SF2. IgG lane, immunoprecipitation of proteins treated with isotype control matched antibody. Myc-DD17-T690 fails to co-immunoprecipitate with both U2AF65 and SRSF1/SF2. Each graph is a representative of three independent experiments done in triplicate ± SEM. Statistical significance: *P < 0.05.
Supplementary tables