

Abstract
Nonalcoholic fatty liver disease (NAFLD) is a major health issue throughout the world. However, no validated treatments for NAFLD are currently available. In‐depth studies have demonstrated the efficacy of (‐)‐epigallocatechin‐3‐gallate (EGCG), a main bioactive chemical extracted from green tea, in treating NAFLD. EGCG exhibits multi‐pronged preventive and therapeutic activities, including promoting lipid and glucose metabolism, anti‐lipid peroxidation and anti‐inflammation activities, anti‐fibrosis, and anti‐NAFLD related tumor, thus contributing to the mitigation of NAFLD occurrence and progression. The objectives of this paper are to review and discuss the currently known targets, signaling pathways and roles of EGCG that interfere with NAFLD pathogenesis, then providing additional experimental evidence and the foundation for the further studies and clinical applications of EGCG in the prevention and treatment of NAFLD.
Keywords: (‐)‐epigallocatechin‐3‐gallate, catechins, fatty liver, nonalcoholic fatty liver disease(NAFLD), nonalcoholic steatohepatitis (NASH)
1. Introduction
Green tea (Camellia sinensis, Theaceae) is the most popular beverage, other than water, especially in East Asian countries. Epigallocatechin‐3‐gallate (EGCG) is the major polyphenolic catechin in green tea, accounting for approximately 50–80% of the catechin content; 200 to 300 mg of EGCG is present in a brewed cup of green tea.1, 2 Although the biological effects of EGCG are not fully understood, studies have indicated that EGCG reduces tumor incidence and multiplicity in different organ sites,3 and plays roles in cardiovascular, neurodegenerative diseases and metabolic syndromes including obesity, insulin resistance and dyslipidemia.4, 5 In addition, EGCG has many beneficial effects on different kinds of liver injuries, such as immune‐mediated liver injury,6 NaF‐induced oxidative hepatic injury,7 and chronic alcohol‐induced liver injury.8
Many of the potential health advantages and beneficial effects of EGCG are primarily a result of its unique structure. EGCG has both a trihydroxy group at carbons 3', 4', and 5' on the B ring and a gallate moiety esterified at carbon 3 on the C ring, which contributes to its ability to scavenge free radicals and chelate transition metal ions4, 9, 10, 11 (Figure 1). EGCG is water soluble, and the stability of the EGCG molecule is not greatly influenced when it is exposed to high temperatures (for instance, boiling water).11 The biological effects and stable characteristics of EGCG may be partly responsible for the popularity of green tea.
Figure 1.
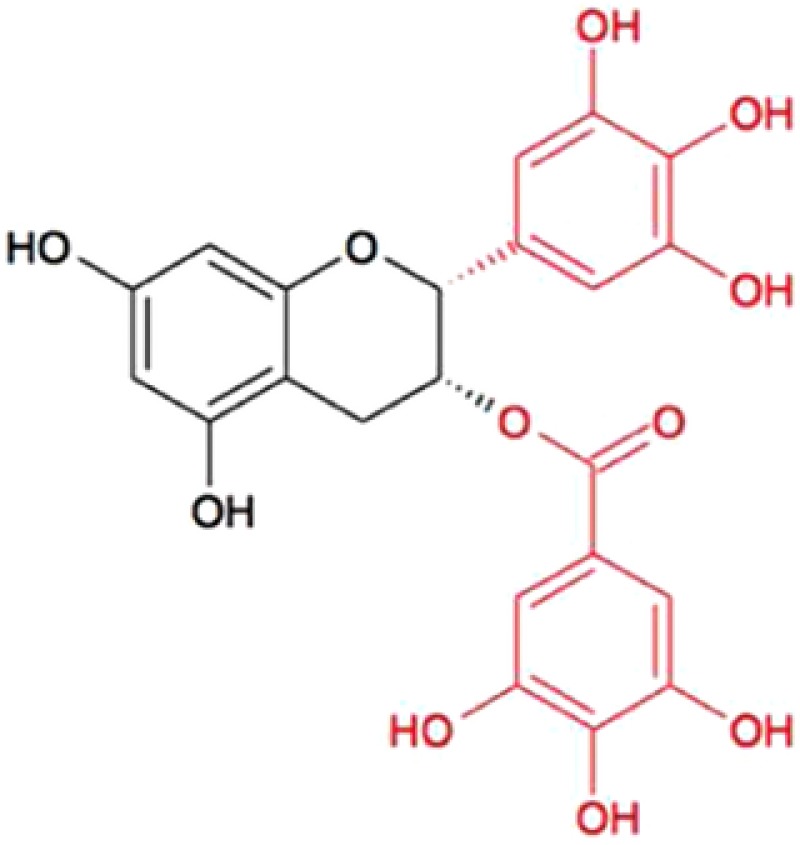
Chemical structure of (‐)‐epigallocatechin‐3‐gallate (from 4).
Nonalcoholic fatty liver disease (NAFLD) is a common hepatic disorder characterized by an abnormal accumulation of triglycerides (TG) in hepatocytes without an obvious history of alcohol use or abuse.9, 12, 13 Although the exact mechanism for the development of NAFLD has not been completely elucidated, metabolic syndromes including obesity and diabetes may contribute to the imbalance between the influx or synthesis of hepatic lipids and their export or oxidation.14, 15 Epidemiological studies have demonstrated that NAFLD affects approximately one‐third of the general population in western countries16, 17 and more frequently occurs in people with diabetes (50.1%) or obesity (70%).13, 14 Furthermore, approximately 20% of patients with simple fatty liver disease (SFLD) develop into chronic nonalcoholic steatohepatitis (NASH).18, 19 40–62% of patients with NASH‐related fibrosis develop into cirrhosis after 5–7 years of follow‐up,20, 21, 22 in which 4–27% of NASH cases progress to hepatocellular carcinoma (HCC).23
NAFLD is a major health issue worldwide, but therapeutic treatment options are limited. Weight loss through lifestyle modifications, including dietary changes and increased physical exercise, remains the backbone of NAFLD management. However, it is challenging for patients to achieve and maintain weight loss goals. Thus, it is often necessary to couple lifestyle changes with another pharmacologic treatment for NASH.24 Despite the numerous options of tested pharmaceuticals, none of them are satisfactory for NAFLD treatment.25, 26 However, natural compounds from food or herbs have been found to be effective in the alleviation of NAFLD. Increasing evidence indicates that EGCG possesses broad biological effects that may be useful in the prevention or treatment of NAFLD.
In this review, studies examining the efficacy of EGCG in treating NAFLD, which were published before March 2017, were retrieved systemically with the goal of discussing the applications of EGCG and its multifaceted bioactivities on NAFLD. Studies that showed fatty degeneration in the liver based on the pathological manifestations or biochemistry test are included. 19 in vivo studies, 7 in vitro studies and 5 relevant clinical trials are summarized in this review.
2. Basic Research
Experimental evidence suggests that EGCG has been associated with benefits in the treatment of NAFLD. These benefits are thought to be related to the regulation of energy metabolism, anti‐oxidation, anti‐inflammatory, anti‐fibrosis, inhibition of NAFLD‐related tumor activities, in addition to other pharmacological mechanisms of EGCG. All these mechanisms will be classified and discussed in detail below. All basic research is summarized in Tables 1 and 2, with related mechanisms presented in Figure 2.
Table 1.
Effects of EGCG in in vivo models of NAFLD
| Models | Period | Dosage and administration | Main results | Reference |
|---|---|---|---|---|
| HFD‐fed maleC57BL/6Jmice | 16 weeks | 3.2 g/kg in the diet |
|
27 |
| 4 weeks | 3.2 g/kg in the diet |
|
||
| HFW‐fed male C57BL/6J mice | 17 weeks | 3.2 g/kg in the diet |
|
28 |
| HFD‐fed male C57BL/6J mice |
|
|
|
29 |
| HFD‐fed male Swiss mice | 16 weeks | 0.1 mL by gavage |
|
30 |
| HFD‐fed male C57BL/6J mice | 8 weeks | 0.2%, 0.5% in the diet |
|
31 |
| HFD‐fed male C57BL/6J mice | 16 weeks | 3.2g/kg in the diet |
|
32 |
| gastric intralipid perfusion male SD rats | 6 weeks | 10, 20, 40 mg/kg 2mL/kg by gavage |
|
33 |
| HFD‐fed male Swiss mice | 16 weeks | 50 mg/kg o.1 mL/day by gavage |
|
42 |
| HFD‐fed male C57BL/6J mice | 24 weeks | 10, 20, 40 mg/kg/d ip. |
|
51 |
| nSREBP‐1C transgenic C57BL6 mice | 12 weeks | 0.05%, 0.1% EGCG in water |
|
53 |
| Male SHRSP‐SF rats | 8 weeks | 0.1% in water |
|
60 |
| HFD‐fed Male SD rats | 6 weeks | 0.1%in water |
|
61 |
| HFD‐fed SD rats | weeks | 10, 50, 100 mg/kg |
|
62 |
| Male ob/ob mice | 5 days+ 48/24/2 h | 85 mg/kg |
|
63 |
| HFW‐fed male C57BL/6 mice | 3 days 17 weeks | 25 mg/kg ip. 3.2 g/kg |
|
65 |
| HFD‐fed female SD rats | 8 weeks | 50 mg/kg ip. 3 times/week |
|
67 |
| MCD‐fed male C57BL/6 mice | 4 weeks | 25, 50, 100 mg/kg ip. |
|
68 |
| HFD‐fed male SD rats | 7 weeks | 0.01%, 0.1% in the water |
|
69 |
| C57BL/KsJ‐db/db mice | 36 weeks | 0.1% in water |
|
70 |
Table 2.
Effects of EGCG in in vitro models of NAFLD
| Models | Period | Dosage and Administration | Main results | Reference |
|---|---|---|---|---|
| HepG2 cells | 24 h | 25 μM |
|
37 |
| HepG2 cells | 24 h | 50, 100, 150, 200μM |
|
38 |
| HepG2 cells | 2/4/8/16/24 h | 5, 10, 50, 100 μM |
|
39 |
| HepG2 cells | 54 h | 0.1, 1, 10 μM |
|
44 |
| HepG2 cells | 24 h | 10, 20 μM |
|
54 |
|
24 h | 40 μM |
|
65 |
| LX‐2 cells | 1 h | 5, 10, 20 uM | 1. p‐Smad2/3 ↓ | 68 |
Figure 2.
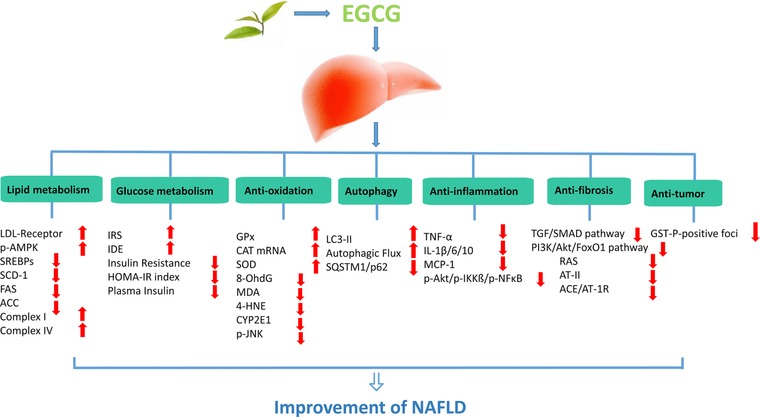
The mechanisms of EGCG on NAFLD.
2.1. The Improvement of Body Weight
Superfluous energy stimulates hepatic lipogenesis and gluconeogenesis, which directly leads to the development of NAFLD.27 Likewise, the whole energy metabolism and energy balance will affect liver energy metabolism. Weight loss, representing the improving of whole energy metabolism remains the backbone therapy in NAFLD patients with overweight and obesity. Furthermore, a 7% weight loss significantly modifies hepatic histology.28 It has been shown that a period of EGCG treatment helps to lose the body weight as well as waist circumference and to improve fecal lipid concentration in most mice experiments. According to those studies, EGCG can make contributions to the improvement of the whole energy and liver metabolism. The relevant basic research is summarized in Table 1.
2.2. The Improvement of Lipid Metabolism
The hepatoprotective mechanisms of EGCG in NAFLD have been examined under various conditions. Yang et al.29, 30 suggested that dietary EGCG supplementation decreases high‐fat‐induced hepatic steatosis. Approximately 69% and 34% liver TG were decreased respectively, by EGCG treatment in two in vivo studies. These changes in biochemical indices in the liver have also been observed in other studies. TG contents, liver total lipid, and liver weight31, 32, 33, 34, 35 have all been shown to decrease significantly following EGCG treatment. Therefore, EGCG may have hepatoprotective effects on NAFLD through regulatory mechanisms that balance lipid metabolism and storage.
2.2.1. Effects on Low Density Lipoprotein Receptors (LDL‐Rs)
LDL‐Rs are critical in mediating cellular LDL uptake and promoting cholesterol metabolism36 in liver tissue. Subramanian et al. demonstrated that LDL receptor‐deficient mice often develop metabolic syndrome and are more likely to develop hepatic steatosis and inflammation when lower and nontoxic doses (0.15%) of cholesterol are administered.37, 38 Recently, several studies showed that EGCG increases LDL‐R activity and affects multiple important biological pathways. Goto et al. performed DNA microarray analysis using HepG2 cells treated with 25 μM EGCG and found that the mRNA expression levels of LDL‐R were strongly up‐regulated by EGCG (2.2‐fold). EGCG also dose‐dependently decreases the level of extracellular apoB protein, which is the major apolipoprotein in LDL.39 Another study found that when HepG2 cells were incubated with the main constituents of green tea, EGCG was the only catechin to increase LDL‐R binding activity (3‐fold) and protein level (2.5‐fold). The mechanism by which EGCG affects LDL‐R appears to be the inhibition of cholesterol synthesis at a lower dose (50 μM) and increases in the efflux of cholesterol from the cells to the medium at the higher dose (200 μM).40 Furthermore, Kuhn et al. reported that 1 and 10 μM EGCG increased LDL‐R protein expression in HepG2 cell models and that LDL‐R expression was increased dramatically with 100 μM EGCG as demonstrated by an immunostaining assay.41 These results suggest that EGCG promotes LDL‐R activities at the gene and protein levels and promotes the translocation of cholesterol from the inside to the outside of the cells.
2.2.2. Effects on AMP‐Activated Protein Kinase (AMPK)
AMPK is considered as a cellular energy sensor. Activation of hepatic AMPK leads to increased fatty acid oxidation in the liver, with simultaneous inhibition of hepatic lipogenesis and cholesterol synthesis as well as effects on other hepatic energy metabolism pathways.42 Furthermore, AMPK also acts on the lipogenesis routes, reducing the activity of enzymes involved in lipid storage in the liver. In 2005, it was shown43 that AMPK inhibits adipocyte differentiation and mature adipocyte apoptosis, and that EGCG activates AMPK and inhibits adipocyte differentiation in 3T3‐L1 cells. A recent study44 found that supplementation with EGCG (50 mg/kg/day) stimulates AMPK activation via LKB1 in HFD‐fed mice and regulates essential enzymes (ACC, FAS) involved in the de novo lipogenesis pathway in the liver. In a study that investigated inhibitory effect of EGCG on FFA‐induced lipid accumulation,45 HepG2 cells were exposed to FFA co‐treated with 50 μM EGCG, and the final results, which were analyzed by proteomic analysis, demonstrated that EGCG reduced cellular lipid accumulation through the activation of AMPK and then shifted some FFA toward oxidation and away from lipid and triglyceride storage. A similar report showed that 10 μM EGCG inhibited hepatic lipogenesis by 65% (p<0.01) by increasing expressions of phospho‐AMPKα (Thr172) and phospho‐ACC (Ser79) in high‐glucose‐treated HepG2 cells.46 These studies suggest that EGCG is a potent activator of the AMPK pathway. By activating AMPK pathway, EGCG may increases the energy expenditure in NAFLD.
2.2.3. Effects on Sterol Regulatory Element‐Binding Proteins (SREBPs) and Related Genes
SREBPs are composed of three subtypes, SREBP‐1a, SREBP‐1c, and SREBP‐2, which regulate the expression of genes that control fatty acid and cholesterol synthesis in the liver.47 SREBPs that are attached to a phosphate group remain inactive in the cytoplasm. The dephosphorylated (active) form has the ability to translocate itself to the nucleus and then stimulate the synthesis of lipogenic enzymes. Studies have shown that EGCG can decrease the level of SREBP‐1 gene expression and reduce its active forms, which may make contribution to the lower level of plasma TG and liver lipids.44 Activated SREBPs can bind to the transcription promoter genes of lipogenic proteins, including ACC and FAS, fatty acid converting enzymes, such as stearoyl‐CoA desaturase (SCD1), and fatty acid elongases, such as fatty acid elongase 6.48, 49 There was also emerging evidence that the expression of lipogenic genes (ACC, FAS, and SCD1) in the liver was dose‐dependently decreased by EGCG treatment.31 These results demonstrate that EGCG can improve lipid metabolism by regulating the gene and protein expression of SREBPs, which in turn regulate the expression of proteins and hepatic enzymes involved in lipogenesis.
2.2.4. Effects on Lipid Oxidation
Mitochondria are the primary cellular organelles for the oxidation and metabolism of fatty acids and glucose. The reduction in mitochondrial function may contribute to the increase in lipid accumulation, which leads to insulin resistance50 and NAFLD. Mitochondrial respiratory chain complexes are responsible for the transport of long chain fatty acids (LCFAs) into mitochondrial and the generation of ATP molecules.51, 52 Complex I and Complex IV deficiencies can interrupt the process of oxidative phosphorylation, thus decreasing the production of energy for the cells to function properly. Santamarina et al.32 found that EGCG appeared to increase the activity of the mitochondrial complex chain, thereby increasing lipid oxidation and preventing the development of hepatic steatosis.
2.3. The Improvement of Glucose Metabolism
Abnormal glucose metabolism, unbalanced insulin clearance and whole‐body insulin resistance (IR) can promote NAFLD. Previous studies have demonstrated significant decline in IR and glucose levels after EGCG treatment.31, 53 It has also been shown that EGCG significantly reduce insulin concentrations and improve insulin sensitivity.29, 32, 53 Other promising results have shown that EGCG can promote glucose metabolism in liver tissue and may contribute to the prevention and treatment of NAFLD.
2.3.1. Effects on Insulin Receptor Substrate‐1 (IRS‐1)
The insulin receptor substrate (IRS) family of docking molecules connects insulin receptor activation to essential downstream kinase cascades and may be the key molecular lesion signature of hepatic insulin resistance.54 Specifically, IRS‐1 is closely related to glucose homeostasis. EGCG has been shown to improve the insulin resistance of liver tissues by promoting the functional recovery of the insulin receptor, IRS‐1, and glycogen synthase kinase (GSK) in nSREBP‐1c transgenic NASH model mice.55 An in vitro study56 investigated the effects of EGCG on insulin signaling under high‐glucose conditions. The results showed that the phosphorylation of IRS‐1 at Ser307 was reduced and tyrosine phosphorylation of IRS‐1 was increased after EGCG treatment. This research demonstrated that EGCG treatment attenuates insulin signaling blockade by reducing IRS‐1 Ser307 phosphorylation through the AMPK activation pathway. These biological effects may improve glucose metabolism in the hepatic cells and contribute to the improvement of NAFLD.
2.3.2. Effects on Insulin‐Degrading Enzyme (IDE)
Insulin‐degrading enzyme (IDE), a rate‐limiting enzyme in the insulin degradation process, plays an important role in maintaining normal plasma insulin levels and insulin sensitivity; approximately 50%–80% of insulin is cleared upon degradation by IDE in the liver.57 Gan et al.53 investigated the effects of EGCG on insulin resistance and insulin clearance in NAFLD mice. EGCG (10, 20 and 40 mg·kg‐1·d‐1, ip) dose‐dependently increased insulin sensitivity, secretion, and up‐regulated IDE protein expression and enzyme activity in the liver of NAFLD mice. However, not much is known about IDE regulation in NAFLD, which might suggest a new action of EGCG in NAFLD treatment and the importance of IDE as a candidate for a targeted therapeutic approach in treating NAFLD. Similarly, other studies demonstrated that the up‐regulation of IDE activity may help to control glucose metabolism in type 2 diabetes mellitus (T2DM).58, 59
2.4. Anti‐Peroxidation
Oxidative stress is known as a primary cause of liver fat accumulation and subsequent liver injury in NAFLD. Peroxidation of plasma and intracellular membranes may cause direct cell necrosis or apoptosis and appears to be responsible for initiating necroinflammation.60 Malondialdehyde (MDA) and other reactive lipid derivatives have the potential to amplify intracellular damage.61
Among the various benefits of EGCG, a great amount of attention has been paid to its use as an anti‐oxidant. The results from two research groups of Japan have shown that EGCG reduces oxidative stress in the livers of NAFLD mice. A marked decrease in immunoreactive 8‐hydroxy‐2'‐deoxyguanosine (8‐OhdG), a marker of oxidative DNA damage, was observed after EGCG treatment (distilled water containing 0.05% or 0.1% EGCG).55, 62 The serum d‐ROM levels, the increased hepatic MDA and 4‐HNE levels, the increased hepatic CYP2E1 and p‐JNK protein levels, and the reduced GPx and CAT mRNA expression levels were all reversed after EGCG treatment.62 Kuzu et al. showed that plasma and liver tissue MDA levels in the HFD+ EGCG (1 g/L EGCG in drinking water) SD rats were significantly lower and that glutathione levels were significantly higher than those in the HFD group.63 Similar results were demonstrated by two studies from China.35, 64 EGCG significantly decreased MDA levels and increased superoxide dismutase (SOD) levels in rat liver tissues. Fiorini et al.65 studied the effects of EGCG on hepatic steatosis in ob/ob mice, demonstrating that EGCG treatment remarkably improved survival rates from 65% survival in vehicle‐treated mice to 100% in orally or ip. EGCG‐treated mice. Animals were pre‐treated with 85 mg/kg EGCG via intraperitoneal injection for two days or oral consumption of treated drinking water for 5 days before 15 minutes of warm ischemia and 24 h of reperfusion. The results indicated that EGCG treatment significantly reduced total hepatic fat content and increased hepatic energy stores and hepatic anti‐oxidant activity through an enhanced glutathione levels. The results of the above‐mentioned studies indicate that EGCG protects the steatotic liver by functioning as an anti‐oxidant. One of them has also reported the dose‐dependent antioxidative effects of EGCG on NAFLD treatment.64
2.5. Regulation of Autophagy
Recent studies have revealed that autophagy, a cell survival mechanism for disposing of excess or defective organelles, is linked to the development of NAFLD and the regulation of autophagy has potential therapeutic effects. Autophagy reduces intracellular lipid droplets by enclosing them and fusing with lysosomes for degradation and is involved in attenuating inflammation and liver injury.66 To investigate whether EGCG regulates autophagy and then improves the lipid clearance in the liver, Zhou et al.67 conducted a series of in vitro and in vivo studies. The results suggest that EGCG (40 mM) improves lipid metabolism through the induction of autophagy by increasing the microtubule‐associated protein light chain (LC3‐II) formation, decreasing SQSTM1/p62 protein levels and increasing AMPK phosphorylation in fat‐loaded HepG2 cells, Huh7 cells, and mouse hepatocytes in primary culture. EGCG treatment was also shown to reduce hepatosteatosis and concomitantly increases autophagy in the livers of mice fed with a high‐fat/Western‐style (HFW) diet. Nevertheless, autophagy is regarded as a double‐edged sword, its potential effects on adipogenesis, adipocyte differentiation, as well as the association between autophagy and NAFLD remain controversial.66
2.6. Anti‐Inflammation
NASH is a severe condition of the inflamed fatty liver. Although the progression of NAFLD to NASH is not well understood, a current line of thinking in the pathogenesis of NASH involves a “multi‐hit hypothesis”,60 in which hepatic inflammation is considered as an important “hit” that accelerates the progression from simple fatty liver to NASH. Research has shown that EGCG can improve the hepatic inflammation in NAFLD. Ding et al.70 showed that EGCG (50 and 100 mg/kg) inhibits methionine‐and choline‐deficient (MCD) diet‐induced steatohepatitis by attenuating inflammatory cell infiltration. Consistent with histological results, EGCG treatment significantly inhibited MCD diet‐induced IL‐1β, IL‐6, TNF‐α and MCP‐1 mRNA expression. However, an in vivo study showed that the concentrations of cytokines including TNF‐α, IL‐6, and IL‐10 and protein expression levels of TLR4, TNFR‐1, IL‐6R, and IL‐10Rα did not change in liver tissue after EGCG treatment.32 More interestingly, another study showed that hepatic mRNA expression levels of TNF‐α, IL‐6, IL‐1β, and monocyte chemoattractant protein‐1 (MCP‐1) were reduced, but the serum levels of TNF‐α and IL‐6 did not change.62 Basing on these results, we hypothesize that EGCG increased the gene expression level of some inflammatory cytokines in liver tissues but may not lead to changes in serum concentration, or did not influence the protein translation of these inflammatory cytokines and related proteins. Kuzu et al.63 examined the preventive role of EGCG in an experimental NASH SD rats model induced by a high‐fat diet. Histopathological results suggested that inflammation, ballooning degeneration, and necrosis decreased significantly in the HFD + EGCG group,55 and the expression levels of inflammatory factors including p‐Akt, p‐IKKß, and p‐NFκB in liver tissues of high‐dose EGCG treatment groups were also decreased. These independent studies clearly demonstrated that the expression of inflammatory mediators and related pathological changes in fatty liver tissues are improved by EGCG treatment.
2.7. Improvement of NAFLD‐Related Hepatic Fibrosis
During the progress of NAFLD, the occurrence of fibrosis is usually considered as a middle‐late event that bridges NASH and cirrhosis. The key factor in hepatic fibrosis is the activation of hepatic stellate cells (HSCs), which are responsible for the excess deposition of extracellular matrix (ECM) and the formation of scars.68 A recent study reported that a series of fibrosis signaling pathways in the livers of obese rats were down‐regulated by EGCG treatment. Both the transforming growth factor (TGF)/SMAD pathway and phosphatidylinositol 3‐kinase(PI3) K/Akt/FoxO1 pathway have a close relationship with the development of hepatic fibrosis, and both were inhibited by EGCG treatment.69 According to another study,70 which included in vivo and in vitro experiments, the mRNA expression of TGF‐β, collagen I‐α1, tissue inhibitor of metalloproteinase 1 (TIMP‐1), α‐smooth muscle actin (a‐SMA), and the phosphorylation of Smad2 and Smad3 were markedly inhibited in the liver tissue following EGCG treatment. In in vitro experiments, EGCG treatment inhibits TGF‐β‐induced Smad2/3 protein phosphorylation in LX‐2 cells in a dose‐dependent manner. Additionally, the activation of the renin‐angiotensin system (RAS), which plays a key role in the liver fibrosis progression, is inhibited as the results of EGCG treatment. EGCG can make contributions to the decrease of serum levels of angiotensin‐II (AT‐II) and suppression of mRNA levels of RAS components, including angiotensin‐converting enzyme (ACE) and AT‐II type 1 receptor (AT‐1R) in the liver.62
2.8. Prevention of NAFLD‐Related Liver Tumorigenesis
The continuous development of NAFLD/NASH can progress to HCC. Oxidative stress, chronic inflammation, and numerous pathophysiological mechanisms are critically involved in NAFLD/NASH‐related liver tumorigenesis. Three in vivo studies62, 71, 72 conducted by a research group from Japan demonstrated that EGCG might be able to prevent NAFLD‐related liver tumorigenesis. In 2011, this research group reported that EGCG (0.1%, for 34 weeks) significantly inhibited the development of liver cell adenomas in the diethylnitrosamine (DEN)‐induced liver tumorigenesis models by inhibiting the phosphorylation of IGF‐1R and related downstream signaling pathways, including the mitogen‐activated protein kinase (MAPK)/ERK and PI3K/Akt pathways, which contribute to the development of HCC. Subsequently, in two other NAFLD/NASH‐related HCC rat models, EGCG administration was shown to inhibit the development of glutathione S‐transferase placental form (GST‐P)‐positive foci (p < 0.01), a hepatic preneoplastic lesions.
3. Clinical Trials
The beneficial effects of EGCG observed in basic studies support further development of EGCG as a potential clinical drug for the treatment of NAFLD. Based on these potential clinical advantages of EGCG, some research groups have conducted a few clinical trials of small sample sizes (Table 3) to examine the efficacy and safety of EGCG or green tea extract (GTE) rich in EGCG on men with NASH, overweight, or obesity. However, at this time, no randomized, controlled, clinical trials have tested the effects of high‐purity EGCG on human NAFLD.
Table 3.
Effects of green tea extract rich in EGCG or EGCG in patients with NAFLD or metabolic syndrome
| Types | Participants | Period | Total Dosage | Main results | Reference |
|---|---|---|---|---|---|
| randomized placebo‐controlled | 22 males 16 females (confirmed NASH by liver biopsy diagnosis) | 6 months | 600 mg/d green tea catechins (52.6% EGCG) |
|
73 |
| double‐blind placebo‐controlled | 102 females (BMI > 27 kg/m2 WC > 80 cm) | 12 weeks | 856.8 mg/d green tea extracts (57.12% EGCG) |
|
74 |
| randomized single‐blind permuted block | 35 subjects (with obesity and metabolic syndrome) | 8 weeks | 4 cup green tea/d (400mg EGCG/d) green tea extracts (460mg EGCG/d) |
|
75 |
| randomized double‐blind placebo‐controlled | 88 females (obese premenopausal) | 12 weeks | EGCG 300 mg/d |
|
72 |
| randomized double‐blind placebo‐controlled | 88 males (overweight or obese) | 8 weeks | EGCG 800 mg/d |
|
71 |
Two research groups73, 74 reported that 300 mg/d of EGCG for 12 weeks and 800 mg capsules of EGCG for 8 weeks on overweight or obese patients (30 kg/m2 <BMI <40 kg/m2, 28 kg/m2<BMI <38 kg/m2), and the biomarkers related to the whole‐body energy metabolism, lipid, and glucose metabolism including BMI, waist circumference, blood lipid levels (TAG, TC, LDL‐C and HDL‐C, et al), and insulin resistance were tested. However, the results were not in line with those from animal research where EGCG improve the overweight‐related fatty liver. Noticeably, the liver function markers (AST, ALT, γ‐GT, et al) were tested in one of the studies, but there was no statistical difference between the EGCG group and control group. Generally, the two studies showed that EGCG failed to decrease the body weight or improve the lipid or glucose metabolism, neither in male nor in female obese patients.
Interestingly, patients with NASH who were diagnosed by a liver biopsy and took 600 mg of green tea catechins (GTC, including52.6% EGCG) per day for 6 months with controlled diets and exercise therapy achieved significant effects, including improved anthropometric parameters and biochemistry levels. GTC treatment resulted in a significant decrease in BMI with higher HLD‐C and lower LDL‐C, and decrease in blood glucose levels, including immunoreactive insulin (IRI), HOMA‐IR, FPG and GA levels, compared with the control group. Meanwhile, the high level of hs‐CRP was decreased, the ALT and AST levels remained under the limits for most of the patients in the GTC treatment group.75 Similarly, subjects with central obesity who underwent 12 weeks of EGCG treatment (a daily dosage of 856.8 mg) showed significant weight loss, reduced waist circumference, and consistent decrease in total cholesterol and LDL plasma levels without any side effects.76 A large dose of GTE rich in EGCG (approximately 50%) significantly decreased body weight and BMI after 8 weeks of treatment.77
Based on these clinical trials, GTE rich in EGCG have beneficial effects on NAFLD, but the pure EGCG does not produce the same effects. According to Masterjohn et al.,78 other catechins in GTE have greater bioavailability in humans, mice, and rats. We hypothesized that other catechins in the GTE may cause synergistic effects on the treatment of NAFLD in certain ways. And this kind of phenomenon is also observed in one clinical trial, where patients in the green tea treatment group exhibited better therapeutic effects than patients in the green tea extracts treatment group.77 Thus, some synergistic effects should be explored and extended basic studies in this direction are needed in the future. Possibly this kind of research can provide more insight on why the functional food exhibits multipronged preventive and therapeutic activities.
Of course, differences in the study sample, design, and treatment dosages could explain the discrepancy among the research results. For example, with the dose change of the treatment groups from the lower dose of EGCG (377 mg daily) to the high‐dose of EGCG (856.8 mg daily), body weight and BMI were decreased after 12‐week treatment and waist circumference was also significantly reduced in the subjects.76, 79 In addition, it is noteworthy that the lengths in clinical trials with vitamin E and obeticholic acid for the treatment of NAFLD are two years and 72 weeks, respectively.80, 81 Perhaps a longer term trial of EGCG is needed to achieve significant effects on NAFLD. Generally, the available data from both animal studies and the clinical trials on obesity and diabetes are promising. But we need further clinical trials to verify the hepatoprotective effects of EGCG on humans before EGCG can be recommended as a useful prophylactic to protect against or for the treatment of NAFLD in humans.
4. Conclusions and Perspectives
Based on the results of in vivo studies, EGCG treatment have beneficial effects on NAFLD because of its effect of anti‐oxidation, anti‐inflammation, and its effect on energy metabolism through up‐regulation of LDL‐R, activation of AMPK, regulating SREBPs, increasing lipid oxidation and improving insulin resistance, etc. More importantly, EGCG can improve NAFLD‐related fibrosis and carcinoma. It seems that EGCG exhibits multipronged preventive and therapeutic effects on the treatment of NAFLD.
Although many biological effects of EGCG in treating NAFLD have been demonstrated in cultured cells and animal models, clinical trials are inadequate, and the clinical efficacy of EGCG in treating NAFLD remains unclear. As is mentioned above, the inconsistent outcomes may be related to the different treating dosages, treating courses, administration routes, enrollment criteria, etc. Further clinical studies with larger sample size and longer treatment period are needed.
Drug safety will be another major concern in clinical practice. According to drug safety analysis in animal studies and clinical trials, low doses (0.2, 0.32 and 0.5% of dietary EGCG) of EGCG, as well as moderate consumption of whole green tea, does not cause liver injury or any other side effects. However, a high dose of EGCG (1500 mg/kg) can lead to mild liver injury in mice models,82 and 500 mM EGCG can be harmful to HepG2 cells.83 Furthermore, with hepatocytes, high dose of EGCG induced a significant increase in ROS formation and caused damage to mitochondria. Several concentrations of EGCG resulted in massive cellular death in an EGCG dose‐dependent manner.84, 85
To fully elucidate the efficacy, safety and mechanisms of EGCG in treating NAFLD, clinical trials with greater sample sizes and a longer follow‐up period and more in‐depth in vitro and in vivo experiments are expected.
Abbreviations
- 8‐OhdG
Immunoreactive8‐hydroxy‐2’‐deoxyguanosine
- ACC
Acetyl‐CoA Carboxylase
- ACE
Angiotensin‐Converting Enzyme
- AKT
Protein kinase B
- AMPK
Adenosine Monophosphate Activated Protein Kinase
- a‐sMA
α‐Smooth Muscle Actin
- AST
Aspartate Aminotransferase
- AT‐1R
AT‐II type 1 receptor
- ATGL
Adipose Triglyceride Lipase
- AT‐II
Angiotensin‐II
- ATL
Alanine Aminotransferase
- BMI
Body Mass Index
- CAT
Catalase
- CD36/FAT
Cluster of Differentiation/Fatty Acid Translocase
- CEBP‐α
CCAAT enhancer‐binding protein‐α
- ChREBP
Carbohydrate Response Element‐Binding Protein
- CYP2E1
Cytochrome P450 2E1
- CYP450
Cytochrome P450
- CYP5
Activating Cytochrome P450
- DEN
Diethylnitrosamine
- ECM
Extracellular Matrix
- EGCG
(‐)‐Epigallocatechin‐3‐gallate
- ERK
Extracellular Signal‐Regulated Kinase
- Fabp‐2
Fatty acid binding protein 2
- FAS
Fatty Acid Synthase
- FATP4
Fatty Acid Transport Protein 4
- FFA
Free Fatty Diet
- FoxO1
Forkhead box protein O1
- FPG
Fasting Plasma Glucose
- GA
Glycoalbumin
- G‐CSF
Granulocyte Colony‐Stimulating Factor
- GOT
Glutamic‐Oxalacetic Transaminase
- GPT
Glutamic‐Pyruvic Transaminase
- GPx
Glutathione Peroxidase
- GSK
Glycogen Synthase Kinase
- GSK3β
Glycogen synthase kinase 3β
- GST
Glutathione S‐Transferase placental form
- GTC
Green Tea Catechins
- GTE
Green Tea Extract
- HCC
Hepatocellular Carcinoma
- HDL
High Density Lipoproteins
- HFD
High‐Fat Diet
- HFW
High‐fat/Western‐style
- HMG‐CoA
3‐hydroxy‐3‐methlglutaryl‐CoA
- HNE
Hydroxynonenals
- HOMA‐IR
Homeostasis Model Assessment of Insulin Resistance
- Hs‐CRP
High sensitive C‐reactive protein
- HSCs
Hepatic stellate Cells
- HSL
Hormone‐Sensitive Lipase
- IDE
Insulin‐degrading Enzyme
- IL‐6/10/18
Interleukin‐6/10/18
- IR
Insulin Resistance
- IRS‐1
Insulin Receptor Substrate‐1
- IκBα
nuclear factor of kappa light polypeptide gene enhancer in B‐cells inhibitor
- JNK
c‐Jun NH2‐terminal kinase
- L/S
Liver to Spleen
- LC3‐II
Microtubule‐Associated protein light chain 3‐II
- LCFAs
Long Chain Fatty Acids
- LDL‐C
Low Density Lipoprotein Cholesterol
- LDL‐R
Low Density Lipoprotein Receptor
- MAPK
Mitogen‐Actived Protein Kinase
- MCAD
Medium Chain acyl coA Decarboxylase
- MCD
Methionine‐and Choline‐Deficient
- MCP‐1
Monocyte Chemoattractant Protein‐1
- MDA
Malondialdehyde
- NAFLD
Nonalcoholic Fatty Liver Disease
- NAS
NAFLD Activity Score
- NASH
Nonalcoholic Steatohepatitis
- NF‐κB
Nuclear Factor‐κB
- NRF1
Nuclear Respiratory Factor 1
- p27
Cyclin‐dependent kinase inhibitor 1B
- p62
ubiquitin‐binding protein p62
- PAI
Plasminogen Activator Inhibitor‐1
- PPAR‐γ
Peroxisome Proliferator‐Activated Receptor‐γ
- RAS
Renin‐Angiotensin System
- RAS
Renin‐Angiotensin System
- ROS
Reactive Oxygen Species
- SCD‐1
Stearoyl CoA desaturase
- Ser307
Anti‐pIRS‐1
- Ser641
Phospho‐glycogen synthase
- Ser79
Phospho‐ACC
- SFLD
Simple Fatty Liver Disease
- SGLT1
Sodium‐dependent Glucose Transporter 1
- SHRSP‐ZE rat
Stroke‐prone spontaneously Hypertensive rats (SHRSP) with Zucker Fatty (ZF) rats
- SOD
superoxide dismutase
- SREBPs
Sterol Regulatory Element‐binding Proteins
- T2DM
Type 2 Diabetes Mellitus
- TAG
Triacylglycerols
- TC
Total Cholesterol
- TG
Triglycerides
- TGF
Transforming Growth Factor
- Thr172
phosphor‐AMPK‐α
- TIMP
Tissue Inhibitor of Metalloproteinases
- TIMP‐1
Tissue Inhibitor of Metalloproteinase 1
- TNF
Tumor Necrosis Factor
- UCP 3
Uncoupling Protein 3
- V/S
Visceral fat to Subcutaneous fat
- γ‐GTP
γ‐glutamyl transpeptidase
The authors would like to thank Professor L.X. Zhu and Tao Wei for proofreading the English‐language manuscript. The copyright line for this article was changed on 17 August 2018 after original online publication.
Conflict of Interest
All authors declare that there are no conflicts of interest in the present paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81573668, No. 81374031, to Q. F.; No. 81673765, to Y.H.)
Chen C., Liu Q., Liu L., Hu Y., Feng Q., Mol. Nutr. Food Res. 2018, 62, 1700483 10.1002/mnfr.201700483
Contributor Information
Yi‐yang Hu, Email: yyhuliver@163.com.
Qin Feng, Email: fengqin1227@163.com.
5. References
- 1. Yang C. S., Wang X., Lu G., Picinich S. C., Nat. Rev. Cancer 2009, 9 (6), 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sae‐tan S., Grove K. A., Lambert J. D., Pharmacol. Res. 2011, 64 (2), 146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Singh B. N., Shankar S., Srivastava R. K., Biochem. Pharmacol. 2011, 82 (12), 1807–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Legeay S., Rodier M., Fillon L., Faure S., Clere N., Nutrients 2015, 7 (7), 5443–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chowdhury A., Sarkar J., Chakraborti T., Pramanik P. K., Chakraborti S., Biomed. Pharmacother. 2016, 78, 50–59. [DOI] [PubMed] [Google Scholar]
- 6. Wang Y., Mei Y., Feng D., Xu L., Clin. Exp. Immunol. 2006, 145 (3), 485–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thangapandiyan S., Miltonprabu S., Can. J. Physiol. Pharmacol. 2013, 91 (7), 528–537. [DOI] [PubMed] [Google Scholar]
- 8. Yuan G., G. Z., Zhou X., Zhang P. et al., Int. J. Mol. Sci. 2006, 7, 204–219. [Google Scholar]
- 9. Higdon J. V., Frei B., Crit. Rev. Food Sci. Nutr. 2003, 43 (1), 89. [DOI] [PubMed] [Google Scholar]
- 10. Wang X., Song K. S., Guo Q. X., Tian W. X., Biochem. Pharmacol. 2003, 66 (10), 2039–2047. [DOI] [PubMed] [Google Scholar]
- 11. Steinmann J., Buer J., Pietschmann T., Steinmann E., Br. J. Pharmacol. 2013, 168 (5), 1059–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu J.‐Y., Zhang L., Li, Z.‐P. Ji G., Curr. Drug Targets 2015, 16, 1347–1355. [DOI] [PubMed] [Google Scholar]
- 13. Sattar N., Forrest E., Preiss D., BMJ 2014, 349, g4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kwon Y. M., Oh S. W., Hwang S. S., Lee C. et al., Am. J. Gastroenterol. 2012, 107 (12), 1852–1858. [DOI] [PubMed] [Google Scholar]
- 15. Kopec K. L., Nut. Clin. Pract. 2011, 10, 565–576. [DOI] [PubMed] [Google Scholar]
- 16. Sayiner M., Koenig A., Henry L., Younossi Z. M., Clin. Liver Dis. 2016, 20 (2), 205–214. [DOI] [PubMed] [Google Scholar]
- 17. Bhala N., Curr. Pharm. Design 2013, 19, 5169–5176. [DOI] [PubMed] [Google Scholar]
- 18. Henao‐Mejia J., Elinav E., Jin C., Hao L., Mehal W. Z., Strowig T., Thaiss C. A., Kau A. L., Eisenbarth S. C., Jurczak M. J., Camporez J.‐P., Shulman G. I., Gordon J. I., Hoffman H. M., Flavell R. A., Nature 2012, 482, 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matteoni C. A., Younossi Z. M., Gramlich T., Boparai N. P., Liu Y. G., McCullough A. J., Gastroenterology 1999, 116, 1413–1419. [DOI] [PubMed] [Google Scholar]
- 20. Hui J. M., K. J. G., Chitturi S., Sud A., et al., Hepatology 2003, 21, 420–427. [DOI] [PubMed] [Google Scholar]
- 21. Adams L. A., Lymp J. F., St. Sauver J., Sanderson S. O. et al., Gastroenterology 2005, 129 (1), 113–121. [DOI] [PubMed] [Google Scholar]
- 22. Starley B. Q., Calcagno C. J., Harrison S. A., Hepatology 2010, 51 (5), 1820–1832. [DOI] [PubMed] [Google Scholar]
- 23. Ratziu V., Bonyhay L., Di Martino V., Charlotte F. et al., Hepatology 2002, 35 (6), 1485–1493. [DOI] [PubMed] [Google Scholar]
- 24. Le Thuy‐Anh R. L., J. Clin. Exp. Hepatol., 2012, 156–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Corrado R. L., Torres D. M., Harrison S. A., Med. Clin. North Am. 2014, 98 (1), 55–72. [DOI] [PubMed] [Google Scholar]
- 26. Beaton M., Can. J. Gastroenterol. 2012, 26(6), 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pan M. H., Lai C. S., Tsai M. L., Ho C. T., Mol. Nutr. Food Res. 2014, 58 (1), 147–171. [DOI] [PubMed] [Google Scholar]
- 28. Marchesini G., Mazzotti A., J. Hepatol. 2015, 62 (1), 15–17. [DOI] [PubMed] [Google Scholar]
- 29. Bose M., Lambert J. D., Ju J., Reuhl K. R., Shapses S. A., Yang C. S., J. Nutr. 2008, 138 (9), 1677–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen Y. K., Cheung C., Reuhl K. R., Liu A. B. et al., J. Agric. Food Chem. 2011, 59 (21), 11862–11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Friedrich M., Petzke K. J., Raederstorff D., Wolfram S., Klaus S., Int. J. Obes. (Lond). 2012, 36 (5), 735–743. [DOI] [PubMed] [Google Scholar]
- 32. Santamarina A. B., Carvalho‐Silva M., Gomes L. M., Okuda M. H. et al., J. Nutr. Biochem. 2015, 26 (11), 1348–1356. [DOI] [PubMed] [Google Scholar]
- 33. Lee M. S., Kim C. T., Kim Y., Ann. Nutr. Metab. 2009, 54 (2), 151–157. [DOI] [PubMed] [Google Scholar]
- 34. Sae‐Tan S., Grove K. A., Kennett M. J., Lambert J. D., Food Funct. 2011, 2 (2), 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu L. I. U., M.‐S. Z., De‐Suo W., Chinese Pharmacol. Bull. 2009, 25(4), 510–514. [Google Scholar]
- 36. GOLDSTEIN M. S. B.A J. L., Nobel lecture. 1985, 9, 284–324. [Google Scholar]
- 37. Subramanian S., Goodspeed L., Wang S., Kim J. et al., J. Lipid Res. 2011, 52 (9), 1626–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Subramanian S., Han C. Y., Chiba T., McMillen T. S., Wang S. A., Haw A., E. A. Kirk, 3rd , O'Brien K. D., Chait A., Arterioscler. Thromb. Vasc. Biol. 2008, 28 (4), 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Goto T., Saito Y., Morikawa K., Kanamaru Y., Nagaoka S., Br. J. Nutr. 2012, 107 (6), 769–773. [DOI] [PubMed] [Google Scholar]
- 40. ROACH C. A. B. A P. D., J. Agric. Food Chem. 2006, 54, 1621−1626. [DOI] [PubMed] [Google Scholar]
- 41. Kuhn D. J., Burns A. C., Kazi A., Dou Q. P., Biochim. Biophys. Acta 2004, 1682 (1‐3), 1–10. [DOI] [PubMed] [Google Scholar]
- 42. Viollet B., Guigas B., Leclerc J., Hebrard S. et al., Acta Physiol. (Oxf). 2009, 196 (1), 81–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hwang J. T., Park I. J., Shin J. I., Lee Y. K., Lee S. K., Baik H. W., Ha J., Park O. J., Genistein E. G. C. G., Biochem. Biophys. Res. Commun. 2005, 338 (2), 694–649. [DOI] [PubMed] [Google Scholar]
- 44. Santamarina A. B., Oliveira J. L., Silva F. P., Carnier J. et al., PLoS One. 2015, 10 (11), e0141227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhonghua Liu Q. L., Jianan Huang, Proteome Sci. 2013, 11, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim J. J., Tan Y., Xiao L., Sun Y. L., Qu X., Biomed. Res. Int. 2013, 2013, 920128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ferré P., Foufelle F., Diabetes Obesity Metab. 2010, 12 (s2), 83–92. [DOI] [PubMed] [Google Scholar]
- 48. Henriksen B. S., Diabetol. Metab. Syndrome 2013, 5, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kohjima M., Higuchi N., Kato M. et al., Int. J. Mol. Med. 2008, 21(4), 507. [PubMed] [Google Scholar]
- 50. Ye J., Front. Med. 2013, 7 (1), 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sverdlov A. L., Elezaby A., Behring J. B., Bachschmid M. M. et al., J. Mol. Cell Cardiol. 2015, 78, 165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Holmstrom M. H., Iglesias‐Gutierrez E., Zierath J. R., Garcia‐Roves P. M., Am. J. Physiol. Endocrinol. Metab. 2012, 302 (6), E731–9. [DOI] [PubMed] [Google Scholar]
- 53. Gan L., Meng Z. J., Xiong R. B., Guo J. Q. et al., Acta Pharmacol. Sin. 2015, 36 (5), 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thirone A. C., Huang C., Klip A., Trends Endocrinol. Metab. 2006, 17 (2), 72–8. [DOI] [PubMed] [Google Scholar]
- 55. Ueno T., Torimura T., Nakamura T., Sivakumar R., Nakayama H., Otabe S., Yuan X., Yamada K., Hashimoto O., Inoue K., Koga H., Sata M., Int. J. Mol. Med. 2009, 24 (01), 17–22. [DOI] [PubMed] [Google Scholar]
- 56. Lin C. L., Lin J. K., Mol. Nutr. Food Res. 2008, 52 (8), 930–939. [DOI] [PubMed] [Google Scholar]
- 57. Fernandez‐Gamba A., Leal M., Morelli L., Castano E., Curr. Pharmaceutical Design 2009, 15 (31), 3644–3655. [DOI] [PubMed] [Google Scholar]
- 58. Abdul‐Hay S. O., Kang D., McBride M., Li L., Zhao J., Leissring M. A., PLoS One 2011, 6 (6), e20818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nazir R.HaA., CNS Neurol. Disorders Drug Targets 2014, 13, 259–264. [DOI] [PubMed] [Google Scholar]
- 60. Takaki A., Kawai D., Yamamoto K., Int. J. Mol. Sci. 2013, 14 (10), 20704–20728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yu J., Marsh S., Hu J., Feng W., Wu C., Gastroenterol. Res. Pract. 2016, 2016, 2862173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kochi T., Shimizu M., Terakura D., Baba A. et al., Cancer Lett. 2014, 342 (1), 60–69. [DOI] [PubMed] [Google Scholar]
- 63. Kuzu N., Bahcecioglu I. H., Dagli A. F., Ozercan I. H. et al., J. Gastroenterol. Hepatol. 2008, 23 (8 Pt 2), e465–70. [DOI] [PubMed] [Google Scholar]
- 64. Yujun Y. A. N., Z. L., Yong L. I. N., jian'an H. U. A. N.G., Bin P. E. N. G., Huajun J. I. A. N.G., J. TEA Sci. 2014, 34 (3), 221–229. [Google Scholar]
- 65. Fiorini R. N., Donovan J. L., Rodwell D., Evans Z. et al., Liver Transplantation 2005, 11 (3), 298–308. [DOI] [PubMed] [Google Scholar]
- 66. Mao Y., Yu F., Wang J., Guo C., Fan X., Hepat. Med. 2016, 8, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhou J., Farah B. L., Sinha R. A., Wu Y. et al., PLoS One. 2014, 9 (1), e87161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen S. L., Zheng M. H., Shi K. Q., Yang T., Chen Y. P., BioDrugs 2013, 27 (1), 25–34. [DOI] [PubMed] [Google Scholar]
- 69. Xiao J., C., Liong E. C., Nanji A. A., Leung T. M., Yue Huen Lau T., Lung Fung M., T. G. L., Eur. J. Nutr. 2014, 53, 187–199. [DOI] [PubMed] [Google Scholar]
- 70. Ding Y., Sun X., Chen Y., Deng Y., Qian K., Eur. J. Pharmacol. 2015, 761, 405–412. [DOI] [PubMed] [Google Scholar]
- 71. Sumi T., S. Y., Shimizu M., Kochi T. et al., SpringerPlus 2013, 2, 690–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shimizu M., Sakai H., Shirakami Y., Yasuda Y. et al., Cancer Prev. Res. (Phila). 2011, 4 (3), 396–403. [DOI] [PubMed] [Google Scholar]
- 73. Mielgo‐Ayuso J., Barrenechea L., Alcorta P., Larrarte E. et al., Br. J. Nutr. 2014, 111 (7), 1263–71. [DOI] [PubMed] [Google Scholar]
- 74. Brown A. L., Lane J., Coverly J., Stocks J. et al., Br. J. Nutr. 2009, 101 (6), 886–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Fukuzawa Y., Kapoor M. P., Yamasaki K., et al., J. Funct. Foods 2014, 9, 48–59. [Google Scholar]
- 76. Chen I. J., Liu C. Y., Chiu J. P., Hsu C. H., Clin. Nutr. 2016, 35 (3), 592–599. [DOI] [PubMed] [Google Scholar]
- 77. Basu A., Sanchez K., Leyva M. J., Wu M. et al., J. Am. Coll. Nutr. 2010, 29 (1), 31–40. [DOI] [PubMed] [Google Scholar]
- 78. Masterjohn C., Bruno R. S., Nutr. Rev. 2012, 70 (1), 41–56. [DOI] [PubMed] [Google Scholar]
- 79. Hsu C. H., Tsai T. H., Kao Y. H., Hwang K. C., Tseng T. Y., Chou P., Clin. Nutr. 2008, 27 (3), 363–370. [DOI] [PubMed] [Google Scholar]
- 80. Sanyal A. J., Chalasani N., Kowdley K. V., McCullough A. et al., N. Engl. J. Med. 2010, 362 (18), 1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Neuschwander‐Tetri B. A., Loomba R., Sanyal A. J., et al., Lancet 2015, 385 (9972), 956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Saleh I. G., Ali Z., Abe N., Wilson F. D. et al., Fitoterapia 2013, 90, 151–159. [DOI] [PubMed] [Google Scholar]
- 83. Saleh I. G., Ali Z., Abe N., Hamada F. M. et al., J. Appl. Biomed. 2014, 12 (4), 291–299. [Google Scholar]
- 84. Hirsch N., Konstantinov A., Anavi S., Aronis A. et al., Mol. Nutr. Food Res. 2016, 60 (12), 2542–2553. [DOI] [PubMed] [Google Scholar]
- 85. Kucera O., Mezera V., Moravcova A., Endlicher R. et al., Oxid. Med. Cell Longev. 2015, 2015, 476180. [DOI] [PMC free article] [PubMed] [Google Scholar]