

ABSTRACT
Delayed healing and nonunion of fractures represent enormous burdens to patients and healthcare systems. There are currently no approved pharmacological agents for the treatment of established nonunions, or for the acceleration of fracture healing, and no pharmacological agents are approved for promoting the healing of closed fractures. Yet several pharmacologic agents have the potential to enhance some aspects of fracture healing. In preclinical studies, various agents working across a broad spectrum of molecular pathways can produce larger, denser and stronger fracture calluses. However, untreated control animals in most of these studies also demonstrate robust structural and biomechanical healing, leaving unclear how these interventions might alter the healing of recalcitrant fractures in humans. This review describes the physiology of fracture healing, with a focus on aspects of natural repair that may be pharmacologically augmented to prevent or treat delayed or nonunion fractures (collectively referred to as DNFs). The agents covered in this review include recombinant BMPs, PTH/PTHrP receptor agonists, activators of Wnt/β‐catenin signaling, and recombinant FGF‐2. Agents from these therapeutic classes have undergone extensive preclinical testing and progressed to clinical fracture healing trials. Each can promote bone formation, which is important for the stability of bridged calluses, and some but not all can also promote cartilage formation, which may be critical for the initial bridging and subsequent stabilization of fractures. Appropriately timed stimulation of chondrogenesis and osteogenesis in the fracture callus may be a more effective approach for preventing or treating DNFs compared with stimulation of osteogenesis alone. © 2016 The Authors. Journal of Orthopaedic Research published by Wiley Periodicals, Inc. on behalf of the Orthopaedic Research Society. J Orthop Res 35:213–223, 2017.
Keywords: fracture healing, osteogenesis, chondrogenesis, delayed union, nonunion fracture
Fracture healing can be a remarkably robust repair process. After union, a fracture callus can achieve structural stability matching or exceeding that of unfractured bone, followed by callus remodeling that restores original bone geometry.1 Yet fracture healing can go awry in 5–10% of cases in the form of delayed or nonunion fractures (referred to collectively as DNFs).2, 3 Recent data specific to open long bone fractures indicated that 17% developed nonunion and another 8% exhibited delayed union.4 With delayed union the initial periosteal response ceases before fracture bridging,5 while nonunion involves persistent lack of bridging after intramembranous and endochondral repair have ceased.6 Nonunions include hypertrophic and atrophic forms. Hypertrophic nonunions have substantial non‐bridging callus that contains cartilage, and these nonunions tend to result from inadequate fracture stability rather than innate biological limitations. Atrophic nonunions have minimal callus or cartilage, and usually exhibit scar tissue within the fracture gap. The time involved in defining nonunion varies depending on the location and type of bone. Longer, larger bones typically need more than 6 months of failed healing before being considered a nonunion.
Several risk factors have been associated with DNFs. Patient‐dependent risk factors include older age, diabetes, smoking, nutritional deficiencies, and the use of anti‐inflammatory agents.3, 7 Local infection can be particularly deleterious to fracture healing,4 and some components of the adaptive immune system may inhibit fracture healing.8 Other local factors associated with DNF include the extent of soft tissue injury,9 and compartment syndrome.10, 11 Certain skeletal sites (e.g., tibia) and types of fractures (e.g., open, comminuted, transverse) are more likely to exhibit delayed healing.2, 4, 12 Most delayed unions eventually heal,5 while others become nonunions in the absence of surgical intervention. The great majority of established nonunions require further surgical intervention to achieve healing.4 DNFs cause substantial morbidity, loss of productivity, decreased quality of life, and extensive health care utilization.12, 13 Long bone nonunions can have a particularly devastating impact on health‐related quality of life, rivalling or exceeding the effects of type 1 diabetes, stroke, or AIDs.14
A variety of pharmacological interventions show potential for improving fracture healing,3 almost all of which promote osteogenesis. For various reasons, less attention is paid to the important role of chondrogenesis, which creates cartilage that provides initial union of most long bone fractures. Perception may account for some of this neglect: DNFs are often associated with persistent callus cartilage, which is problematic when it results from delayed or impaired conversion to bony callus.15 However, persistent callus cartilage does not necessarily impair healing when bony callus development is undiminished.16 Another factor is that analysis of chondrogenesis requires time‐consuming histomorphometry, and compared with osteogenesis, the contribution of chondrogenesis to functional outcomes (e.g., callus strength) is less obvious and less amenable to quantification. While often overlooked, chondrogenesis represents a potentially important therapeutic target for healing recalcitrant fractures.
PRECLINICAL FRACTURE HEALING MODELS
The most commonly used preclinical fracture healing model involves young rodents subjected to closed and internally stabilized long bone fractures that heal through intramembranous and endochondral bone formation.17 Intramembranous bone is that which forms directly on existing bone surfaces, while endochondral bone forms from a cartilage scaffold. The “standard” closed fracture model is very useful for studying the biology of fracture healing18 and for identifying agents that impair bone healing,19 but may not be ideal for understanding whether an agent might prevent or treat DNFs. The standard model typically excludes complicated (e.g., comminuted) fractures that may exhibit delayed healing,16 and spontaneous nonunions are rare.1 With the standard model, full biomechanical recovery of fractured bones can occur within 3–4 weeks in young mice16 and rats20 in the absence of active treatments. Even agents that do not promote osteogenesis or chondrogenesis can increase callus strength in the standard closed fracture model by delaying callus remodeling.16
Other models or model adaptations that mimic aspects of DNFs include local ischemia,21 adjacent muscle crushing,22 surgically open fractures,15 periosteal damage,15, 23 externally fixated critical‐size segmental defects,24, 25 and non‐rigidly fixated osteotomy.26 Some of these ‘higher hurdle’ models require greater surgical intervention and expertise, which limits their use. But delayed healing can also be induced by simple modifications of the standard model, such as the use of aged mice,27 aged ovariectomized rats (Fig. 1)20 or administering glucocorticoids.28
Figure 1.
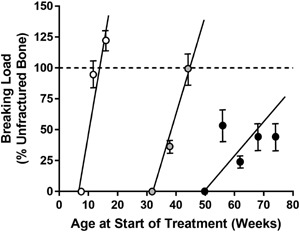
Biomechanical strength recovery of internally stabilized closed femoral fractures created in normal gonad‐intact female rats at 8 weeks of age (white circles), and in OVX rats at 32 (gray circles) or 50 (black circles) weeks of age. Animals were sacrificed at various time points after fracture for biomechanical testing of callus breaking load by a 3‐point bending test. Data represent percentage of the non‐fractured contralateral femur diaphysis, n = 3–10 per group per time point. Fractured femurs from older OVX rats showed slower regain of strength compared with young gonad‐intact rats. Reproduced with permission from Meyer et al.20
THE CLINICAL LANDSCAPE FOR FRACTURE HEALING TRIALS AND INDICATIONS
Numerous clinical trials examined pharmacological approaches to accelerate fracture healing,3, 29 with only a handful of successes, as described below. No pharmacological agent or combination product (drug plus delivery device) is FDA approved for “accelerated fracture healing,” although an ultrasound device, Exogen® was approved for this indication.30 In addition to biological challenges, clinical trial settings can pose practical challenges to establishing efficacy. The window for accelerating radiographic union or return‐to‐function can be narrow, with most placebo patients reaching those endpoints within a few weeks of each other.31, 32 Improved radiographic union rate is not always accompanied by clinical improvements such as return‐to‐function or reduced fracture healing complications,33 which are increasingly relied upon as co‐primary endpoints. Regulatory approval pathways for acceleration of fracture healing remain unclear,29 and no drugs are currently approved for the prevention or treatment of DNFs. The only medications approved for fracture healing are bone morphogenetic proteins (BMPs) mixed with osteoconductive carriers and administered to open fractures, as described below. There remains a clear unmet need for non‐surgical and non‐biophysical interventions that prevent or treat DNFs.
THE BASICS OF FRACTURE HEALING
Fracture healing begins with an injury‐induced hematoma and inflammation, which promotes the condensation of mesenchymal cells from the periosteum, endosteum, and bone marrow and their subsequent differentiation along chondrocyte and osteoblast lineages (Fig. 2). The earliest anabolic response involves intramembranous bone formation on the periosteum. This response rarely bridges a fracture by itself,34 and fractures can heal without it,5 but this periosteal bone creates a more robust bony scaffold adjacent to the fracture gap upon which the endochondral phase can act.23 Bridging of the fractured ends usually occurs through endochondral bone formation, starting with chondrocyte production of a cartilaginous scaffold (soft callus) that expands to bridge the fractured ends and then mineralizes to form the rigid hard callus (Fig. 2). The hard callus is then gradually remodeled by osteoclasts and osteoblasts until normal bone geometry is re‐established.1 When rigid fixation is achieved in humans after a near‐perfect reduction, fractures can heal by osteoclasts that create tunnels across the fracture site that refill with new bone. This healing, called osteonal, direct, or primary healing, occurs without a cartilage intermediate or significant callus.12 Metaphyseal fractures can heal via bone formation on existing trabecular elements, which represents another form of primary healing with minimal cartilage or callus. But most fractures heal by the combined effects of intramembranous and endochondral bone formation.
Figure 2.
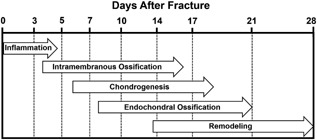
Stages of fracture healing in rodents subjected to internally stabilized experimental long bone fractures, as described by Bonnarens and Einhorn.17 Reproduced with permission from Hadjiargyrou et al.111
Deficiencies or dysregulation of numerous processes intrinsic to fracture repair can lead to impaired healing, including inadequate intramembranous bone formation adjacent to the fractured ends, insufficient cartilage formation within the marrow space and fracture gap, delayed or premature transition from cartilage to bone, and premature callus remodeling. Angiogenesis plays an important role during the endochondral response, and angiogenesis inhibitors can severely impair callus formation,19 as can ischemia by other means.21 An insufficient early endochondral response, leading to inadequate cartilaginous callus, can compromise bony union and also lead to a smaller, less stable hard callus post‐union.21, 35, 36
NATURAL ANALOGIES TO FRACTURE HEALING AND THE IMPORTANCE OF CHONDROGENESIS AND OSTEOGENESIS
Some fundamental aspects of fracture healing, particularly the endochondral phase, have clear parallels with long bone development.3, 37 The common reliance on endochondral bone formation for fracture healing and long bone development points to natural processes that may be amplified to prevent or treat DNFs. Maintaining chondrocytes in an undifferentiated and proliferative state is important for creating cartilaginous scaffolds upon which osteoblasts create trabeculae in the developing growth plate as well as the hard callus during fracture healing. During development, the acceleration of chondrocyte differentiation was associated with reduced growth plate cartilage, inappropriate regional cartilage mineralization, and impaired bone growth.38 And during fracture healing, accelerated chondrocyte differentiation was associated with reduced soft callus that can lead to a deficient hard callus.36 Such parallels are also evident in the abnormal skeletal phenotype38 and impaired fracture healing39, 40 of mice deficient in PTHrP, a key factor in long bone development (Fig. 3). Conversely, rat studies showed that activation of the PTH/PTHrP receptor (PTHR1) led to increased cartilage in the growth plate of unfractured femurs, and in the callus of fractured femurs.41
Figure 3.
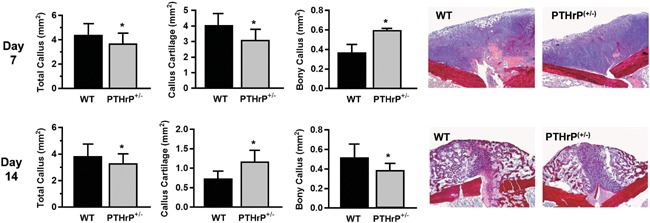
Impaired long bone fracture healing in PTHrP haplo‐insufficient mice compared with wild‐type (WT) mice. PTHrP(+/−) mice exhibited an early transient deficit in callus cartilage a more sustained deficit in callus size and bone content. Day 7 and 14 refer to the time after creation of internally stabilized closed femoral fracture. Data represent means ± SD, n = 6/group. *Significant differences versus wildtype (WT) controls, p < 0.05. Reproduced with permission from Wang et al.39
The importance of chondrocytes and the cartilage they produce is evident in high hurdle fracture healing models. Delayed fracture healing in aged mice was associated with reduced chondrogenesis compared with young mice.27 Insulin‐resistant mice with impaired fracture healing exhibited premature chondrocyte differentiation36 and accelerated soft callus resorption,35 leading to smaller calluses and prolonged nonunion. In sheep, reduced callus cartilage caused by unstable fixation also led to delayed union.26 The impressive healing potential of cartilage was shown by the ability of pure cartilage grafts to heal critical size segmental defects in mice. This healing was accomplished by cartilage graft mineralization and bone formation, which may have been mediated by the transdifferentiation of graft‐derived chondrocytes into osteoblasts.25
While cartilage can be important for initial union, no amount of cartilage will successfully heal fractures without its timely mineralization and replacement with bone matrix. Prolongation of the endochondral repair phase led to biomechanically deficient calluses in sheep,42 and persistent callus cartilage and its delayed conversion to bone reduced fracture union in rats.43 Drug‐induced impairment of callus mineralization can also reduce callus strength.44 An ideal pharmacologic profile for meeting the unmet needs of fracture healing may involve the promotion or maintenance of adequate cartilage to achieve union, while permitting or promoting its timely conversion to mineralized bone once union is achieved. Agents that only stimulate osteogenesis can improve the structural integrity of united fractures, but this effect alone may not improve outcomes for established or pending nonunions.
AGENTS AND BIOLOGICAL PATHWAYS THAT PROMOTE FRACTURE HEALING
The scope of biological modifiers in this review is limited to therapeutic classes that improved fracture healing in multiple preclinical studies and progressed clinically to fracture healing trials.
Bone Morphogenetic Proteins (BMPs)
BMPs are bone‐derived osteoinductive agents that induce bone formation when implanted subcutaneously or within bone defects.45 BMPs act by promoting the differentiation of mesenchymal stem cells into chondroblasts and osteoblasts.46 The predominant effect of BMPs is to stimulate bone formation,47 which increases hard callus development and callus strength.48, 49 However, BMPs also stimulate chondrogenesis during the early stages of fracture healing (Fig. 4).48, 50 Several BMPs and BMP receptors are expressed in fractured bone,18, 40 and their functional involvement is suggested by the inability of mice lacking BMP‐2 to initiate fracture healing.51
Figure 4.
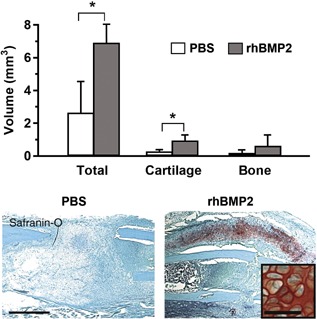
Recombinant human BMP2 (rhBMP2) induced cartilage formation during the repair of stabilized closed long bone fractures in mice. Mice received PBS (control) or 10 μg of rhBMP2 by direct injection into the fresh fracture site, and were sacrificed 10 days later. Upper graph: Total callus volume and callus cartilage volume were significantly greater in the rhBMP2 group versus PBS controls when measured by histomorphometry 10 days post‐fracture (**P < 0.05). Lower images: Safranin‐O/Fast Green staining of callus sections, with a higher‐magnification image showing chondrocytes spanning the fracture line that is near the left side of the inset. Reproduced with permission from Yu et al.49
A single clinical trial of BMP‐7 (OP‐1) was the basis for its approval by the FDA for the treatment of recalcitrant long bone nonunions where use of autograft is unfeasible and alternative treatments have failed.52 The trial tested BMP‐7 mixed with a type I collagen carrier, delivered to open fractures in 63 patients with tibial nonunions of at least 9 months duration.53 An active comparator group comprised 61 similar patients treated with autologous bone graft. All patients were treated with a locked intramedullary nail. After 9 months, similar and high percentages of patients in each group exhibited healing based on radiographic assessments and fracture site pain. BMP‐7 was FDA‐approved in 2001 under a humanitarian device exemption as an alternative to autograft.29
BMP‐2 was approved for fracture healing in the form of BMP‐2 mixed with an absorbable collagen sponge (ACS) delivered locally to open fractures. This combination product “device,” known as INFUSE® Bone Graft, was FDA‐approved in 2004 for use in acute tibial fractures treated with an intramedullary nail within 14 days of injury,54 based on data from a randomized controlled trial of 450 patients with open tibial fracture. A standard of care (SOC) group was treated with reamed or unreamed intramedullary nail fixation, while the BMP‐2 group received SOC plus BMP‐2/ACS. The BMP‐2 group exhibited reduced risk of secondary interventions and an increased rate of clinical and radiographic healing.55 However, subsequent clinical trials of BMP‐2 in tibial fractures have not demonstrated efficacy. One trial of BMP‐2/ACS in patients with open tibial fracture treated with reamed intramedullary nailing did not show improved healing, and there was a trend toward a higher infection rate.56 A trial testing BMP‐2 in a calcium/phosphate matrix, delivered percutaneously to closed tibial fractures, also failed to show improvements in healing.31
The use of BMPs carries some identified safety risks. BMPs can promote regional bone loss by recruiting osteoclasts, which can lead to clinical complications.57 BMPs are also associated with other complications, including the risk of heterotopic ossification.57 Despite their prevalent use in orthopedics through limited indications,58 there remains a need for alternative therapies with different biological effects and pharmaceutical presentations. In particular, agents that can be administered to closed fractures without special carriers, grafts or graft substitutes would represent a major advance.
PTH Receptor (PTHR1) Agonists
PTHR1 agonists have been widely investigated for fracture healing.3 The endogenous PTHR1 agonists include parathyroid hormone (PTH) and PTH‐related protein (PTHrP), which have some distinct effects on PTH1R activation59 and clear differences in their endogenous roles. The fundamental role of PTH is to maintain calcium, which it does by stimulating bone resorption and by promoting renal calcium reabsorption and intestinal calcium absorption. Endogenous PTH generally reduces bone mass, as shown by the high bone mass of mice lacking endogenous PTH(1‐84)60 and humans with hypoparathyroidism.61 Yet paradoxically, intermittent administration of exogenous PTH(1‐84) or its active fragment PTH(1‐34) increases bone mass by stimulating bone formation in excess of bone resorption. PTHrP and PTHrP analogs share this interesting pharmacodynamic property.62, 63
PTH is not expressed in bone and is not induced after fracture, but its receptor PTHR1 is expressed by many cell types, including chondrocytes and osteoblasts.40, 64 Systemic PTH deficiency in mice was associated with impaired fracture healing,65 and exogenous PTH and PTH fragments can increase fracture callus density and strength.41, 66, 67 PTH promotes early chondrogenesis and also osteogenesis in fracture models,41, 66, 67 which may have favorable implications for treatment of DNFs. Indeed, several case reports suggest PTH(1‐34) may promote healing of DNFs.68, 69, 70 A non‐placebo‐controlled trial of elderly osteoporotic women with pelvic factures showed an increased rate of radiographic healing and some clinical improvements in those treated with PTH(1‐84).71 A placebo‐controlled trial of PTH(1‐34) in patients with closed distal radius fractures failed to achieve its primary endpoint, but accelerated radiographic healing was observed at the lower dose tested.32 A non‐placebo‐controlled study of PTH(1‐34) in patients with proximal humerus fractures failed to show improvements in radiographic or clinical healing.72 No forms of PTH are currently approved for fracture healing, and no controlled clinical trials formally studied whether PTH or PTH(1‐34) can prevent or treat DNFs.
PTHrP has some biological effects that overlap with those of PTH, along with some unique attributes that suggest promise for promoting fracture healing. Unlike PTH, PTHrP has fundamental endogenous roles in bone development and fracture healing. Whereas the primary role of PTH is to maintain serum calcium, often at the expense of bone mass,60 PTHrP is considered an endogenous bone anabolic factor that acts by promoting osteoblast differentiation, survival, and activity.73 PTHrP also inhibits terminal chondrocyte differentiation, maintains chondrocyte proliferation, inhibits chondrocyte apoptosis, and prevents premature cartilage mineralization.38, 74, 75, 76 PTHrP is expressed by osteoblasts during the intramembranous phase of fracture healing, and by mesenchymal cells, proliferating chondrocytes and osteoblasts during the endochondral healing phase.77 PTHrP is functionally expressed in the intact and the fractured periosteum of rodents.78, 79 Periosteal PTHrP mRNA was rapidly upregulated in fractured mouse bone, and ablation of periosteal PTHrP impaired fracture healing by reducing callus cartilage, bony callus, and callus size.40 It is tempting to speculate that the loss of periosteal PTHrP contributes to the strong inhibitory effects on fracture healing that can result from periosteal damage.15, 23
Consistent with its endogenous role in the maintenance of bone mass, exogenous PTHrP administration has strong bone anabolic effects in humans and animals. An early PTHrP study in rats indicated dose‐dependent BMD gains that exceeded the effects of PTH(1‐34).62 Recently, abaloparatide, a selective PTH1R activator with homology to PTHrP, was reported to cause significantly greater BMD gains compared with PTH(1‐34) in postmenopausal women.80 PTHrP and PTHrP analogs have yet to be clinically tested in fracture healing trials, but preclinical studies indicate positive effects of PTHrP in models of impaired bone healing. A PTHrP analog improved the density and strength of rabbit osteotomy sites, overcoming the detrimental effects of glucocorticoids.81 The negative impacts of glucocorticoids on fracture healing82 may relate at least in part to their ability to suppress bone cell production of PTHrP.83 Exogenous PTHrP enhanced the size and density of calluses in diabetic mice with impaired fracture healing84, and PTHrP administration also upregulated pro‐angiogenic factors in a diabetic bone regeneration model.85 The latter finding suggests the potential for PTHrP to overcome the suppression of angiogenesis that can impair bone healing in diabetes. Endogenous PTHrP expression, which was markedly reduced in the regenerate bone of diabetic mice, was largely restored by exogenous PTHrP.86 In healthy mice, PTHrP mRNA expression was upregulated throughout the post‐fracture healing process, and PTH administration further increased PTHrP expression in the healing fractures in association with enhanced chondrogenesis and osteogenesis (Fig. 5).67 Thus it appears that bone anabolism through PTH1R activation can increase endogenous PTHrP expression in fracture sites, which could perhaps lead to a virtuous cycle that promotes chondrogenesis and osteogenesis. An important role for endogenous PTHrP in chondrogenesis and osteogenesis was indirectly demonstrated by impaired fracture healing in PTHrP‐deficient mice (Fig. 3).39
Figure 5.
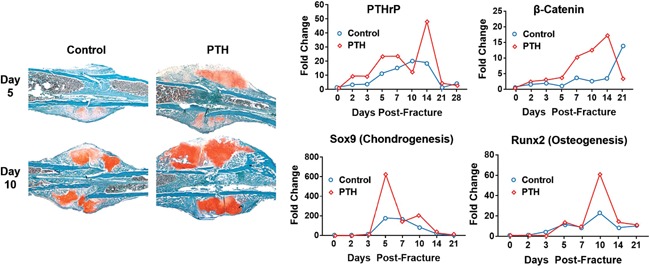
Changes in PTHrP, Wnt signaling (i.e., beta‐catenin), chondrogenesis and osteogenesis in normal mice after creation of internally stabilized closed femur fractures. Mice were treated with PTH(1‐34) (30 μg/kg/day) for 14 days post‐fracture. Healing fractures were harvested at various times and assessed by histology (left panels) or by RNA analyses (right panels). Histology sections obtained 5 and 10 days post‐fracture were stained with Safranin O/fast green, which labels cartilage and chondrogenic cells (red color). The right panels show that PTH(1‐34) increased mRNA expression of PTHrP, the chondrogenic marker Sox9, the osteogenic marker Runx2, and the Wnt signaling marker beta‐catenin. Reproduced with permission from Kakar et al.67
These findings indicate that PTHR1 agonists exert diverse biological effects that may favor fracture healing. Endogenous PTHrP and exogenous PTHrP and PTH can maintain chondrocytes in an undifferentiated and proliferative state while stimulating osteogenesis by osteoblasts. In low‐ and high‐hurdle fracture healing models, PTHrP and PTH promoted the production and maintenance of cartilaginous callus for bridging and union, while stimulating bone formation to enhance callus strength. Clinical data with PTH(1‐34) show some beneficial effects in accelerating fracture healing, but it remains to be tested whether PTH, PTHrP or analogs thereof can prevent or treat DNFs.
Activators of Wnt/β‐Catenin Signaling
Recent comprehensive reviews have described Wnt/β‐catenin signaling pathways86 and how they may be leveraged in orthopedic settings.87 Wnts represent a large family of secreted factors that activate signaling pathways, with prominent roles in embryonic development and tissue regeneration.86 Wnts stimulate bone formation by activating low density lipoprotein receptor related protein‐5 (LRP5) or LRP6 and Frizzled co‐receptors on the cell surface, which leads to activation of β‐catenin signaling.88 When endogenous Wnt inhibitors prevent Wnt from activating these co‐receptors, bone formation is suppressed.89 Two prominent Wnt inhibitors are sclerostin and DKK1.89 Inhibition of sclerostin or DKK1 via loss‐of‐function mutations or inhibitory antibodies can increase bone formation and bone mass in intact animals.89 Antibodies that inhibit DKK189 or sclerostin91, 92 can also increase callus strength in animals with experimental fractures, and their subcutaneous route of administration would potentially allow for the treatment of open or closed fractures.
While other anabolic agents covered in this review are direct receptor agonists, sclerostin, and DKK1 antibodies function as inhibitors of inhibitors. This may be an important distinction because the timing, location and magnitude of effects of sclerostin or DKK1 antibodies will depend on the presence and levels of endogenous sclerostin, DKK1, and free Wnts. Sclerostin antibody administration to rats increased skeletal mRNA expression of the Wnt inhibitor Dkk1 by nearly 10‐fold,93 and endogenous DKK1 appears to be one factor that limits the ability of sclerostin antibodies to increase bone mass and callus strength.94 There are additional Wnt inhibitors besides sclerostin and DKK186 that may also inhibit Wnt signaling despite the administration of DKK1 or sclerostin antibodies. BMPs have their own endogenous inhibitors that can reduce BMP receptor activation.95 PTH and PTHrP have no validated endogenous inhibitors that impair PTH/PTHrP receptor activation.
The remainder of this section will focus on sclerostin antibodies because, unlike DKK1 antibodies, their anabolic effects on bone have been corroborated in humans.96 Sclerostin antibodies increased bone formation in a variety of orthopedic models, including implants,97 osteotomies,91, 98 bone defects,24 and long bone fractures.99, 100 However, sclerostin antibodies have not been shown to have pro‐chondrogenic effects in bone healing models,24, 91, 98, 99, 100 and several studies showed that sclerostin inhibition reduced the volume or balance of cartilage in fracture calluses (Fig. 6).90, 92, 98, 99, 100 These findings align with developmental biology wherein Wnt/β‐catenin signaling appears to inhibit differentiation of mesenchymal stem cells along the chondrocyte lineage, while promoting their differentiation to the osteoblast lineage.101 Interestingly though, inhibition of Wnt/β‐catenin signaling in the chondrocyte lineage of transgenic mice with tibial fractures was associated with a marked suppression of early callus chondrogenesis, along with reduced bony callus formation and decreased callus strength.102 Thus while endogenous PTHrP appears to have similar anabolic roles during bone development and fracture healing, the roles of Wnt/β‐catenin signaling in these two processes may have some divergence. It is important to note that reductions in callus cartilage with sclerostin inhibition has not been associated with any untoward effects on healing, presumably because bony callus formation remains robust. However there is little evidence that sclerostin inhibition promotes cartilage formation. Similarly, Wnt3a, a direct activator of wnt/β‐catenin signaling, has been shown to promote skeletal repair via intramembranous rather than endochondral bone formation.103 The effect of sclerostin antibodies or other activators of Wnt/β‐catenin signaling in bone may be limited to increased osteogenesis, and for those fractures destined to achieve union, this effect should increase callus strength. But for a pending or established nonunion, it is not clear that increased bone formation alone would improve outcomes. In a bony defect model characterized by frequent nonunions, sclerostin antibody provided minimal efficacy beyond the modest addition of non‐bridging bone at the defect margins.104
Figure 6.
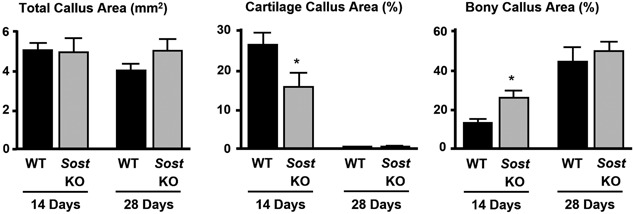
Histomorphometry of callus composition for normal wild‐type (WT) mice and Sost knockout (KO) mice subjected to internally stabilized closed femoral fractures. Data were generated on samples collected 14 or 28 days post‐fracture. Data represent means and SEM, n = 6–10/group/time point. *P < 0.05 for SOST KO versus WT. Reproduced with permission from Li et al.100
Two placebo‐controlled clinical trials assessed the effects of a sclerostin antibody (romosozumab; AMG 785/CDP7851) on fracture healing. One study (http://clinicaltrials.gov: NCT 00907296) involved adults with fresh tibial shaft fractures treated with an intramedullary nail, with the hypothesis that romosozumab would reduce radiographic healing time. The other study (http://clinicaltrials.gov: NCT01081678) involved adults with a fresh unilateral hip fracture treated with post‐surgical fixation, with the hypothesis that romosozumab would accelerate healing of hip fractures and improve physical functioning. Results of these studies had not been presented publically as of early 2016, but the sponsors indicated they would not pursue fracture healing indications due to the nature of the efficacy results, as well as recent regulatory guidance that suggested challenges in achieving registration for accelerated fracture healing.105 When fracture union and callus strength were assessed in animals prior to complete bridging and biomechanical recovery, sclerostin inhibition did not lead to acceleration of fracture healing.94, 99
Fibroblast Growth Factor‐2 (FGF‐2)
FGF‐2 is a potent mitogen with pleiotropic skeletal and extraskeletal effects. Systemically injected recombinant FGF‐2 increased bone formation in ovariectomized rats,106 and FGF‐2 inhibited terminal differentiation of cultured chondrocytes,107 suggesting FGF‐2 may promote osteogenesis and chondrogenesis. In a nonhuman primate study, internally fixated ulnar osteotomies that were subjected to periosteal stripping had a 40% nonunion rate in a control group treated with empty hydrogel carrier, whereas all animals in a group treated with FGF‐2 in hydrogel achieved union.108 Biomechanical assessments of calluses that achieved union showed significantly greater strength in the FGF‐2 group versus controls, consistent with increased osteogenesis. Histology was not assessed until the healed animals had stable calluses, so effects of FGF‐2 on cartilage could not be properly assessed. Nonetheless, the combined effects of increased union and increased strength of the post‐union calluses represents a high standard of preclinical evidence, and suggests FGF‐2 has the potential to prevent or treat DNFs. A placebo‐controlled clinical trial was conducted for FGF‐2 delivered percutaneously in hydrogel to closed tibial fractures.109 Inclusion criteria included transverse or short‐oblique fracture morphology, which tend to heal more slowly than oblique or spiral fractures. Over a 24‐week period, a greater percentage of patients in the FGF‐2 group achieved radiographic union (bridging of all four cortices). However there was no treatment effect on the rate at which fractures showed bridging of three of four cortices, and no difference in clinical healing endpoints. FGF‐2 is not currently indicated for fracture healing, and it is unclear whether registration trials for FGF‐2 will be conducted.
SUMMARY
The ultimate fates of FGF‐2, sclerostin antibody, PTH, PTHrP and other bone‐active factors for fracture healing are unknown, but experiences with these agents and with BMPs provide clues as to the attributes of therapeutic agents that may someday prevent or treat DNFs. All agents summarized in this review can increase bone formation, volume, density and strength. Some agents (PTH, PTHrP, FGF‐2) regulate the proliferation and differentiation of chondrocytes in ways that maintain their ability to produce cartilage, whereas sclerostin inhibitors and other activators of Wnt/β‐catenin signaling appear to favor osteogenesis over chondrogenesis. Some agents (BMPs and PTHrP) are expressed locally and functionally during fracture healing, suggesting physiological roles during repair that could be pharmacological augmented. Some agents are delivered to open fractures in special carriers (BMP‐2, BMP‐7), while others can be administered systemically (PTH, PTHrP, sclerostin antibody) or percutaneously to closed fractures (FGF‐2, and potentially PTH, PTHrP or sclerostin antibody). With the possible exception of sclerostin antibody, for which clinical fracture healing data have yet to be published, each agent produced some benefits in patients with fractures, though none has been tested in controlled clinical trials of patients with existing DNFs or with closed fractures at high risk of DNF.
DNFs have varying etiology and pathophysiology, and it is unlikely that one therapy will have universal utility in all at‐risk patients. Genetic or serum‐based factors have been explored as a way of identifying patients at risk for nonunion,110 and such biomarkers might someday help tailor adjuvant therapies based on patient‐ and fracture‐specific characteristics. In the meantime, there seems to be a critical interplay during fracture healing between cartilaginous and osteogenic processes that may be exploited to favorably influence outcomes for a range of DNFs.
A final proposal relates to translational aspects of preclinical fracture models. Many agents increase callus density and strength in the standard rodent closed fracture model, where union and biomechanical recovery is likely without treatments, but agents under development for DNFs require a higher level of preclinical evidence for proof of concept. Increased callus strength above and beyond that of unfractured bone is a common finding in the literature that does not by itself indicate potential to improve DNF outcomes. For research programs focused on DNF indications, preclinical efficacy in a true non‐union model represents the strongest proof of concept. Alternatively, modifications to the standard closed fracture model can lead to delayed healing, and treatment‐related increases in callus strength in these higher‐hurdle models may suggest the potential for improving DNF outcomes. When relying on the standard rodent fracture model, hints of therapeutic potential for DNFs may require biomechanical assessments at early post‐fracture time points, when strength recovery in untreated controls is far from complete. Histologic assessments conducted before full strength recovery is also recommended to allow proper evaluation of treatment effects on cartilage. For programs directed toward DNFs, a promising histological profile may include an early increase in callus cartilage, followed (post‐bridging) by an increase in its mineralization and conversion to bone. This profile should lead to improved strength of fractures that would have healed on their own, and may also promote union and strength recovery of fractures that would not.
AUTHORS’ CONTRIBUTIONS
Dr. Kostenuik drafted the original manuscript and contributed to its revisions. Dr. Mirza critically reviewed the original manuscript and contributed to revisions. Drs. Kostenuik and Mirza have read and approved the final submitted manuscript.
CONFLICT OF INTEREST
Dr. Kostenuik serves or has served as a consultant for Allergan, Amgen, Daiichi‐Sankyo, Kuros Biosurgery, Grunenthal, Merck Research Laboratories, Regeneron, SetPoint Medical, Sofinnova, The Column Group, and Ultragenyx. Dr. Kostenuik has served as a paid speaker for Amgen Inc. and owns stock in Amgen and Radius Health. Dr. Kostenuik has provided medical writing support to Merck, Radius Health, and Amgen. Dr. Mirza serves or has served as a consultant and or advisory board member for the American Medical Association, Conmed Linvatec, ActiveLife Scientific, Siena Biologics, Oculapps, Acumed, Tensegrity Technologies, Newry Corp, Select Equity Group, and MD Covered. Dr. Mirza owns stock in Amgen, Organovo, Cara Therapeutics, and GW Pharmaceuticals. Dr. Mirza serves or has served as a paid speaker for Siena Biologics and Acumed.
The copyright line for this article was changed on August 7, 2018, after original online publication.
REFERENCES
- 1. Mosekilde L, Bak B. 1993. The effects of growth hormone on fracture healing in rats: a histological description. Bone 14:19–27. [DOI] [PubMed] [Google Scholar]
- 2. Tzioupis C, Giannoudis PV. 2007. Prevalence of long‐bone non‐unions. Injury 38:S3–9. [DOI] [PubMed] [Google Scholar]
- 3. Einhorn TA, Gerstenfeld LC. 2015. Fracture healing: mechanisms and interventions. Nat Rev Rheumatol 11:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Westgeest J, Weber D, Dulai SK, et al. 2016. Factors associated with development of nonunion or delayed healing after an open long bone fracture: a prospective cohort study of 736 subjects. J Orthop Trauma 30:149–155. [DOI] [PubMed] [Google Scholar]
- 5. Marsh D. 1998. Concepts of fracture union, delayed union, and nonunion. Clin Orthop Relat Res S22–30. [DOI] [PubMed] [Google Scholar]
- 6. Andrew J, Hoyland J, Andrew S, et al. 1993. Demonstration of TGF‐b1 mRNA by in situ hybridization in normal human fracture healing. Calcif Tissue Int 52:74–78. [DOI] [PubMed] [Google Scholar]
- 7. Giannoudis PV, MacDonald DA, Matthews SJ, et al. 2000. Nonunion of the femoral diaphysis. The influence of reaming and non‐steroidal anti‐inflammatory drugs. J Bone Joint Surg Br 82:655–658. [DOI] [PubMed] [Google Scholar]
- 8. Reinke S, Geissler S, Taylor WR, et al. 2013. Terminally differentiated CD8(+) T cells negatively affect bone regeneration in humans. Sci Transl Med 5:177ra136. [DOI] [PubMed] [Google Scholar]
- 9. Gustilo RB, Mendoza RM, Williams DN. 1984. Problems in the management of type III (severe) open fractures: a new classification of type III open fractures. J Trauma 24:742–746. [DOI] [PubMed] [Google Scholar]
- 10. Reverte MM, Dimitriou R, Kanakaris NK, et al. 2011. What is the effect of compartment syndrome and fasciotomies on fracture healing in tibial fractures? Injury 42:1402–1407. [DOI] [PubMed] [Google Scholar]
- 11. Court‐Brown C, McQueen M. 1987. Compartment syndrome delays tibial union. Acta Orthop Scand 58:249–252. [DOI] [PubMed] [Google Scholar]
- 12. Hak DJ, Fitzpatrick D, Bishop JA, et al. 2014. Delayed union and nonunions: epidemiology, clinical issues, and financial aspects. Injury 45:S3–S7. [DOI] [PubMed] [Google Scholar]
- 13. Kanakaris NK, Giannoudis PV. 2007. The health economics of the treatment of long‐bone non‐unions. Injury 38:S77–S84. [DOI] [PubMed] [Google Scholar]
- 14. Schottel PC, O'Connor DP, Brinker MR. 2015. Time trade‐off as a measure of health‐related quality of life: long bone nonunions have a devastating impact. J Bone Joint Surg Am 97:1406–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tagil M, McDonald MM, Morse A, et al. 2010. Intermittent PTH(1‐34) does not increase union rates in open rat femoral fractures and exhibits attenuated anabolic effects compared to closed fractures. Bone 46:852–859. [DOI] [PubMed] [Google Scholar]
- 16. Gerstenfeld LC, Sacks DJ, Pelis M, et al. 2009. Comparison of effects of the bisphosphonate alendronate versus the RANKL inhibitor denosumab on murine fracture healing. J Bone Miner Res 24:196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bonnarens F, Einhorn T. 1984. Production of a standard closed fracture in laboratory animal bone. J Orthop Res 2:97–101. [DOI] [PubMed] [Google Scholar]
- 18. Bais M, McLean J, Sebastiani P, et al. 2009. Transcriptional analysis of fracture healing and the induction of embryonic stem cell‐related genes. PLoS ONE 4:e5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hausman M, Schaffler M, Majeska R. 2001. Prevention of fracture healing in rats by an inhibitor of angiogenesis. Bone 29:560–564. [DOI] [PubMed] [Google Scholar]
- 20. Meyer R, Tsahakis P, Martin D, et al. 2001. Age and ovariectomy impair both the normalization of mechanical properties and the accretion of mineral by the fracture callus in rats. J Orthop Res 19:428–435. [DOI] [PubMed] [Google Scholar]
- 21. Lu C, Miclau T, Hu D, et al. 2007. Ischemia leads to delayed union during fracture healing: a mouse model. J Orthop Res 25:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hanratty BM, Ryaby JT, Pan XH, et al. 2009. Thrombin related peptide TP508 promoted fracture repair in a mouse high energy fracture model. J Orthop Surg Res 4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kokubu T, Hak DJ, Hazelwood SJ, et al. 2003. Development of an atrophic nonunion model and comparison to a closed healing fracture in rat femur. J Orthop Res 21:503–510. [DOI] [PubMed] [Google Scholar]
- 24. Virk MS, Alaee F, Tang H, et al. 2013. Systemic administration of sclerostin antibody enhances bone repair in a critical‐sized femoral defect in a rat model. J Bone Joint Surg Am 95:694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bahney CS, Hu DP, Taylor AJ, et al. 2014. Stem cell‐derived endochondral cartilage stimulates bone healing by tissue transformation. J Bone Miner Res 29:1269–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lienau J, Schmidt‐Bleek K, Peters A, et al. 2009. Differential regulation of blood vessel formation between standard and delayed bone healing. J Orthop Res 27:1133–1140. [DOI] [PubMed] [Google Scholar]
- 27. Naik AA, Xie C, Zuscik MJ, et al. 2009. Reduced COX‐2 expression in aged mice is associated with impaired fracture healing. J Bone Miner Res 24:251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jirarattanaphochai K, Kiat TS, Chua D, et al. 1993. The effect of methylprednisolone on porcine bone morphogenetic protein in fracture healing. An experimental allograft model in rabbits. Clin Orthop Relat Res 292:366–375. [PubMed] [Google Scholar]
- 29. Dent‐Acosta RE, Storm N, Steiner RS, et al. 2012. The tactics of modern‐day regulatory trials. J Bone Joint Surg Am 94:39–44. [DOI] [PubMed] [Google Scholar]
- 30. http://www.fda.gov/ohrms/dockets/dailys/00/mar00/031300/aav0001.pdf. 2000. FDA Approval Order for Exogen.
- 31. Lyon T, Scheele W, Bhandari M, et al. 2013. Efficacy and safety of recombinant human bone morphogenetic protein‐2/calcium phosphate matrix for closed tibial diaphyseal fracture: a double‐blind, randomized, controlled phase‐II/III trial. J Bone Joint Surg Am 95:2088–2096. [DOI] [PubMed] [Google Scholar]
- 32. Aspenberg P, Genant HK, Johansson T, et al. 2010. Teriparatide for acceleration of fracture repair in humans: a prospective, randomized, double‐blind study of 102 postmenopausal women with distal radial fractures. J Bone Miner Res 25:404–414. [DOI] [PubMed] [Google Scholar]
- 33. Raschke M, Rasmussen MH, Govender S, et al. 2007. Effects of growth hormone in patients with tibial fracture: a randomised, double‐blind, placebo‐controlled clinical trial. Eur J Endocrinol 156:341–351. [DOI] [PubMed] [Google Scholar]
- 34. Simon AM, Manigrasso MB, O'Connor JP. 2002. Cyclo‐oxygenase 2 function is essential for bone fracture healing. J Bone Miner Res 17:963–976. [DOI] [PubMed] [Google Scholar]
- 35. Kayal RA, Tsatsas D, Bauer MA, et al. 2007. Diminished bone formation during diabetic fracture healing is related to the premature resorption of cartilage associated with increased osteoclast activity. J Bone Miner Res 22:560–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shimoaka T, Kamekura S, Chikuda H, et al. 2004. Impairment of bone healing by insulin receptor substrate‐1 deficiency. J Biol Chem 279:15314–15322. [DOI] [PubMed] [Google Scholar]
- 37. Kronenberg HM. 2003. Developmental regulation of the growth plate. Nature 423:332–336. [DOI] [PubMed] [Google Scholar]
- 38. Amizuka N, Warshawsky H, Henderson JE, et al. 1994. Parathyroid hormone‐related peptide‐depleted mice show abnormal epiphyseal cartilage development and altered endochondral bone formation. J Cell Biol 126:1611–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang YH, Qiu Y, Han XD, et al. 2013. Haploinsufficiency of endogenous parathyroid hormone‐related peptide impairs bone fracture healing. Clin Exp Pharmacol Physiol 40:715–723. [DOI] [PubMed] [Google Scholar]
- 40. Wang M, Nasiri AR, Broadus AE, et al. 2015. Periosteal PTHrP regulates cortical bone remodeling during fracture healing. Bone 81:104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nakazawa T, Nakajima A, Shiomi K, et al. 2005. Effects of low‐dose, intermittent treatment with recombinant human parathyroid hormone (1‐34) on chondrogenesis in a model of experimental fracture healing. Bone 37:711–719. [DOI] [PubMed] [Google Scholar]
- 42. Epari DR, Schell H, Bail HJ, et al. 2006. Instability prolongs the chondral phase during bone healing in sheep. Bone 38:864–870. [DOI] [PubMed] [Google Scholar]
- 43. McDonald MM, Morse A, Mikulec K, et al. 2013. Matrix metalloproteinase‐driven endochondral fracture union proceeds independently of osteoclast activity. J Bone Miner Res 28:1550–1560. [DOI] [PubMed] [Google Scholar]
- 44. Lenehan T, Balligand M, Nunamaker D, et al. 1985. Effect of EHDP on fracture healing in dogs. J Orthop Res 3:499–507. [DOI] [PubMed] [Google Scholar]
- 45. Urist MR. 1965. Bone: formation by autoinduction. Science 150:893–899. [DOI] [PubMed] [Google Scholar]
- 46. Susperregui AR, Vinals F, Ho PW, et al. 2008. BMP‐2 regulation of PTHrP and osteoclastogenic factors during osteoblast differentiation of C2C12 cells. J Cell Physiol 216:144–152. [DOI] [PubMed] [Google Scholar]
- 47. Iwasaki M, Nakahara H, Nakase T, et al. 1994. Bone morphogentic protein 2 stimulates osteogenesis but does not affect chondrogenesis in osteochondrogenic differentiation of periosteum‐derived cells. J Bone Miner Res 9:1195–1204. [DOI] [PubMed] [Google Scholar]
- 48. Lu C, Xing Z, Yu YY, et al. 2010. Recombinant human bone morphogenetic protein‐7 enhances fracture healing in an ischemic environment. J Orthop Res 28:687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu YY, Lieu S, Lu C, et al. 2010. Bone morphogenetic protein 2 stimulates endochondral ossification by regulating periosteal cell fate during bone repair. Bone 47:65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang EA, Rosen V, D'Alessandro JS, et al. 1990. Recombinant human bone morphogenetic protein induces bone formation. Proc Natl Acad Sci USA 87:2220–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Intini G, Nyman JS. 2015. Dkk1 haploinsufficiency requires expression of Bmp2 for bone anabolic activity. Bone 75:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. http://www.accessdata.fda.gov/cdrh_docs/pdf/h010002a.pdf. 2001. FDA Approval of OP‐1 Implant.
- 53. Friedlaender GE, Perry CR, Cole JD, et al. 2001. Osteogenic protein‐1 (bone morphogenetic protein‐7) in the treatment of tibial nonunions. J Bone Joint Surg Am 83‐A Suppl 1:S151–S158. [PMC free article] [PubMed] [Google Scholar]
- 54. http://www.accessdata.fda.gov/cdrh_docs/pdf/P000054b.pdf. 2004. FDA Summary of Safety and Effectiveness Data.
- 55. Govender S, Csimma C, Genant HK, et al. 2002. Recombinant human bone morphogenetic protein‐2 for treatment of open tibial fractures: a prospective, controlled, randomized study of four hundred and fifty patients. J Bone Joint Surg Am 84A:2123–2134. [DOI] [PubMed] [Google Scholar]
- 56. Aro HT, Govender S, Patel AD, et al. 2011. Recombinant human bone morphogenetic protein‐2: a randomized trial in open tibial fractures treated with reamed nail fixation. J Bone Joint Surg Am 93:801–808. [DOI] [PubMed] [Google Scholar]
- 57. Carragee EJ, Hurwitz EL, Weiner BK. 2011. A critical review of recombinant human bone morphogenetic protein‐2 trials in spinal surgery: emerging safety concerns and lessons learned. Spine J 11:471–491. [DOI] [PubMed] [Google Scholar]
- 58. Ong KL, Villarraga ML, Lau E, et al. 2010. Off‐label use of bone morphogenetic proteins in the United States using administrative data. Spine (PhilaPa 1976) 35:1794–1800. [DOI] [PubMed] [Google Scholar]
- 59. Cupp ME, Nayak SK, Adem AS, et al. 2013. Parathyroid hormone (PTH) and PTH‐related peptide domains contributing to activation of different PTH receptor‐mediated signaling pathways. J Pharmacol Exp Ther 345:404–418. [DOI] [PubMed] [Google Scholar]
- 60. Miao D, He B, Lanske B, et al. 2004. Skeletal abnormalities in Pth‐null mice are influenced by dietary calcium. Endocrinology 145:2046–2053. [DOI] [PubMed] [Google Scholar]
- 61. Chan FK, Tiu SC, Choi KL, et al. 2003. Increased bone mineral density in patients with chronic hypoparathyroidism. J Clin Endocrinol Metab 88:3155–3159. [DOI] [PubMed] [Google Scholar]
- 62. Weir EC, Terwilliger G, Sartori L, et al. 1992. Synthetic parathyroid hormone‐like protein (1‐74) is anabolic for bone in vivo. Calcif Tissue Int 51:30–34. [DOI] [PubMed] [Google Scholar]
- 63. Chouinard L, Lesage E, Smith SY, et al. 2014. The long term effects of abaloparatide (BA058) on bone histomorphometry in osteopenic rats. J Bone Miner Res 29:S468. [Google Scholar]
- 64. Amizuka N, Karaplis AC, Henderson JE, et al. 1996. Haploinsufficiency of parathyroid hormone‐related peptide (PTHrP) results in abnormal postnatal bone development. Dev Biol 175:166–176. [DOI] [PubMed] [Google Scholar]
- 65. Ren Y, Liu B, Feng Y, et al. 2011. Endogenous PTH deficiency impairs fracture healing and impedes the fracture‐healing efficacy of exogenous PTH(1–34). PLoS ONE 6:e23060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Alkhiary YM, Gerstenfeld LC, Krall E, et al. 2005. Enhancement of experimental fracture‐healing by systemic administration of recombinant human parathyroid hormone (PTH 1‐34). J Bone Joint Surg Am 87:731–741. [DOI] [PubMed] [Google Scholar]
- 67. Kakar S, Einhorn TA, Vora S, et al. 2007. Enhanced chondrogenesis and Wnt signaling in PTH‐treated fractures. J Bone Miner Res 22:1903–1912. [DOI] [PubMed] [Google Scholar]
- 68. Borges JL, Freitas A, Bilezikian JP. 2013. Accelerated fracture healing with teriparatide. Arq Bras Endocrinol Metabol 57:153–156. [DOI] [PubMed] [Google Scholar]
- 69. Oteo‐Alvaro A, Moreno E. 2010. Atrophic humeral shaft nonunion treated with teriparatide (rh PTH 1‐34): a case report. J Shoulder Elbow Surg 19:e22–28. [DOI] [PubMed] [Google Scholar]
- 70. Uemura T, Okada M, Yokoi T, et al. 2015. Successful bone healing of nonunion after ulnar shortening osteotomy for smokers treated with teriparatide. Orthopedics 38:e733–737. [DOI] [PubMed] [Google Scholar]
- 71. Peichl P, Holzer LA, Maier R, et al. 2011. Parathyroid hormone 1‐84 accelerates fracture‐healing in pubic bones of elderly osteoporotic women. J Bone Joint Surg Am 93:1583–1587. [DOI] [PubMed] [Google Scholar]
- 72. Johansson T. 2016. PTH 1‐34 (teriparatide) may not improve healing in proximal humerus fractures. A randomized, controlled study of 40 patients. Acta Orthop 87:79–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Martin T. 2005. Osteoblast‐derived PTHrP is a physiological regulator of bone formation. J Clin Invest 115:2322–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Miao D, He B, Karaplis A, et al. 2002. Parathyroid hormone is essential for normal fetal bone formation. J Clin Invest 109:1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tsukazaki T, Ohtsuru A, Namba H, et al. 1996. Parathyroid hormone‐related protein (PTHrP) action in rat articular chondrocytes: comparison of PTH(1‐34), PTHrP(1‐34), PTHrP(1‐141), PTHrP(100‐114) and antisense oligonucleotides against PTHrP. J Endocrinol 150:359–368. [DOI] [PubMed] [Google Scholar]
- 76. Zerega B, Cermelli S, Bianco P, et al. 1999. Parathyroid hormone [PTH(1‐34)] and parathyroid hormone‐related protein [PTHrP(1‐34)] promote reversion of hypertrophic chondrocytes to a prehypertrophic proliferating phenotype and prevent terminal differentiation of osteoblast‐like cells. J Bone Miner Res 14:1281–1289. [DOI] [PubMed] [Google Scholar]
- 77. Okazaki K, Jingushi S, Ikenoue T, et al. 2003. Expression of parathyroid hormone‐related peptide and insulin‐like growth factor I during rat fracture healing. J Orthop Res 21:511–520. [DOI] [PubMed] [Google Scholar]
- 78. Kartsogiannis V, Moseley J, McKelvie B, et al. 1997. Temporal expression of PTHrP during endochondral bone formation in mouse and intramembranous bone formation in an in vivo rabbit model. Bone 21:385–392. [DOI] [PubMed] [Google Scholar]
- 79. Wang M, VanHouten JN, Nasiri AR, et al. 2014. Periosteal PTHrP regulates cortical bone modeling during linear growth in mice. J Anat 225:71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Leder BZ, O'Dea LS, Zanchetta JR, et al. 2015. Effects of abaloparatide, a human parathyroid hormone‐related peptide analog, on bone mineral density in postmenopausal women with osteoporosis. J Clin Endocrinol Metab 100:697–706. [DOI] [PubMed] [Google Scholar]
- 81. Bostrom MP, Gamradt SC, Asnis P, et al. 2000. Parathyroid hormone‐related protein analog RS‐66271 is an effective therapy for impaired bone healing in rabbits on corticosteroid therapy. Bone 26:437–442. [DOI] [PubMed] [Google Scholar]
- 82. Waters RV, Gamradt SC, Asnis P, et al. 2000. Systemic corticosteroids inhibit bone healing in a rabbit ulnar osteotomy model. Acta Orthop Scand 71:316–321. [DOI] [PubMed] [Google Scholar]
- 83. Walsh CA, Birch MA, Fraser WD, et al. 1995. Expression and secretion of parathyroid hormone‐related protein by human bone‐derived cells in vitro: effects of glucocorticoids. J Bone Miner Res 10:17–25. [DOI] [PubMed] [Google Scholar]
- 84. Liu A, Li Y, Wang Y, et al. 2015. Exogenous parathyroid hormone‐related peptide promotes fracture healing in lepr(−/−) mice. Calcif Tissue Int 97:581–591. [DOI] [PubMed] [Google Scholar]
- 85. Lozano D, de Castro LF, Dapia S, et al. 2009. Role of parathyroid hormone‐related protein in the decreased osteoblast function in diabetes‐related osteopenia. Endocrinology 150:2027–2035. [DOI] [PubMed] [Google Scholar]
- 86. Monroe DG, McGee‐Lawrence ME, Oursler MJ, et al. 2012. Update on Wnt signaling in bone cell biology and bone disease. Gene 492:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Agholme F, Aspenberg P. 2011. Wnt signaling and orthopedics, an overview. Acta Orthop 82:125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Baron R, Kneissel M. 2013. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med 19:179–192. [DOI] [PubMed] [Google Scholar]
- 89. Ke HZ, Richards WG, Li X, et al. 2012. Sclerostin and Dickkopf‐1 as therapeutic targets in bone diseases. Endocr Rev 33:747–783. [DOI] [PubMed] [Google Scholar]
- 90. Li X, Grisanti M, Fan W, et al. 2011. Dickkopf‐1 regulates bone formation in young growing rodents and upon traumatic injury. J Bone Miner Res 26:2610–2621. [DOI] [PubMed] [Google Scholar]
- 91. Ominsky MS, Li C, Li X, et al. 2011. Inhibition of sclerostin by monoclonal antibody enhances bone healing and improves bone density and strength of nonfractured bones. J Bone Miner Res 26:1012–1021. [DOI] [PubMed] [Google Scholar]
- 92. Suen PK, He YX, Chow DH, et al. 2014. Sclerostin monoclonal antibody enhanced bone fracture healing in an open osteotomy model in rats. J Orthop Res 32:997–1005. [DOI] [PubMed] [Google Scholar]
- 93. Taylor S, Ominsky MS, Hu R, et al. 2016. Time‐dependent cellular and transcriptional changes in the osteoblast lineage associated with sclerostin antibody treatment in ovariectomized rats. Bone 84:148–159. [DOI] [PubMed] [Google Scholar]
- 94. Florio M, Gunasekaran K, Stolina M, et al. 2016. A bispecific antibody targeting sclerostin and DKK‐1 promotes bone mass accrual and fracture repair. Nat Commun 7:11505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. van Bezooijen RL, Roelen BA, Visser A, et al. 2004. Sclerostin is an osteocyte‐expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med 199:805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. McClung MR, Grauer A, Boonen S, et al. 2014. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med 370:412–420. [DOI] [PubMed] [Google Scholar]
- 97. Agholme F, Isaksson H, Kuhstoss S, et al. 2011. The effects of Dickkopf‐1 antibody on metaphyseal bone and implant fixation under different loading conditions. Bone 48:988–996. [DOI] [PubMed] [Google Scholar]
- 98. Feng G, Chang‐Qing Z, Yi‐Min C, et al. 2015. Systemic administration of sclerostin monoclonal antibody accelerates fracture healing in the femoral osteotomy model of young rats. Int Immunopharmacol 24:7–13. [DOI] [PubMed] [Google Scholar]
- 99. Morse A, Yu NY, Peacock L, et al. 2015. Endochondral fracture healing with external fixation in the Sost knockout mouse results in earlier fibrocartilage callus removal and increased bone volume fraction and strength. Bone 71:155–163. [DOI] [PubMed] [Google Scholar]
- 100. Li C, Ominsky MS, Tan HL, et al. 2011. Increased callus mass and enhanced strength during fracture healing in mice lacking the sclerostin gene. Bone 49:1178–1185. [DOI] [PubMed] [Google Scholar]
- 101. Hill TP, Spater D, Taketo MM, et al. 2005. Canonical Wnt/beta‐catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell 8:727–738. [DOI] [PubMed] [Google Scholar]
- 102. Huang Y, Zhang X, Du K, et al. 2012. Inhibition of beta‐catenin signaling in chondrocytes induces delayed fracture healing in mice. J Orthop Res 30:304–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Minear S, Leucht P, Jiang J, et al. 2010. Wnt proteins promote bone regeneration. Sci TranslMed 2:29ra 30. [DOI] [PubMed] [Google Scholar]
- 104. Alaee F, Virk MS, Tang H, et al. 2014. Evaluation of the effects of systemic treatment with a sclerostin neutralizing antibody on bone repair in a rat femoral defect model. J Orthop Res 32:197–203. [DOI] [PubMed] [Google Scholar]
- 105. http://www.sec.gov/Archives/edgar/data/318154/000144530513000364/amgn-1231201210k.htm
- 106. Power RA, Iwaniec UT, Magee KA, et al. 2004. Basic fibroblast growth factor has rapid bone anabolic effects in ovariectomized rats. Osteoporos Int 15:716–723. [DOI] [PubMed] [Google Scholar]
- 107. Wroblewski J, Edwall‐Arvidsson C. 1995. Inhibitory effects of basic fibroblast growth factor on chondrocyte differentiation. J Bone Miner Res 10:735–742. [DOI] [PubMed] [Google Scholar]
- 108. Kawaguchi H, Nakamura K, Tabata Y, et al. 2001. Acceleration of fracture healing in nonhuman primates by fibroblast growth factor‐2. J Clin Endocrinol Metab 86:875–880. [DOI] [PubMed] [Google Scholar]
- 109. Kawaguchi H, Oka H, Jingushi S, et al. 2010. A local application of recombinant human fibroblast growth factor 2 for tibial shaft fractures: a randomized, placebo‐controlled trial. J Bone Miner Res 25:2735–2743. [DOI] [PubMed] [Google Scholar]
- 110. Pountos I, Georgouli T, Pneumaticos S, et al. 2013. Fracture non‐union: can biomarkers predict outcome? Injury 44:1725–1732. [DOI] [PubMed] [Google Scholar]
- 111. Hadjiargyrou M, Lombardo F, Zhao S, et al. 2002. Transcriptional profiling of bone regeneration. Insight into the molecular complexity of wound repair. J Biol Chem 277:30177–30182. [DOI] [PubMed] [Google Scholar]