

Abstract
Listeria monocytogenes causes listeriosis, a potentially fatal food-borne disease. The condition is especially harmful to pregnant women. Listeria outbreaks can originate from diverse foods, highlighting the need for novel strategies to improve food safety. The first step in Listeria invasion is internalization of the bacteria, which is mediated by the interaction of the internalin family of virulence factors with host cell receptors. A crucial interaction for Listeria invasion of the placenta, and thus a target for therapeutic intervention, is between internalin B (InlB) and the receptor c-Met. Single-domain antibodies (VHH, also called nanobodies, or sdAbs) from camel heavy-chain antibodies are a novel solution for preventing Listeria infections. The VHH R303, R330, and R326 all bind InlB with high affinity; however, the molecular mechanism behind their mode of action was unknown. We demonstrate that despite a high degree of sequence and structural diversity, the VHH bind a single epitope on InlB. A combination of gentamicin protection assays and florescent microscopy establish that InlB-specific VHH inhibit Listeria invasion of HeLa cells. A high-resolution X-ray structure of VHH R303 in complex with InlB showed that the VHH binds at the c-Met interaction site on InlB, thereby acting as a competitive inhibitor preventing bacterial invasion. These results point to the potential of VHH as a novel class of therapeutics for the prevention of listeriosis.
Keywords: antibody; single-domain antibody (sdAb, nanobody); structural biology; protein-protein interaction; virulence factor; Listeria; receptor endocytosis; virulence factor
Introduction
Listeriosis is a potentially lethal food-borne disease caused by the Gram-positive bacteria Listeria monocytogenes. Although infections have a low rate of incidence in the general population, the disease has an unusually high mortality rate of 20–30% (1). Listeria is transmitted by consumption of contaminated foods. Soft cheeses, deli meats, and ready-to-eat foods have historically been considered at high risk of Listeria contamination. Clinical presentation of listeriosis includes severe gastroenteritis; however, invasive infections can cross the blood–brain barrier, leading to central nervous system infections and fatal meningitis (2). Pregnant women are especially susceptible to Listeria infection due to T-cell suppression (3). The danger during pregnancy is further compounded by the capacity of the bacteria to cross the placental barrier, which can result in termination of the developing fetus (4). The pathogenesis of L. monocytogenes infection and invasion is well characterized (5) and points to potential avenues for the generation of novel therapeutic interventions.
The invasion of nonphagocytic cells by L. monocytogenes occurs through the action of a complex set of virulence factors that allow the bacteria to enter host cells, escape the vacuole, and hijack the actin network to spread from cell to cell (5). Listeria host cell entry is the initial step in pathogenesis, and it is mediated by two members of the internalin family of virulence factors (InlA2 and InlB) (6, 7). Binding of InlA and InlB to host cell receptors activates signaling cascades that trigger receptor-mediated endocytosis and internalization of the bacteria. InlA and InlB have different receptors and are responsible for mediating entry into different cell types and biological barriers.
The interaction of InlA with the host receptor E-cadherin is important for Listeria penetration of the intestinal barrier and invasion of several epithelial cell types (8). On the other hand, InlB binds the receptor tyrosine kinase c-Met (9), which permits Listeria internalization into a variety of cell types, including HeLa, Vero, and hepatocyte cell lines (7, 10–13). c-Met functions as the receptor for the hepatocyte growth factor and is required for normal embryonic development, pointing to the importance of InlB in pregnancy-related listeriosis. Indeed, synergistic action of InlA and InlB is required for L. monocytogenes to cross and penetrate the placental barrier (14). Given the importance of InlB receptor interaction in fetal listeriosis, disruption of this interaction may represent a target for therapeutic intervention.
The mature, cell surface form of InlB consists of an N-terminal internalin domain (residues 31–321), a B repeat (residues 322–397), and three GW domains (residues 398–630). The N-terminal internalin domain is primarily responsible for c-Met receptor binding and activation. The domain is composed of an α-helical cap (residues 31–85), seven leucine-rich repeats (LRRs) (residues 86–239), and an interrepeat (IR) (residues 240–321) (15). A fragment consisting of the cap and the LRR (residues 31–241, InlB241) is the minimum unit that binds the c-Met receptor (9). However, c-Met activation and endocytosis also requires the entire N-terminal internalin domain (residues 31–321, InlB321), including the IR region (16).
Although interruption of the InlB–c-Met interaction is an intriguing approach for preventing Listeria cellular invasion, one potential pitfall is that the protein is buried in the peptidoglycan layer. One innovative solution is to use single-domain antibodies (VHH), derived from the antigen-binding fragment of the heavy-chain antibodies found in camelids (17). VHH are 10 times smaller (12–15 kDa) than conventional IgG antibodies (150 kDa) and may be able to penetrate the Listeria cell wall to bind InlB.
Previously, four VHH (R303, R326, R330, and R419) that bind the LRR domain of InlB with nanomolar affinity were isolated from a nonimmune phage display library (18, 19). As the InlB-LRR domain is crucial for interaction with c-Met, we hypothesized that these VHH could inhibit bacterial endocytosis and protect the cells from Listeria invasion. We demonstrate that InlB-specific VHH effectively neutralize Listeria invasion in vitro. Furthermore, high-resolution X-ray structures reveal that the mechanism behind VHH-mediated Listeria inhibition is competitive inhibition of the InlB–c-Met interaction.
Results
VHH bind overlapping epitopes on InlB
Previous work had identified several VHH (R303, R330, R419, and R326) from a preimmune (naive) phage display library that bound the LRR domain of InlB (residues 36–249; InlB249) (18). Using an indirect ELISA the relative affinity of the VHH for the LRR domain of InlB was compared (Fig. 1A). A variable concentration of immobilized InlB249 was detected using a fixed concentration of biotinylated VHH. Consistent with previously reported surface plasmon resonance results (18), VHH R303, R326, and R330 bound to immobilized InlB249 with a similar apparent affinity (Fig. 1A). However, no binding of VHH R419 to InlB249 was observed.
Figure 1.

Binding of VHH to InlB. A, indirect ELISA measuring VHH affinity for InlB249. The plate was coated with InlB, and biotinylated VHH were used as primary antibodies. Data were fit to a four-parameter logistic curve, and the EC50 value was calculated. VHH R303 (EC50 = 0.4 ng/μl), R326 (EC50 = 0.4 ng/μl), and R330 (EC50 = 0.5 ng/μl) bound to the protein with similar affinity. R419 and an irrelevant VHH control did not bind. Error bars, S.D. of three separate trials. B, indirect ELISA measuring VHH affinity for InlB321. A plate was coated with InlB, and biotinylated VHH were used as primary antibodies. Data were fit to a four-parameter logistic curve, and the EC50 value was calculated. VHH R303 (EC50 = 0.5 ng/μl) and R330 (EC50 = 0.6 ng/μl) bound to the protein with similar affinity. R326 bound with reduced affinity (EC50 = 1.5 ng/μl), and R419 and an irrelevant VHH control did not bind. Error bars, S.D. of three separate trials. C, epitope mapping by competitive ELISA. InlB249 and InlB321 were immobilized and detected with a mixture of biotinylated VHH (R303, R326, and R330) and unlabeled R303 as a competitive inhibitor. VHH R303 inhibited the binding of both R330 and R326, suggesting that the VHH all bind overlapping epitopes on InlB. Error bars, S.D. of three separate trials.
Because the VHH were originally generated by screening a phage display library against a truncated version of InlB with only the cap and the LRR domain (InlB249), we next investigated whether the VHH would also bind InlB if the IR domain was also present (InlB321) (Fig. 1B). R303 and R330 both bound to InlB321 with similar affinity; however, R326 bound with ∼2-fold lower affinity to InlB321 when compared with R303 and R330 (Fig. 1B).
The VHH R303, R330, and R326 displayed variability in their CDR sequences and canonical CDR cluster classification (Table 1). Furthermore, based on nucleotide sequence alignments with antibody germ line segments, the VHH may be derived from different species of Camelid. This was expected, as the phage display library used to isolate these VHH is originated from the immune repertoire of three species of Camelid (18, 19). R303 is unquestionably from Camelus dromedaries, whereas R330, R326, and R419 are derived from either Llama glama or Vicugna pacos. Given this diversity, we next investigated the epitope specificity of the VHH.
Table 1.
CDR amino acid sequences and canonical clusters of InlB-specific VHH
| VHH | CDR-1 |
CDR-2 |
CDR-3 |
|||
|---|---|---|---|---|---|---|
| Sequence | Clustera | Sequence | Cluster | Sequence | Cluster | |
| R303 | AASGHTYSTYCMG | 13-6 | RINVGGSSTW | 10-3 | TLHRFCNTWSLGTLNV | 16-1 |
| R326 | VTSGRIEGILLVG | 13-5 | SIDRNGNTR | 9-1 | GALSSGVNPWA | 11-1 |
| R330 | AASGSSIYTMG | NDb | DISWNGGSTY | 10-2 | NADDLMIDRDY | 11-1 |
| R419 | AASGRTYSTYAMG | 13-4 | AINWSGGNTH | 10-2 | AAPKGHTGDHY | 11-1 |
a CDR definitions and structural clusters defined in North et al. (22).
b Not determined; CDR loop could not be classified.
The possibility that the VHH bound distinct InlB epitopes was investigated using a competitive ELISA. A single fixed concentration of InlB249 or InlB321 was immobilized, and a mixture of biotinylated R303, R326, or R330 was added along with an 80-fold higher concentration of unlabeled R303 to act as an inhibitor (Fig. 1C).
Assuming the VHH bound to spatially distinct epitopes, the expectation was that R303 would not act as an inhibitor for the other VHH. On the other hand, if the VHH bound to overlapping epitopes, R303 should inhibit binding. In all cases, R303 acted as an inhibitor to binding, suggesting that all three VHH (R303, R330, and R326) bound overlapping epitopes (Fig. 1C).
InlB-specific VHH inhibit Listeria invasion of HeLa cells in vitro
Interaction of InlB with the host cell c-Met receptor is essential for Listeria invasion of epithelial cells (11, 12). Interference of this interaction may provide a site for therapeutic intervention by preventing Listeria colonization and invasion. Gentamicin protection assays were employed to determine whether InlB-specific VHH could inhibit Listeria invasion in vitro.
L. monocytogenes were treated with the four VHH (R303, R330, R326, and R419) and allowed to invade HeLa cells in vitro. InlB249 was used as a positive control, as it has been previously shown to inhibit Listeria invasion of HeLa cells (10), and an irrelevant anti-GFP VHH (20) was used as a negative control. Following protein treatment, gentamicin was added to eradicate noninternalized Listeria. HeLa cells were lysed, the internalized bacteria were counted, and the efficiency of Listeria invasion was calculated (Fig. 2).
Figure 2.

Gentamicin protection assay measuring VHH-mediated inhibition of Listeria invasion. HeLa cells were infected with L. monocytogenes in the presence of PBS buffer control, InlB (positive control to inhibit Listeria invasion), irrelevant VHH negative control (20), and anti-InlB VHH R303, R326, R330, and R419. Invasion was quantified by counting the number of intracellular L. monocytogenes released from infected HeLa cells following gentamicin treatment. The percentage inhibition was calculated relative to the PBS buffer control. VHH R303 and R330 inhibited L. monocytogenes similar to the InlB control treatment, R326 was modestly effective at inhibiting invasion, and R419 was not effective. The center horizontal bars represent the mean, and error bars represent the S.D. (n = 3).
As R303, R326, and R330 bound overlapping epitopes on InlB249 and InlB321 (Fig. 1), it was hypothesized that these three VHH would perform similarly in the invasion assay. However, there were some differences in the ability of VHH to inhibit Listeria invasion. R303 and R330 were both highly effective at inhibiting Listeria internalization of HeLa cells (94 ± 1.4 and 75 ± 1.9%, respectively; Fig. 2). However, R326 exhibited a reduced ability to inhibit Listeria invasion of HeLa cells (36 ± 5.5%). Given that R419 did not bind InlB (Fig. 1), it was not surprising that the VHH resulted in a level of invasion inhibition similar to that of the irrelevant VHH control (Fig. 2).
As a second line of evidence to evaluate VHH neutralization of Listeria, a fluorescence microscopy–based invasion assay was conducted (21). A strain of constitutively expressing GFP Listeria was constructed, followed by biotinylation and subsequent invasion of the strain into HeLa cells. Listeria cells were treated with PBS (negative control), an irrelevant GFP-specific VHH (negative control), InlB249 (positive control), and each of the InlB-specific VHH. Following invasion, the cells were treated with streptavidin conjugated to DyLight550. If the GFP-expressing Listeria invaded the HeLa cells, they would not be detected by the red-labeled streptavidin; however, if the Listeria were impeded from cell invasion, they would be available for detection by the labeled streptavidin and would thus be stained red.
The negative controls, PBS and irrelevant VHH, resulted in minimal red staining of the GFP-expressing Listeria, indicating that the strain had invaded the HeLa cells (Fig. 3). When treated with the InlB249-positive control and with R303, R330, and R326, the majority of the Listeria cells were stained red, indicating that they remained extracellular to the HeLa cells and had been inhibited from invasion (Fig. 3). R419 did not inhibit Listeria invasion, consistent with the results of the ELISA and gentamicin protection assays (Figs. 1–3).
Figure 3.
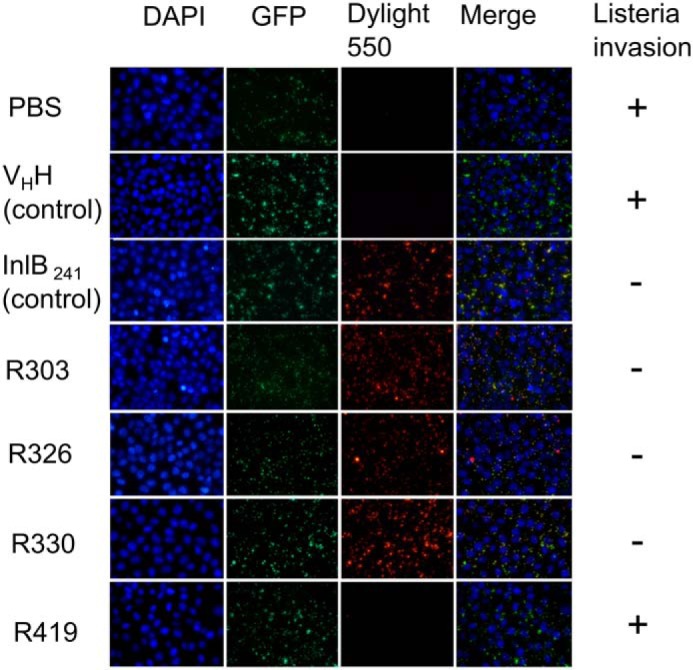
Fluorescence microscopy–based assay measuring VHH-mediated inhibition of Listeria invasion. HeLa cells were infected with biotinylated GFP-expressing L. monocytogenes in the presence of PBS buffer control and protein treatments: InlB (positive control to inhibit Listeria invasion), irrelevant VHH negative control (20), and anti-InlB VHH R303, R326, R330, and R419. Streptavidin conjugated to Dylight550 was used to detect extracellular bacteria. Three anti-InlB VHH (R303, R326, and R330) inhibited Listeria invasion of HeLa cells.
Structures of VHH R303, R326, and R330
X-ray structures of VHH R303, R326, and R330 were determined at resolutions ranging from 1.3 to 1.8 Å (Table 2). Consistent with the divergent amino acid sequences of the VHH CDRs (Table 1), the X-ray structures revealed variability in the antigen-binding sites. The CDRs were assigned using the definitions reported by North et al. (22). The CDR loop conformations were assigned from the X-ray structures using the PyIgClassify CDR loop database (23) (Table 1).
Table 2.
Data collection and refinement statistics
| R303 | R326 | R330 | R303-InlB249 | R303-InlB321 | |
|---|---|---|---|---|---|
| Beamline | 08ID CLS | 08ID CLS | 08ID CLS | 08ID CLS | 08B1-1 CLS |
| Wavelength | 0.97950 | 0.97950 | 0.97950 | 0.97950 | 0.97950 |
| Resolution range (Å) | 38.24–1.30 (1.35–1.30) | 47.9–1.76 (1.82–1.76) | 25.32–1.65 (1.71–1.65) | 41.48–1.55 (1.61–1.55) | 36.42–1.51 (1.56–1.51) |
| Space group | P21 | P6522 | P3121 | P43 | P1 |
| a, b, c (Å) | 46.44, 31.19, 74.75 | 97.70, 97.70, 243.8 | 58.62, 58.62, 100.53 | 82.96, 82.96, 64.22 | 46.89, 66.96, 73.83 |
| α, β, γ (degrees) | 90, 93.81, 90 | 90,90,120 | 90,90,120 | 90,90,90 | 116.67, 97.73, 95.09 |
| Total reflection | 157,298 (12,840) | 1,199,511 (108,483) | 109,804 (9497) | 473,463 (45,070) | 358,050 (36,591) |
| No. of unique reflections | 52,542 (5027) | 68,308 (6329) | 24,195 (2285) | 62,224 (6093) | 122,420 |
| Multiplicity | 3.0 (2.6) | 17.6 (17.1) | 4.5 (4.1) | 7.6 (7.4) | 2.9 (3.0) |
| Completeness (%) | 98.88 (95.90) | 97.89 (91.92) | 97.80 (94.12) | 98.23 (96.60) | 93.51 (93.91) |
| Mean 〈I/σI〉 | 10.36 (2.66) | 19.85 (4.50) | 8.74 (3.16) | 17.31 (4.37) | 15.91 (2.81) |
| Wilson B-factor | 14.32 | 25.12 | 21.37 | 17.74 | 16.63 |
| Rmerge | 0.048 (0.3515) | 0.076 (0.7292) | 0.096 (0.494) | 0.067 (0.4513) | 0.038 (0.3838) |
| Rpim | 0.033 (0.2644) | 0.018 (0.166) | 0.051 (0.2786) | 0.02437 (0.1642) | 0.0278 (0.2704) |
| Rwork/Rfree (%) | 14.76/17.74 | 16.70/18.57 | 18.29/22.41 | 15.32/16.84 | 16.09/18.06 |
| No. of non-hydrogen atoms | |||||
| Protein | 1945 | 3401 | 1761 | 2536 | 6257 |
| Solvent | 358 | 405 | 228 | 368 | 1085 |
| Root mean square deviations | |||||
| Bond lengths (Å) | 0.014 | 0.022 | 0.011 | 0.005 | 0.012 |
| Bond angles (degrees) | 1.58 | 1.72 | 1.10 | 1.06 | 1.10 |
| Average B factor (Å) | 22.15 | 33.55 | 30.53 | 23.48 | 24.37 |
| Protein | 19.71 | 32.52 | 29.12 | 21.59 | 22.79 |
| Solvent | 35.41 | 41.36 | 41.42 | 36.52 | 33.47 |
| Ramachandran plot (%) | |||||
| Favored region | 97 | 98.6 | 91 | 97 | 97.3 |
| Allowed | 3 | 1.4 | 0.9 | 3 | 2.7 |
| Outliers | 0 | 0 | 0 | 0 | 0 |
| Protein Data Bank code | 6DBA | 6DBD | 6DBE | 6DBF | 6DBG |
R303 was solved to a resolution of 1.3 Å, and the structure contains two molecules in the asymmetric unit arranged in a head-to-tail fashion. R303 had the longest CDR-3 of the three VHH with a length of 16 amino acids (Table 1 and Fig. 4A). A noncanonical disulfide bond was formed between CDR-1 and CDR-3 (residues 33–102) that linked the long 16-residue CDR-3 loop against the framework region of the antibody (Fig. 4A). CDR-3 formed a short helical segment (residues 102–107) in proximity to the noncanonical disulfide bond. The fixing of CDR-3 against the framework region resulted in a large solvent-accessible surface area (1970 Å2) available for antigen recognition. The CDR-1 loop bisects the antibody paratope, creating two relatively flat interaction surfaces on either side of the loop. The paratope region between CDR-1 and CDR-3 showed a positively charged electrostatic surface, with a wide pocket-like structure forming (Fig. 4A).
Figure 4.
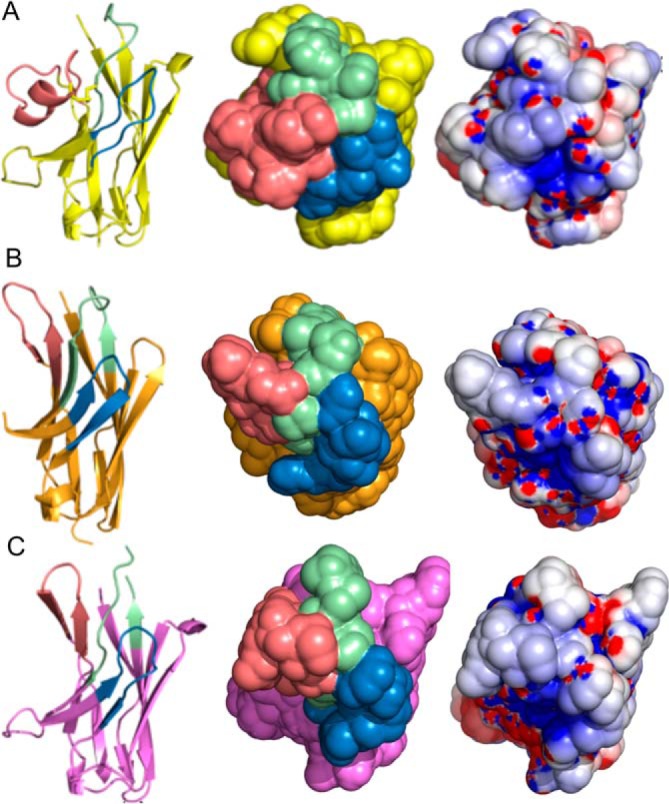
Crystal structures of anti-InlB VHH. A, R303; B, R330; C, R326. Left, ribbon diagram with different CDR structures. CDR-1 is colored green, CDR-2 is colored blue, and CDR-3 is colored salmon. Center, surface representation of CDR loops. Right, electrostatic surface (positively charged (blue) and negatively charged (red)).
The structure of VHH R326 was solved to a resolution of 1.8 Å and contained a tetramer in the asymmetric unit. Unlike R303, R326 had no disulfide bond connecting CDR-3 to CDR-1. Structurally, R326 was distinct from the other two VHH with the three CDR loops protruding from the framework region, forming a convex paratope structure (Fig. 4B). The paratope was a large solvent-accessible surface area (1650 Å2) with a positively charged electrostatic surface (Fig. 4B).
The structure of VHH R330 was solved to a resolution of 1.6 Å and contained a dimer in the asymmetric unit. Similar to R326, the paratope of R330 was a wide, roughly convex shape with a positively charged solvent-accessible surface area of 2050 Å2 (Fig. 4C). Interestingly, the structure of CDR-1 of R330 did not fall into one of the previously characterized structural clusters identified by North et al. (22) (Table 1). CDR-1 also was disordered at the apex of the loop (residue 28 in chain A; residues 29 and 30 in chain B).
Structure of R303–InlB249 and R303–InlB321
To determine the molecular mechanism behind VHH neutralization of Listeria invasion, the structures of VHH R303 in complex with the LRR domain of InlB (InlB249) and the longer InlB fragment of the LRR domain linked to the IR region (InlB321) were both determined to a resolution of ∼1.5 Å. The two complex structures crystallized in different space groups (Table 2). R303 in complex with InlB249 crystallized as a monomer, whereas R303 with InlB321 was a dimer in the asymmetric unit.
The overall binding interactions between R303 and InlB249 and InlB321 were identical (Fig. 5A), indicating that the IR domain of InlB321 played no role in binding. This finding is consistent with the observation that R303 binds to both proteins (InlB249 and InlB321) with similar affinity (Fig. 1, A and B).
Figure 5.
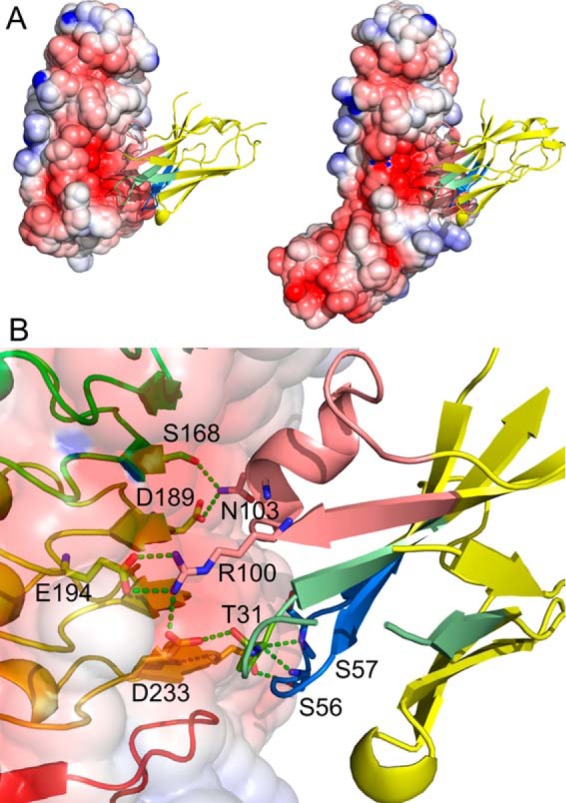
Structure of VHH R303 in complex with InlB. A, VHH R303 (ribbon diagram) binds at an electronegative cavity on both InlB249 (left) and InlB321 (right). B, binding interactions of VHH R303 (right) and InlB (left). VHH R303 is color-coded by CDR, with CDR-1 colored green, CDR-2 colored blue, and CDR-3 colored salmon.
The entire interaction between R303 and InlB occurs on an electronegative cavity on the concave face of the InlB-LRR domain, resulting an approximate buried surface area of 1400 Å2. The bulk of the binding interactions are mediated by CDR-3 and CDR-2, with CDR-1 displaying only limited contact with InlB (Fig. 5B).
Consistent with the picomolar affinity of R303 for the InlB-LRR domain (18), there were extensive polar and nonpolar contacts between the antibody and InlB. Interactions originating from CDR-3 on R303 are of central importance and form the majority of the binding interactions (Fig. 5). There were a series of salt bridges that likely contribute significantly to the high-affinity binding of the VHH. The salt bridges are formed between Arg-100vhh on CDR-3 of R303 and Glu-194inl and Glu-236inl on InlB (where the superscript “vhh” denotes residues on VHH R303 and the superscript “inl” denotes residues on InlB) (Fig. 5B). This central arginine residue on R303 also forms a hydrogen bond to Tyr-214inl. Additional polar interactions include 12 hydrogen bonds between the antibody and InlB. On CDR-3, Asn-103vhh hydrogen-bonds to Ser-168inl, Asp-189inl, and Thr-190inl. The adjacent residue on CDR-3, Thr-104vhh, hydrogen-bonds to the hydroxyl side chain of Tyr-170inl (Fig. 5B). On CDR-2, Ser-56vhh and Ser-57vhh form hydrogen bonds to Asp-233inl (Fig. 5B). In addition to the polar contacts, there are aromatic stacking interactions, with the side chain of Phe-104vhh on CDR-3 inserting between Tyr-214inl and Tyr-170inl (Fig. 5B).
Discussion
Specificity of VHH isolated from a nonimmune library
The anti-InlB VHH (R303, R330, and R326) used in this study were isolated from a preimmune phage display library from the naive immune repertoires of camels, alpacas, and llamas (18). Each of the isolated VHH was unique in terms of primary sequence diversity and CDR canonical structure (Table 1). Furthermore, based on alignment with germ line gene segments, the VHH originate from different species of Camelid (R303 (camel), R326 (llama or alpaca), and R330 (llama or alpaca)). However, despite this structural and sequence diversity, the specificity of the VHH converged onto a single epitope (Fig. 1C). This epitope was centralized to a negatively charged cavity on the concave face of the LRR domain of InlB (Fig. 5A).
The specific structural features of the InlB antigen and the particular binding properties associated with VHH in general may be responsible for the observed VHH specificity. It has been observed previously that VHH often bind concave features on protein antigens due to the convex shape of the paratope formed on the three CDR loops (24, 25). Given this preference, the VHH specificity toward the InlB-LRR electronegative cavity may be the result of the protein only having this one concave surface feature.
VHH properties facilitate neutralization of Listeria
The biophysical and binding properties of VHH are distinct compared with traditional monoclonal antibodies. VHH are small and stable, and their convex shape allows VHH to bind protein cavities, which are frequently inaccessible to traditional monoclonal antibodies (17). This combination of properties provides several advantages that may have contributed to the effectiveness of VHH R303, R330, and R326 for the in vitro neutralization of L. monocytogenes (Figs. 2 and 3). In particular, the small size and preferential binding of VHH toward protein cavities may explain the success of VHH at Listeria neutralization compared with traditional antibody formats.
Several mouse anti-InlB antibodies displayed variable effectiveness at inhibiting Listeria invasion of Vera cells, suggesting that specific epitopes must be recognized for neutralization to occur (10). In some cases, InlB epitopes may be inaccessible; an InlB-specific ScFv was only able to bind InlB following enzymatic digestion of the bacterial cell wall, suggesting that the epitopes were buried in the cell wall (26). As VHH R303, R330, and R326 are all able to neutralize Listeria invasion (Figs. 2 and 3), it can be inferred that the immunodominant epitope must be accessible to the VHH. The small size of the VHH may facilitate penetration of the bacterial peptidoglycan layer to access the protein–protein interaction surface on InlB. This further highlights the specific advantages of using VHH in targeting difficult-to-access cell surface epitopes.
InlB-specific VHH inhibit Listeria invasion through competitive inhibition
The neutralization of Listeria invasion by VHH R303, R330, and R326 could potentially be mediated by two different mechanisms. The VHH could bind InlB and inhibit its interaction with c-Met simply through steric effects, or the VHH could competitively inhibit the native interaction of InlB with c-Met. The X-ray structure of R303 in complex with InlB (Fig. 5) permits an analysis of the molecular mechanism behind the anti-bacterial activity of the VHH.
c-Met is a receptor tyrosine kinase whose ectodomain consists of six domains: Sema, Psi, and four Ig-like domains (Ig1–4) (27). The natural ligand for c-Met is the hepatocyte growth factor/scatter factor (HGF/SF). In healthy cells, the c-Met–HGF/SF interaction mediates cell signaling related to embryogenesis and tissue regeneration, and deregulation of c-Met is also important in carcinogenesis (28). Interestingly, whereas L. monocytogenes hijacks c-Met as a vehicle for bacterial entry, the interaction of InlB with c-Met does not mimic the natural HGF/SF ligand, as the two proteins bind c-Met at distinct sites (9, 27).
InlB–c-Met receptor binding and subsequent cell signaling events that ultimately result in bacterial internalization are mediated by different domain–domain interactions. A fragment comprising the cap region and LRR domain of InlB (InlB241) is the minimum unit for c-Met receptor binding (9). The binding of the InlB241 fragment to the Ig1 domain of c-Met occurs at the electronegative cavity on the concave face of the InlB-LRR domain (Fig. 6A) (27). However, c-Met receptor activation and cell invasion by L. monocytogenes require a larger fragment of InlB, consisting of the cap region, LRR domain, and interrepeat (InlB321) (16). The secondary, weaker interaction of the InlB-IR domain with the c-Met Sema domain (Fig. 6A) is required for receptor activation and not binding (27).
Figure 6.
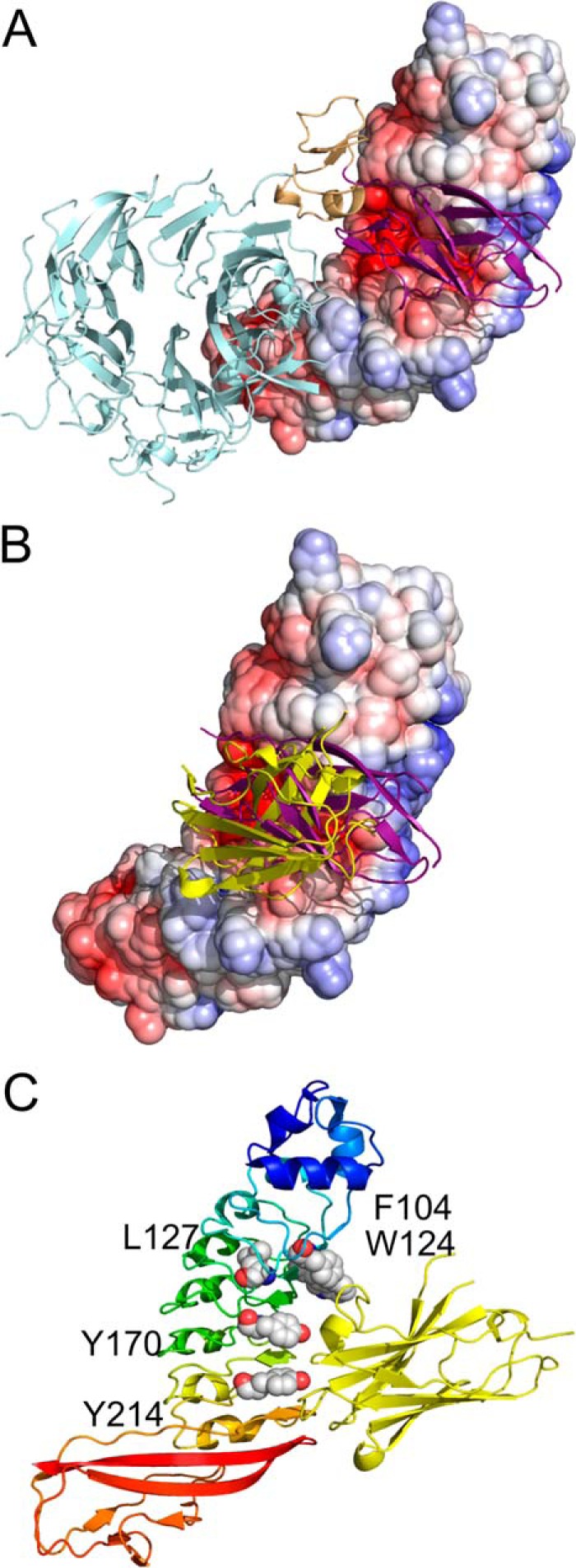
VHH R303 inhibits InlB interaction with c-Met through competitive inhibition. A, X-ray structure of InlB (electrostatic surface) and c-Met ectodomain (ribbon diagram) (Protein Data Bank code 2UZX) (27). The Ig1 domain of c-Met (purple) binds to an electronegative cavity on the surface of InlB. B, VHH R303 (yellow) bind to InlB (electrostatic surface) in a nearly identical fashion as the Ig1 domain (purple) of c-Met. C, the InlB residues involved in binding the Ig1 domain of c-Met (Phe-104, Trp-124, Leu-127, Tyr-170, and Tyr-214) are buried upon complex formation with R303 (yellow ribbon).
VHH R303 binds InlB directly at the c-Met receptor-binding site: the electronegative cavity on the concave face of InlB (Figs. 5A and 6A). Overlap of the structure of R303–InlB321 with that of the c-Met ectodomain in complex with InlB321 (27) demonstrated that R303 would directly occupy the same physical space as the c-Met Ig1 domain, mimicking the interaction with the Ig1 domain (Fig. 6B).
The binding of the c-Met Ig1 domain to InlB is mediated by many of the same residues involved in the R303–InlB interaction. There are five residues on InlB that are important for c-Met receptor binding: Asp-128inl, Glu-150inl, Tyr-170inl, Tyr-214inl, and Trp-124inl (27). Of these five InlB residues, four are either interacting with R303 directly through hydrogen bond interactions (Tyr-170inl and Tyr-214inl; Figs. 5B and 6C) or are buried upon complex formation (Glu-150inl and Trp-124inl; Fig. 6C).
The high-affinity binding of R303 to the c-Met receptor-binding site on InlB provides a clear molecular mechanism for the neutralization of L. monocytogenes by the VHH used in this study. By mimicking the interactions of c-Met, the natural ligand of InlB, the VHH are acting as high-affinity competitive inhibitors, neutralizing bacterial invasion.
Therapeutic potential of Listeria-specific VHH
Listeria infections are a particular challenge facing pregnant women. Maternal infection is frequently asymptomatic or displays nonspecific symptoms, making diagnosis a serious challenge during prenatal care (29, 30). Even in cases with diagnosis, antibiotic treatment is not always successful, presumably due to the intracellular nature of the pathogen (31).
Prevention of Listeria infection is currently the most effective strategy for safeguarding women from the disease during pregnancy. Typically, pregnant women are advised to avoid consumption of foods at high risk of Listeria contamination. However, a series of deadly Listeria outbreaks in fresh produce, fruit, and other foods traditionally at low risk of Listeria contamination, highlight the need for alternative and novel approaches to safeguarding the food supply (32–34).
A prophylactic strategy of blocking Listeria entry into nonphagocytic cells by inhibiting the interaction of InlB with the c-Met receptor is a potential venue of Listeria treatment or prophylactic. A recent report using the c-Met inhibitor tanespimycin as a Listeria antibiotic suggests that this approach may represent a viable therapeutic strategy (35).
The ability of InlB-specific VHH to neutralize Listeria invasion in vitro points to a therapeutic potential for the prevention or treatment of listeriosis. There have been several recent reports of using VHH as anti-bacterial agents against a variety of bacterial pathogens, including Clostridium difficile, Bacillus anthracis, Shigella, botulism, and Bordetella pertussis (36–40). In each of these cases, the anti-bacterial strategy was to employ the high-affinity binding of VHH to neutralize secreted bacterial toxins. The use of InlB-specific VHH represents a novel approach to combating bacterial disease using VHH. The dependence of the internalin–host cell receptor interaction in Listeria pathogenesis provides a novel mechanism of VHH-mediated therapeutic intervention by inhibiting host cell invasion (Figs. 2 and 3). Although further in vivo studies are required to validate the therapeutic potential of VHH for the treatment and prevention of listeriosis, the results presented here highlight the future potential of VHH as anti-bacterial agents.
Experimental procedures
Expression and purification of VHH
The plasmids (pSJF2H) for VHH R303 and R330 were a generous gift of Dr. Roger MacKenzie (National Research Council, Ottawa, Canada). Genes for VHH R419 and R326 were codon-optimized and synthesized as double-stranded gene blocks (GenScript, Piscataway, NJ). R326 and R419 were cloned into the plasmid pET22b using the restriction enzyme sites NcoI and XhoI.
Plasmids for R303 and R330 were transformed into Escherichia coli TG1, whereas R419 and R326 were transformed into E. coli BL21 (DE3) for protein expression. All of the InlB-specific VHH were extracted from the periplasm using an osmotic shock procedure and purified using Ni-NTA chromatography and size-exclusion chromatography, as described previously (41). The control anti-GFP VHH was expressed and purified as described previously (20).
Expression and purification of InlB
The sequences for InlB-LRR (InlB249) and InlB-LRR-IR (InlB321) were codon-optimized for E. coli expression and synthesized as gene blocks (GenScript). InlB249 was cloned into pET-15-TEV-NESG, and InlB321 was cloned into pET28a. Both plasmids were transformed into E. coli BL21 (DE3) for expression.
Cultures were grown overnight (30 °C, 225 rpm) in 2× YT medium with ampicillin (100 μg/ml). The overnight culture was transferred to 6 × 1 liter of 2× YT-amp and incubated (30 °C, 225 rpm) until A600 of 0.5. The culture was then induced with isopropyl β-d-thiogalactopyranoside (0.4 mm) and incubated overnight (20 °C, 225 rpm). Bacteria were harvested by centrifugation (6 °C, 5000 × g, 10 min). The pellet was suspended in TBS buffer (20 mm Tris-HCl, pH 8, 150 mm NaCl, 1 mm phenylmethanesulfonyl fluoride) and lysed using sonication. The cytoplasmic fraction (supernatant) was isolated by centrifugation (6 °C, 10,000 rpm, 30 min). Both proteins were purified by Ni-NTA affinity and size-exclusion chromatography.
Indirect and competitive ELISA
For the indirect ELISA, a 96-well plate was coated with serial dilutions of InlB (5–0.02 ng/μl) in PBS overnight at 4 °C. The wells were blocked for 1 h with BSA (3% in PBS). Biotinylated VHH (R303, R330, R326, and R419) were used as a primary antibody (15 μg/ml, 1 h). The plate was washed three times with PBS-Tween (0.05% Tween 20) followed by the addition of streptavidin horseradish peroxidase (Fisher) (1:50,000 dilution in 3% BSA 1 h). Finally, detection was carried out by the addition of 3,3,5,5-tetramethyl benzidine (15 min). The reaction was stopped by the addition of 0.18 m H2SO4, and the absorbance was measured at 450 nm using a plate reader (BioTek Instruments Inc., Winooski, VT).
A similar procedure was carried out for the competitive ELISA except that InlB was immobilized at a fixed concentration (10 μg/ml), and a mixture of biotinylated VHH (15 μg/ml) and unlabeled R303 (80 μg/ml) was added as a competitor.
Gentamicin protection assay
HeLa cells were cultured in 1× RPMI 1640 culture medium (HyClone) containing 2.05 mm l-glutamine, 10% FBS, and penicillin/streptomycin and incubated at 37 °C with 5% CO2. For infection, log phase L. monocytogenes (A600 = 0.3–0.5) were grown at 37 °C in 2× YT medium agitated at 225 rpm.
Treatment solutions of InlB249 and VHH diluted to 100 μg/ml in unsupplemented RPMI 1640 were added to a 24-well cell plate containing 1 × 105 HeLa cells/well and incubated for 30 min at 37 °C, 5% CO2. Log phase L. monocytogenes (MOI of 50:1) were then added to the wells, and the plate was centrifuged (1000 rpm for 3 min) and incubated at 37 °C with 5% CO2 for 1 h. Infected cells were washed twice with PBS to remove nonadherent bacteria. To kill extracellular bacteria, RPMI 1640 containing 100 μg/ml gentamicin was added and incubated for 60 min (37 °C, 5% CO2). To enumerate intracellular bacteria, wells were washed once with PBS and then lysed with 1% Triton X (Sigma) in PBS at the appropriate times. Recovered intracellular bacteria were quantified by plating serial dilutions on LB agar plates and enumerating colony counts.
Replicate wells were included in which total and surface-adherent Listeria were enumerated by harvesting the supernatant immediately after incubation of bacteria with HeLa cells (total) or collecting the Triton X-100 lysate before treatment with gentamicin (adherent). Each experiment was done in duplicates, and duplicates were performed at least three times independently.
Fluorescence microscopy
GFP-expressing L. monocytogenes were created as described previously (42). HeLa cells were cultured in 1× RPMI 1640 culture medium (HyClone) containing 2.05 mm l-glutamine, 10% FBS, and penicillin/streptomycin and incubated at 37 °C with 5% CO2. HeLa cells were seeded at a density of 4 × 105 cells/ml onto a microscope coverglass placed in each well of a 24-well plate. GFP-Listeria was grown overnight in BHI broth containing antibiotics, and the concentration was measured at A600. The bacteria were washed three times with sterile 1× PBS (pH 7.4) and labeled with 0.5 mg/ml EZ-Link Sulfo-NHS-LC-Biotin (Thermo Scientific). After quenching excess biotin by washing three times with 1% BSA, the bacteria were incubated with 100 μg/ml nanobodies at 37 °C for 30 min. HeLa cells were stained with 1 μl of 10 μg/ml 4′,6-diamidino-2-phenylindole and infected with biotinylated GFP-Listeria at an MOI of 50:1. After centrifuging for 15 min at 300 rpm, the plate was incubated for 1 h at 37 °C with 5% CO2 followed by three washes with unsupplemented RPMI 1640. Biotinylated GFP-Listeria were detected by the addition of 2.5 μl of 1 mg/ml Streptavidin-Dylight550 (Thermo Scientific) to each well, and the plate was incubated for 30 min at 37 °C with 5% CO2. The wells were washed with RPMI 1640, and the coverslips were fixed with 4% p-formaldehyde for 30 min at 4 °C. After washing the wells three times with 1× PBS (pH 7.4), the coverslips were removed from the plate, and Fluoromount-G (SouthernBiotech) was added to mount them onto slides. The slides were analyzed in a Leica DMI3000 B fluorescence microscope at ×63 magnification.
Crystallization of VHH and R303–InlB complexes
The crystallization and preliminary X-ray diffraction for R303 were reported previously (41). For complex formation, R303 and InlB were incubated (1:1.2 (w/w), 30 min, 25 °C) and purified by gel filtration chromatography (Bio-Rad NGC quest system using Enrich Sec70 column). The complexes (R303–InlB249 and R303–InlB321) and purified VHH (R330 and R326) were dialyzed against 10 mm HEPES, pH 7.4, and concentrated to 10 mg/ml.
Crystallization trials were carried out in Intelli 96-well sitting-drop plates using a Gryphon crystallization robot (Art Robbins Instruments). Sitting-crystal drops were set up using 1 μl of protein and 1 μl of reservoir solution. The proteins were screened using the PEGs, PEG II, and PACT crystallization suites (Qiagen Inc.). Crystal optimizations were carried out in 24-well Limbro plates (Hampton Research) using hanging-drop vapor diffusion and variable drop sizes. Optimal crystal conditions for VHH R330 were 0.1 m HEPES, pH 7.5, 25% PEG 3350. For VHH R326, the optimal crystal conditions were 0.2 m ammonium sulfate, 0.1 m sodium acetate, 22% PEG 3350. Crystals of R303–InlB249 grew in 0.2 m disodium tartrate, 20% PEG 3350. Finally, crystals of R303–InlB321 grew in 0.1 m sodium citrate tribasic dihydrate, pH 5.0, 34% Jeffamine ED-2001.
Data collection and X-ray structure determination
Crystals were dipped in cryoprotectant (mother liquor supplemented with 25% glycerol) and flash-frozen in liquid nitrogen. X-ray data were collected at the Canadian Light Source on beamline 08ID-1 (43). Diffraction data were processed using Xia2 (44). All structures were solved by molecular replacement using Phaser as implemented in Phenix (45). For molecular replacement, the previously solved structures of R303 (41) and InlB241 (46) and InlB321 (27) were used as search models. The structure was automatically built and refined using Phenix. Manual fitting of σA-weighted Fo − Fc electron density maps was carried out using Coot (47). The final model and refinement statistics are given in Table 2.
Author contributions
M. T. K., I. H., and C. L. B. formal analysis; M. T. K., I. H., A. S., T. M. B., and C. L. B. investigation; M. T. K., I. H., A. S., and C. L. B. methodology; M. T. K., A. S., T. M. B., and C. L. B. writing-review and editing; I. H. and C. L. B. writing-original draft; C. L. B. conceptualization; C. L. B. supervision; C. L. B. funding acquisition; C. L. B. project administration.
Acknowledgments
We thank Dr. Roger Mackenzie, Robert Gene, and Jyothi Kumaran (National Research Council, Ottawa, Canada) for the R303 and R330 plasmids and Dr. Brett Collins (University of Queensland, Australia) for the gift of the anti-GFP VHH plasmid. The Listeria GFP plasmid pNF8 was a gift of Dr. M. P. Doyle (Oklahoma State University).
This work was supported by NIGMS, National Institutes of Health, Grant SC3GM112532. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- InlA
- internalin A
- InlB
- internalin B
- LRR
- leucine-rich repeat
- VHH
- variable region of camelid heavy-chain antibody
- IR
- interrepeat
- CDR
- complementarity-determining region
- Ni-NTA
- nickel-nitrilotriacetic acid
- MOI
- multiplicity of infection
- HGF/SF
- hepatocyte growth factor/scatter factor.
References
- 1. Allerberger F. (2003) Listeria: growth, phenotypic differentiation and molecular microbiology. FEMS Immunol. Med. Microbiol. 35, 183–189 10.1016/S0928-8244(02)00447-9 [DOI] [PubMed] [Google Scholar]
- 2. Dussurget O., Pizarro-Cerda J., and Cossart P. (2004) Molecular determinants of Listeria monocytogenes virulence. Annu. Rev. Microbiol. 58, 587–610 10.1146/annurev.micro.57.030502.090934 [DOI] [PubMed] [Google Scholar]
- 3. Madjunkov M., Chaudhry S., and Ito S. (2017) Listeriosis during pregnancy. Arch. Gynecol. Obstet. 296, 143–152 10.1007/s00404-017-4401-1 [DOI] [PubMed] [Google Scholar]
- 4. Janakiraman V. (2008) Listeriosis in pregnancy: diagnosis, treatment, and prevention. Rev. Obstet. Gynecol. 1, 179–185 [PMC free article] [PubMed] [Google Scholar]
- 5. Radoshevich L., and Cossart P. (2018) Listeria monocytogenes: towards a complete picture of its physiology and pathogenesis. Nat. Rev. Microbiol. 16, 32–46 [DOI] [PubMed] [Google Scholar]
- 6. Cossart P., Pizarro-Cerdá J., and Lecuit M. (2003) Invasion of mammalian cells by Listeria monocytogenes: functional mimicry to subvert cellular functions. Trends Cell Biol. 13, 23–31 10.1016/S0962-8924(02)00006-5 [DOI] [PubMed] [Google Scholar]
- 7. Dramsi S., Biswas I., Maguin E., Braun L., Mastroeni P., and Cossart P. (1995) Entry of Listeria monocytogenes into hepatocytes requires expression of inIB, a surface protein of the internalin multigene family. Mol. Microbiol. 16, 251–261 10.1111/j.1365-2958.1995.tb02297.x [DOI] [PubMed] [Google Scholar]
- 8. Mengaud J., Ohayon H., Gounon P., Mege R.-M., and Cossart P. (1996) E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell 84, 923–932 10.1016/S0092-8674(00)81070-3 [DOI] [PubMed] [Google Scholar]
- 9. Shen Y., Naujokas M., Park M., and Ireton K. (2000) InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell 103, 501–510 10.1016/S0092-8674(00)00141-0 [DOI] [PubMed] [Google Scholar]
- 10. Braun L., Nato F., Payrastre B., Mazié J. C., and Cossart P. (1999) The 213-amino-acid leucine-rich repeat region of the Listeria monocytogenes InlB protein is sufficient for entry into mammalian cells, stimulation of PI 3-kinase and membrane ruffling. Mol. Microbiol. 34, 10–23 10.1046/j.1365-2958.1999.01560.x [DOI] [PubMed] [Google Scholar]
- 11. Braun L., Ohayon H., and Cossart P. (1998) The InIB protein of Listeria monocytogenes is sufficient to promote entry into mammalian cells. Mol. Microbiol. 27, 1077–1087 10.1046/j.1365-2958.1998.00750.x [DOI] [PubMed] [Google Scholar]
- 12. Greiffenberg L., Goebel W., Kim K. S., Weiglein I., Bubert A., Engelbrecht F., Stins M., and Kuhn M. (1998) Interaction of Listeria monocytogenes with human brain microvascular endothelial cells: InlB-dependent invasion, long-term intracellular growth, and spread from macrophages to endothelial cells. Infect. Immun. 66, 5260–5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lingnau A., Domann E., Hudel M., Bock M., Nichterlein T., Wehland J., and Chakraborty T. (1995) Expression of the Listeria monocytogenes EGD inlA and inlB genes, whose products mediate bacterial entry into tissue culture cell lines, by PrfA-dependent and -independent mechanisms. Infect. Immunity 63, 3896–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Disson O., Grayo S., Huillet E., Nikitas G., Langa-Vives F., Dussurget O., Ragon M., Le Monnier A., Babinet C., Cossart P., and Lecuit M. (2008) Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature 455, 1114–1118 10.1038/nature07303 [DOI] [PubMed] [Google Scholar]
- 15. Schubert W. D., and Heinz D. W. (2003) Structural aspects of adhesion to and invasion of host cells by the human pathogen Listeria monocytogenes. Chembiochem 4, 1285–1291 10.1002/cbic.200300624 [DOI] [PubMed] [Google Scholar]
- 16. Banerjee M., Copp J., Vuga D., Marino M., Chapman T., van der Geer P., and Ghosh P. (2004) GW domains of the Listeria monocytogenes invasion protein InlB are required for potentiation of Met activation. Mol. Microbiol. 52, 257–271 10.1111/j.1365-2958.2003.03968.x [DOI] [PubMed] [Google Scholar]
- 17. Muyldermans S. (2013) Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 82, 775–797 10.1146/annurev-biochem-063011-092449 [DOI] [PubMed] [Google Scholar]
- 18. Gene R. W., Kumaran J., Aroche C., van Faassen H., Hall J. C., MacKenzie C. R., and Arbabi-Ghahroudi M. (2015) High affinity anti-Internalin B VHH antibody fragments isolated from naturally and artificially immunized repertoires. J. Immunol. Methods 416, 29–39 10.1016/j.jim.2014.10.009 [DOI] [PubMed] [Google Scholar]
- 19. Kumaran J., Mackenzie C. R., and Arbabi-Ghahroudi M. (2012) Semiautomated panning of naive camelidae libraries and selection of single-domain antibodies against peptide antigens. Methods Mol. Biol. 911, 105–124 [DOI] [PubMed] [Google Scholar]
- 20. Kubala M. H., Kovtun O., Alexandrov K., and Collins B. M. (2010) Structural and thermodynamic analysis of the GFP:GFP-nanobody complex. Protein Sci. 19, 2389–2401 10.1002/pro.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Agerer F., Waeckerle S., and Hauck C. R. (2004) Microscopic quantification of bacterial invasion by a novel antibody-independent staining method. J. Microbiol. Methods 59, 23–32 10.1016/j.mimet.2004.05.008 [DOI] [PubMed] [Google Scholar]
- 22. North B., Lehmann A., and Dunbrack R. L. Jr. (2011) A new clustering of antibody CDR loop conformations. J. Mol. Biol. 406, 228–256 10.1016/j.jmb.2010.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adolf-Bryfogle J., Xu Q., North B., Lehmann A., and Dunbrack R. L. Jr. (2015) PyIgClassify: a database of antibody CDR structural classifications. Nucleic Acids Res. 43, D432–D438 10.1093/nar/gku1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De Genst E., Silence K., Decanniere K., Conrath K., Loris R., Kinne J., Muyldermans S., and Wyns L. (2006) Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. U.S.A. 103, 4586–4591 10.1073/pnas.0505379103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lauwereys M., Arbabi Ghahroudi M., Desmyter A., Kinne J., Hölzer W., De Genst E., Wyns L., and Muyldermans S. (1998) Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. 17, 3512–3520 10.1093/emboj/17.13.3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jonquières R., Bierne H., Fiedler F., Gounon P., and Cossart P. (1999) Interaction between the protein InlB of Listeria monocytogenes and lipoteichoic acid: a novel mechanism of protein association at the surface of Gram-positive bacteria. Mol. Microbiol. 34, 902–914 10.1046/j.1365-2958.1999.01652.x [DOI] [PubMed] [Google Scholar]
- 27. Niemann H. H., Jäger V., Butler P. J., van den Heuvel J., Schmidt S., Ferraris D., Gherardi E., and Heinz D. W. (2007) Structure of the human receptor tyrosine kinase Met in complex with the Listeria invasion protein InlB. Cell 130, 235–246 10.1016/j.cell.2007.05.037 [DOI] [PubMed] [Google Scholar]
- 28. Zhang Y., Xia M., Jin K., Wang S., Wei H., Fan C., Wu Y., Li X., Li X., Li G., Zeng Z., and Xiong W. (2018) Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 17, 45 10.1186/s12943-018-0796-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Charlier C., Perrodeau E., Leclercq A., Cazenave B., Pilmis B., Henry B., Lopes A., Maury M. M., Moura A., Goffinet F., Dieye H. B., Thouvenot P., Ungeheuer M. N., Tourdjman M., Goulet V., et al. (2017) Clinical features and prognostic factors of listeriosis: the MONALISA national prospective cohort study. Lancet Infect. Dis. 17, 510–519 10.1016/S1473-3099(16)30521-7 [DOI] [PubMed] [Google Scholar]
- 30. Mylonakis E., Paliou M., Hohmann E. L., Calderwood S. B., and Wing E. J. (2002) Listeriosis during pregnancy: a case series and review of 222 cases. Medicine 81, 260–269 10.1097/00005792-200207000-00002 [DOI] [PubMed] [Google Scholar]
- 31. Hof H., Nichterlein T., and Kretschmar M. (1997) Management of listeriosis. Clin. Microbiol. Rev. 10, 345–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Angelo K. M., Conrad A. R., Saupe A., Dragoo H., West N., Sorenson A., Barnes A., Doyle M., Beal J., Jackson K. A., Stroika S., Tarr C., Kucerova Z., Lance S., Gould L. H., et al. (2017) Multistate outbreak of Listeria monocytogenes infections linked to whole apples used in commercially produced, prepackaged caramel apples: United States, 2014–2015. Epidemiol. Infect. 145, 848–856 10.1017/S0950268816003083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Garner D., and Kathariou S. (2016) Fresh produce-associated listeriosis outbreaks, sources of concern, teachable moments, and insights. J. Food Prot. 79, 337–344 10.4315/0362-028X.JFP-15-387 [DOI] [PubMed] [Google Scholar]
- 34. McCollum J. T., Cronquist A. B., Silk B. J., Jackson K. A., O'Connor K. A., Cosgrove S., Gossack J. P., Parachini S. S., Jain N. S., Ettestad P., Ibraheem M., Cantu V., Joshi M., DuVernoy T., Fogg N. W. Jr., et al. (2013) Multistate outbreak of listeriosis associated with cantaloupe. N. Engl. J. Med. 369, 944–953 10.1056/NEJMoa1215837 [DOI] [PubMed] [Google Scholar]
- 35. Puthiyakunnon S., He X., Boddu S., Huang S. H., and Cao H. (2017) C-Met inhibitors are potential novel therapeutic agents against Listeria monocytogenes infection through blocking the bacteria entry into nonphagocytic cells. Curr. Top. Med. Chem. 17, 278–289 [DOI] [PubMed] [Google Scholar]
- 36. Lo A. W., Moonens K., De Kerpel M., Brys L., Pardon E., Remaut H., and De Greve H. (2014) The molecular mechanism of Shiga toxin Stx2e neutralization by a single-domain antibody targeting the cell receptor-binding domain. J. Biol. Chem. 289, 25374–25381 10.1074/jbc.M114.566257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Malik A. A., Imtong C., Sookrung N., Katzenmeier G., Chaicumpa W., and Angsuthanasombat C. (2016) Structural characterization of humanized nanobodies with neutralizing activity against the Bordetella pertussis CyaA-hemolysin: implications for a potential epitope of toxin-protective antigen. Toxins 8, 99 10.3390/toxins8040099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shali A., Hasannia S., Gashtasbi F., Abdous M., Shahangian S. S., and Jalili S. (2018) Generation and screening of efficient neutralizing single domain antibodies (VHHs) against the critical functional domain of anthrax protective antigen (PA). Int. J. Biol. Macromol. 114, 1267–1278 10.1016/j.ijbiomac.2018.03.034 [DOI] [PubMed] [Google Scholar]
- 39. Sulea T., Hussack G., Ryan S., Tanha J., and Purisima E. O. (2018) Application of Assisted Design of Antibody and Protein Therapeutics (ADAPT) improves efficacy of a Clostridium difficile toxin A single-domain antibody. Sci. Rep. 8, 2260 10.1038/s41598-018-20599-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yao G., Lam K. H., Weisemann J., Peng L., Krez N., Perry K., Shoemaker C. B., Dong M., Rummel A., and Jin R. (2017) A camelid single-domain antibody neutralizes botulinum neurotoxin A by blocking host receptor binding. Sci. Rep. 7, 7438 10.1038/s41598-017-07457-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huh I., Gene R., Kumaran J., MacKenzie C. R., and Brooks C. L. (2014) In situ proteolysis, crystallization and preliminary X-ray diffraction analysis of a VHH that binds Listeria internalin B. Acta Crystallogr. F Struct. Biol. Commun. 70, 1532–1535 10.1107/S2053230X1402010X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ma L., Zhang G., and Doyle M. P. (2011) Green fluorescent protein labeling of Listeria, Salmonella, and Escherichia coli O157:H7 for safety-related studies. PLoS One 6, e18083 10.1371/journal.pone.0018083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grochulski P., Fodje M. N., Gorin J., Labiuk S. L., and Berg R. (2011) Beamline 08ID-1, the prime beamline of the Canadian Macromolecular Crystallography Facility. J. Synchrotron Radiat. 18, 681–684 10.1107/S0909049511019431 [DOI] [PubMed] [Google Scholar]
- 44. Winter G., Lobley C. M., and Prince S. M. (2013) Decision making in xia2. Acta Crystallogr. D Biol. Crystallogr. 69, 1260–1273 10.1107/S0907444913015308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marino M., Braun L., Cossart P., and Ghosh P. (1999) Structure of the lnlB leucine-rich repeats, a domain that triggers host cell invasion by the bacterial pathogen L. monocytogenes. Mol. Cell 4, 1063–1072 10.1016/S1097-2765(00)80234-8 [DOI] [PubMed] [Google Scholar]
- 47. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]