

Abstract
Hedgehog (Hh) signaling plays a key role in the development and maintenance of animal tissues. This signaling is mediated by the atypical G protein–coupled receptor (GPCR) Smoothened (Smo). Smo activation leads to signaling through several well-characterized effectors to activate Hh target gene expression. Recent studies have implicated activation of the heterotrimeric G protein subunit Gαi and the subsequent decrease in cellular cAMP levels in promoting the Hh response in flies and mammals. Although Hh stimulation decreases cAMP levels in some insect cell lines, here using a bioluminescence resonance energy transfer (BRET)-based assay we found that this stimulation had no detectable effect in Drosophila S2-R+ cells. However, we observed an unexpected and significant Gαs-dependent increase in cAMP levels in response to strong Smo activation in Smo-transfected cells. This effect was mediated by Smo's broadly conserved core, and was specifically activated in response to phosphorylation of the Smo C-terminus by GPCR kinase 2 (Gprk2). Genetic analysis of heterotrimeric G protein function in the developing Drosophila wing revealed a positive role for cAMP in the endogenous Hh response. Specifically, we found that mutation or depletion of Gαs diminished low-threshold Hh responses in Drosophila, whereas depletion of Gαi potentiated them (in contrast to previous findings). Our analysis suggested that regulated cAMP production is important for controlling the sensitivity of cellular responses to Hh in Drosophila.
Keywords: Hedgehog signaling pathway, heterotrimeric G protein, cyclic AMP (cAMP), G protein-coupled receptor (GPCR), Drosophila, G-protein–coupled receptor kinase, Smoothened
Introduction
The seven-transmembrane–spanning receptor protein Smo is an atypical member of the G protein–coupled receptor (GPCR)3 protein family that activates cytoplasmic signaling in response to secreted Hedgehog (Hh) family proteins. Insufficient or excessive Smo activity can have severe consequences for the formation and maintenance of tissues (1, 2). In fact, activating mutations in Smo have been shown to drive tumor formation in several tissues (1, 3). This suggests that Smo activity must be tightly regulated in cells. Consistent with this, several complex and intersecting regulatory mechanisms for finely controlling Smo activity have been identified. Many of the details are best understood in Drosophila melanogaster, where ubiquitination (4, 5), sumoylation (6, 7), and phosphorylation (8–13) of the Smo cytoplasmic C-terminus (SmoCT) all play key roles in controlling Smo protein levels, localization, and activity.
SmoCT phosphorylation seems to be a particularly important means of controlling Smo activity. Phosphorylation of the Smo autoinhibitory domain (SAID) region of the SmoCT by the cAMP-regulated protein kinase A (PKA) is the principal mechanism for activating Drosophila Smo. PKA phosphorylation primes the SAID for phosphorylation at adjacent sites by CkI, and the combined effects are sufficient to stabilize Smo, promote its accumulation at the plasma membrane, and drive it into an active conformation (8–10, 14). However, other kinases, including GPCR kinase 2 (Gprk2) (11), CkIγ (12), and Fu (13), act after PKA to modulate Drosophila Smo activity by phosphorylating other sites in the SmoCT. Like the PKA phosphorylation sites, most of these sites are not conserved between flies and vertebrates. Interestingly, however, Gprk2 and a homologous mammalian kinase, GRK2 (together with casein kinase I), phosphorylate conserved sites in the proximal SmoCT in Drosophila and mice, respectively (11, 15). In both organisms, phosphorylation of these sites promotes Smo signaling activity (11, 15, 16), indicating that GRK phosphorylation of the SmoCT is an evolutionarily conserved mechanism for Smo activation (although it may not be critical in all species) (17). However, precisely how GRK phosphorylation enhances Smo activity is not known.
Once activated, Smo signals through a series of downstream canonical effectors that includes the atypical kinesin Costal-2/KIF7 and (in some organisms like Drosophila and zebrafish (18, 19)) the protein kinase Fused, which are components of the so-called Hh Signaling Complex (HSC), as well as Suppressor of Fused (Sufu) (1, 2). Canonical signaling ultimately converges on the transcription factor cubitus interruptus (Ci)/GLI. In the absence of Hh, Ci/GLI2/3 are converted through limited proteolysis to transcriptional repressors. Hh-dependent activation of canonical signaling prevents this processing, causing Ci/GLI2/3 to accumulate in full-length transcriptional activator forms that drive transcription of Hh target genes (1, 2). Phosphoregulation of Ci/GLI2/3 processing is another key point of control in the Hh pathway. PKA again plays the key role, although in this case it acts as a negative rather than positive regulator. Ci/GLI2/3 phosphorylation by PKA primes the proteins for further phosphorylation by other kinases, ultimately promoting ubiquitination and subsequent partial proteolysis to the repressor forms (1, 2).
The central role of PKA in Drosophila and vertebrate Hh signaling pathways has long suggested that regulation of the second messenger cAMP, the activator of PKA, could be important for pathway function. Indeed, Smo shares some structural and functional similarities with classical GPCRs (20) and there is evidence that Smo can couple to heterotrimeric G proteins, specifically to Gαi. Activation of Gαi by Smo suppresses cAMP production and can promote target gene expression in flies (21, 22) and mammals (23, 24), and is involved in GLI-independent noncanonical responses in mammals as well (25–28). Recent studies have demonstrated that certain other GPCRs can influence Hh signaling by activating Gαs or Gαi. For example, Gpr161 is a constitutively active GPCR that signals through Gαs to keep cAMP levels high in the absence of ligand, ensuring efficient PKA-dependent processing of GLI proteins and silencing of target gene expression (29). Conversely, the Gαi-coupled GPCR Gpr175 suppresses cAMP production and promotes Hh pathway activation (30).
The physiological importance of heterotrimeric G protein signaling in the Hh response has been controversial. Experiments using a cAMP-insensitive form of the PKA catalytic subunit initially shed doubt upon the importance of regulated PKA activity in the Hh response (31, 32). However, several studies cited above have now shown clear links between Gα proteins, cAMP, and Hh pathway activity. The link between decreased cAMP production and increased Hh target gene expression seems fairly straightforward in mammals, where the principal role of PKA is to inhibit GLI proteins (2). However, there is conflicting evidence showing that PKA can also promote Shh signaling in vertebrate cells and tissues (33, 34). The situation is similarly complex in Drosophila, where PKA functions both as an activator and inhibitor of the Hh response. For example, complete loss of PKA activity causes constitutive Ci activation and ligand-independent target gene expression (31, 35), but more moderate PKA inhibition or reduction of cAMP levels can actually decrease Hh-dependent target gene expression (8, 36). Similarly, increased cAMP levels or PKA activity can either prevent (31, 37) or enhance (8, 36, 38) expression of Hh target genes. The evidence suggests that control of cAMP levels within certain upper and lower limits is important for proper functioning of the pathway. Some of the conflicting evidence may result from cell-type–specific effects that could reflect differences in basal cellular cAMP levels in different systems (39).
To address some of the conflicting evidence about the implication of heterotrimeric G proteins in the Hh response and to examine how G protein signaling could be regulated at the level of Smo, we sought to characterize the effects of Hh signaling on cAMP levels and target gene expression using cell-based assays in Drosophila S2 cells. To our surprise, we did not observe changes in cAMP levels in Hh-treated cells. However, stronger activation of Smo caused a substantial Gαs-dependent increase in cAMP levels specifically in response to Gprk2 phosphorylation of the SmoCT. This effect on cAMP production was mediated by the evolutionarily conserved core of Smo, and occurred independently of the canonical signaling effectors Fu and Cos2. Although not essential for signaling, Gαs and Gαi depletion experiments in vivo both suggest that heterotrimeric G protein-mediated regulation of cAMP levels is important for adjusting the sensitivity of cells to endogenous Hh ligand levels.
Results
Smo can activate Gαs-dependent signaling
Drosophila Smo has been shown to activate Gαi in transfected Sf9 cells and Hh-treated Clone-8 cells (21, 40). Initially to see if we could observe the same response, we used a bioluminescence resonance energy transfer (BRET) assay based on the EPAC-BRET biosensor (41) to measure changes in cAMP levels in live S2 cells. The BRET signal produced by this biosensor is inversely correlated to cAMP levels. In control experiments, stimulation of cAMP production by expression of a partially activated mutant form of Gαs (GαsQ215L) (42) caused the expected decrease in the EPAC-BRET signal (Fig. 1A). Conversely, inhibiting cAMP production with the comparable activated form of Gαi (GαiQ205L) significantly increased the EPAC-BRET signal (Fig. 1A). Thus this assay can reliably detect both increases and decreases in cellular cAMP levels in S2 cells.
Figure 1.

Strong Smo activation stimulates cAMP production in S2 cells. Graphed data represent mean ± S.D. A, EPAC-BRET assay to measure changes in cAMP levels in S2-R+ cells in response to expression of constitutively active forms of Gαs (GαsQ215L) or Gαi (GαiQ205L). t test versus cells transfected with empty vector (−): **, p < 0.01; ***, p < 0.001. B, ptc-luc reporter assay monitoring target gene activation in mock- and HhN-transfected S2-R+ cells. t test: ***, p < 0.001. C, measurement of cAMP levels by EPAC-BRET assay in mock- and HhN-transfected S2-R+ cells. t test: n.s., not significant. D, schematic diagram of truncated/mutated Smo variants used in this study (not to scale). The relative locations of the 7-transmembrane domains (black boxes), the fours clusters of Gprk2 phosphorylation sites (green boxes), and the SAID containing three clusters of PKA/CkI phosphorylation sites (blue box) are indicated. Numbers refer to amino acid positions in Smo. E, ptc-luc reporter assay monitoring target gene activation in control and SmoWT-GFP transfected cells, with and without HhN co-transfection. t test: ***, p < 0.001. F, measurement of cAMP levels by EPAC-BRET assay in cells transfected with empty vector (−), SmoWT-GFP, or SmoWT-GFP plus HhN. t test: ***, p < 0.001; n.s., not significant. G, ptc-luc reporter activity in cells transfected with empty vector (−) or SmoSD-GFP. H, EPAC-BRET assay of cAMP levels in cells transfected with empty vector (−) or SmoSD-GFP. t test: ***, p < 0.001. I, EPAC-BRET assay of cAMP levels in SmoWT-GFP-expressing cells treated for 0.5, 1, 2, or 4 h with medium conditioned by control cells or cells expressing HhN, or co-transfected with HhN expression plasmid (Smo + Hh). t test: **, p < 0.01; ***, p < 0.001; n.s., not significant. J, immunoblot (IB) analysis of SmoWT-GFP in cells treated as in I.
We then conducted EPAC-BRET assays to test for Hh-dependent changes in cellular cAMP levels. Transfection of S2 cells with an expression plasmid for HhN, a secreted and biologically active N-terminal fragment of Hh (43) did not yield the expected decrease in cellular cAMP levels, although it did activate target gene expression (assessed using a ptc-luciferase reporter assay) (44) (Fig. 1, B and C). Even when using a myristoylated, membrane-targeted form of the EPAC-BRET biosensor to more specifically assess changes in cAMP pools at the plasma membrane (45), we did not observe the anticipated decrease (Fig. S1).
To see if enhancing Smo activity would lead to an effect in this assay, we expressed GFP-tagged, WT and mutant forms of Smo (see Fig. 1D). ptc-luc reporter activity was similarly low in WT Smo (SmoWT)-transfected cells compared with mock transfected controls (Fig. 1E), indicating that the level of Smo overexpression was moderate. Co-expression with HhN caused strong activation of ptc-luc reporter transcription (Fig. 1E). Unexpectedly, we observed a substantial Hh-dependent increase in cellular cAMP levels under these conditions (Fig. 1F). We obtained the same effect when we strongly activated signaling by expressing the PKA- and CkI-phosphomimetic, constitutively active mutant form of Smo, SmoSD123 (hereafter referred to as SmoSD) (8) (Fig. 1, G and H). Thus strong activation of the Hh pathway increased both target gene expression and cAMP levels in S2 cells.
To determine whether the increase in cAMP levels we observed is an acute response to Smo activation rather than a gradual or adaptive response to days-long pathway activation in our HhN co-transfection experimental setup, we carried out a time course analysis on SmoWT-expressing cells treated with medium conditioned by control- or HhN-expressing cells. Treatment with control-conditioned medium had no effect on cAMP levels at any time point tested (Fig. 1I). In short-term treatments (between 1 and 10 min), we did not observe HhN-dependent changes in cAMP levels (results not shown). Increases in cAMP levels were observed after 30 and 60 min of exposure to HhN, but they did not reach statistical significance (Fig. 1I). After 2 h of HhN treatment, cAMP levels were significantly increased, and by 4 h the magnitude of increase was similar to that observed in control cells that had expressed co-transfected HhN for 2–3 days (Fig. 1I). In Western blot analysis at each of these time points, HhN-dependent stabilization of Smo-GFP was just detectable after 30 min of treatment, and the shifted, hyperphosphorylated active form of Smo-GFP first became readily apparent after 2 h of HhN exposure and increased at 4 h (Fig. 1J). Thus the time course of cAMP regulation closely matched the profile of Smo activation. Although the cAMP increase was similar in cells expressing HhN for days and those treated for only 4 h, the transiently exposed cells had only a small percentage of the levels of Smo protein observed in the continuously exposed cells (Fig. 1J). This suggests that the effect does not require extremely high levels of Smo protein.
The most straightforward interpretation of these results is that strong activation of Smo activates Gαs, either directly or indirectly (e.g. by regulating another GPCR). If this is true, we expected that double-stranded RNA (dsRNA)-mediated depletion of Gαs would block the increase in cAMP levels. We first confirmed that treatment of Gαs-transfected cells with dsRNA targeting the gαs transcript efficiently blocked production of the protein (Fig. 2A). Consistent with our expectation, the ability of Smo to increase cAMP levels was strictly dependent upon Gαs. dsRNA-mediated depletion of Gαs had no significant effect on basal cAMP levels in control cells (Fig. 2B). However, the SmoSD-dependent increase in cAMP levels was almost completely inhibited (Fig. 2B). We confirmed that SmoSD protein levels were unchanged by Gαs depletion (Fig. 2C). We conclude that strong activation of Smo activates Gαs, either directly or indirectly, to stimulate cAMP production in S2 cells.
Figure 2.

The increase in cAMP downstream of Smo is Gαs-dependent. Graphed data represent mean ± S.D. A, immunoblot (IB) analysis of myc-tagged Gαs-transfected S2-R+ cells, treated with dsRNAs targeting β-gal (−) or Gαs. The blot was probed with anti-myc tag antibody to show efficient depletion of transfected Gαs, and with anti-α-tubulin as a loading control. B, EPAC-BRET assay of cAMP levels in control (−) or SmoSD-GFP-transfected S2-R+ cells treated with dsRNA targeting β-gal (control) or Gαs. t test: ***, p < 0.001; n.s., not significant. C, immunoblot analysis of cells treated as in B. Blot was probed with anti-GFP antibody to reveal SmoSD-GFP expression and anti-α-tubulin as a loading control.
GRK phosphorylation of SmoCT triggers Gαs-dependent downstream signaling
Although PKA/CkI phosphorylation is the principal trigger for Smo activation, more than half of the transcriptional response observed in ptc-luc reporter assays depends upon phosphorylation of Smo by Gprk2 (11). To see if Gprk2 affects heterotrimeric G protein–dependent signaling downstream of Smo, we treated S2 cells with a gprk2-specific dsRNA. The ability of Smo to activate Gαs depended upon Gprk2, as efficient depletion of the kinase strongly impaired the ability of SmoSD to stimulate cAMP production (Fig. 3, A and B). To confirm that this is due to a direct effect of Gprk2 phosphorylation on Smo activity, and not to an indirect effect of Gprk2 on cAMP regulation (as described previously in Ref. 36), we targeted the Gprk2 phosphorylation sites in SmoCT rather than the kinase. Consistent with the depletion experiments, mutating the four clusters of Gprk2 phosphorylation sites to Ala (in SmoSDc1–4A) prevented SmoSD from stimulating cAMP production (Fig. 3C). However, SmoSDc1–4A was well-expressed and retained substantial ability to stimulate target gene expression (∼20-fold over baseline levels) (Fig. 3, D and E). Although phosphorylation by PKA and CkI is sufficient to stabilize Smo and activate some signaling, we conclude that it is phosphorylation by Gprk2 that specifically triggers Gαs-dependent downstream regulation of cAMP levels.
Figure 3.

Gprk2 phosphorylation of the SmoCT activates Gαs-dependent signaling. Graphed data represent mean ± S.D. t test: ***, p < 0.001. A, EPAC-BRET assay measuring cAMP levels in mock (−) or SmoSD-transfected S2-R+ cells treated with dsRNA targeting β-gal (control) or gprk2. B, immunoblot (IB) analysis of cells treated as in A. Blot was probed with anti-GFP antibody to reveal SmoSD-GFP expression, anti-Gprk2 to show efficient depletion of Gprk2 protein, and anti-α-tubulin as a loading control. C and D, EPAC-BRET assay measuring cAMP levels (C) and ptc-luc reporter assay (D) in control (−), SmoSD-GFP, and SmoSD.c1–4A-GFP-transfected cells. E, immunoblot analysis of cells treated as in C. Blot was probed with anti-GFP antibody to reveal SmoSD-GFP expression and anti-α-tubulin as a loading control.
Gαs-dependent signaling is mediated via the conserved core of Smo
We previously showed that a C-terminally–truncated form of Smo that retains only the broadly conserved core of the protein (amino acids 1–663; Smocore) is constitutively active and capable of activating Hh target gene expression, although less effectively than SmoSD (Fig. 4A) (11). This suggested that Smo proteins from different species share common aspects in their signaling mechanisms (2, 46). We found that Smocore possesses all the sequences necessary to promote Gαs-dependent signaling, in a constitutive manner. In Smocore-expressing cells, cAMP levels increased to a similar extent as in SmoSD-expressing cells (Fig. 4, B and C), both throughout cells and more specifically at the membrane (Fig. S1), and in a Gαs-dependent manner (Fig. 4, D and E). In Smocore-expressing cells, co-expression of Ptc almost completely suppressed both ptc-luc reporter activation and the increase in cAMP production (Fig. 4, A–C). This suggests that the stimulation of cAMP production is a specific, Hh-regulatable signaling activity of Smocore.
Figure 4.

cAMP regulation is mediated by the evolutionarily conserved core of Smo. Graphed data represent mean ± S.D. t test: **, p < 0.01; ***, p < 0.001; n.s., not significant. A and B, ptc-luc reporter assay (A) and EPAC-BRET assay measuring cAMP levels (B) in mock-transfected cells (−), cells expressing full-length (SmoSD-GFP), and cells expressing the C-terminally truncated (at amino acid 663) conserved core form of Smo (Smocore-GFP) with or without Ptc co-expression. C, immunoblot (IB) analysis of cells treated as in A and B. Blot was probed with anti-GFP antibody to reveal SmoSD-GFP or Smocore-GFP expression and anti-α-tubulin as a loading control. D, EPAC-BRET assay of cAMP levels in mock (−) or Smocore-GFP-transfected cells treated with dsRNA targeting β-gal (control) or Gαs. E, immunoblot analysis of cells treated as in D. Blot was probed with anti-GFP antibody to reveal Smocore-GFP expression and anti-α-tubulin as a loading control. F, ptc-luc reporter assay in mock-transfected cells (−) and cells expressing Smocore-GFP or the Gprk2 phosphosite mutant Smocore.c1–3A-GFP. G, EPAC-BRET assay of mock transfected cells (−) and cells expressing Smocore-GFP, treated with dsRNA targeting β-gal (control) or gprk2. H, immunoblot analysis of cells treated as in G. Blot was probed with anti-GFP antibody to reveal Smocore-GFP expression and anti-α-tubulin as a loading control. I, EPAC-BRET assay measuring cAMP levels in mock-transfected cells (−) and cells expressing Smocore-GFP or Smocore.c1–3A-GFP. J, immunoblot analysis of cells treated as in I. Blot was probed with anti-GFP antibody to reveal Smocore-GFP expression and anti-α-tubulin as a loading control. K, EPAC-BRET assay of cAMP levels in cells transfected with the indicated C-terminally truncated GFP-tagged Smo variants. L, immunoblot analysis of cells treated as in K. Blot was probed with anti-GFP antibody to reveal GFP-tagged Smo variants and anti-α-tubulin as a loading control.
We tested additional mutants to identify the determinants in Smocore required to activate Gαs-dependent signaling. Unlike full-length Smo, Smocore activity is entirely dependent upon Gprk2 phosphorylation (Fig. 4F) (11). Its ability to drive Gαs-dependent signaling was similarly reliant upon Gprk2 phosphorylation, as stimulation of cAMP production by Smocore was strongly impaired by either depletion of Gprk2 (Fig. 4, G and H) or mutation of the Gprk2 phosphorylation sites (Smocore.c1–3A) (Fig. 4, I and J). We previously showed that the C-terminus of Smocore (between amino acid 626 and 663) is required for its activity in ptc-luc reporter assays (11). This same region is also required for Gαs activation (Fig. 4, K–L). Thus the 7-transmembrane domain region of Smo is not sufficient to activate Gαs-dependent signaling; sequences in the broadly-conserved membrane-proximal portion of the cytoplasmic tail are also required.
Activation of Gαs-dependent signaling downstream of Smo is distinct from HSC signaling
Phosphorylation of Smo by Gprk2 affects canonical signaling through the HSC in complex ways. For example, it promotes Smo C-terminal dimerization, which itself promotes Fu activation (11, 47). It also promotes interaction of Costal-2 with Smocore (11). We wondered to what extent the Gαs-dependent signaling we observed is linked to activation of these more typical effectors of Smo. To assess this, we examined the effects of manipulating Fu and Cos2 activity on the ability of Smo to stimulate cAMP production. Fu is recruited to the plasma membrane and activated by Smo and Cos2, and in turn promotes high-threshold target gene expression (47, 48). Neither deletion of the C-terminal Fu-binding domain (SmoSDΔFu) (49) (Fig. 5, A and B) nor dsRNA-mediated depletion of Fu (Fig. 5, D and E) had any effect on the ability of SmoSD to stimulate cAMP production, although in both conditions canonical pathway signaling was partially reduced (by ∼30–50%) as expected (Fig. 5, C and F).
Figure 5.

HSC-dependent and Gαs-dependent signaling downstream of Smo are separable and distinct. All data represent mean ± S.D. t test: ***, p < 0.001; n.s., not significant. A, EPAC-BRET assay of cAMP levels in mock-transfected cells (−) and cells expressing SmoSD-GFP or a mutated form of SmoSD from which the C-terminal Fu-binding domain was deleted (SmoSDΔFu-GFP). B, immunoblot (IB) analysis of cells treated as in A. Blot was probed with anti-Smo antibody to reveal SmoSD-GFP variant expression and anti-α-tubulin as a loading control. C, ptc-luc reporter assay of cells as in A. D, EPAC-BRET assay of cAMP levels in control (−) or SmoSD-GFP expressing cells treated with β-gal (control) or fu dsRNA. E, immunoblot analysis of cells treated as in D. Blot was probed with anti-Smo antibody to reveal SmoSD-GFP expression and anti-α-tubulin as a loading control. F, ptc-luc reporter assay of cells as in D. G, ptc-luc reporter assay of mock- (−) or SmoSD-transfected cells, co-transfected with either empty vector (control) or mycCSBD. H, immunoblot analysis of cells treated as in G. Blot was probed with anti-GFP antibody to reveal SmoSD-GFP expression, with anti-myc tag to reveal mycCSBD, and anti-α-tubulin as a loading control. I, EPAC-BRET assay of cAMP levels in cells treated as in G. J, EPAC-BRET assay of cAMP levels in mock-transfected cells (−) and cells transfected with GαsQ215L, SmoSD-GFP, SmoSD.c14A-GFP, or SmoSD.c14A-GFP + GαsQ215L. K, ptc-luc reporter assay of cells as in J.
Activation of signaling through the HSC depends upon interaction of activated Smo with Cos2 (50, 51). To test if Cos2 also directly participates in Gαs-dependent signaling downstream of Smo, we made use of a dominant-negative form of Cos2 consisting of just the cargo domain, termed the C-terminal Smo-binding domain (CSBD). This protein targets the interaction between Smo and Cos2 and thereby attenuates Hh signaling (40). As expected, CSBD expression almost completely inhibited ptc-luc reporter activation downstream of SmoSD (Fig. 5G), and this was associated with a strong reduction in Smo protein levels (as others have seen) (40) (Fig. 5H). Interestingly, however, CSBD had no significant effect on the ability of SmoSD to stimulate cAMP production despite the decrease in SmoSD protein levels (Fig. 5I). This suggests that the pool of Smo that signals to Gαs may be different from the Cos2-associated pool signaling through the HSC. Together, these results suggest that Gαs-dependent signaling downstream of Smo is separable and distinct from signaling through the HSC components Cos2 and Fu.
Our analysis to this point suggested that Gαs may act in parallel to the HSC downstream of Smo, with both cooperating to promote Hh target gene expression. If so, then the loss of Gαs-mediated signaling might explain the decrease in target gene expression we observe when Gprk2 phosphorylation of Smo is blocked. If this were true, then increasing cAMP levels should rescue target gene expression downstream of the Gprk2 nonphosphorylatable Smo variant. We used the activated GαsQ215L mutant to test this. Expression of either GαsQ215L or SmoSD increased cellular cAMP levels (Fig. 5J), but only SmoSD drove ptc-reporter expression as expected (Fig. 5K). Both effects depended upon Gprk2 phosphorylation of the SmoCT, as expected (Fig. 5, J and K). Although GαsQ215L restored the increase in cAMP levels in SmoSDc1–4A-expressing cells (Fig. 5J), it did not rescue ptc-reporter activation (Fig. 5K). These results suggest that phosphorylation of Smo by Gprk2 does something else to downstream signaling besides promoting Gαs activity.
Gαs enhances the response to low levels of Hh
Having not seen any obvious role for heterotrimeric G protein signaling in the endogenous response of S2 cells to HhN-conditioned medium, we turned to an in vivo system to see if Gαs plays any physiological role in Hh signaling. First, we generated clones of cells homozygous for a null allele of gαs (gαsR60) in wing discs using the FLP-FRT system (52). These clones were small when generated in an otherwise WT background. Even when generating them in a Minute heterozygous background to give them a growth advantage (53), gαsR60 clones were only about half the size of WT clones generated in parallel (Fig. 6, A–C). We did not observe changes in intermediate or high-threshold Hh responses (expression of Ptc and Engrailed, respectively) in gαsR60 clones at the anterior/posterior (A/P) boundary (not shown). However, we did observe an effect on expression of the low threshold target gene dpp (assessed using a dpp-LacZ enhancer trap line). Normally, dpp-LacZ staining is fairly symmetrical in dorsal and ventral compartments of the wing pouch (see below) (Fig. 6D). However, in some larger gαsR60 clones located away from the A/P boundary, dpp-LacZ expression was reduced (Fig. 6, E and F). As the concentration of Hh is thought to decline with increased distance from the P compartment where it is produced, this suggests that Gαs may play a role in the normal response to Hh where ligand levels are low.
Figure 6.
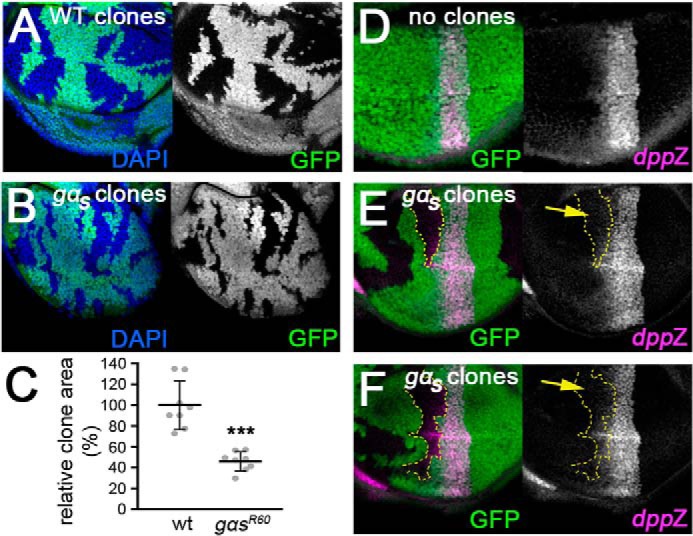
gαs mutant clones impair low threshold Hh target gene expression. A, wing imaginal disc bearing clones of WT cells (marked by lack of GFP expression (green)), generated in a Minute heterozygous background. Nuclei are stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). B, wing imaginal disc bearing clones of gαsR60 homozygous mutant cells generated and analyzed in parallel to WT clones in A. C, measurements of wing pouch clone areas from discs as in A and B, expressed as the percentage of the wing pouch area that is composed of clone cells (normalized to the value for WT clones). t test: ***, p < 0.001. D, disc in which no gαsR60 homozygous mutant clones (marked by absence of GFP (green)) were generated. The disc was immunostained with an anti-β-Gal antibody to reveal dorsal/ventral symmetry of expression of a dpp-LacZ enhancer trap (pink). E and F, discs bearing larger gαsR60 homozygous mutant clones, stained as in D. Clone outlines are traced in yellow.
We were concerned that this approach might underestimate the role of Gαs because of the potential for nonautonomous rescue in mosaic discs. Wing discs cells have been demonstrated to be highly connected through gap junctions, with small dyes able to diffuse over many cell diameters at late third instar stage (54). cAMP has a lower molecular weight than these dyes, suggesting that it too may be able to diffuse in discs. Diffusion of cAMP from neighboring WT cells might limit the cell-autonomous effects in gαsR60 clones.
To circumvent this potential complication, we used expression of a dsRNA transgene to deplete Gαs throughout a much larger territory in the disc. Expression of Gαs dsRNA throughout the developing wing pouch (using nubbin-GAL4) caused a failure in wing inflation (Fig. 7, A and B), a phenotype that has been linked to a failure in Gαs activation downstream of the GPCR Rickets (55). This suggests that the dsRNA transgene does in fact deplete Gαs.
Figure 7.

Depletion of heterotrimeric G protein α subunits reveals a positive role for cAMP in cells responding to low levels of Hh. A, wing from a nub-GAL4/+ fly, showing the WT wing pattern. B, wing from an animal expressing a dsRNA targeting gαs under the control of nub-GAL4, which failed to properly inflate. C, wing from an animal expressing a dsRNA targeting gαi under the control of nub-GAL4. D, quantification of the average proportion of total wing area that is bounded by longitudinal wing veins 3 and 4 (L3-L4:total wing area) for control and gαi-depleted wings (as in A and C). Data represent mean ± S.D. of six wings. t test: ***, p < 0.001. E, ap-GAL4/+ wing disc immunostained for full-length Ci155 (red), dpp-LacZ (blue), and Ptc (green). Yellow dotted line indicates the boundary between GAL4-expressing dorsal (D) and WT ventral (V) compartments. F, plots of fluorescence intensity along the anterior-posterior axis of dorsal compartment Ci155 (red), dpp-LacZ (blue), and Ptc (green) immunostaining from discs as in E. The corresponding ventral compartment values for each are plotted in gray. Location of anterior/posterior boundary is indicated by a dotted line. Data represent the mean ± S.D. of four wing discs. Error bars are indicated every 10 pixels. G, wing disc with dsRNA targeting gαs expressed throughout the dorsal compartment using ap-GAL4, immunostained for full-length Ci155 (red), dpp-LacZ (blue), and Ptc (green). Ventral compartment serves as an internal WT control. H, plots of fluorescence intensity as in F for ap-GAL4>gαs dsRNA discs. I, wing disc with dsRNA targeting gαi expressed throughout the dorsal compartment using ap-GAL4, immunostained for full-length Ci155 (red), dpp-LacZ (blue), and Ptc (green). Ventral compartment serves as an internal WT control. J, plots of fluorescence intensity as in F for ap-GAL4>gαi dsRNA discs.
Consistent with the clonal analysis, depletion of Gαs selectively impaired low threshold dpp expression. We used apterous (ap)-GAL4 to drive expression of Gαs dsRNA throughout the dorsal compartment of the wing disc, leaving the ventral compartment as an internal WT control. In control ap-GAL4/+ wing discs, low threshold Hh responses (such as stabilization of full-length Ci (Ci155) and expression of dpp) and intermediate threshold responses (expression of Ptc) appeared similar in the GAL4-expressing dorsal and WT ventral compartments (Fig. 7E). Quantification and averaging of the relative fluorescence intensities from several discs confirmed that both compartments show virtually identical Hh responses both in terms of intensity and distance from the A/P boundary (Fig. 7F). ap-GAL4-driven expression of Gαs dsRNA had little effect on Ci stabilization or Ptc expression (Fig. 7, G and H). The most striking effect was a strong and statistically significant reduction in dpp expression (Fig. 7, G and H). Most notably, the A-P distance over which dpp was expressed was substantially less in the Gαs-depleted dorsal compartment, indicating that cells exposed to low levels of Hh were no longer able to mount an effective transcriptional response. This fits well with our observations of gαsR60 mutant clones, and suggests that Gαs plays a positive signaling role in situations where Hh levels are low and limiting for a response.
Consistent with this interpretation, we observed a Hh concentration-dependent requirement for Gαs in Smo-transfected S2 cells. In a HhN dose-response test, we found that the standard conditions we used for testing Hh induction of the pathway (100 ng of transfected HhN plasmid) produced approximately a 60% maximal response in ptc-luc reporter assays (Fig. 8A). Under these conditions, we did not observe a significant effect of Gαs depletion on ptc-luc reporter expression (Fig. 8B). However, at a lower level of Hh (20 ng of HhN plasmid), which was sufficient for ∼25% maximal response in ptc reporter assays (Fig. 8A) and for stimulating cAMP production (Fig. 8C), depletion of Gαs decreased Hh-dependent ptc-luc reporter activation by Smo-GFP (Fig. 8D). The effect was modest (∼25% decrease), suggesting that signaling through Gαs is not essential for the response. Rather, this result together with our in vivo analysis suggest that Gαs enhances responsiveness to low levels of Hh in vivo.
Figure 8.

Gαs enhances signaling when Hh levels are low. All data represent mean ± S.D. t test: **, p < 0.01; ***, p < 0.001; n.s., not significant. A, ptc-luc reporter assay of SmoWT-GFP-expressing cells co-transfected with increasing amounts of HhN expression plasmid (in ng). B, ptc-luc reporter assay of cells transfected with empty vector (−) or with SmoWT-GFP and 100 ng of a HhN expression plasmid, and treated with dsRNAs targeting β-gal (control) or Gαs. C, EPAC-BRET assay of cAMP levels in cells co-transfected with SmoWT-GFP and either empty vector (0) or 20 or 100 ng of HhN expression plasmid. D, ptc-luc reporter assay of cells transfected with empty vector (−) or with SmoWT-GFP and 20 ng of a HhN expression plasmid, and treated with dsRNAs targeting β-gal (control) or Gαs.
Gαi depletion reveals an inhibitory role for this protein in the Hh response
Our analysis of Gαs function appears to contradict previous studies in several systems showing that inhibition (rather than stimulation) of cAMP production, through Gαi, promotes Hh target gene expression (21, 22). In particular, gαi mutant clones in wing discs, which appeared fairly small, failed to express dpp (22). We tested whether depletion of Gαi over a larger territory would produce a similar effect. Expression of a dsRNA targeting Gαi throughout the developing wing pouch (using the nubbin-GAL4 driver) impaired growth, causing the resulting adult wings to be undergrown, similar to those of gαi hypomorphic mutants (Fig. 7C) (40). This suggests that the dsRNA did target Gαi. Interestingly, the wings were not uniformly smaller. Instead, the size of the central region bounded by longitudinal wing veins 3 and 4 (L3 and L4) as a proportion of total wing area was significantly increased upon Gαi depletion (Fig. 7D). This is a characteristic feature of increased Hh signaling in the wing disc. Consistent with this interpretation, Gαi depletion throughout the dorsal compartment significantly increased the level and extent of dorsal Ci155 stabilization and expression of dpp and Ptc (Fig. 7, I and J). Compared with control discs, cells further from the A/P boundary (and thus exposed to lower levels of ligand) in the Gαi-depleted discs were able to activate robust target gene transcription. This suggests that Gαi depletion enhanced sensitivity of cells to low levels of Hh, which is the opposite of the effect of Gαs depletion. Our analyses of Gαi and of Gαs function in wing discs are thus consistent in revealing a positive effect of cAMP on sensitivity to Hh in vivo.
Discussion
Despite conflicting evidence about the ability and necessity of Smo itself activating Gα signaling, heterotrimeric G proteins have increasingly been linked to Hh signaling under normal and pathological circumstances (e.g. Refs. 29 and 56). The emerging model (57), based largely on observations in mammalian cells, suggests that cAMP plays primarily an inhibitory role in the pathway. In the absence of Hh, a Gαs-coupled GPCR (like Gpr161 in mammals) signals to maintain cAMP at high levels, thereby ensuring efficient PKA phosphorylation and processing of GLI proteins to their repressor forms. In response to Hh ligands, decreased Gαs signaling and/or increased Gαi signaling by Smo or certain other Gαi-coupled GPCRs lowers cAMP levels, allowing GLI proteins to avoid PKA phosphorylation and accumulate in their full-length activator forms. However, the situation must be more complex, as cAMP/PKA cannot only inhibit but also promote Hh signaling in both flies and mammals (8, 33, 34, 36, 38), most directly in the case of Drosophila by phosphorylating and activating Smo. Consistent with this, we have observed a Gαs-dependent increase in cellular cAMP levels downstream of Smo activation, and identified a positive role for Gαs in Hh signaling in vivo.
We expected to see Gαi activation and a decrease in cAMP levels in Hh-treated S2 cells, similar to what was previously observed in other insect cell lines (21, 22). However, we observed no consistent effect on total cAMP levels in S2 cells exposed to HhN. Studies in mouse cerebellar granule cell progenitors suggested that Hh signaling is controlled by a relatively small pool of cAMP that is restricted in its localization, likely near the cilium (58). Hh-dependent changes in cAMP levels may similarly be restricted to a smaller pool in S2 cells such that they do not significantly impact the total cAMP pool; in fact, using a membrane-anchored, myristoylated FRET biosensor of PKA activity in S2 cells, Li et al. (45) observed an ∼2-fold increase in PKA activity in response to Hh. However, we saw no change in membrane cAMP levels with a comparable myristoylated cAMP biosensor. It may be that the EPAC-BRET biosensor is not sensitive enough to detect relatively small changes in cAMP levels in response to activation of endogenous signaling, where negative feedback may also limit the magnitude of the response. Alternatively, these cells may differ in some other way from the other cell lines previously tested (discussed below). Nevertheless, hyperactivation of the pathway at the level of Smo produced a substantial cAMP response. The fact that Gαs depletion blocked the increase in cAMP levels strongly suggests that it is due to increased cAMP production rather than a decreased rate of degradation by phosphodiesterases. The relatively slow accumulation of cAMP over several hours is somewhat unusual, as GPCRs typically activate heterotrimeric G protein signaling on a time scale of seconds to minutes. However, it fits with the previously characterized prolonged time frame for Smo activation (59), which involves phosphorylation by PKA and CkI as well as trafficking to the plasma membrane.
At the level of Smo, we identified several features required for this response. The ability of Smo to trigger Gαs-dependent cAMP production lies within its highly conserved core. The seven-transmembrane domain is not sufficient, as the C-terminal 26 amino acids of Smocore (between amino acids 626 and 651), situated at the end of its cytoplasmic tail, are required for the effect. This same region is required for Smocore to interact with Cos2 and to promote expression of Hh target genes (11). However, we did not observe a link between HSC-dependent signaling and the cAMP response. In fact the ability of CSBD, which interferes with the interaction between Smo and Cos2 (50), to destabilize SmoSD and block target gene expression and yet not affect the cAMP response suggests that the two responses may be mediated by separate pools of Smo protein. Interestingly, mutation of the N-linked glycosylation sites in mouse Smo impaired its ability to signal through Gαi without affecting canonical signaling, pointing to the possible existence of a specific Gα-signaling state of Smo in mammalian cells (60). Like signaling through the HSC, the cAMP response is Hh-activated and inhibited by Ptc, suggesting that activation of Smo by PKA and CkI is required. Crucially, however, whereas PKA/CkI phosphorylation is sufficient for activation of Hh target gene expression, the cAMP response is strictly dependent upon SmoCT phosphorylation by Gprk2. This is reminiscent of the phenomenon of phosphorylation barcoding that has been described for some GPCRs, where phosphorylation at different sets of sites in the receptor tail by different kinases dictates distinct signaling responses (61).
Given these constraints, how could Smo activity lead to signaling through Gαs? The most straightforward model would be that Smo directly couples to Gαs, like a typical GPCR. If so, then we could explain the difference between our observations and others' demonstration of Smo–Gαi coupling as a cell-type or context-dependent ability of Smo to couple to different Gα proteins, as observed for many GPCRs (62). However, we have been unable to obtain compelling evidence for a direct interaction between Smo and Gαs. We favor the alternative explanation that Smo regulates a GPCR that couples to Gαs in S2 cells. Gpr161 provides a precedent for this in mammalian cells. The ability of Gpr161 to localize to the cilium where it can couple to Gαs is determined by the activity of Smo in the cilium (63), thus providing an indirect link between Smo and Gαs. Interestingly, there is some evidence that Smo is trafficked to the cilium in response to phosphorylation by GRK2 in mammalian cells (15) (although this has been disputed) (16). In Drosophila, Gprk2 is required for internalization of Smo from the plasma membrane in response to Hh (64). Thus it could be that the regulation of Smo–Gαs signaling by Gprk2 that we observe in S2 cells reflects GRK-dependent trafficking of Smo to a subcellular location where it regulates a Gαs-coupled GPCR, perhaps on endosomes (from which mammalian GPCRs have increasingly been shown to signal through heterotrimeric G proteins after GRK-dependent internalization) (65). If this is the case, the difference in Gα signaling between S2 cells and other insect cell lines could reflect differences in the GPCRs they express.
Regardless of whether Smo directly or indirectly regulates Gαs in S2 cells, we find that Gαs does play a role in the endogenous Hh response. Because Smo activation leads to localized PKA activity at the plasma membrane (45), we imagined that a localized burst of cAMP production might be required to enhance Smo phosphorylation to allow high-threshold target gene expression. However, our results suggest that such a mechanism is not required for high-threshold signaling. Instead, we consistently see selective impairment of low-threshold responses when Gαs is depleted or mutated in S2 cells, in wing disc clones, and most clearly, throughout an entire compartment. The narrowing of the domain of dpp expression in response to gαs-depletion suggests that the sensitivity of cells to low levels of Hh was reduced, consistent with the impairment of ptc-luc reporter activation we observed in S2 cells in response to low but not high levels of Hh. Thus, rather than boosting Smo activity toward its maximum, Gαs-driven cAMP production may be most important for enhancing Smo activation above a signaling threshold under limiting ligand conditions. Interestingly, while this manuscript was under review, Pusapati et al. (66) reported similar findings in vertebrates. They showed that GPCR signaling through Gαs, downstream of both Smo and GRKs, regulates the sensitivity of responding cells to Sonic Hedgehog. The parallel between these findings and ours indicates that regulation of cAMP in target cells is an evolutionarily conserved mechanism for fine-tuning Hh responsiveness.
Finally, in contrast to previously published work, we observe an inhibitory rather than stimulatory role for Gαi in the Hh response. Specifically, Gαi depletion led to ectopic expression of Hh target genes in cells located away from the A/P boundary, where ligand levels are lower. This difference from previous work may be due to a difference in the extent of signaling impairment in Gαi-depleted cells versus gαi mutant clones, similar to the way that PKA inhibition can suppress the Hh response (8), whereas complete elimination of the catalytic subunit leads to constitutive target gene expression (31). In any case, these results are consistent with our Gαs depletion experiments and other work showing a positive role for Gαs/cAMP in the Hh response (36, 67, 68). Together they clearly suggest a positive role for regulated cAMP production in controlling the sensitivity of cells to Hh.
Experimental procedures
Expression constructs
Expression plasmids encoding C-terminally–tagged GFP Smo mutants and Smo truncations under control of the metallothionein promoter (in pRmHa3.puro) were previously described (11). The expression construct for SmoΔFu (lacking the C-terminal-most 52 amino acids containing a Fu-binding domain) (49) was generated by PCR amplification of a portion of the Smo coding sequence spanning the EcoRI site at codon 797–798 to the Leu-984 codon. Primers were designed to introduce a 3′ stop codon and NotI site immediately following codon 984. The resulting EcoRI-NotI fragment was used to replace the corresponding C-terminus–encoding fragment in a pRmHa3.puro backbone containing the SmoSD coding sequence fused to C-terminal GFP (engineered as a cassette flanked by NotI and KpnI restriction sites). To create the Cos2 Smo-binding domain (CSBD) expression construct (40), sequences encoding amino acids 1001–1201 of Cos2 were PCR amplified, introducing a 5′ EcoRI and 3′ KpnI site. The resulting EcoRI-KpnI restriction fragment was cloned into a modified pRmHa3.puro plasmid encoding an in-frame N-terminal Myc epitope tag. For expressing Ptc, an EcoRI-KpnI fragment encoding full-length Ptc fused to a C-terminal GFP tag (obtained from Stephen Cohen) was cloned into pRmHa3.puro. The plasmids encoding the EPAC-BRET biosensor or the same protein lacking the GFP10 moiety (used as a control for background emission), as well as plasmids encoding constitutively active GαsQ215L and GαiQ205L, were prepared as described (36). pRmHa3.puro/HhN, which encodes an active N-terminal fragment of Drosophila Hh plasmid (59), was used in the preparation of HhN-conditioned media and for co-transfection of cells transfected with Smo expression plasmids in some experiments. An empty pRmHa3.puro vector (−) was used for transfection as a control.
BRET and ptc-luciferase reporter assays, dsRNA treatment
EPAC-BRET assays were performed essentially as described (36). Briefly, for BRET experiments not involving dsRNA treatment, S2-R+ cells were seeded in 24-well plates and transfected on day 1. Each well was transiently transfected with 100–150 ng of pMT.puro/GFP10-EPAC-RLucII_T781A,F782A encoding the EPAC-BRET cAMP biosensor protein (described previously in Refs. 36 and 41) consisting of the cAMP-binding domain of the EPAC protein fused to GFP10 and Renilla luciferase II (RlucII) at the amino and carboxyl termini, respectively. cAMP binding promotes a more open conformation of the EPAC-BRET protein, causing the BRET signal to decrease (41). Cells were also transfected with pMT.puro expression constructs for Smo variants (with C-terminal GFP tag) or HhN, depending on the experiment. On day 2, transgene expression was induced and cells were transferred to 3–4 wells of white-walled, clear-bottomed 96-well plates. BRET measurements were performed on day 4 or 5 as described previously (36).
For experiments involving dsRNA treatment, dsRNA was prepared as described (11). dsRNAs targeted β-gal (11), gprk2 (11), fu (nucleotides 1043–1547 of fu-RA (FBtr0074602), gαs (nucleotides 282–697 of Gαs-RA (FBtr0072144)), or gαi (nucleotides 533–986 of gαi-RA (FBtr0076934)). Cells were transfected as above on day 1. On day 2, cells were split and plated into 96-well plates and each well was treated with 0.5–2 μg of dsRNA. On days 4 or 5, a second dose of dsRNA was administered and transgene expression was induced. Cells were processed for BRET measurements on day 7. BRET signals were determined by calculating the ratio of the light emitted by GFP10 (515 nm) and RlucII (410 nm). The net BRET value represents the BRET signal from which the background (represented by the BRET signal generated by cells expressing a biosensor lacking the GFP10 acceptor moiety) was subtracted. All graphs represent the composite results of at least two independent experiments, each performed in triplicate or quadruplicate measurements, with the exception of the experiment in Fig. 1I, which was performed once in triplicate. Statistical significance was determined by two-tailed Student's t tests.
ptc-luc reporter assays were performed essentially as described (11). Assays were performed at least two times each with triplicate or quadruplicate measurements and data compiled, with the exception of the experiment in Fig. 8A, which was performed once in quadruplicate. Due to interference with the normalization plasmid encoding Renilla luciferase in some dsRNA experiment setups, all data are presented as measurements of firefly luciferase reporter activity. Statistical significance was assessed using two-tailed Student's t tests.
HhN treatment, Smo activation time course experiments, and immunoblotting
For time course experiments, Smo-expressing S2-R+ cells were treated with medium conditioned by either control cells or cells expressing HhN as previously described (64). Cells were transfected with 150 ng each of pMT.puro/GFP10-EPAC-RLucII_T781A,F782A, pMT.puro/SmoWT-GFP, and (in the control cells) pMT.puro/HhN and processed as above. On day 4, growth medium was gently removed and 0.1 ml of either control or HhN-conditioned media was added to each well at the appropriate time. Cells treated at different intervals were processed simultaneously by adding DeepBlueC directly to the conditioned media and BRET values were determined as above. Time-dependent changes in SmoWT phosphorylation following exposure to control- or HhN-conditioned media were determined by immunoblotting with rabbit α-GFP antibody (Torres Pines Scientific) as described (11). In other experiments, immunoblotting was performed with mouse monoclonal anti-myc (9E10; Santa Cruz Biotechnology), mouse monoclonal anti-α-tubulin (12G10; obtained from the Developmental Studies Hybridoma Bank (DSHB)), guinea pig anti-Gprk2 (64), or guinea pig anti-Smo.
Drosophila crosses, immunostainings, and image analysis
For generating gαs mutant clones, we used the gαsR60 mutant allele (stock obtained from the Bloomington Drosophila Stock Centre). This allele contains a mutation that converts Tyr-231 to a stop codon, deleting the C-terminal ∼one-third of the protein, and should be a null allele. To make gαs clones in a Minute background, w;dpp10638,FRT42D,gαsR60/CyO,Kr-GAL4,UAS-GFP males were crossed at 25 °C to w,hsFLP;FRT42D,Ub::GFP,M(2R)531/CyO virgins. For WT clones, w;dpp10638,FRT42D males were used. Offspring from the two crosses were heat shocked at 37.5 °C and dissected in parallel. For no clones control, the heat shock was omitted. Wing disc-bearing anterior halves were dissected from wandering third instar larvae and processed for immunofluorescence staining with rabbit anti-β-gal antibody (Santa Cruz Biotechnology). To quantify clonal growth, the total area of the wing pouch and of the GFP-negative regions contained in the wing pouch for 10 discs were measured using the Histogram function of Photoshop. Clone size was expressed as the total GFP-negative clonal area over total wing pouch area.
For Gαs and Gαi depletion experiments w,UAS-Dcr;ap-GAL4,dpp10638/CyO or w,UAS-Dcr;nub-GAL4,dpp10638/CyO females were crossed at 27 °C to w,UAS-GFP males (control), w;UAS-Gαs.dsRNA males (from Vienna Drosophila Resource Centre; stock number v24958), or w;UAS-Gαi.dsRNA males (69) (kindly provided by J. Knoblich). Wandering larvae were dissected and anterior halves processed for immunofluorescence with rabbit anti-β-gal, rat anti-Ci155 (2A1 from DSHB), and mouse anti-Ptc (Apa-1 from DSHB) and discs were imaged by confocal microscopy as described (11). To quantify fluorescence, boxes of equal size spanning the A/P boundary were drawn in the dorsal and ventral regions of each wing disc image and fluorescence values along the anterior-posterior axis were calculated using the Plot Profile function of Image J. Dorsal and ventral data were normalized by dividing each data point by the average maximum intensity value from the ventral compartment of that disc (defined as the average of the 10 highest values), effectively converting them to % maximum WT response. Data were then arranged to align the A/P boundaries and pixel-by-pixel data from four discs was averaged to yield a mean intensity plot.
Author contributions
S. D. P., D. M., and D. R. H. conceptualization; S. D. P., F. S., and D. M. formal analysis; S. D. P., F. S., D. M., and P. I. investigation; S. D. P. and D. R. H. visualization; S. D. P. and D. R. H. writing-review and editing; D. R. H. supervision; D. R. H. funding acquisition; D. R. H. writing-original draft.
Supplementary Material
Acknowledgments
We thank Karen Oh for expert technical assistance and Stephen Cohen (University of Copenhagen, Denmark), Jurgen Knoblich (Institute of Molecular Biotechnology, Austria), and Michel Bouvier (Université de Montréal, Canada) for generously providing reagents. Fly stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study, as were antibodies from the Developmental Studies Hybridoma Bank (created by the NICHD, National Institutes of Health, and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242).
This work was supported by Canadian Institutes of Health Research (CIHR) Grant 106426 (to D. R. H.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Fig. S1.
- GPCR
- G protein–coupled receptor
- Smo
- Smoothened
- PKA
- protein kinase A
- HSC
- Hh signaling complex
- SAID
- Smo autoinhibitory domain
- Sufu
- Suppressor of Fused
- Ci
- Cubitus interruptus
- Hh
- Hedgehog
- BRET
- bioluminescence resonance energy transfer
- CSBD
- C-terminal Smo-binding domain
- GFP
- green fluorescent protein
- β-gal
- β-galactosidase
- DSHB
- Developmental Studies Hybridoma Bank
- CT
- C terminus.
References
- 1. Jiang J., and Hui C. C. (2008) Hedgehog signaling in development and cancer. Dev. Cell 15, 801–812 10.1016/j.devcel.2008.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Briscoe J., and Thérond P. P. (2013) The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 14, 416–429 10.1038/nrm3598 [DOI] [PubMed] [Google Scholar]
- 3. Pak E., and Segal R. A. (2016) Hedgehog signal transduction: key players, oncogenic drivers, and cancer therapy. Dev. Cell 38, 333–344 10.1016/j.devcel.2016.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li S., Chen Y., Shi Q., Yue T., Wang B., and Jiang J. (2012) Hedgehog-regulated ubiquitination controls smoothened trafficking and cell surface expression in Drosophila. PLos Biol. 10, e1001239 10.1371/journal.pbio.1001239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xia R., Jia H., Fan J., Liu Y., and Jia J. (2012) USP8 promotes smoothened signaling by preventing its ubiquitination and changing its subcellular localization. PLos Biol. 10, e1001238 10.1371/journal.pbio.1001238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma G., Li S., Han Y., Li S., Yue T., Wang B., and Jiang J. (2016) Regulation of smoothened trafficking and Hedgehog signaling by the SUMO pathway. Dev. Cell 39, 438–451 10.1016/j.devcel.2016.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qi Y., Liu H., and Lin X. (2016) Sumoylation stabilizes smoothened to promote Hedgehog signaling. Dev. Cell 39, 385–387 10.1016/j.devcel.2016.11.001 [DOI] [PubMed] [Google Scholar]
- 8. Jia J., Tong C., Wang B., Luo L., and Jiang J. (2004) Hedgehog signalling activity of Smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 432, 1045–1050 10.1038/nature03179 [DOI] [PubMed] [Google Scholar]
- 9. Zhang C., Williams E. H., Guo Y., Lum L., and Beachy P. A. (2004) Extensive phosphorylation of Smoothened in Hedgehog pathway activation. Proc. Natl. Acad. Sci. U.S.A. 101, 17900–17907 10.1073/pnas.0408093101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Apionishev S., Katanayeva N. M., Marks S. A., Kalderon D., and Tomlinson A. (2005) Drosophila Smoothened phosphorylation sites essential for Hedgehog signal transduction. Nat. Cell Biol. 7, 86–92 10.1038/ncb1210 [DOI] [PubMed] [Google Scholar]
- 11. Maier D., Cheng S., Faubert D., and Hipfner D. R. (2014) A broadly conserved G-protein-coupled receptor kinase phosphorylation mechanism controls Drosophila smoothened activity. PLoS Genet. 10, e1004399 10.1371/journal.pgen.1004399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li S., Li S., Han Y., Tong C., Wang B., Chen Y., and Jiang J. (2016) Regulation of smoothened phosphorylation and high-level Hedgehog signaling activity by a plasma membrane associated kinase. PLos Biol. 14, e1002481 10.1371/journal.pbio.1002481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sanial M., Bécam I., Hofmann L., Behague J., Argüelles C., Gourhand V., Bruzzone L., Holmgren R. A., and Plessis A. (2017) Dose-dependent transduction of Hedgehog relies on phosphorylation-based feedback between the G-protein-coupled receptor Smoothened and the kinase Fused. Development 144, 1841–1850 10.1242/dev.144782 [DOI] [PubMed] [Google Scholar]
- 14. Zhao Y., Tong C., and Jiang J. (2007) Hedgehog regulates smoothened activity by inducing a conformational switch. Nature 450, 252–258 10.1038/nature06225 [DOI] [PubMed] [Google Scholar]
- 15. Chen Y., Sasai N., Ma G., Yue T., Jia J., Briscoe J., and Jiang J. (2011) Sonic Hedgehog dependent phosphorylation by CK1α and GRK2 is required for ciliary accumulation and activation of smoothened. PLos Biol. 9, e1001083 10.1371/journal.pbio.1001083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao Z., Lee R. T., Pusapati G. V., Iyu A., Rohatgi R., and Ingham P. W. (2016) An essential role for Grk2 in Hedgehog signalling downstream of Smoothened. EMBO Rep. 17, 739–752 10.15252/embr.201541532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen Y., Li S., Tong C., Zhao Y., Wang B., Liu Y., Jia J., and Jiang J. (2010) G protein-coupled receptor kinase 2 promotes high-level Hedgehog signaling by regulating the active state of Smo through kinase-dependent and kinase-independent mechanisms in Drosophila. Genes Dev. 24, 2054–2067 10.1101/gad.1948710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Preat T., Thérond P., Limbourg-Bouchon B., Pham A., Tricoire H., Busson D., and Lamour-Isnard C. (1993) Segmental polarity in Drosophila melanogaster: genetic dissection of fused in a Suppressor of fused background reveals interaction with costal-2. Genetics 135, 1047–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wilson C. W., Nguyen C. T., Chen M. H., Yang J. H., Gacayan R., Huang J., Chen J. N., and Chuang P. T. (2009) Fused has evolved divergent roles in vertebrate Hedgehog signalling and motile ciliogenesis. Nature 459, 98–102 10.1038/nature07883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ayers K. L., and Thérond P. P. (2010) Evaluating Smoothened as a G-protein-coupled receptor for Hedgehog signalling. Trends Cell Biol. 20, 287–298 10.1016/j.tcb.2010.02.002 [DOI] [PubMed] [Google Scholar]
- 21. Riobo N. A., Saucy B., Dilizio C., and Manning D. R. (2006) Activation of heterotrimeric G proteins by Smoothened. Proc. Natl. Acad. Sci. U.S.A. 103, 12607–12612 10.1073/pnas.0600880103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ogden S. K., Fei D. L., Schilling N. S., Ahmed Y. F., Hwa J., and Robbins D. J. (2008) G protein Gαi functions immediately downstream of Smoothened in Hedgehog signalling. Nature 456, 967–970 10.1038/nature07459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shen F., Cheng L., Douglas A. E., Riobo N. A., and Manning D. R. (2013) Smoothened is a fully competent activator of the heterotrimeric G protein Gi. Mol. Pharmacol. 83, 691–697 10.1124/mol.112.082511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carbe C. J., Cheng L., Addya S., Gold J. I., Gao E., Koch W. J., and Riobo N. A. (2014) Gi proteins mediate activation of the canonical hedgehog pathway in the myocardium. Am. J. Physiol. Heart Circ. Physiol. 307, H66–72 10.1152/ajpheart.00166.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chinchilla P., Xiao L., Kazanietz M. G., and Riobo N. A. (2010) Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle 9, 570–579 10.4161/cc.9.3.10591 [DOI] [PubMed] [Google Scholar]
- 26. Belgacem Y. H., and Borodinsky L. N. (2011) Sonic hedgehog signaling is decoded by calcium spike activity in the developing spinal cord. Proc. Natl. Acad. Sci. U.S.A. 108, 4482–4487 10.1073/pnas.1018217108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Polizio A. H., Chinchilla P., Chen X., Manning D. R., and Riobo N. A. (2011) Sonic Hedgehog activates the GTPases Rac1 and RhoA in a Gli-independent manner through coupling of smoothened to Gi proteins. Sci. Signal. 4, pt7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Villanueva H., Visbal A. P., Obeid N. F., Ta A. Q., Faruki A. A., Wu M. F., Hilsenbeck S. G., Shaw C. A., Yu P., Plummer N. W., Birnbaumer L., and Lewis M. T. (2015) An essential role for Gα(i2) in Smoothened-stimulated epithelial cell proliferation in the mammary gland. Sci. Signal. 8, ra92 10.1126/scisignal.aaa7355 [DOI] [PubMed] [Google Scholar]
- 29. Mukhopadhyay S., Wen X., Ratti N., Loktev A., Rangell L., Scales S. J., and Jackson P. K. (2013) The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell 152, 210–223 10.1016/j.cell.2012.12.026 [DOI] [PubMed] [Google Scholar]
- 30. Singh J., Wen X., and Scales S. J. (2015) The Orphan G protein-coupled receptor Gpr175 (Tpra40) enhances Hedgehog signaling by modulating cAMP levels. J. Biol. Chem. 290, 29663–29675 10.1074/jbc.M115.665810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li W., Ohlmeyer J. T., Lane M. E., and Kalderon D. (1995) Function of protein kinase A in hedgehog signal transduction and Drosophila imaginal disc development. Cell 80, 553–562 10.1016/0092-8674(95)90509-X [DOI] [PubMed] [Google Scholar]
- 32. Briscoe J., Chen Y., Jessell T. M., and Struhl G. (2001) A hedgehog-insensitive form of patched provides evidence for direct long-range morphogen activity of sonic hedgehog in the neural tube. Mol. Cell 7, 1279–1291 10.1016/S1097-2765(01)00271-4 [DOI] [PubMed] [Google Scholar]
- 33. Tiecke E., Turner R., Sanz-Ezquerro J. J., Warner A., and Tickle C. (2007) Manipulations of PKA in chick limb development reveal roles in digit patterning including a positive role in Sonic Hedgehog signaling. Dev. Biol. 305, 312–324 10.1016/j.ydbio.2007.02.017 [DOI] [PubMed] [Google Scholar]
- 34. Milenkovic L., Scott M. P., and Rohatgi R. (2009) Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. J. Cell Biol. 187, 365–374 10.1083/jcb.200907126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jiang J., and Struhl G. (1995) Protein kinase A and hedgehog signaling in Drosophila limb development. Cell 80, 563–572 10.1016/0092-8674(95)90510-3 [DOI] [PubMed] [Google Scholar]
- 36. Cheng S., Maier D., and Hipfner D. R. (2012) Drosophila G-protein-coupled receptor kinase 2 regulates cAMP-dependent Hedgehog signaling. Development 139, 85–94 10.1242/dev.068817 [DOI] [PubMed] [Google Scholar]
- 37. Zhou Q., Apionishev S., and Kalderon D. (2006) The contributions of protein kinase A and smoothened phosphorylation to hedgehog signal transduction in Drosophila melanogaster. Genetics 173, 2049–2062 10.1534/genetics.106.061036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ohlmeyer J. T., and Kalderon D. (1997) Dual pathways for induction of wingless expression by protein kinase A and Hedgehog in Drosophila embryos. Genes Dev. 11, 2250–2258 10.1101/gad.11.17.2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maier D., Cheng S., and Hipfner D. R. (2012) The complexities of G-protein-coupled receptor kinase function in Hedgehog signaling. Fly (Austin) 6, 135–141 [DOI] [PubMed] [Google Scholar]
- 40. Ogden S. K., Casso D. J., Ascano M. Jr., Yore M. M., Kornberg T. B., and Robbins D. J. (2006) Smoothened regulates activator and repressor functions of Hedgehog signaling via two distinct mechanisms. J. Biol. Chem. 281, 7237–7243 10.1074/jbc.M510169200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang L. I., Collins J., Davis R., Lin K. M., DeCamp D., Roach T., Hsueh R., Rebres R. A., Ross E. M., Taussig R., Fraser I., and Sternweis P. C. (2007) Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem. 282, 10576–10584 10.1074/jbc.M609695200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Quan F., Thomas L., and Forte M. (1991) Drosophila stimulatory G protein α subunit activates mammalian adenylyl cyclase but interacts poorly with mammalian receptors: implications for receptor-G protein interaction. Proc. Natl. Acad. Sci. U.S.A. 88, 1898–1902 10.1073/pnas.88.5.1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Porter J. A., von Kessler D. P., Ekker S. C., Young K. E., Lee J. J., Moses K., and Beachy P. A. (1995) The product of hedgehog autoproteolytic cleavage active in local and long-range signalling. Nature 374, 363–366 10.1038/374363a0 [DOI] [PubMed] [Google Scholar]
- 44. Chen C. H., von Kessler D. P., Park W., Wang B., Ma Y., and Beachy P. A. (1999) Nuclear trafficking of Cubitus interruptus in the transcriptional regulation of Hedgehog target gene expression. Cell 98, 305–316 10.1016/S0092-8674(00)81960-1 [DOI] [PubMed] [Google Scholar]
- 45. Li S., Ma G., Wang B., and Jiang J. (2014) Hedgehog induces formation of PKA-Smoothened complexes to promote Smoothened phosphorylation and pathway activation. Sci. Signal. 7, ra62 10.1126/scisignal.2005414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen Y., and Jiang J. (2013) Decoding the phosphorylation code in Hedgehog signal transduction. Cell Res. 23, 186–200 10.1038/cr.2013.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shi Q., Li S., Jia J., and Jiang J. (2011) The Hedgehog-induced Smoothened conformational switch assembles a signaling complex that activates Fused by promoting its dimerization and phosphorylation. Development 138, 4219–4231 10.1242/dev.067959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou Q., and Kalderon D. (2011) Hedgehog activates fused through phosphorylation to elicit a full spectrum of pathway responses. Dev. Cell 20, 802–814 10.1016/j.devcel.2011.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Malpel S., Claret S., Sanial M., Brigui A., Piolot T., Daviet L., Martin-Lannerée S., and Plessis A. (2007) The last 59 amino acids of Smoothened cytoplasmic tail directly bind the protein kinase Fused and negatively regulate the Hedgehog pathway. Dev. Biol. 303, 121–133 10.1016/j.ydbio.2006.10.042 [DOI] [PubMed] [Google Scholar]
- 50. Ruel L., Rodriguez R., Gallet A., Lavenant-Staccini L., and Thérond P. P. (2003) Stability and association of Smoothened, Costal2 and Fused with Cubitus interruptus are regulated by Hedgehog. Nat. Cell Biol. 5, 907–913 10.1038/ncb1052 [DOI] [PubMed] [Google Scholar]
- 51. Lum L., Zhang C., Oh S., Mann R. K., von Kessler D. P., Taipale J., Weis-Garcia F., Gong R., Wang B., and Beachy P. A. (2003) Hedgehog signal transduction via Smoothened association with a cytoplasmic complex scaffolded by the atypical kinesin, Costal-2. Mol. Cell 12, 1261–1274 10.1016/S1097-2765(03)00426-X [DOI] [PubMed] [Google Scholar]
- 52. Dang D. T., and Perrimon N. (1992) Use of a yeast site-specific recombinase to generate embryonic mosaics in Drosophila. Dev. Genet. 13, 367–375 10.1002/dvg.1020130507 [DOI] [PubMed] [Google Scholar]
- 53. Blair S. S. (2003) Genetic mosaic techniques for studying Drosophila development. Development 130, 5065–5072 10.1242/dev.00774 [DOI] [PubMed] [Google Scholar]
- 54. Fraser S. E., and Bryant P. J. (1985) Patterns of dye coupling in the imaginal wing disk of Drosophila melanogaster. Nature 317, 533–536 10.1038/317533a0 [DOI] [PubMed] [Google Scholar]
- 55. Baker J. D., and Truman J. W. (2002) Mutations in the Drosophila glycoprotein hormone receptor, rickets, eliminate neuropeptide-induced tanning and selectively block a stereotyped behavioral program. J. Exp. Biol. 205, 2555–2565 [DOI] [PubMed] [Google Scholar]
- 56. He X., Zhang L., Chen Y., Remke M., Shih D., Lu F., Wang H., Deng Y., Yu Y., Xia Y., Wu X., Ramaswamy V., Hu T., Wang F., Zhou W., et al. (2014) The G protein α subunit Gαs is a tumor suppressor in Sonic hedgehog-driven medulloblastoma. Nat. Med. 20, 1035–1042 10.1038/nm.3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mukhopadhyay S., and Rohatgi R. (2014) G-protein-coupled receptors, Hedgehog signaling and primary cilia. Semin. Cell Dev. Biol. 33, 63–72 10.1016/j.semcdb.2014.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Niewiadomski P., Zhujiang A., Youssef M., and Waschek J. A. (2013) Interaction of PACAP with Sonic hedgehog reveals complex regulation of the hedgehog pathway by PKA. Cell Signal 25, 2222–2230 10.1016/j.cellsig.2013.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Denef N., Neubüser D., Perez L., and Cohen S. M. (2000) Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell 102, 521–531 10.1016/S0092-8674(00)00056-8 [DOI] [PubMed] [Google Scholar]
- 60. Marada S., Navarro G., Truong A., Stewart D. P., Arensdorf A. M., Nachtergaele S., Angelats E., Opferman J. T., Rohatgi R., McCormick P. J., and Ogden S. K. (2015) Functional divergence in the role of N-linked glycosylation in Smoothened signaling. PLoS Genet. 11, e1005473 10.1371/journal.pgen.1005473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nobles K. N., Xiao K., Ahn S., Shukla A. K., Lam C. M., Rajagopal S., Strachan R. T., Huang T. Y., Bressler E. A., Hara M. R., Shenoy S. K., Gygi S. P., and Lefkowitz R. J. (2011) Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal. 4, ra51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Masuho I., Ostrovskaya O., Kramer G. M., Jones C. D., Xie K., and Martemyanov K. A. (2015) Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Sci. Signal. 8, ra123 10.1126/scisignal.aab4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pal K., Hwang S. H., Somatilaka B., Badgandi H., Jackson P. K., DeFea K., and Mukhopadhyay S. (2016) Smoothened determines β-arrestin-mediated removal of the G protein-coupled receptor Gpr161 from the primary cilium. J. Cell Biol. 212, 861–875 10.1083/jcb.201506132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cheng S., Maier D., Neubueser D., and Hipfner D. R. (2010) Regulation of Smoothened by Drosophila G-protein-coupled receptor kinases. Dev. Biol. 337, 99–109 10.1016/j.ydbio.2009.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vilardaga J. P., Jean-Alphonse F. G., and Gardella T. J. (2014) Endosomal generation of cAMP in GPCR signaling. Nat. Chem. Biol. 10, 700–706 10.1038/nchembio.1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pusapati G. V., Kong J. H., Patel B. B., Gouti M., Sagner A., Sircar R., Luchetti G., Ingham P. W., Briscoe J., and Rohatgi R. (2018) G protein-coupled receptors control the sensitivity of cells to the morphogen Sonic Hedgehog. Sci. Signal. 11, eaao5749 10.1126/scisignal.aao5749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nybakken K., Vokes S. A., Lin T. Y., McMahon A. P., and Perrimon N. (2005) A genome-wide RNA interference screen in Drosophila melanogaster cells for new components of the Hh signaling pathway. Nat. Genet. 37, 1323–1332 10.1038/ng1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. DasGupta R., Nybakken K., Booker M., Mathey-Prevot B., Gonsalves F., Changkakoty B., and Perrimon N. (2007) A case study of the reproducibility of transcriptional reporter cell-based RNAi screens in Drosophila. Genome Biol. 8, R203 10.1186/gb-2007-8-9-r203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schaefer M., Petronczki M., Dorner D., Forte M., and Knoblich J. A. (2001) Heterotrimeric G proteins direct two modes of asymmetric cell division in the Drosophila nervous system. Cell 107, 183–194 10.1016/S0092-8674(01)00521-9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.