

Abstract
Cancer stem cells (CSCs) are unique populations of cells that can self-renew and generate different cancer cell lineages. Although CSCs are believed to be a promising target for novel therapies, the specific mechanisms by which these putative therapeutics could intervene are less clear. Nitric oxide (NO) is a biological mediator frequently up-regulated in tumors and has been linked to cancer aggressiveness. Here, we search for targets of NO that could explain its activity. We find that it directly affects the stability and function of octamer-binding transcription factor 4 (Oct4), known to drive the stemness of lung cancer cells. We demonstrated that NO promotes the CSC-regulatory activity of Oct4 through a mechanism that involves complex formation between Oct4 and the scaffolding protein caveolin-1 (Cav-1). In the absence of NO, Oct4 forms a molecular complex with Cav-1, which promotes the ubiquitin-mediated proteasomal degradation of Oct4. NO promotes Akt-dependent phosphorylation of Cav-1 at tyrosine 14, disrupting the Cav-1:Oct4 complex. Site-directed mutagenesis and computational modeling studies revealed that the hydroxyl moiety at tyrosine 14 of Cav-1 is crucial for its interaction with Oct4. Both removal of the hydroxyl via mutation to phenylalanine and phosphorylation lead to an increase in binding free energy (ΔGbind) between Oct4 and Cav-1, destabilizing the complex. Together, these results unveiled a novel mechanism of CSC regulation through NO-mediated stabilization of Oct4, a key stem cell transcription factor, and point to new opportunities to design CSC-related therapeutics.
Keywords: cancer biology; cancer stem cells; caveolin; cell signaling; lung cancer; proteasome; GSK3β; OCT4; nitric oxide, caveolin-1; protein degradation; protein–protein interaction; differentiation; regulation
Introduction
Cancer stem cells (CSCs)2 or cancer stem-like cells are specific cell populations that can differentiate and generate cancer cells in various types of cancer, including lung cancer (1, 2). These cells have characteristics similar to those of normal stem cells, in particular, an ability to self-renew and generate different cell lineages (3). Such ability is believed to facilitate the progression of cancers and their aggressive behaviors (4). Increasing evidence also suggests that CSCs possess several virulent features that make them invasive and resistant to chemo/radiotherapy (5).
The successful establishment of induced pluripotent stem cells that form fully differentiated cells has revealed that the differentiated cells retain the capacity to revert to stem cells, and certain regulatory transcription factors, such as octamer-binding transcription factor 4 (Oct4), Nanog, c-Myc, Klf4, Sox2, and Lin28, are essential for somatic cell reprogramming (6, 7). Likewise, cancer dedifferentiation is evident and has been linked to cancer progression and aggressive behaviors (8, 9). Similar to somatic cell reprogramming, tumor dedifferentiation is a reversal of cell development to a more immature state. Such discovery has led to an interest in uncovering the origin of CSCs and targeting these cells for therapy. In cancer, Oct4, Sox2, and Nanog have been shown to associate with the stemness of cells by sustaining the level of stem cell–related proteins, such as CD133, ALDH1A1, and ABCG2 (10). Induced expression of the Oct4 gene and transmembrane delivery of the Oct4 protein were shown to induce the dedifferentiation of melanoma cells to CSC-like cells with a strong reduction in melanocytic markers and acquisition of spheroid-forming ability (11). In lung cancer, the expression of Oct4 correlates with aggressive behaviors and CSC properties (12–14). Moreover, in various cancers, the overexpression of Oct4 and its downstream targets is associated with CSCs and the progression of disease (15–17), although the underlying mechanisms remain obscure.
In recent years, the role of the microenvironment in cancer progression has increasingly been recognized (18). Nitric oxide (NO) is a key mediator frequently up-regulated in lung cancer microenvironment (19). It is also known to play a pivotal role in controlling several cellular processes in both normal and pathological conditions (20). In lung cancer, NO and its producing enzymes, nitric-oxide synthases (NOSs), are highly up-regulated (19, 21). NO has also been shown to promote anoikis or cell survival after detachment (22), motility (23), CSC-like phenotypes (24), and chemotherapeutic resistance (25, 26).
Caveolin-1 (Cav-1) is a 21-kDa scaffolding protein found in caveolae (27). Cav-1 is required for the formation of caveolae as well as its function, including endocytosis, lipid homeostasis, and signal transduction (28). The tumor suppressor function and scaffolding capacity of Cav-1 have been extensively studied in various cancers (29–32), but its role in the regulation of CSCs remains obscure. Furthermore, the co-expression of Cav-1 and NOS in certain cancers has been reported (33). In addition, the caveolin-scaffolding domain of Cav-1 exhibits protein–protein interactions with NOS, which resulted in a decrease of the cellular function of NOS (34, 35). These findings suggest that Cav-1 and NO may play an opposite role in regulating CSCs. However, detailed mechanisms and their cross-talk remain to be investigated.
Oct4 is a key CSC mediator that promotes the expression of other CSC factors, and its introduction into cancer cells induces cancer stemness (36). We found that Oct4 interacts with Cav-1 and that such interaction is under the regulation of NO. In this study, we hypothesized that Cav-1 regulates the cellular level of Oct4, and therefore it influences CSCs in lung cancer via a NO-dependent mechanism. Using molecular techniques and computer modeling approaches, we demonstrated for the first time that Cav-1 forms a molecular complex with Oct4 and regulates its expression through ubiquitin–proteasome-mediated degradation. We also investigated the roles of NO in complex formation and stem properties of lung cancer cells, and unveiled a novel mechanism of CSC regulation that may lead to a better understanding of cancer cell biology under nitrosative oxidative stress conditions, which may be exploited for CSC-targeted therapy.
Results
Nitric oxide increases CSC-like phenotypes of human lung cancer cells
To study the potential regulation of CSCs by NO, we first characterized the optimal dose response to NO in human lung cancer cells. Subconfluent monolayers of human lung cancer H460 cells were exposed to various concentrations of DPTA NONOate, a well-established NO donor, for 24 h, and cell viability was determined by MTT assay. At the concentration range of 0–40 μm, the NO donor was found to have no significant effect on cell viability; however, at 80 μm or higher, a significant decrease in cell viability was observed in the treated cells (Fig. 1A). Apoptosis and necrosis studies by Hoechst 33342/propidium iodide (PI) assays also showed that the low-dose (<40 μm) DPTA NONOate had no significant effect on DNA condensation/fragmentation or nuclear PI fluorescence (Fig. 1, B and C), indicating the lack of apoptotic and necrotic cell death under the test conditions.
Figure 1.

NO donor promotes human lung CSC-like phenotypes. A, H460 cells were treated with NO donor (DPTA NONOate) for 24 h and analyzed for cell viability by MTT assay. Apoptotic and necrotic cell death after the treatment was analyzed by Hoechst 33342/PI co-staining assays. B, percentages of apoptotic and necrotic nuclei in NO-treated cells were analyzed and calculated as relative to the control cells. C, ×10 immunofluorescence images of the treated and nontreated cells stained with Hoechst 33342/PI. After being treated with DPTA NONOate (0–40 μm) for 5 days, H460 (D), H23 (G), and H292 (J) cells were suspended and subjected to spheroid formation assay. Spheroid number H460 (E), H23 (H), H292 (K), and size H460 (F), H23 (I), H292 (L) were analyzed and calculated as relative to the control after 10, 20, and 40 days. Plots are means ± S.D. (n = 3). *, p < 0.05 versus nontreated cells.
To determine the effect of NO on the spheroid-forming ability of lung cancer cells under nonattachment conditions, H460, H23, and H292 cells were treated with 0–40 μm DPTA NONOate for 5 days, and their spheroid-forming capacity was evaluated by seeding them at low density on ultralow attached plates. Time courses of spheroid formation for H460, H23, and H292 cells at 10, 20, and 40 days post-seeding are depicted in Fig. 1, D, G, and J, whereas quantitative analyses of the spheroid number and size are shown in Fig. 1, E/F, H/I, and K/L, respectively. A significant increase in the number of spheroids formed at 20 days was recorded for the H460 and H292 cells treated with 10 μm or higher concentrations of DPTA NONOate, whereas a similar increase was observed for H23 cells at 5 μm or higher concentrations. At 40 days post-seeding, a significant increase in spheroid number was observed in H460, H23, and H292 cells treated with 5 μm or higher concentrations of NONOate. However, none of the treated cells showed a significant change in the spheroid number at 10 days. Fig. 1, F, I, and L, shows the relative spheroid size of the treated and untreated H460, H23, and H292 cells, respectively. Although the spheroid size of the treated cells was smaller than that of the control cells at 10 days, a significant increase in the spheroid size was observed at 20 and 40 days for all cell lines tested. At 20 days post-seeding, the increase in spheroid size was observed at the treatment dose of 10 μm or higher concentration for H460 and H292 cells and at 5 μm or higher concentration for H23 cells. At 40 days post-seeding, all cell lines exhibited a significant increase in spheroid size at the treatment dose of 5 μm or higher concentration. These results indicate that nontoxic concentrations of DPTA NONOate promote spheroid formation of lung cancer H460, H23, and H292 cells.
Nitric oxide increases the expression of stemness-related proteins
Stem cell markers such as CD133, ALDH1A1, and ABCG2 and stemness transcription factors such as Oct4, Sox2, and Nanog are commonly accepted as key drivers or markers of CSCs (10). We used these proteins to verify the CSC-inducing effect of NO in the tested lung cells. H460 cells were cultivated in the presence or absence of DPTA NONOate (5–40 μm) for 1, 3, and 5 days, and the expression level of these markers was assessed by Western blotting. Fig. 2, A–C, shows a dose- and time-dependent expression of CD133, ABCG2, ALDH1A1, and Oct4 in response to the NO treatment. No significant changes in the expression level of these proteins were observed 1 day after the treatment (Fig. 2D). At 3 days, an increased expression of Oct4 was seen at the treatment dose of 10 μm, whereas CD133 expression was up-regulated at 20 and 40 μm (Fig. 2E). Surprisingly, we observed a reduction in Sox2 expression at 3 days after the treatment with 20 and 40 μm DPTA NONOate. Fig. 2F further shows the up-regulation of CD133, ALDH1A1, and Oct4 at 5 days post-treatment with 10 μm or higher concentrations of DPTA NONOate and a decrease in Sox2 expression at the dose of 10 μm or higher.
Figure 2.

NO donor increases CSC markers in NSCLC cell lines. H460 cells were treated with DPTA NONOate for 1 day (A), 3 days (B), and 5 days (C) after which they were analyzed for CSC markers (CD133, ALDH1A1, and ABCG2) and CSC transcription factors (Oct4, Nanog, and Sox2) by Western blotting. The blots were reprobed with tubulin to confirm equal loading of the protein samples. The immunoblot signals at 1 day (D), 3 days (E), and 5 days (F) after the treatment were quantified by densitometry, and mean data from independent experiments were normalized and presented. The bars are means ± S.D. (n = 3). *, p < 0.05 versus nontreated cells. G, expression of CD133 and Oct4 in H460 cells treated with DPTA NONOate (40 μm) for 5 days were analyzed by fluorescence microscopy (×10). Immunofluorescence was performed using mouse anti-CD133 mAb followed by Alexa-Fluor568–labeled secondary antibody to visualize CD133 expression and using mouse anti-Oct4 mAb followed by Alexa-Fluor488-labeled secondary antibody to visualize Oct4 expression in separated experiments. Cells were stained with Hoechst 33342 dye to aid visualization of the cell nucleus. The CD133 and Oct4 proteins were appeared as red and green fluorescence, respectively. H23 (H) and H292 (I) were treated with DPTA NONOate for 5 days and then were analyzed for CSC markers (CD133, ABCG2, and ALDH1A1) and Oct4. The blots were reprobed with tubulin to confirm equal loading of the protein samples. The immunoblot signals of H23 (J) and H292 (K) were quantified by densitometry, and mean data from independent experiments were normalized and presented. The bars are means ± S.D. (n = 3). *, p < 0.05 versus nontreated cells.
To confirm the above finding, we performed immunofluorescence experiments assessing the expression of CSC markers CD133 and Oct4 in H460 cells following NO exposure. The cells were treated with 40 μm DPTA NONOate for 5 days and analyzed for CD133 and Oct4 expression by immunofluorescence staining. Consistent with the Western blotting results, the immunofluorescence results indicate the up-regulation of CD133 and Oct4 expression in the treated lung cancer cells (Fig. 2G).
The expression levels of stemness-associated proteins in response to the NO donor treatment were also assessed in H23 and H292 cells. Fig. 2, H and J, shows that treatment of H23 cells with DPTA NONOate for 5 days induced an up-regulation of CD133 and ABCG2 at the treatment dose of 5 μm or higher, whereas an increase in Oct4 and ALDH1A1 was observed at the dose of 20 μm or higher. Fig. 2, I and K, shows the results of NO treatment in H292 cells. A significant increase in Oct4 expression was seen at the treatment dose of 5 μm or higher and at 20 μm or higher for CD133, ABCG2, and ALDH1A1. These results along with the spheroid formation results support the regulatory role of NO in controlling the stemness of lung cancer cells.
Microarray gene profiles of nitric oxide–mediated gene expression
Two different passages of H460 cells treated with DPTA NONOate and their nontreated counterparts were subjected to microarray analysis as mentioned under “Experimental procedures.” The gene probe list of the chip is shown in Table S1. The CU-DREAM (37) program was used to analyze the raw data, and gene probes with significant differences (p < 0.05) were chosen for further evaluation using the Bioconductor R statistic program. The heat map generated from the Bioconductor R statistics program (Fig. 3A) illustrates the intensity detected for different gene probes. H460 control cell samples were labeled control 1 and control 2, and the DPTA NONOate–treated cells were labeled as DPTA NONOate 1 and DPTA NONOate 2. The high-resolution microarray heat map image is available in Fig. S1. The computer program bracket links two samples that have similar genotypes. As expected, control 1 and control 2 displayed expression profiles distinct from those of NO-treated 1 and 2. The probed gene with significant differences (p < 0.01) (Table S2) was selected as the next interpretation. Although the mRNA level of Oct4 analyzed by microarray in NO-treated cells was not significantly altered (Table S2), the Oct4 protein expression was significantly up-regulated (Fig. 2), suggesting that the NO may not regulate Oct4 up-regulation via transcriptional means but may affect the protein stability.
Figure 3.

Microarray analysis of the effect of nitric oxide on H460 cells. H460 cells treated with DPTA NONOate and untreated control were subjected to analyzing the mRNA expression profile by Affymetrix chips as per the manufacture's procedure. A, gene probes with a significant difference (p < 0.05) between DPTA NONOate-treated and the control group analyzed by CU-DREAM program generated heat maps of genes by the Bioconductor R statistic program. B, results form Enrichr-EhEA-2016 demonstrated the gene list of nitric oxide-mediated gene expression matched with targeted genes of transcription factors evaluated by ChIP-seq assay. C, qRT-PCR was used to verify the microarray results. The mRNA expression of Oct4 downstream targets, including COLEC12, LAMP1, MYH3, PER3, ROS26, and UBE2S, were subjected to RT-PCT analysis. The bars are means ± S.D. (n = 3). *, p < 0.05 versus nontreated cells; **, p < 0.001–0,01; and ***, p < 0.001. D, NO-inducing gene alteration (only form Oct4 axis) and its primer sequences for RT-PCR.
The genes displayed significant differences (p < 0.01) (Table S2) between treatments, and control groups were subjected for Gene Ontology (GO) using Enrichr (an integrative web-based and mobile software application; http://amp.pharm.mssm.edu/Enrichr/)3 (38, 39), providing the gene set enrichment analysis. We used the Enrichr-EhEA-2016 feature (39), which analyzed and indicated the responsible transcription factors (TFs) for the set of gene expression changed with ChIP databases (PMID: 20709693) (40). Fig. 3B shows the analyzed results by Enrichr-EhEA-2016 indicating the TF names and PubMed IDs of ChIP-seq experiments; a list of NO-mediated gene expression changes that matched the gene list in each ChIP-seq; and the p value calculated from matched and unmatched genes in each ChIP-seq. The results showed that genes altered by NO treatment are the regulated target genes of various TFs, including Ap2-α, Ets1, Jar1d1A, Oct4, Myc, Sall4, Klf4, E2A, Top2B, Maf, and Stat3 (Fig. 3B). To verify the effect of NO in regulation of CSCs through Oct4 activity, the Oct4-targeted genes were analyzed by RT-PCR (Fig. 3C). The mRNA expression of COLEC12, LAMP1, MYH3, PER3, ROS26, and UBE2S genes were up-regulated after treatment with NO in a concentration-dependent manner. The RT-PCR results were consistent with the results from the microarray analysis. Fig. 3D indicates the selected NO-mediated gene alteration (only genes of which their mRNA expression is controlled by Oct4) and its primer sequences.
Nitric oxide suppresses the degradation of Oct4 by diminishing its ubiquitin proteasomal degradation
Based on previous studies showing the role of Oct4 in controlling stem cell properties and expression of CSC markers CD133 and ALDH1A1 (41, 42), our finding demonstrates that the effects of NO on Oct4 were not involved in the transcription process of Oct4. Thus, we investigated the potential regulation of NO on Oct4 post-translational modifications. Because NO has previously been shown to promote the stability of various proteins (43, 44), we tested whether NO could stabilize Oct4, which may contribute to its up-regulation after the NO donor treatment.
The cells were treated with 40 μm DPTA NONOate at various times in the presence or absence of cycloheximide (CHX), a protein synthesis inhibitor, and the expression of Oct4 was monitored by Western blotting (Fig. 4A). The results showed that treatment of the cells with DPTA NONOate significantly increased the expression of Oct4 over time as compared with the untreated control (Fig. 4B), suggesting the stabilizing effect of NO on Oct4.
Figure 4.

NO donor increases Oct4 stability through Akt-dependent mechanisms. A, H460 cells were treated with 40 μm DPTA NONOate and 50 μg/ml CHX for the indicated times and analyzed for Oct4 levels by Western blotting. The blots were reprobed with GAPDH to confirm equal loading of the samples. B, immunoblot signals of Oct4 were quantified by densitometry, and mean data from independent experiments were normalized and presented. The bars are means ± S.D. (n = 3). *, p < 0.05 versus nontreated cells. C, H460 cells were treated with DPTA NONOate (0–40 μm) and lactacystin (10 μm) for 12 h and subjected to immunoprecipitation (IP) of Oct4. The immunoprecipitation complexes were analyzed for ubiquitin level by Western blotting. D, immunoblot signals of ubiquitin were quantified by densitometry, and mean data from independent experiments were normalized and presented. The bars are means ± S.D. (n = 3). *, p < 0.05 versus nontreated cells. E, H460 cells were treated with DPTA NONOate (0–40 μm) for 5 days and analyzed for phosphorylated Akt (Ser-473), Akt, phosphorylated Src (Tyr-416), Src, phosphorylated ERK1/2, Eek (1/2), and Cav-1 by Western blotting. The blots were reprobed with tubulin to confirm equal loading of the samples. H23(F) and H292 (G) were treated with DPTA NONOate (0–40 μm) for 5 days and analyzed for phosphorylated Akt (Ser-473), Akt, and Cav-1 by Western blotting. The blots were reprobed with tubulin to confirm equal loading of the samples. The immunoblot signals H460 (H), H23 (I), and H292 (J) were quantified by densitometry, and mean data from independent experiments were normalized and presented. The bars are means ± S.D. (n = 3). *, p < 0.05 versus nontreated cells. K, H460 cells were treated with 40 μm DPTA NONOate, 40 μm NONOate + 2.5 μm perifosine (specific Akt inhibitor), or 2.5 μm perifosine for 5 days and subjected to Western blot analysis for Oct4 detection. L, Western blotting bands of Oct4 were quantified by densitometry, and mean data from independent experiments were normalized and presented. The bars are means ± S.D. (n = 3). *, p < 0.05 versus control cells.
Because previous studies have shown that the cellular level of Oct4 is regulated by the ubiquitin–proteasomal degradation pathway (45), we next investigated whether Oct4 up-regulation by NO may occur through this pathway. Immunoprecipitation studies were carried out to evaluate the effect of NO on Oct4 ubiquitination. H460 cells were treated with various concentrations of DPTA NONOate in the presence of lactacystin, a highly specific proteasome inhibitor, for 12 h, after which cell lysates were prepared and immunoprecipitated using an anti-Oct4 antibody. The resulting immune complexes were then analyzed for protein ubiquitination by Western blotting using an anti-ubiquitin antibody. The results showed that Oct4 ubiquitination was dose-dependently reduced by the NO donor treatment (Fig. 4, C and D). This study indicates that NO exerts a promoting effect on Oct4 expression by inhibiting its ubiquitin–proteasomal degradation.
Nitric oxide increases phosphorylated Akt (Ser-473) and caveolin-1 levels
Phosphorylation of Oct4 by cellular kinases is crucial to its biological activity (46). We next tested whether NO could alter the activity of key cellular kinases, including Akt, Src, and Erk in lung cancer H460, H23, and H292 cells. The cells were cultivated in the presence or absence of various concentrations of DPTA NONOate, and the expression levels of these proteins as well as their phosphorylated forms were determined by Western blotting. The results showed that NO treatment significantly increased the expression ratio of p-Akt (Ser-473) to Akt in a dose-dependent manner in H460 (Fig. 4, E and H), H23 (Fig. 4, F and I), and H292 (Fig. 4, G and J) cells.
We further investigated whether the effect of NO-mediated Oct4 up-regulation is an Akt-dependent mechanism, and H460 cells were treated with 40 μm DPTA NONOate in the presence or absence of 2.5 μm perifosine (selective Akt inhibitor) for 5 days. As control, cells were treated with 2.5 μm perifosine alone for 5 days and analyzed for Oct4 expression by Western blotting (Fig. 4, K and L). Treatment of the cells with DPTA NONOate strongly up-regulated the Oct4 level, whereas the perifosine treatment alone dramatically down-regulated the Oct4 level. In addition, the up-regulation of Oct4 by DPTA NONOate could be blocked by the perifosine treatment, suggesting that the up-regulation of Oct4 by NO occurs through an Akt-dependent pathway.
Cav-1 was shown to exhibit an inhibitory effect on inducible NO synthases (34, 35). We therefore investigated whether NO could affect Cav-1 expression. H460, H23, and H292 cells were cultivated in the presence or absence of various concentrations of DPTA NONOate, and the expression level of Cav-1 was examined by Western blotting. The results showed that Cav-1 expression was strongly up-regulated in the NO-treated cells in a dose-dependent manner in H460 (Fig. 4, E and H), H23 (Fig. 4, F and I), and H292 (Fig. 4, G and J) cells.
Caveolin-1 enhances the degradation of Oct4 via ubiquitin–proteasome pathway
Cav-1 is known to regulate several cancer cell behaviors, including migration (47), invasion (48), and anoikis resistance (49). Because Cav-1 is up-regulated by NO, we tested whether Cav-1 could be involved in the regulation of lung CSCs. Cav-1 expression was ectopically expressed or knocked down by FLAG–Cav-1 or shRNA–Cav-1, respectively, and their effects on spheroid formation and CSC marker expression were examined in H460 cells. Fig. 5A shows microscopic images of the spheroids formed by the genetically modified and control cells. Quantitative analyses of the spheroid number and size showed a significant increase in the spheroid number in shRNA–Cav-1 cells and a decrease in FLAG–Cav-1 cells as compared with controls (Fig. 5B). Spheroid size was also found to decrease in FLAG–Cav-1 cells; however, no significant change was observed in shRNA–Cav-1 cells. These results indicate that Cav-1 is a negative regulator of CSCs in the tested cell system. Western blot analysis of CSC-related proteins, including CD133, ALDH1A1, and Oct4, showed that the expression of these proteins was inversely correlated with Cav-1 expression (Fig. 5, C and D), thus supporting the suppressive role of Cav-1 in CSC regulation. These results also suggest that certain downstream targets of Cav-1, such as Oct4, may be involved in the regulation of CSCs by Cav-1. For verification, the data were provided in Fig. S2. The expression of Cav-1 protein in nontreated H460 cells was comparable with mock control transfectants (Fig. S2A). The effect of shRNA–Cav-1 plasmid on the alteration of Cav-1, Oct4, and CD133 proteins was verified with two other shRNA–Cav-1 sequences, and the results were consistent (Fig. S2B).
Figure 5.

Caveolin-1 diminishes human lung CSC-like phenotypes. A, stable transfection of H460 with short hairpin plasmid (sh-cav1), Cav-1–overexpressing plasmid (FLAG-cav1), and control were suspended and subjected to spheroid formation assay. B, relative spheroid number and size were analyzed and calculated as relative to mock-transfected cells on days 10 and 20 of the assay. All plots are means ± S.D. (n = 3). *, p < 0.05 versus mock cells. C, stable transfections of H460 cells with sh-cav1, FLAG-cav1, or control were analyzed for Cav-1, CSC markers (CD133 and ALDH1A), and CSC transcription factor (Oct4) by Western blotting. Blots were reprobed with GAPDH as a loading control. D, immunoblot signals were quantified by densitometry, and mean data from independent experiments were normalized and presented. The bars are means ± S.D. (n = 3). *, p < 0.05 versus mock cells. E, stable transfections of H460 cells with sh-cav1 and FLAG-cav1 were treated with 50 μg/ml CHX at indicated times and analyzed for Oct4 level by Western blotting. The blots were reprobed with GAPDH to confirm equal loading of the samples. F, immunoblot signals were quantified by densitometry, and mean data from independent experiments were normalized and presented. The bars are means ± S.D. (n = 3). #, p < 0.05 versus FLAG-cav1-transfected cells. G, stable transfected H460 cells were treated with 10 μm lactacystin for 12 h and subjected to Oct4 immunoprecipitation (IP) of Oct4. The immune complexes were analyzed for ubiquitin by Western blotting. H, immunoblot signals were quantified by densitometry, and mean data from independent experiments were normalized and presented. The bars are means ± S.D. (n = 3). *, p < 0.05 versus mock cells.
Cav-1 is known to regulate several targeted proteins by controlling their stability post-translationally (50, 51). To test whether Cav-1 could down-regulate Oct4 (Fig. 5, C and D) in a post-translational modification manner, we performed a CHX protein synthesis inhibition study in which Cav-1 knockdown and overexpressing cells were treated with 50 μg/ml CHX, a protein synthesis inhibitor, over time and analyzed for Oct4 expression by Western blotting. The results showed that Oct4 protein levels were significantly reduced in the Cav-1–overexpressing cells as compared with the knockdown cells at 24 and 36 h post-treatment (Fig. 5, E and F). This result suggests that Cav-1 regulates the stability of Oct4 post-translationally.
Oct4 was previously shown to be degraded via the ubiquitin–proteasomal pathway (52, 53). To test whether Cav-1 could regulate ubiquitination of Oct4, the ubiquitination level of Oct4 in Cav-1–overexpressing and knockdown cells was determined. Cells were treated with lactacystin, a proteasome inhibitor, and analyzed for Oct4 ubiquitination by immunoprecipitation and Western blotting (Fig. 5G). The results showed that Oct4 ubiquitination substantially increased in the Cav-1–overexpressing cells but decreased in the knockdown cells relative to controls (Fig. 4, G and H). Together, these results suggest Cav-1 mediated down-regulation of Oct4 via ubiquitin–proteasomal degradation as a mechanism of CSC regulation by Cav-1.
Caveolin-1 interacts with Oct-4
The scaffolding function of Cav-1 has been shown to be important in the stability and function of several proteins (35, 51, 54). To test whether Cav-1 could interact and form a molecular complex with Oct4, H460 cells were immunoprecipitated with an anti-Oct4 or anti-Cav-1 antibody and analyzed for specific protein-binding partners by Western blotting. Fig. 6A shows that when Oct4 was immunoprecipitated with anti-Oct4 antibody, the resulting immune complexes were positive with anti-Cav-1 antibody but not with anti-p-Cav-1 (Tyr-14) antibody, indicating that Oct4 forms a molecular complex with Cav-1 but not with p-Cav-1 (Tyr-14). To confirm, Akt activity in the cells was enhanced by SC79, a specific Akt activator, and the alteration in level of p-Cav-1 (Tyr-14) and its interaction with Oct4 were analyzed (Fig. S2C). Under active Akt status (10 μm SC79 treatment), we observed the significant up-regulation of p-Cav-1 (Tyr-14) and the decrease of the Oct4:Cav-1 complex (Fig. S2C).
Figure 6.

Caveolin-1 exhibits direct protein–protein interaction with Oct4. A, H460 cell lysates were prepared and immunoprecipitated (IP) with anti-Oct4 antibody or control IgG and then analyzed Cav-1 and phosphorylated Cav-1 (Tyr-14) by Western blotting assay. B, H460 cell lysates were prepared and immunoprecipitated with anti-Cav-1 antibody or control IgG and then analyzed Oct4 with Western blotting assay. C, H460 cell lysates were prepared and immunoprecipitated with anti-phosphorylated Oct4 (Ser-236) antibody or control IgG and then analyzed the Cav-1 expression by Western blotting assay. D, H460 cell lysates were prepared and immunoprecipitated with anti-phosphorylated Cav-1(Tyr-14) antibody or control IgG, then evaluated Oct4 protein level by Western blotting. In all experiments, immunoblots were performed on cell lysates used as input for immunoprecipitation (IP) using GAPDH antibody to confirm equal loading of the samples. E, protein–protein interaction of Cav-1 and Oct4 was confirmed by PLA in H460 cells. After plating H460 cells for 24 h, the cells were permeabilized and stained with rabbit anti-Cav-1 and mouse anti-Oct4 for 24 h. The stained cells were subjected to PLA following the manufacturer's instruction. The interaction between Cav-1 and Oct4 was visualized by fluorescence microscopy (×20). Cells were stained with Hoechst 33342 dye to aid visualization of the cell nucleus. The Cav-1:Oct4 complexes appear as green fluorescence.
The results showed that Cav-1 was immunoprecipitated with anti-Cav-1 antibody, and the immune complexes were similarly positive for anti-Oct4 antibody (Fig. 6B). A previous study (46) showed that Akt activity could facilitate phosphorylation of Oct4 and subsequently promoted proliferation of CSCs. To test whether phosphorylated Oct4 could bind with Cav-1, anti-p-Oct4 antibody was used for immunoprecipitation, and the immunoprecipitation complexes were subjected to evaluation for Cav-1 expression. Fig. 6C shows that p-Oct4 (Ser-236) formed complexes only with the Cav-1-β form (21 kDa mass). As expected, p-Cav-1(Tyr-14) formed no immune complexes with Oct4 (Fig. 6D). Taken together, we found the Cav-1:Oct4 protein:protein complex can be inhibited by the phosphorylation of Cav-1 at tyrosine 14.
To substantiate the Oct4:Cav-1 interaction, in-cell co-localization experiments were conducted using proximity ligation assay. In this study, cells were treated with anti-Oct4 and anti-Cav-1, followed by an addition of plus and minus proximity ligation assay (PLA) probes with attached specific DNA strands. The two DNA strands in proximity were ligased, amplified, and visualized under a fluorescence microscope. Fig. 6E shows the in-cell Oct4:Cav-1 protein:protein complexes (green fluorescence) with Hoechst 33342 (blue fluorescence) serving as a nuclear stain. These results support the immunoprecipitation results and indicate the direct interaction between Oct4 and Cav-1 in the tested lung cancer cells.
NO-mediated phosphorylation of Akt promotes Oct4 expression by dissociating it from caveolin-1 complex
To investigate the effect of NO on the binding interaction between Oct4 and Cav-1, immunoprecipitation studies were conducted in cells treated with DPTA NONOate.
After the treatment, cell lysates were prepared and immunoprecipitated with an anti-Oct4 antibody. The immune complexes were then analyzed for Cav-1 by Western blotting. Fig. 7, A and B, shows that as compared with the untreated control, the NO donor treatment dramatically decreased the binding interaction between Oct4 and Cav-1. We also investigated the potential role of Akt in regulating the Oct4:Cav-1 complex formation because Akt is a known target of NO (22) and is involved in many cancer cell behaviors, including cell invasiveness, anoikis, chemotherapy resistance, and tumorigenesis (56). In this study, H460 cells were treated with DPTA NONOate in the presence or absence of perifosine, a specific Akt inhibitor, and after the treatment, cells lysates were prepared and immunoprecipitated with an anti-Oct4 antibody. The resulting immune complexes were then probed for Cav-1 by Western blotting. The results showed that the Akt inhibitor (perifosine) significantly increased the Cav-1:Oct4 complex formation, whereas the NO donor decreased the complex formation (Fig. 7, C and D). These results indicate that the complex formed by Cav-1 and Oct4 is regulated by NO through an Akt-dependent process. Together with the results from Fig. 4, NO increased stability of Oct4 by inducing Akt phosphorylation (Ser-473), which diminished the interaction of Oct4 to Cav-1.
Figure 7.
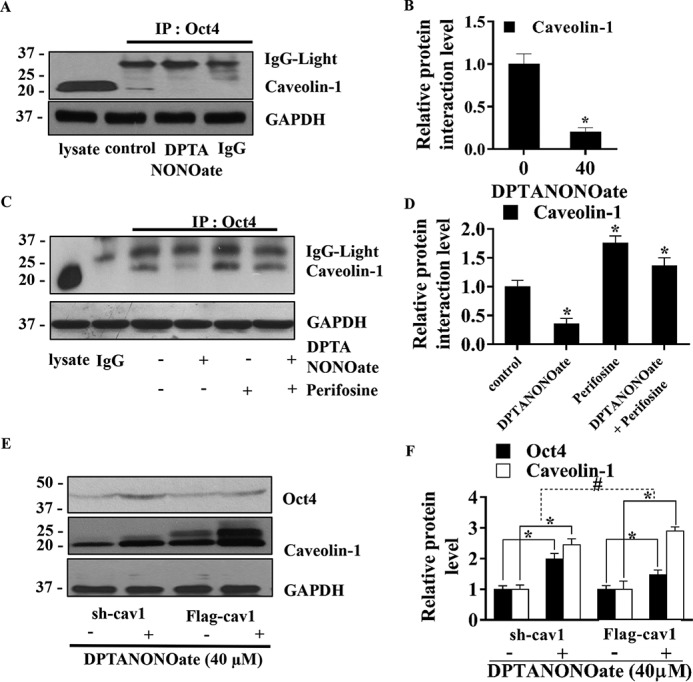
NO-mediated Akt diminishes the interaction between Cav-1 and Oct4. A, after treating with DPTA NONOate (40 μm) for 12 h, the H460 cell lysates were prepared and subjected to immunoprecipitation (IP) with anti-Oct4 antibody or control IgG and then analyzed the Cav-1 level by Western blotting. B, interaction signals were quantified by densitometry, and mean data from independent experiments were normalized and presented. The bars are means ± S.D. (n = 3). *, p < 0.05 versus control cells. C, H460 cells were treated with 40 μm DPTA NONOate, 2.5 μm perifosine (specific Akt inhibitor), or 40 μm NONOate + 2.5 μm perifosine for 12 h, subjected to immunoprecipitation with anti-Oct4 antibody or control IgG, and then evaluated the Cav-1 protein level by Western blotting. D, interaction signals were quantified by densitometry, and mean data from independent experiments were normalized and presented. The bars are means ± S.D. (n = 3). *, p < 0.05 versus control cells. In all experiments, the immunoblots were performed on cell lysates used as input for immunoprecipitation using GAPDH antibody to confirm equal loading of the samples. E, stable transfections of H460 cells with sh-cav1 or FLAG-cav1 were treated with 40 μm NONOate for 5 days, and cell lysates were performed, and measurement of Oct4 and Cav-1 levels was by Western blotting. F, immunoblot signals were quantified by densitometry, and mean data from independent experiments were normalized and presented. The bars are means ± S.D. (n = 3). *, p < 0.05 versus nontreated cells. #, p < 0.05 versus the sh-cav1 means difference of nontreated and treated cells.
Cav-1 was earlier shown to decrease the cellular level of Oct4 by forming a repression complex. To investigate whether Cav-1 diminishes the activity of NO on Oct4, Cav-1 knockdown (shRNA–Cav-1) or overexpressing (FLAG–Cav-1) cells were treated with DPTA NONOate, and the expression level of Oct4 in these cells was determined by Western blotting. Fig. 7, E and F, shows that the NO treatment significantly increased Oct4 levels in the both Cav-1 knockdown and overexpressing cells. As expected, the difference in Oct4 expression between the treated and untreated cells was significantly higher in the knockdown cells compared with the overexpressing cells. These results suggest that Cav-1 not only decreases the Oct4 level but also diminishes the efficacy of NO to up-regulate Oct4.
Tyrosine 14 of caveolin-1 is required for its interaction with Oct4
Our immunoprecipitation study demonstrated that phosphorylated Cav-1 (Tyr-14) was unable to form a complex with Oct4 (Fig. 6, A and D). To determine whether tyrosine 14 of Cav-1 is essential for the complex formation, site-directed mutation of tyrosine to phenylalanine at position 14 of Cav-1 (Y14F) was performed in pcDNA3.1 His-Myc plasmid. The mutated plasmid was transfected into H460 cells, and its effect on Cav-1:Oct4 complex formation was determined compared with WT FLAG–Cav-1 transfection. Cell lysates of His–Myc–Cav-1 (Y14F)-transfected cells and control FLAG–Cav-1-transfected cells were prepared and immunoprecipitated with anti-His and anti-FLAG antibody, respectively. The immune complexes were analyzed for Oct4 expression by Western blotting. As expected, the tyrosine to phenylalanine mutation at position 14 of Cav-1 prevented its interaction with Oct4 (Fig. 8A). These results clearly indicate that tyrosine 14 of Cav-1 is essential to the complex formation with Oct4 and suggest that phosphorylation of the tyrosine residue may interrupt the complex formation.
Figure 8.

Effect of Cav-1 (Y14F) mutation on the interaction between Cav-1 and Oct4. A, H460 cells were transfected with Cav1-(Y14F)–Myc–His or FLAG–Cav1. The transfected cells were subjected to co-immunoprecipitation by pulling down the tagged proteins (IP:His for Cav1(Y14F)Myc-His and IP:FLAG for FLAG-Cav1) and measuring the level of Oct4 by Western blotting. B, 3D model of WT Cav-1 (wtCav1) (green) bound with Oct4 (purple), where the Tyr-14 residue is presented as a green stick. C, close-up of the Cav-1 residue 14 on intermolecular hydrogen bond interaction with the Oct4 residues (Gly-308) (dashed line) for the WT, pTyr-14, and Y14F Cav-1:Oct4 complexes.
Cav-1:Oct4 complex models
Most Cav-1–interacting proteins contain a caveolin-binding motif (CBM) comprising φXφXXXXφ, φXXXXφXXφ, or φXφXXXXφXXφ, where φ is an aromatic amino acid, and X is any amino acid (50), but Oct4 does not contain this domain (42). To investigate how Cav-1 interacts with Oct4 and what specific amino acid residue(s) may be responsible for this interaction, we conducted a computational modeling study of the Cav-1:Oct4 complex. The optimized structures of all model complexes are depicted in Fig. 8, B and C, and the intermolecular hydrogen binding (H-bond) interactions formed between the two proteins are summarized in Table 1. Note that the H-bond was defined by the following criteria: (i) the distance between the H-bond donor (D) and acceptor (A) atoms is less than 3.5 Å, and (ii) the angle between D-H-A is larger than 120°. From Table 1, it appears that most H-bonds between Cav-1 and Oct4 proteins are conserved, even though Tyr-14 is either phosphorylated or mutated to phenylalanine. Interestingly, the Y14F substitution in Cav-1 introduced a loss of H-bond at the mutated residue, whereas an additional H-bond between the Cav-1 pTyr-14 residue and the Oct4 Ala-74 residue was detected (see also Fig. 8C).
Table 1.
Hydrogen bond pairs detected at the Cav-1–Oct4 interface for the three studied systems
@ means “at” the specific functional group.
| CAV1 | OCT4 | WT CAV1–OCT4 | pTyr-14 CAV1–OCT4 | Y14F CAV1–OCT4 |
|---|---|---|---|---|
| Y6@OH | D20@O | √ | √ | |
| Y6@N | √ | |||
| H12@ND1 | S12@OG | √ | √ | √ |
| H12@N | √ | √ | √ | |
| Y14@OH | G308@O | √ | ||
| pY14@O1P | A74@N | √ | ||
| pY14@O3P | G308@N | √ | ||
| V41@O | Y325@OH | √ | ||
| Y325@N | √ | √ | ||
| H45@NE2 | G17@N | √ | ||
| H45@O | H329@NE2 | √ | √ | √ |
| T46@OG | G18@N | √ | √ | √ |
| V52@O | K254@NZ | √ | √ | √ |
| N53@O | √ | √ | √ | |
| R54@O | √ | √ | √ | |
| N53@OD1 | R282@NE | √ | √ | √ |
| R282@NH2 | √ | √ | √ | |
| D61@OD1 | Y292@N | √ | √ | |
| Y292@OH | √ | |||
| F68@O | R295@NH1 | √ | √ | √ |
| E69@OE2 | Q294@N | √ | √ | √ |
The effect of phosphorylation (pTyr-14) and single mutation (Y14F) of Cav-1 on its binding interaction with Oct4 was investigated using the molecular modeling/Poisson-Boltzmann surface area (MM/PBSA) method. The binding free energy (ΔGbind) and its energetic components on the minimized structures of the WT, pTyr-14, and Y14F Cav-1 complexed with Oct4 are summarized in Table 2. The overall structures of the three complexes are relatively similar, and thus the entropic term can be ignored. Table 2 shows that both pTyr-14 and Y14F Cav-1 induce an increase in ΔGbind ∼30 and 3 kcal/mol, respectively. Although the negative charge of pTyr-14 leads to a more favorable electrostatic interaction (by ∼78 kcal/mol), the large positive polar solvation free energy (ΔGsol-ele of 602.4 kcal/mol) reduces the binding efficiency between pTyr-14 Cav-1 and Oct4 proteins. Meanwhile, both van der Waals and electrostatic contributions are slightly reduced in the case of Y14F Cav-1 mutant. These computational models are consistent with our experimental data that could indicate that NO-mediated Akt liberates Oct4 from the Cav-1:Oct4 complex through Tyr-14 phosphorylation of Cav-1. The released Oct4 is accumulated in the cells and promotes the expression of stemness-related genes as depicted in Fig. 9, A and B.
Table 2.
The binding free energy and its components (kcal/mol) calculated from MM/PBSA method
| WT CAV1–OCT4 | pTyr-14 CAV1–OCT4 | Y14F CAV1–OCT4 | |
|---|---|---|---|
| MM | |||
| ΔEele | −372.8 | −450.9 | −362.9 |
| ΔEvdW | −161.4 | −156.0 | −160.6 |
| ΔEMM | −534.2 | −610.9 | −523.5 |
| PBSA | |||
| ΔGsol-ele | 494.8 | 602.4 | 487.0 |
| ΔGsol-np | −25.4 | −26.2 | −25.4 |
| ΔGsol | 469.4 | 576.2 | 461.6 |
| ΔEele + ΔGsol-ele | 122.0 | 151.5 | 124.1 |
| ΔEvdW + ΔGsol-np | −186.8 | −182.2 | −186.0 |
| ΔGbind | −64.8 | −34.8 | −62.0 |
Figure 9.

Schematic overview of NO-promoted CSC-like phenotypes through stabilization of Oct4 cellular level by dissociating it from the Cav-1:Oct4 degradation complex. A, in the absence of NO, Cav-1 binds to Oct4 and enhances its degradation through the ubiquitin–proteasome pathway. The reduction of cellular Oct4 level by Cav-1 leads to a decrease in stemness-related gene expression, which diminishes CSC-like phenotypes. B, in the presence of NO, NO promotes phosphorylation of Cav-1 (tyrosine 14) through the activation of Akt signaling. Because Cav-1 is phosphorylated, Oct4 is dissociated from the Cav-1 complex. The liberated Oct4 accumulates in the nucleus and enhances the expression of stemness-related genes, which promote CSC-like phenotypes.
Discussion
In this study, we provided new evidence showing that NO promotes CSC-like phenotypes in lung cancer cells in part through its stabilizing effect on Oct4. CSCs are the specific population of cancer cells that resides in solid tumors and in leukemia (1, 2). The regulation of CSCs is believed to control the aggressiveness of cancers (4). Various intrinsic and extrinsic stimuli and their cross-talk were documented as the important factors controlling the stemness of cancer cells (57, 58). The intrinsic factors include tumor suppressor proteins, oncoproteins, and epigenetic regulators, whereas the extrinsic factors include microenvironmental mediators, cytokines, and paracrines in the circulating system. NO is a microenvironmental mediator that is elevated in several types of cancer, including lung cancer, colon cancer, and breast cancer (19, 59, 60). An aberrantly high expression of NO was also shown to be correlated with poor prognosis of several cancers. These clinical data are consistent with previous experimental data by our groups and others showing the stimulating effect of NO on cancer cell aggressiveness (22–26).
Oct-4 is recognized as a key transcription factor controlling the stemness of both normal and cancer cells. In lung cancer cells, the expression of Oct4 is highly correlated with the aggressiveness and stemness of the cells (12, 13, 41). Moreover, in various cancer types, an increased expression of Oct4 was found to be highly correlated with CSC level and disease progression (16, 61). The level of Oct4 is primarily regulated by post-translational modifications that control both the expression and activity of Oct4 (52). Alterations in this process could result in Oct4 dysregulation and malfunctions. The stability of Oct4 is largely controlled by its degradation that occurs mainly through the ubiquitin–proteasome degradation pathway (52). We found that NO exerts its effect on Oct4 by interfering with this degradation process, thereby increasing its cellular level. In conformity with this finding, we also observed an increase in stem cell marker expression and CSC-like behaviors in the treated cells with elevated Oct4 levels.
In addition, Sox2 dramatically decreased in dose- and time-dependent manners after NO treatment. The up-regulation of Oct4 with down-regulation of Sox2 were also found in cervical cancer, and such a pattern was found to be strongly associated with a poor prognosis (62). Besides, a previous study found that the concomitant increase of Oct4, Klf4, and MYC with activated status of Akt can deplete endogenous Sox2 (63). Likewise, our microarray analysis revealed that NO promoting transcriptional signaling of Oct4, Kfl4, and Myc cascades and Akt activity may negatively influence Sox2 expression in our system. Previous studies indicated that Sox2 functions as a differentiation enhancer in several cells, including lung cells (64–67), suggesting that NO not only increased stem cell transcription factor Oct4, but also decreased differentiation signals by decreasing the expression of Sox2.
Akt has been reported to regulate Oct4 (46), and its activation due to genetic instability is frequently observed in lung cancer (68). Enhanced Akt activation has also been shown to be correlated with chemo- and radiotherapy resistance (69) and metastasis (70), and both are key characteristics of CSCs. Moreover, the levels of Akt and Oct4 are correlated with poor prognosis of patients. In this study, we found that NO regulates CSC-like phenotypes of lung cancer cells by increasing the stability of Oct4 in an Akt-dependent manner.
We demonstrated that Cav-1 has a negative regulatory effect on Oct4 stability through its scaffolding property. Indeed, the Cav-1:Oct4 complex facilitates Oct4 degradation via the ubiquitin–proteasome pathway. The disruption of such a complex diminishes the degradation of Oct4 and increases its cellular level, thus promoting the CSC-like behaviors. Moreover, we found that Oct4, which lacks the CBM (42), can form a molecular complex with Cav-1 through H-bonding. To our knowledge, this is the first demonstration of the protein–protein interaction of Cav-1 and its binding partner that does not occur through the interaction between CBM and caveolin-scaffolding domain (50). We further found that the tyrosine residue at position 14 of Cav-1 is required for its interaction with Oct4, i.e. via H-bonding possibly between the H-bond donor OH-tyrosine 14 and H-bond acceptor that resides in Oct4. Such binding interaction is disrupted when the Tyr-14 of Cav-1 is phosphorylated or mutated.
Although Cav-1 is highly co-expressed with NOS in cancer (33), it can negatively regulate NOS. For example, Cav-1 was shown to inhibit the function of NO synthases that produce NO by interacting via its caveolin-scaffolding domain (34, 35). This suggests that Cav-1 and NO may play an opposite role in the regulation of CSCs. We found that Cav-1 forming the complex with Oct-4 and facilitate Oct4 degradation. Together with results from Fig. 4 indicated that NO contradicts the role of Cav-1 through Akt-dependent manner by Akt-mediated diminish the interaction of Oct4 to Cav-1.
Surprisingly, we found that treatment of the cells with NO also increased the Cav-1 level. Although there is no clear explanation, in our opinion such a phenomenon may be due to the compensation mechanism of the cells for balancing control of Oct4. Besides, we previously showed that NO could increase the Cav-1 level by directly inhibiting its degradation through S-nitrosylation (71).
As Oct4 (72–74) and Cav-1 (75–78) were found in various types of normal and cancerous cells, and the function of Oct4 in the regulation of stem cell properties was widely demonstrated in both CSC and noncancerous stem cells (72–74), it is interesting to investigate for the Cav-1:Oct4 suppressor complex in normal stem cells. Also, the abundance of Oct4 in cancer cells may be enhanced by the fact that the cancer cells frequently reside in a high NO concentration environment (19) or gain of Akt function (79).
In summary, our data demonstrated that NO positively regulates CSC markers, including CD133, ALDH1A1, and ABCG2, and promotes tumor spheroid formation, a key feature of CSCs, through its stabilizing effect on Oct4. Such up-regulation depends on Akt activation and complex formation between Oct4 and Cav-1. By forming a molecular complex with Oct4, Cav-1 facilitates ubiquitin–proteasome-mediated degradation of Oct4, which lowers its cellular level and thus bioactivity. NO promotes the release of Oct4 from the complex, decreasing its degradation and enhancing its activity on CSCs. In forming the complex, the tyrosine 14 residue of Cav-1 is crucial for its interaction with Oct4. Mutation or phosphorylation of this residue, i.e. by tyrosine to phenylalanine substitution or by NO-mediated Akt activation, diminishes the interaction. Together, our findings unveil a novel mechanism of CSC regulation, which could be important in understanding the aggressive behaviors of cancer cells and in identifying potential drug targets for CSC therapy.
Experimental procedures
Cells and reagents
Human nonsmall cell lung cancer (NSCLC)-derived cell lines H460, H23, and H292 were obtained from the American Type Culture Collection ATCC® (Manassas, VA). The cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mm l-glutamine, and 100 units/ml each of penicillin and streptomycin (Gibco) at 37 °C in a 5% CO2-humidified incubator. Cells were routinely passaged at pre-confluent density using a 0.25% trypsin solution with 0.53 mm EDTA.
RPMI 1640 medium, FBS, l-glutamine, penicillin/streptomycin, phosphate-buffered saline (PBS), trypsin, and EDTA were purchased from Gibco. NO donor dipropylenetriamine (DPTA) NONOate was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). MTT, dimethyl sulfoxide (DMSO), Hoechst 33342, PI, and bovine serum albumin (BSA) were purchased from Sigma. Perifosine (Akt inhibitor), antibodies for Akt (catalog no. 9272), phosphorylated Akt (Ser-473) (catalog no. 4060), ERK (catalog no. 4695), phosphorylated ERK (Thr-202/Tyr-204) (catalog no. 4370), Src (catalog no. 2109), phosphorylated Src (Tyr-416) (catalog no. 6943), ABCG2 (catalog no. 42078), ALDH1A1 (catalog no. 54135), Oct4 (catalog no. 4286), Sox2 (catalog no. 3579), Nanog (catalog no. 8822), Caveolin-1 (Cav-1) (catalog no. 3267), phosphorylated Cav-1 (Tyr-14) (catalog no. 3251), β-actin (catalog no. 3700), α-tubulin (catalog no. 2125), GAPDH (catalog no. 2118), and secondary antibodies conjugated with horseradish peroxidase (HRP) were purchased from Cell Signaling (Danvers, MA). Antibody for CD133 (catalog no. 130-090-422) was purchased from NewEast Bioscience (Miltenyi Biotec GmbH, Germany). Antibody for ubiquitin (catalog no. u0508) was purchased from Sigma. Protein G-agarose bead was purchased from GE Healthcare (Uppsala, Sweden).
Cytotoxicity assay
For cytotoxicity assay, cells were seeded onto 96-well plates at a density of 1 × 104 cells/well and were allowed to incubate overnight. Cells were then treated with various concentrations of NO donor (DPTA NONOate 0–200 μm) and analyzed for cell viability using MTT assay according to the manufacturer's protocol (Sigma). In brief, the medium was replaced, and the treated cells were treated with MTT (500 μg/ml) for 4 h at 37 °C. After that, the supernatant was removed, and 100 μl of DMSO was added to solubilized crystal formazan product. The intensity of the formazan product was determined by spectrophotometry at 570 nm using a microplate reader (Anthros, Durham, NC). The cytotoxicity index was calculated by dividing the absorbance of the treated cells by that of the control cells.
Cell death assay
H460 cells was plated on 96-well plates at a density 1 × 104 cells/well and allowed to attach overnight, after which they were subjected to treatment with DPTA NONOate (0–200 μm) for 24 h. After treatment, the cells were washed with PBS and incubated with 10 μg/ml Hoechst 33342 and 5 μg/ml PI for 30 min at 37 °C. Nuclear condensation or DNA fragmentation of apoptotic cells and PI-positive necrotic cells was visualized and scored under a fluorescence microscope (Olympus IX51 with DP70 digital camera system, Tokyo, Japan).
Spheroid formation assay
After treatment, the cells were trypsinized with 2.5% trypsin/EDTA and seeded into a 24-well ultralow attachment plate at the density of 2.5 × 103 cells/well in RPMI 1640 serum-free medium. Phase-contrast images of formed spheroids were taken at days 10, 20 and 40 post-seeding using an inverted phase-contrast microscope (Olympus 1X51 with DP70).
Microarray analysis
After specific treatment, the cells were harvested; mRNA was collected, and cRNA was prepared as per Affymetrix's protocol. The gene probes list is shown in Table S1. Gene expression profiles of two independent experiments were analyzed by the CU-DREAM program (37). The gene probes with significant differences (p < 0.05) between control and treatment groups were chosen to evaluate and generate a heat map by using the Bioconductor R statistic programs. The gene probes with a strongly significant difference (p < 0.01) (Table S2) between control and treatment groups were Enrichr (http://amp.pharm.mssm.edu/Enrichr/)3 (38, 39) for analyzing gene set enrichment.
RNA isolation and RT-PCR
Total RNA was prepared using TRIzol reagent (Invitrogen), and cDNA was prepared using SuperScript III first-strand synthesis system and oligo(dT) primer (First BASE Laboratories Sdn Bhd) (primer sequences shown in Fig. 3). Quantitative PCR analysis was performed on a 7500 Fast real-time PCR using a Power SYBR Green PCR master mix (Applied Biosystems). 95 °C for 10 min was used for initial enzyme activation and was followed by 40 cycles of denaturation at 95 °C for 15 s and then at 60 °C for 1 min for annealing and extension processes. Relative expression of each gene was normalized with GAPDH gene product.
Plasmid construction and transfection
Plasmid construction
Cav-1 shRNA (sc-29241-SH) was purchased from Santa Cruz Biotechnology, Inc. A full-length WT Cav-1–expression vector was generated by subcloning a pGex-Cav1 (a gift from Ambra Pozzi (Addgene plasmid catalog no. 33364) into pCANw-FLAG to generate the pCANw-FLAG–Cav-1. The plasmid containing site-directed mutation of Cav-1 (Y14F) was purchased as pcDNA3.1(−)myc–His–Cav-1(Y14F) and verified by GeneArt® gene synthesis (quality assurance documentation 16AAX3YC).
Plasmid transfection
Subconfluent (70%) monolayers of cells were transfected with pCANw–FLAG–cav-1 or Cav-1 shRNA or pcDNA3.1(−)myc–His–cav(Y14F) plasmid in Opti-MEM reduced serum media (Gibco®, Life Technology, Inc.) using Lipofectamine 3000 reagent, according to the manufacture's protocol (Invitrogen). After 48 h, the medium was replaced with RPMI 1460 medium containing 10% FBS, and the transfected cells were used as transient transfection. For stable transfection, the transfected cells were further cultured and selected for antibiotic resistance for 30 days. The expression of the targeted protein or tagged protein were verified by Western blotting. The cells were cultured in antibiotic-free RPMI 1640 medium for at least two passages before further study.
Co-immunoprecipitation
After specific treatments, cells were washed with PBS and lysed with lysis buffer (20 mm Tris-HCl, 1 mm MgCl2, 150 mm NaCl, 1% Nonidet P-40, 10% glycerol, supplemented with protease inhibitor mixture (Merck, Calbiochem)) and phosphatase inhibitor mixture (Sigma) at 4 °C for 20 min. The lysate was pre-cleared by incubating with beads at 4 °C for 1 h and then centrifuged at 12,000 × g for 20 min at 4 °C. Cell supernatant was determined for protein content using the Bradford method (PierceTM, BCA protein assay kit, ThermoFisher Scientific). Cell lysates (500 μg of protein) were incubated with specific antibody at 4 °C overnight. After that, the lysate:immune complexes were incubated with protein G-conjugated agarose beads for 4 h at 4 °C. The immune:protein:bead complexes were washed five times with cold lysis buffer and suspended in 6× Laemmli sample buffer. The protein complexes were separated by SDS-PAGE and analyzed for protein expression by immunoblotting assay.
Western blot analysis
After specific treatments, cells were incubated with lysis buffer containing 20 mm Tris-HCl (pH 7.5), 1% Triton X-100, 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm sodium orthovanadate, 50 mm sodium fluoride, 1 μg/ml leupeptin, 100 mm phenylmethylsulfonyl fluoride, and protease inhibitor mixture (Roche Applied Science) for 30 min on ice. Cellular lysates were collected and determined for protein content using BCA protein assay kit (Pierce). Equal amounts of protein from each sample were resolved by SDS-PAGE. After separation, proteins were transferred onto 0.45-μm nitrocellulose membranes (Bio-Rad). The blots were blocked for 1 h in 5% nonfat dry milk in TBST (Tris-buffered saline with 0.1% Tween containing 25 mm Tris-HCl (pH 7.5), 125 mm NaCl, and 0.1% Tween 20) and incubated with appropriate primary antibodies at 4 °C overnight. After three washes with TBST, the membrane blots were incubated with HRP-conjugated secondary antibodies for 2 h at room temperature. The immune blots were detected by enhanced chemiluminescence (Supersignal West Pico; Pierce) and quantified using ImageJ software.
Immunofluorescence
Cells were washed with PBS, fixed with 4% paraformaldehyde in PBS at room temperature for 10 min, and permeabilized with 0.1% Triton X-100 in PBS for 5 min. They were then washed with PBS and blocked for nonspecific binding by incubation with 3% BSA in PBS for 30 min. The blocked cells were incubated with specific antibody overnight, and after washing with PBS, they were incubated with species-specific Alexa-Fluor488–conjugated antibody or Alexa-Fluor569–conjugated antibody for 2 h. The stained cells were washed and mounted with an antifade mounting medium. Hoechst 33342 was used to stain the cell nucleus. The cells were visualized by fluorescence imaging (Olympus IX5).
Proximity ligation assay
Cells were fixed with 4% paraformaldehyde in PBS at room temperature for 10 min and permeabilized with 0.1% Triton X-100 in PBS for 5 min. Then the fixed cells were incubated with primary antibodies (against Cav-1 and Oct4) for 24 h at 4 °C. Excess primary antibodies were removed by washing the cells with cold PBS, and oligonucleotide-conjugated secondary antibodies (PLA probe MINUS and PLA probe PLUS) were added to the cells and allowed to be incubated for 1 h at 37 °C. The samples were washed with a buffer containing 0.01 m Tris (pH 7.4), 0.15 m NaCl, and 0.05% Tween 20 and treated with a ligation solution for 30 min at 37 °C. After that, the ligate nucleotides were amplified and labeled with a fluorescence probe by added amplification solution (fluorescently labeled oligonucleotides) and polymerase solution in pre-heated humidity 37 °C incubator for 100 min. After washing and drying at room temperature in the dark, the samples were mounted using an anti-fade mounting medium containing 4,6-diamidino-2-phenylindole. Specific protein–protein interactions were analyzed by fluorescence microscopy (Olympus IX5).
Computational method
To date, the three-dimensional structures of both CAV1 and OCT4 have yet to be resolved; therefore, in this study, homology modeling, a promising tool to predict protein structure based on a template containing a similar sequence, was applied using Phrye2 (80). The sequences of CAV1 and OCT4 were taken from Uniprot entry codes Q03135 (81) and Q01860 (82), respectively. According to the sequence alignment by BLAST (83, 84), the crystal structure of lipoprotein MtsA (PDB code 3HH8 (52) with 27% similarity) was used as a template for CAV1 WT, whereas OCT4 was built from POU protein (PDB code 3L1P (85) with 88% similarity) and OTX2 homeobox protein (PDB code 2DMS with 23% similarity).
Each modeled protein was performed in an aqueous solution by all-atom molecular dynamics (MD) simulations using AMBER14 package with standard procedures (86–88) as summarized below. The two predicted structures were individually minimized using steepest descent and conjugated gradient based on the AMBER ff12SB force fields (89) to reduce the steric hindrance. Next, the 10-ns MD simulation with the NPT ensemble at 300 K and 1 atm was carried out by the PMEMD module. To restrain the covalent bond involved with hydrogen atoms, the SHAKE algorithm was applied (90). The particle mesh Ewald summation was used to treat the long-range electrostatic interactions, whereas 10 Å of a cutoff value was defined for nonbonded interactions (91). The last snapshot of the CAV1- and OCT4-simulated systems was further used for structure prediction of the WT CAV1:OCT4 complex.
Molecular protein–protein docking was performed using ZDOCK 3.0.2 (92) to predict the complex structure of OCT4 binding to WT CAV1 based on the parameters used in previous studies (55, 93). From the 2000 docked structures, the WT CAV1:OCT4 complex with the highest ZDOCK score was retrieved and subsequently used to construct the phosphorylated Tyr-14 CAV1 (pTyr-14 CAV1) and Y14F CAV1 mutant in complex with OCT4. To reduce the bad contact, all complexes were then minimized by steepest descent and conjugated gradient approaches using AMBER14 with the AMBER ff12SB force fields (91). Furthermore, the intermolecular hydrogen bonding interaction and MM/PBSA binding free energy of CAV1:OCT4 complexes were analyzed to investigate the effect of the phosphorylation and the single mutation at Tyr-14 position using MMPBSA.py module.
Statistical analysis
The data from three or more independent experiments are presented as the mean ± S.D. Multiple comparisons were examined for significant difference of multiple groups, using analysis of variance (ANOVA), followed by individual comparisons with Scheffe's post hoc test. Statistical significance was considered at p < 0.05.
Author contributions
A. Maiuthed and P. C. data curation; A. Maiuthed, A. Meeprasert, and P. C. formal analysis; A. Maiuthed, N. B., S. L., A. Mutirangura, C. A., A. Meeprasert, T. R., and P. C. investigation; A. Maiuthed, T. R., and P. C. methodology; A. Maiuthed and P. C. writing-original draft; Y. R. and P. C. conceptualization; Y. R. and P. C. supervision; Y. R. and P. C. writing-review and editing; P. C. resources; P. C. funding acquisition; P. C. validation; P. C. project administration.
Supplementary Material
This work was supported by Grant PHD/0063/2557 from Thailand Research Fund through The Royal Golden Jubilee Ph.D. program and Grant RSA6180036 from the Thailand Research Fund. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S2 and Tables S1–S2.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- CSC
- cancer stem cell
- DPTA
- dipropylenetriamine
- NOS
- nitric-oxide synthase
- PLA
- proximity ligation assay
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PI
- propidium iodide
- NSCLC
- non-small cell lung cancer
- FBS
- fetal bovine serum
- HRP
- horseradish peroxidase
- CBM
- caveolin-binding motif
- NOS
- NO synthase
- TF
- transcription factor
- -seq
- -sequencing
- CHX
- cycloheximide
- MM/PBSA
- molecular modeling/Poisson-Boltzmann surface area
- PDB
- Protein Data Bank
- MD
- molecular dynamics.
References
- 1. Bonnet D., and Dick J. E. (1997) Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, 730–737 10.1038/nm0797-730 [DOI] [PubMed] [Google Scholar]
- 2. Salcido C. D., Larochelle A., Taylor B. J., Dunbar C. E., and Varticovski L. (2010) Molecular characterisation of side population cells with cancer stem cell-like characteristics in small-cell lung cancer. Br. J. Cancer 102, 1636–1644 10.1038/sj.bjc.6605668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shackleton M. (2010) Normal stem cells and cancer stem cells: similar and different. Semin. Cancer Biol. 20, 85–92 10.1016/j.semcancer.2010.04.002 [DOI] [PubMed] [Google Scholar]
- 4. Lobo N. A., Shimono Y., Qian D., and Clarke M. F. (2007) The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 23, 675–699 10.1146/annurev.cellbio.22.010305.104154 [DOI] [PubMed] [Google Scholar]
- 5. Chang J. C. (2016) Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine 95, S20–25 10.1097/MD.0000000000004766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., and Yamanaka S. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 10.1016/j.cell.2007.11.019 [DOI] [PubMed] [Google Scholar]
- 7. Yu J., Hu K., Smuga-Otto K., Tian S., Stewart R., Slukvin I. I., and Thomson J. A. (2009) Human induced pluripotent stem cells free of vector and transgene sequences. Science 324, 797–801 10.1126/science.1172482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gabbert H., Wagner R., Moll R., and Gerharz C.-D. (1985) Tumor dedifferentiation: An important step in tumor invasion. Clin. Exp. Metastasis 3, 257–279 10.1007/BF01585081 [DOI] [PubMed] [Google Scholar]
- 9. Friedmann-Morvinski D., and Verma I. M. (2014) Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO Rep. 15, 244–253 10.1002/embr.201338254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu A., Yu X., and Liu S. (2013) Pluripotency transcription factors and cancer stem cells: small genes make a big difference. Chin. J. Cancer 32, 483–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kumar S. M., Liu S., Lu H., Zhang H., Zhang P. J., Gimotty P. A., Guerra M., Guo W., and Xu X. (2012) Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene 31, 4898–4911 10.1038/onc.2011.656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karoubi G., Gugger M., Schmid R., and Dutly A. (2009) OCT4 expression in human non-small cell lung cancer: implications for therapeutic intervention. Interact. Cardiovasc. Thorac. Surg. 8, 393–397 10.1510/icvts.2008.193995 [DOI] [PubMed] [Google Scholar]
- 13. Kobayashi I., Takahashi F., Nurwidya F., Nara T., Hashimoto M., Murakami A., Yagishita S., Tajima K., Hidayat M., Shimada N., Suina K., Yoshioka Y., Sasaki S., Moriyama M., Moriyama H., and Takahashi K. (2016) Oct4 plays a crucial role in the maintenance of gefitinib-resistant lung cancer stem cells. Biochem. Biophys. Res. Commun. 473, 125–132 10.1016/j.bbrc.2016.03.064 [DOI] [PubMed] [Google Scholar]
- 14. Chen Y.-C., Hsu H.-S., Chen Y.-W., Tsai T.-H., How C.-K., Wang C.-Y., Hung S.-C., Chang Y.-L., Tsai M.-L., Lee Y.-Y., Ku H.-H., and Chiou S.-H. (2008) Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS ONE 3, e2637 10.1371/journal.pone.0002637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu C. G., Lu Y., Wang B. B., Zhang Y. J., Zhang R. S., Lu Y., Chen B., Xu H., Jin F., and Lu P. (2011) Clinical implications of stem cell gene Oct-4 expression in breast cancer. Ann. Surg. 253, 1165–1171 10.1097/SLA.0b013e318214c54e [DOI] [PubMed] [Google Scholar]
- 16. Hatefi N., Nouraee N., Parvin M., Ziaee S.-A., and Mowla S. J. (2012) Evaluating the expression of Oct4 as a prognostic tumor marker in bladder cancer. Iran. J. Basic Med. Sci. 15, 1154–1161 [PMC free article] [PubMed] [Google Scholar]
- 17. Kosaka T., Mikami S., Yoshimine S., Miyazaki Y., Daimon T., Kikuchi E., Miyajima A., and Oya M. (2016) The prognostic significance of OCT4 expression in patients with prostate cancer. Hum. Pathol. 51, 1–8 10.1016/j.humpath.2015.12.008 [DOI] [PubMed] [Google Scholar]
- 18. Quail D. F., and Joyce J. A. (2013) Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437 10.1038/nm.3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu P. F., Zhao D. H., Qi Y., Wang J. G., Zhao M., Xiao K., and Xie L. X. (2018) The clinical value of exhaled nitric oxide in patients with lung cancer. Clin. Respir. J. 12, 23–30 DOI not found. [DOI] [PubMed] [Google Scholar]
- 20. Hirst D. G., and Robson T. (2011) Nitric oxide physiology and pathology. Methods Mol. Biol. 704, 1–13 10.1007/978-1-61737-964-2_1 [DOI] [PubMed] [Google Scholar]
- 21. Masri F. (2010) Role of nitric oxide and its metabolites as potential markers in lung cancer. Ann. Thorac. Med. 5, 123–127 10.4103/1817-1737.65036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Powan P., and Chanvorachote P. (2014) Nitric oxide mediates cell aggregation and mesenchymal to epithelial transition in anoikis-resistant lung cancer cells. Mol. Cell. Biochem. 393, 237–245 10.1007/s11010-014-2066-7 [DOI] [PubMed] [Google Scholar]
- 23. Saisongkorh V., Maiuthed A., and Chanvorachote P. (2016) Nitric oxide increases the migratory activity of non-small cell lung cancer cells via AKT-mediated integrin αv and β1 upregulation. Cell Oncol. 39, 449–462 10.1007/s13402-016-0287-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yongsanguanchai N., Pongrakhananon V., Mutirangura A., Rojanasakul Y., and Chanvorachote P. (2015) Nitric oxide induces cancer stem cell-like phenotypes in human lung cancer cells. Am. J. Physiol. Cell Physiol 308, C89–C100 10.1152/ajpcell.00187.2014 [DOI] [PubMed] [Google Scholar]
- 25. Chanvorachote P., Nimmannit U., Stehlik C., Wang L., Jiang B. H., Ongpipatanakul B., and Rojanasakul Y. (2006) Nitric oxide regulates cell sensitivity to cisplatin-induced apoptosis through S-nitrosylation and inhibition of Bcl-2 ubiquitination. Cancer Res. 66, 6353–6360 10.1158/0008-5472.CAN-05-4533 [DOI] [PubMed] [Google Scholar]
- 26. Matsunaga T., Yamaji Y., Tomokuni T., Morita H., Morikawa Y., Suzuki A., Yonezawa A., Endo S., Ikari A., Iguchi K., El-Kabbani O., Tajima K., and Hara A. (2014) Nitric oxide confers cisplatin resistance in human lung cancer cells through upregulation of aldo-keto reductase 1B10 and proteasome. Free Radic. Res. 48, 1371–1385 10.3109/10715762.2014.957694 [DOI] [PubMed] [Google Scholar]
- 27. Williams T. M., and Lisanti M. P. (2004) The caveolin proteins. Genome Biol. 5, 214–214 10.1186/gb-2004-5-3-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu P., Rudick M., and Anderson R. G. (2002) Multiple functions of caveolin-1. J. Biol. Chem. 277, 41295–41298 10.1074/jbc.R200020200 [DOI] [PubMed] [Google Scholar]
- 29. Bélanger M. M., Roussel É., Couet J. (2004) Caveolin-1 is down-regulated in human lung carcinoma and acts as a candidate tumor suppressor gene. Chest 125, 106S–107S 10.1378/chest.125.5_suppl.106S [DOI] [PubMed] [Google Scholar]
- 30. Wiechen K., Diatchenko L., Agoulnik A., Scharff K. M., Schober H., Arlt K., Zhumabayeva B., Siebert P. D., Dietel M., Schäfer R., and Sers C. (2001) Caveolin-1 is down-regulated in human ovarian carcinoma and acts as a candidate tumor suppressor gene. Am. J. Pathol. 159, 1635–1643 10.1016/S0002-9440(10)63010-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shyamsunder P., Vidyasekar P., Shukla A. R., Mohan S., and Verma R. S. (2013) Lowered expression levels of a tumor suppressor gene–caveolin-1 within dysregulated gene networks of Fanconi anemia. Gene 527, 521–528 10.1016/j.gene.2013.06.051 [DOI] [PubMed] [Google Scholar]
- 32. Sun M.-Z., Guan Z., Liu S., Zhou X., Wang N., Shao S., and Lin D. (2012) Caveolin-1 interferes cell growth of lung cancer NCI-H446 cell through the interactions with phospho-ERK1/2, estrogen receptor and progestin receptor. Biomed. Pharmacother. 66, 242–248 10.1016/j.biopha.2011.11.003 [DOI] [PubMed] [Google Scholar]
- 33. Yokomori H., Oda M., Yoshimura K., Nomura M., Wakabayashi G., Kitajima M., and Ishii H. (2003) Elevated expression of caveolin-1 at protein and mRNA level in human cirrhotic liver: relation with nitric oxide. J. Gastroenterol. 38, 854–860 10.1007/s00535-003-1161-4 [DOI] [PubMed] [Google Scholar]
- 34. Ju H., Zou R., Venema V. J., and Venema R. C. (1997) Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J. Biol. Chem. 272, 18522–18525 10.1074/jbc.272.30.18522 [DOI] [PubMed] [Google Scholar]
- 35. Felley-Bosco E., Bender F. C., Courjault-Gautier F., Bron C., and Quest A. F. (2000) Caveolin-1 down-regulates inducible nitric oxide synthase via the proteasome pathway in human colon carcinoma cells. Proc. Natl. Acad. Sci. U.S.A. 97, 14334–14339 10.1073/pnas.250406797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen Y. C., Hsu H. S., Chen Y. W., Tsai T. H., How C. K., Wang C. Y., Hung S. C., Chang Y. L., Tsai M. L., Lee Y. Y., Ku H. H., and Chiou S. H. (2008) Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS ONE 3, e2637 10.1371/journal.pone.0002637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aporntewan C., and Mutirangura A. (2011) Connection up- and down-regulation expression analysis of microarrays (CU-DREAM): a physiogenomic discovery tool. Asian Biomed. 5, 257–262 10.5372/1905-7415.0502.034 [DOI] [Google Scholar]
- 38. Chen E. Y., Tan C. M., Kou Y., Duan Q., Wang Z., Meirelles G. V., Clark N. R., and Ma'ayan A. (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kuleshov M. V., Jones M. R., Rouillard A. D., Fernandez N. F., Duan Q., Wang Z., Koplev S., Jenkins S. L., Jagodnik K. M., Lachmann A., McDermott M. G., Monteiro C. D., Gundersen G. W., and Ma'ayan A. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lachmann A., Xu H., Krishnan J., Berger S. I., Mazloom A. R., and Ma'ayan A. (2010) ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26, 2438–2444 10.1093/bioinformatics/btq466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jin G. P., Chang Z. Y., Schöler H. R., and Pei D. (2002) Stem cell pluripotency and transcription factor Oct4. Cell Res. 12, 321–329 10.1038/sj.cr.7290134 [DOI] [PubMed] [Google Scholar]
- 42. Zeineddine D., Hammoud A. A., Mortada M., and Boeuf H. (2014) The Oct4 protein: more than a magic stemness marker. Am. J. Stem Cells 3, 74–82 [PMC free article] [PubMed] [Google Scholar]
- 43. Li F., Sonveaux P., Rabbani Z. N., Liu S., Yan B., Huang Q., Vujaskovic Z., Dewhirst M. W., and Li C.-Y. (2007) Regulation of HIF-1α stability through S-nitrosylation. Mol. Cell 26, 63–74 10.1016/j.molcel.2007.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Matsumoto A., Comatas K. E., Liu L., and Stamler J. S. (2003) Screening for nitric oxide-dependent protein–protein interactions. Science 301, 657–661 10.1126/science.1079319 [DOI] [PubMed] [Google Scholar]
- 45. Xu H., Wang W., Li C., Yu H., Yang A., Wang B., and Jin Y. (2009) WWP2 promotes degradation of transcription factor OCT4 in human embryonic stem cells. Cell Res. 19, 561–573 10.1038/cr.2009.31 [DOI] [PubMed] [Google Scholar]
- 46. Zhao Q.-W., Zhou Y.-W., Li W.-X., Kang B., Zhang X.-Q., Yang Y., Cheng J., Yin S.-Y., Tong Y., He J.-Q., Yao H.-P., Zheng M., and Wang Y.-J. (2015) Akt-mediated phosphorylation of Oct4 is associated with the proliferation of stem-like cancer cells. Oncol. Rep. 33, 1621–1629 10.3892/or.2015.3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chanvorachote P., Chunhacha P., and Pongrakhananon V. (2014) Caveolin-1 induces lamellipodia formation via an Akt-dependent pathway. Cancer Cell Int. 14, 52–52 10.1186/1475-2867-14-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Diaz-Valdivia N., Bravo D., Huerta H., Henriquez S., Gabler F., Vega M., Romero C., Calderon C., Owen G. I., Leyton L., and Quest A. F. (2015) Enhanced caveolin-1 expression increases migration, anchorage-independent growth and invasion of endometrial adenocarcinoma cells. BMC Cancer 15, 463 10.1186/s12885-015-1477-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chanvorachote P., Pongrakhananon V., and Chunhacha P. (2014) Prolonged nitric oxide exposure enhances anoikis resistance and migration through epithelial-mesenchymal transition and caveolin-1 upregulation. BioMed Res. Int. 2014, 941359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Byrne D. P., Dart C., and Rigden D. J. (2012) Evaluating caveolin interactions: do proteins interact with the caveolin scaffolding domain through a widespread aromatic residue-rich motif? PLoS ONE 7, e44879 10.1371/journal.pone.0044879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Galbiati F., Volonte D., Brown A. M., Weinstein D. E., Ben-Ze'ev A., Pestell R. G., and Lisanti M. P. (2000) Caveolin-1 expression inhibits Wnt/β-catenin/Lef-1 signaling by recruiting β-catenin to caveolae membrane domains. J. Biol. Chem. 275, 23368–23377 10.1074/jbc.M002020200 [DOI] [PubMed] [Google Scholar]
- 52. Xu H. M., Liao B., Zhang Q. J., Wang B. B., Li H., Zhong X. M., Sheng H. Z., Zhao Y. X., Zhao Y. M., and Jin Y. (2004) Wwp2, an E3 ubiquitin ligase that targets transcription factor Oct-4 for ubiquitination. J. Biol. Chem. 279, 23495–23503 10.1074/jbc.M400516200 [DOI] [PubMed] [Google Scholar]
- 53. Liao B., and Jin Y. (2010) Wwp2 mediates Oct4 ubiquitination and its own auto-ubiquitination in a dosage-dependent manner. Cell Res. 20, 332–344 10.1038/cr.2009.136 [DOI] [PubMed] [Google Scholar]
- 54. Chen S. F., Liou J. Y., Huang T. Y., Lin Y. S., Yeh A. L., Tam K., Tsai T. H., Wu K. K., and Shyue S. K. (2010) Caveolin-1 facilitates cyclooxygenase-2 protein degradation. J. Cell. Biochem. 109, 356–362 [DOI] [PubMed] [Google Scholar]
- 55. Tantong S., Pringsulaka O., Weerawanich K., Meeprasert A., Rungrotmongkol T., Sarnthima R., Roytrakul S., and Sirikantaramas S. (2016) Two novel antimicrobial defensins from rice identified by gene coexpression network analyses. Peptides 84, 7–16 [DOI] [PubMed] [Google Scholar]
- 56. Altomare D. A., and Testa J. R. (2005) Perturbations of the AKT signaling pathway in human cancer. Oncogene 24, 7455–7464 10.1038/sj.onc.1209085 [DOI] [PubMed] [Google Scholar]
- 57. Zon L. I. (2008) Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature 453, 306–313 10.1038/nature07038 [DOI] [PubMed] [Google Scholar]
- 58. de Haan G., and Van Zant G. (1997) Intrinsic and extrinsic control of hemopoietic stem cell numbers: mapping of a stem cell gene. J. Exp. Med. 186, 529–536 10.1084/jem.186.4.529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rao C. V. (2004) Nitric oxide signaling in colon cancer chemoprevention. Mutat. Res. 555, 107–119 10.1016/j.mrfmmm.2004.05.022 [DOI] [PubMed] [Google Scholar]
- 60. Ridnour L. A., Barasch K. M., Windhausen A. N., Dorsey T. H., Lizardo M. M., Yfantis H. G., Lee D. H., Switzer C. H., Cheng R. Y., Heinecke J. L., Brueggemann E., Hines H. B., Khanna C., Glynn S. A., Ambs S., and Wink D. A. (2012) Nitric oxide synthase and breast cancer: role of TIMP-1 in NO-mediated Akt activation. PLoS ONE 7, e44081 10.1371/journal.pone.0044081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hayashi H., Arao T., Togashi Y., Kato H., Fujita Y., De Velasco M. A., Kimura H., Matsumoto K., Tanaka K., Okamoto I., Ito A., Yamada Y., Nakagawa K., and Nishio K. (2015) The OCT4 pseudogene POU5F1B is amplified and promotes an aggressive phenotype in gastric cancer. Oncogene 34, 199–208 10.1038/onc.2013.547 [DOI] [PubMed] [Google Scholar]
- 62. Kim B. W., Cho H., Choi C. H., Ylaya K., Chung J.-Y., Kim J.-H., and Hewitt S. M. (2015) Clinical significance of OCT4 and SOX2 protein expression in cervical cancer. BMC Cancer 15, 1015 10.1186/s12885-015-2015-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ormsbee Golden B. D., Wuebben E. L., and Rizzino A. (2013) Sox2 expression is regulated by a negative feedback loop in embryonic stem cells that involves AKT signaling and FoxO1. PLoS ONE 8, e76345 10.1371/journal.pone.0076345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kempfle J. S., Turban J. L., and Edge A. S. (2016) Sox2 in the differentiation of cochlear progenitor cells. Sci. Rep. 6, 23293 10.1038/srep23293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gontan C., de Munck A., Vermeij M., Grosveld F., Tibboel D., and Rottier R. (2008) Sox2 is important for two crucial processes in lung development: branching morphogenesis and epithelial cell differentiation. Dev. Biol. 317, 296–309 10.1016/j.ydbio.2008.02.035 [DOI] [PubMed] [Google Scholar]
- 66. Tompkins D. H., Besnard V., Lange A. W., Keiser A. R., Wert S. E., Bruno M. D., and Whitsett J. A. (2011) Sox2 activates cell proliferation and differentiation in the respiratory epithelium. Am. J. Respir. Cell Mol. Biol. 45, 101–110 10.1165/rcmb.2010-0149OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tompkins D. H., Besnard V., Lange A. W., Wert S. E., Keiser A. R., Smith A. N., Lang R., and Whitsett J. A. (2009) Sox2 is required for maintenance and differentiation of bronchiolar Clara, ciliated, and goblet cells. PLoS ONE 4, e8248 10.1371/journal.pone.0008248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tang J. M., He Q. Y., Guo R. X., and Chang X. J. (2006) Phosphorylated Akt overexpression and loss of PTEN expression in non-small cell lung cancer confers poor prognosis. Lung Cancer 51, 181–191 10.1016/j.lungcan.2005.10.003 [DOI] [PubMed] [Google Scholar]
- 69. Cassinelli G., Zuco V., Gatti L., Lanzi C., Zaffaroni N., Colombo D., and Perego P. (2013) Targeting the Akt kinase to modulate survival, invasiveness and drug resistance of cancer cells. Curr. Med. Chem. 20, 1923–1945 10.2174/09298673113209990106 [DOI] [PubMed] [Google Scholar]
- 70. Qiao M., Sheng S., and Pardee A. B. (2008) Metastasis and AKT activation. Cell Cycle 7, 2991–2996 10.4161/cc.7.19.6784 [DOI] [PubMed] [Google Scholar]
- 71. Chanvorachote P., Nimmannit U., Lu Y., Talbott S., Jiang B. H., and Rojanasakul Y. (2009) Nitric oxide regulates lung carcinoma cell anoikis through inhibition of ubiquitin–proteasomal degradation of caveolin-1. J. Biol. Chem. 284, 28476–28484 10.1074/jbc.M109.050864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Villodre E. S., Kipper F. C., Pereira M. B., and Lenz G. (2016) Roles of OCT4 in tumorigenesis, cancer therapy resistance and prognosis. Cancer Treat. Rev. 51, 1–9 10.1016/j.ctrv.2016.10.003 [DOI] [PubMed] [Google Scholar]
- 73. Trosko J. E. (2006) From adult stem cells to cancer stem cells: Oct-4 Gene, cell-cell communication, and hormones during tumor promotion. Ann. N.Y. Acad. Sci. 1089, 36–58 10.1196/annals.1386.018 [DOI] [PubMed] [Google Scholar]
- 74. Shi G., and Jin Y. (2010) Role of Oct4 in maintaining and regaining stem cell pluripotency. Stem Cell Res. Ther. 1, 39 10.1186/scrt39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cohen A. W., Schubert W., Brasaemle D. L., Scherer P. E., and Lisanti M. P. (2005) Caveolin-1 expression is essential for proper nonshivering thermogenesis in brown adipose tissue. Diabetes 54, 679–686 10.2337/diabetes.54.3.679 [DOI] [PubMed] [Google Scholar]
- 76. Wang H., Wang A. X., and Barrett E. J. (2011) Caveolin-1 is required for vascular endothelial insulin uptake. Am. J. Physiol. Endocrinol. Metab. 300, E134–E144 10.1152/ajpendo.00498.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kogo H., Aiba T., and Fujimoto T. (2004) Cell type-specific occurrence of caveolin-1α and -1β in the lung caused by expression of distinct mRNAs. J. Biol. Chem. 279, 25574–25581 10.1074/jbc.M310807200 [DOI] [PubMed] [Google Scholar]
- 78. Kato T., Miyamoto M., Kato K., Cho Y., Itoh T., Morikawa T., Okushiba S., Kondo S., Ohbuchi T., and Katoh H. (2004) Difference of caveolin-1 expression pattern in human lung neoplastic tissue. Atypical adenomatous hyperplasia, adenocarcinoma and squamous cell carcinoma. Cancer Lett. 214, 121–128 10.1016/j.canlet.2004.04.017 [DOI] [PubMed] [Google Scholar]
- 79. Carnero A. (2010) The PKB/AKT pathway in cancer. Curr. Pharm. Des. 16, 34–44 10.2174/138161210789941865 [DOI] [PubMed] [Google Scholar]
- 80. Kelley L. A., Mezulis S., Yates C. M., Wass M. N., and Sternberg M. J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Glenney J. R., Jr. (1992) The sequence of human caveolin reveals identity with VIP21, a component of transport vesicles. FEBS Lett. 314, 45–48 10.1016/0014-5793(92)81458-X [DOI] [PubMed] [Google Scholar]
- 82. Takeda J., Seino S., and Bell G. I. (1992) Human Oct3 gene family: cDNA sequences, alternative splicing, gene organization, chromosomal location, and expression at low levels in adult tissues. Nucleic Acids Res. 20, 4613–4620 10.1093/nar/20.17.4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Boratyn G. M., Camacho C., Cooper P. S., Coulouris G., Fong A., Ma N., Madden T. L., Matten W. T., McGinnis S. D., Merezhuk Y., Raytselis Y., Sayers E. W., Tao T., Ye J., and Zaretskaya I. (2013) BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 41, W29–W33 10.1093/nar/gkt282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Johnson M., Zaretskaya I., Raytselis Y., Merezhuk Y., McGinnis S., and Madden T. L. (2008) NCBI BLAST: a better web interface. Nucleic Acids Res. 36, W5–W9 10.1093/nar/gkn201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Esch D., Vahokoski J., Groves M. R., Pogenberg V., Cojocaru V., Vom Bruch H., Han D., Drexler H. C., Araúzo-Bravo M. J., Ng C. K., Jauch R., Wilmanns M., and Schöler H. R. (2013) A unique Oct4 interface is crucial for reprogramming to pluripotency. Nat. Cell Biol. 15, 295–301 10.1038/ncb2680 [DOI] [PubMed] [Google Scholar]
- 86. Kaiyawet N., Rungrotmongkol T., and Hannongbua S. (2013) Effect of halogen substitutions on dUMP to stability of thymidylate synthase/dUMP/mTHF ternary complex using molecular dynamics simulation. J. Chem. Inf. Model. 53, 1315–1323 10.1021/ci400131y [DOI] [PubMed] [Google Scholar]
- 87. Meeprasert A., Khuntawee W., Kamlungsua K., Nunthaboot N., Rungrotmongkol T., and Hannongbua S. (2012) Binding pattern of the long acting neuraminidase inhibitor laninamivir towards influenza A subtypes H5N1 and pandemic H1N1. J. Mol. Graph. Model. 38, 148–154 10.1016/j.jmgm.2012.06.007 [DOI] [PubMed] [Google Scholar]
- 88. Meeprasert A., Hannongbua S., and Rungrotmongkol T. (2014) Key binding and susceptibility of NS3/4A serine protease inhibitors against hepatitis C virus. J. Chem. Inf. Model. 54, 1208–1217 10.1021/ci400605a [DOI] [PubMed] [Google Scholar]
- 89. Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., and Simmerling C. (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ryckaert J.-P., Ciccotti G., and Berendsen H. J. C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Physics 23, 327–341 10.1016/0021-9991(77)90098-5 [DOI] [Google Scholar]
- 91. York D. M., Darden T. A., and Pedersen L. G. (1993) The effect of long-range electrostatic interactions in simulations of macromolecular crystals: a comparison of the Ewald and truncated list methods. J. Chem. Physics 99, 8345–8348 10.1063/1.465608 [DOI] [Google Scholar]
- 92. Pierce B. G., Wiehe K., Hwang H., Kim B. H., Vreven T., and Weng Z. (2014) ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 30, 1771–1773 10.1093/bioinformatics/btu097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kongsune P., Rungrotmongkol T., Nunthaboot N., Yotmanee P., Sompornpisut P., Poovorawan Y., Wolschann P., and Hannongbua S. (2012) Molecular insights into the binding affinity and specificity of the hemagglutinin cleavage loop from four highly pathogenic H5N1 isolates towards the proprotein convertase furin. Monatshefte für Chemie Chemical Monthly 143, 853–860 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.