

Abstract
Genomes of phages, mitochondria, and chloroplasts are transcribed by a diverse group of transcriptional machineries with structurally related single-subunit RNA polymerases (RNAPs). Our understanding of transcription mechanisms of these enzymes is predominantly based on biochemical and structural studies of three most-studied members, transcription factor–independent phage T7 RNAP, transcription factor–dependent phage N4 virion-encapsidated RNAP, and transcription factor–dependent mitochondrial RNAPs (mtRNAP). Although these RNAPs employ completely different mechanisms for promoter recognition and transcription termination, these enzymes are relatively large and formed by single polypeptides. Historically being a model enzyme for studying the mechanisms of transcription by T7-like RNAPs, however, T7 RNAP represents only a small group of RNAPs in this family. The vast majority of T7-like RNAPs are transcription factor–dependent, and several of them are heterodimeric enzymes. Here, we report X-ray crystal structures of transcription complexes of the smallest and heterodimeric form of T7-like RNAP, bacteriophage N4 RNAPII, providing insights into the structural organization of a minimum RNAP in this family. We analyze structural and functional aspects of heterodimeric architecture of N4 RNAPII concerning the mechanisms of transcription initiation and transition to processive RNA elongation. Interestingly, N4 RNAPII maintains the same conformation in promoter-bound and elongation transcription complexes, revealing a novel transcription mechanism for single-subunit RNAPs. This work establishes a structural basis for studying mechanistic aspects of transcription by factor-dependent minimum RNAP.
Keywords: RNA polymerase, DNA transcription, X-ray crystallography, viral transcription, RNA synthesis, N4 phage, transcription complex, X-ray structure, transcription
Introduction
DNA-dependent RNA polymerases (RNAPs)3 that belong to the T7-like family are found in phages, mitochondria, and chloroplasts (1). The family is named after the first isolated (2) and the most extensively studied member, bacteriophage T7 RNAP, formed by a 98 kDa single polypeptide (3). Understanding of structural organizations of T7-like RNAPs had been established by a series of studies that captured the multiple functional states of the enzyme, including the apo-form (4), promoter DNA–bound (5), initiation (6), early elongation (7), and elongation (8, 9) complexes, as well as a complex with transcription inhibitor lysozyme (10). It was later significantly expanded by determining structures of other members such as N4 virion-encapsidated RNAP (vRNAP) (11–13) and human mitochondrial RNAP (hmtRNAP) (14–17). All T7-like RNAPs, whose crystal structures have been determined so far, consist of the amino (N)-terminal and the polymerase domains. The polymerase domain contains highly conserved elements and motifs that participate in the basic function of RNA synthesis such as NTP binding and selection as well as catalysis of nucleotidyl transfer reaction. However, the N-terminal domains and inserts found in T7-like RNAPs show surprising diversity, resulting in wide range of molecular masses (70–110 kDa) (18, 19). Because synthesis of a faithful RNA copy of DNA remains the priority function for all T7-like RNAPs, their size differences may stem from the optimization of gene expression in their working environments. For this reason, structural studies of distantly related members of this family are important for understanding the link between architecture of RNAPs and their functional fitness. A comparative structure–function analysis of the family members requires a suitable reference, a minimum functional RNAP that possesses only a basic set of functional elements.
Studies of several members of T7-like RNAPs have shown that, indeed, aside from the catalysis of RNA synthesis, their capabilities in RNA transcription-related functions during the initiation, elongation, and termination vary dramatically. The most drastic variations occur during the transcription initiation step. Based on the abilities to recognize promoter and unwind double-strand DNA, members of T7-like family can be grouped into either the transcription factor–independent or –dependent RNAPs (20). RNAPs of T7 and other closely related phages are transcription factor–independent and capable of recognizing, binding, and unwinding promoter DNA to initiate RNA synthesis on their own (21). The other members of the family from phages, eukaryotic organelles mitochondria, and chloroplasts are transcription factor–dependent and are unable to start RNA synthesis from promoters without their specific transcription initiation factors (20, 22).
Coliphage N4 genome encodes two members of T7-like enzymes including the vRNAP and the RNAPII for expression of the early and middle genes of N4 phage genome, respectively (23, 24). The N4 RNAPII has a heterodimeric architecture comprising the gp15 and gp16 subunits and represents a small group of heterodimeric enzymes within the family of T7-like RNAPs (20). Several features distinguish RNAPII from other members, making it an interesting subject for structure–function studies. First, the molecular mass of N4 RNAPII (70 kDa) is one of the smallest in the T7-like enzymes; accordingly, studying its structure could reveal minimum structural requirements for performing basic RNAP functions such as DNA binding, catalysis of RNA synthesis, and transcript elongation. Second, RNAPII is a heterodimeric enzyme containing gp15 and gp16 subunits. Studying structure and function of the naturally heterodimeric RNAPII addresses the long-standing question of the way of splitting a single-subunit RNAP to functional modules, i.e. separating parts of the enzyme required for catalysis and formation of promoter-bound initiation complex. Third, promoter-specific transcription of N4 RNAPII requires transcription factors gp1 and gp2 for unwinding promoter DNA and recruiting RNAPII to single-stranded DNA, respectively (20). These factors are not related to any transcription factors in the mtRNAP transcription system (TFA and TFB2 in human and Mtf1 in yeast) (25). In this study, we report the X-ray crystal structures of N4 RNAPII in complex with promoter DNA (PDB ID: 6DT7) and engaged in the transcript elongation (PDB IDs: 6DT8 and 6DTA) for expanding our understanding of the evolution of the T7-like RNAPs.
Results
N4 RNAPII binds to a single-stranded promoter DNA to form a functional initiation complex
We initially attempted to crystallize the apo-form N4 RNAPII but could not find conditions that produce crystals. We therefore aimed to crystallize RNAPII in complex with promoter DNA. RNAPII cannot bind and initiate transcription from double-stranded promoter DNA without transcription factors gp1 and gp2. However, RNAPII can bind and initiate transcription without these factors from single-stranded DNA (25). We designed and tested a consensus N4 middle promoter DNA template (“Experimental Procedures”) that lacks a fragment of the nontemplate strand to mimic a melted DNA bubble around the transcription start site (Fig. 1A) (26). Specific binding of RNAPII to this DNA was confirmed by a native gel mobility shift assay (Fig. S1A). In vitro transcription assay shows that the RNAPII initiates transcription with this template from two separated locations: at the major and the minor sites, the latter is 3 bp upstream from the major site (5′-GTCCACCC-3′, where start sites are underlined) (Fig. S1B). Transcription from the major site resulted in synthesis of 3-mer GGG and longer poly-G transcripts, produced by transcription slippage as dominant RNA products from this template even in the presence of both GTP and UTP (Fig. S1B, lanes 1 and 2). The minor site produces RNA transcripts containing UMP residues at their third positions (GGU, GGUG, or GGUGG) and, thereby, having different mobility as compared with the transcripts initiated from the major site (Fig. S1B, lane 1). In the structure of N4 RNAPII and DNA complex described later, a DNA base responsible for the transcription initiation at the major site positions at the i site of RNAP active site. Hereafter, DNA bases located downstream and upstream from the major transcription start site DNA base are counted as +1, +2, etc., and −1, −2, etc., respectively.
Figure 1.

The overall crystal structure of N4 RNAPII–promoter DNA complex. A, schematic representation of the N4 consensus middle promoter DNA template used for crystallization of the complex. The hairpin-like template lacks the nontemplate region from −3 to + 6. The region of the TS DNA solved in the crystal structure is shown by a bracket. The arrows indicate the identified transcription start sites. B, the overall view of the N4 RNAPII-promoter DNA structure compared with the T7 RNAP–promoter DNA structure. The protein structures are shown as surface models with DNAs shown as ribbon models. N4 RNAPII subunits gp15 and gp16 are shown in light blue and light green, correspondingly; DNA is shown in magenta. C, domain organization of N4 RNAPII. Two views of the complex are represented as ribbon models overlaid on surface models with subdomains and structural motifs labeled by different colors. The NTD, Thumb, Palm, Fingers, and Palm insertion subdomains are shown in gray, green, red, marine, and wheat, correspondingly; DNA is shown in magenta. Structural homologs of RNAPII promoter recognition elements, the specificity loop, intercalating β-hairpin, AT-rich recognition motif are shown in light blue, yellow, and blue, correspondingly. Structural analog of the RNA exit pore is shown in the right panel. D, schematic secondary structure map of N4 RNAPII. α-helices and β-strands are depicted as rectangles and arrows, correspondingly, colored according to their subdomain localization as in panel C. The structural motifs conserved in T7-like RNAPs are underlined and signed.
Overall crystal structure of N4 RNAPII and DNA complex
We crystallized the RNAPII–DNA binary complex and determined its structure at 2.35 Å resolution (Table S1). The high-quality electron density map completely covers both subunits of RNAPII and the segment of DNA located in the DNA-binding channel of RNAPII. The structure of N4 RNAPII resembles a “right hand” in a grasping conformation that accommodates the single-stranded region of DNA. Approximate dimensions of the RNAPII–DNA complex are 80 Å × 71 Å × 65 Å. Although RNAPII is composed of two subunits, its overall shape is similar to that of T7 RNAP (Fig. 1B).
The structural alignment between the N4 RNAPII and the T7 RNAP (PDB ID: 1CEZ) (6) allowed for clear identification of the N-terminal domain (NTD) and the polymerase domain, including the Thumb, Palm and Finger subdomains (Fig. 1, C and D, and Fig. S2). We also identified the structural elements of RNAPII such as the specificity loop in the Fingers as well as the AT-rich DNA sequence recognition motif and the intercalating β-hairpin in the NTD based on their structural homologies with those elements in the T7 RNAP (Fig. 1, C and D, and Fig. S2).
In the crystal structure, almost all traceable residues of DNA (−8 to +2) locate inserted into the catalytic cleft of the enzyme (Fig. 2, A and B). The upstream duplex and the 5′ terminal residue are disordered. The orientation of DNA in the complex suggests that the upstream duplex may not interact with the specificity loop or AT-rich recognition motif of RNAPII (Fig. 1C and Fig. S2). The majority of RNAPII-DNA interactions are not DNA sequence specific.
Figure 2.
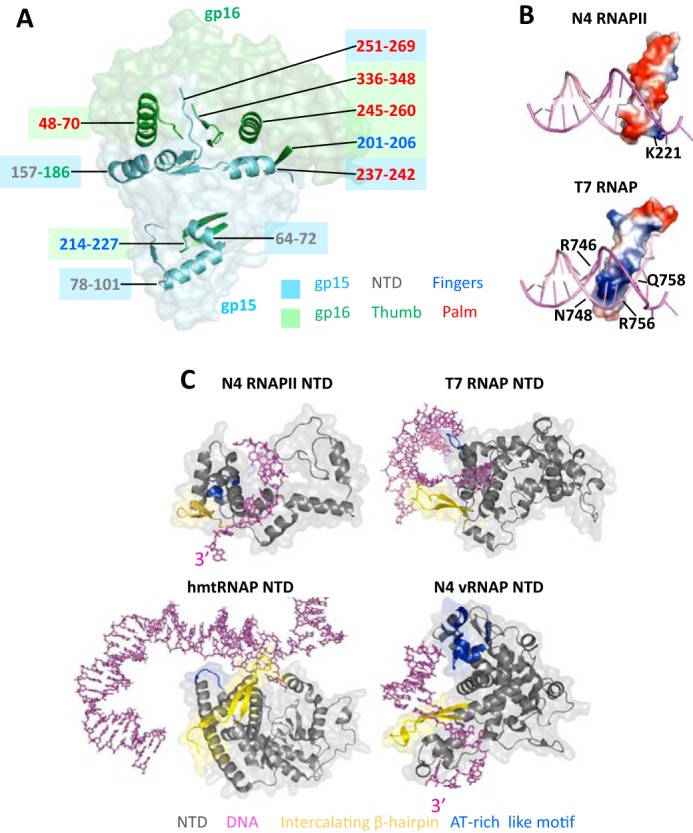
The mechanism of RNAPII heterodimerization and structural organization of the N4 RNAPII promoter recognition motifs. A, surface model of RNAPII-promoter DNA complex with structural elements of gp15 and gp16 subunits that form heterodimerization surfaces overlaid as ribbon models. Subunits and their structural elements are colored as in Fig. 1B. Positions of the elements involved in heterodimerization are colored as corresponding subunits and subdomains in Fig. 1. B, distribution of electrostatic potentials on the surfaces of specificity loops of N4 RNAPII and T7 RNAP. Blue, red, and white colors depict areas with positive, negative, and neutral charges, correspondingly. Residues that form basic patches on the specificity loop tips are shown. C, structural organizations of NTDs in T7-like RNAPs and interaction of their promoter recognition motifs with DNA. NTDs of N4 RNAPII, T7 RNAP, human mitochondrial RNAP, and N4 vRNAP are shown as ribbon models overlaid on their surface models and colored as in Fig. 1. Promoter templates are shown as stick models in magenta.
Dimerization of N4 RNAPII subunits
The T7 RNAP can be physically split at the junction between the NTD (residues 1–179) and the polymerase domains (residues 180–880), and the functional recombinant T7 RNAP can be assembled in vivo and in vitro by mixing these two recombinant polypeptides (27). Unlike the synthetic split version of T7 RNAP, the naturally split N4 RNAPII uses an alternative approach to form a functional RNAP with two polypeptides. Thus, gp15 subunit comprises the NTD together with the Thumb and a short segment of the Palm, whereas gp16 contains the rest of subdomains including the Palm and Fingers (Fig. 1, B and C, and Fig. S2). We also note that the binding interface between gp15 and gp16 is represented mostly by charged patches (Fig. S3A), which is, likely, important for enabling solubility of the subunits before dimerization. Splitting T7 RNAP in a manner of RNAPII results in exposing hydrophobic patches on their dimerization surfaces, making such synthetic subunits prone to aggregation (Fig. S3B).
Dimerization of subunits gp15 and gp16 involves two binding interfaces. The main dimerization surface is formed by a bundle of structural elements, including α-helices, short β-strands, and segments of unstructured loops (Fig. 2A), making nine pairs of polar contacts (Asp162–Arg257, Arg166–Asp260, Asp177–Tyr53, Arg238–His245, Gly239–Arg253, Arg240–Asp248, Gln251–Thr339, Glu264–His336, and Glu266–Ile68, between gp15 and gp16 residues, correspondingly). Other residues on the dimerization interfaces also engage in a number of hydrophobic and van der Waals interactions. The orientation of constituent elements of the gp15 dimerization surface relative to those in the corresponding dimerization surface of gp16 is close to perpendicular. The second smaller dimerization site lies on the interface between the NTD and the specificity loop. This interaction includes four polar contacts (Arg72–Glu214, Gln79–His217, Asp101–Lys221, and Asp101–Tyr223, in gp15 and gp16, correspondingly) and a hydrophobic interaction between the side chains of Phe96 and Phe103 in the NTD and Phe218 in the specificity loop.
The determinants for promoter recognition and melting are functionally disabled
The specificity loops of T7 RNAP and N4 vRNAP are positively charged and insert into the major groove of DNA to recognize DNA sequences during the RNAP–promoter DNA complex formation (Fig. 2B) (5, 11). DNA recognition requires substantial flexibility of the specificity loop and in part depends on its length. Accordingly, the specificity loops of the T7 RNAP and N4 vRNAP are long and flexible, suitable for establishing the DNA base-specific interaction. In contrast, the specificity loop of the N4 RNAPII is mostly negatively charged (Fig. 2B) and rigid because of interaction with the NTD (Fig. 2A), arguing against its role in DNA sequence recognition during the promoter DNA complex formation.
The NTDs of T7 RNAP and N4 vRNAP function as platforms for promoter DNA recognition and unwinding (5, 13, 14). Shapes and electrostatic potentials of the NTD provide complementary surfaces for specific binding of the double-stranded and hairpin forms of promoter DNA in T7 RNAP and N4 vRNAP, respectively. Particularly, the AT-rich recognition motif and the intercalating β-hairpin of these RNAPs are separated by a significant distance for recognizing promoter sequence and DNA unwinding. In contrast, the NTD of N4 RNAPII (gp15 residues 1–168) is substantially smaller in size and the AT-rich recognition motif and the intercalating β-hairpin locate closer to each other, indicating that these elements are not suitable for the promoter DNA binding or unwinding (Fig. 2C and Fig. S2).
Polymerase domain and RNA exit pore
The polymerase domain of RNAPII harbors a deep cleft for template DNA binding; the bottom of the cleft possesses conserved motifs (A, B, and DXXGR motifs), including two conserved Asp residues of the Palm (gp15 residues 226–269; gp16 residues 1–90 and 245–404) for coordinating catalytic Mg ions (Fig. 3A). The Palm has a 71-residue insertion (Palm insertion) located in the back of the active site (Fig. 1, C and D), but its function is unknown.
Figure 3.

N4 RNAPII–DNA contacts in the promoter binary complex. A, overall view of RNAPII–DNA contacts in the binary complex. DNA and modeled initiating nucleotides are presented as stick models in magenta and red, correspondingly. The O- and Y-helices of RNAPII are shown as ribbon models with side chains involved in positioning the +2 RNA nucleotide shown as stick models. Two catalytic aspartates are also shown as stick models with a magnesium ion modeled as a yellow sphere. Side chains of amino acid residues involved in specific contacts with DNA are shown as stick models colored as corresponding structural elements in Fig. 1C. The TS DNA residues are labeled. B, two views of surface electrostatic potentials of N4 RNAPII. The basic surface patch in the catalytic cleft that interacts with the TS DNA is indicated in the left panel. The basic surface patch of the proposed RNA exit pore that may interact with the separated 5′ end of transcript is indicated in the right panel. C, schematic representation of the RNAPII–DNA contacts in the binary complex. DNA residues that were solved in the structure are shown in magenta; unresolved DNA bases are shown in pink. Rectangles represent DNA bases; spheres represent sugar-phosphate DNA backbone. Solid lines contour the solved DNA residues, dotted lines depict unresolved residues. Protein residues contacting DNA are shown in colors as in Fig. 1C with background colors of corresponding RNAPII subunits. Dotted lines connecting DNA and protein residues depict contacts with DNA bases; solid lines depict protein contacts with the sugar-phosphate backbone of DNA.
The N-terminal part of the Fingers subdomain contains seven α-helices and four β-strands (Fig. 1D), including the motif B (RX3KX7YG) for binding NTP during the RNA synthesis. A structural analysis of T7-like RNAPs revealed that the enzymes fall into two classes based on their sizes of O/Y-helices in the Fingers. T7 RNAP and N4 vRNAP contain long O/Y-helices touching Thumbs, whereas N4 RNAPII and mtRNAP have short O/Y-helices unable to interact with Thumbs. In case of hmtRNAP, transcription factor TFB2M binds in between the Fingers and Thumb, trapping the nontemplate DNA in the initiation complex (17). Because the N4 transcription factor gp2 plays a similar role in the open complex formation of the N4 RNAPII transcription, it may also locate in between the Fingers and Thumb of the N4 RNAPII.
The C-terminal part of the Fingers contains the specificity loop, which contacts the NTD to form a circle of about 20 Å in diameter. The opening is positively charged, and it locates suitable for passing single-stranded RNA, suggesting its function as the RNA exit pore (Fig. 3B, right).
N4 RNAPII–DNA contacts in the promoter binary complex
The template-strand (TS) DNA enters the catalytic cleft through a narrow passage formed by the NTD and the Thumb (Fig. 1C). The RNAPII surfaces around the passage and along the entire length of TS DNA inside the catalytic cleft are positively charged (Fig. 3B, left). The catalytic cleft fits eight DNA bases (−6 to +2) (Fig. 1C). The −6C base at the 3′ end of TS DNA forms a stacking interaction with His59 residue of the NTD (Fig. 3, A and B) and following bases until −3 position faces toward the RNA exit pore. There is a sharp turn between bases −3 and −2, which sets the base stacking profile further downstream to the residue +2 (Fig. 3A). There are extensive interactions between RNAP and DNA from −2 to +2 positions (Fig. 3C) that place DNA bases toward the NTP-binding sites (i and i+1 sites) at the active site of RNAP. Particularly, DNA bases from −1 to +2 adopt an A-form helical conformation ready for base pairing with incoming nucleotides (Fig. 3A).
Organization of N4 RNAPII elongation complexes
We reconstituted functional elongation complexes of RNAPII by binding DNA:RNA scaffolds that mimic natural nucleic acid components of an elongation complex to the enzyme (scaffolds 1 and 2, “Experimental Procedures”). Initially, we tested the assembly of an elongation complex using the DNA:RNA scaffold made of the N4 promoter template (Fig. 1A) and an 8-mer RNA primer annealed to the single-stranded region of the TS DNA. However, the resulting elongation complex failed to produce crystals. To overcome this problem, we first crystallized binary complexes of RNAPII with DNA templates from scaffolds 1 and 2, and then soaked corresponding RNA primers into preformed crystals. Binding of RNA primers to DNA templates in crystals was confirmed by 5′ end labeling of nucleic acids in washed crystals with 32P (not shown). To explore the mechanism of RNA extension in RNAPII elongation complex, crystals containing the scaffold 2 were additionally soaked in a solution supplemented with Mg2+ and the next incoming nucleotide GTP.
The overall geometry and conformation of RNAPII in the elongation complexes remains essentially unchanged as compared with the binary complex (Fig. 4A) except for disordering of the Thumb's tip (Fig. 4B). The catalytic cleft of RNAPII is occupied by the DNA:RNA hybrid with the 5′ end of RNA located near the proposed RNA exit pore (Figs. 4C and 5, A and C) and the 3′ end of RNA located at the i site of the active site, indicating that the elongation complexes are in the posttranslocated state. (Fig. 5, A and C). The reconstituted RNAPII elongation complexes contained DNA:RNA hybrids of different traceable lengths (Fig. 5). In the elongation complex assembled with the scaffold 1 and 12-mer RNA (EC1), the electron density is traceable for 6 bp of the DNA:RNA hybrid (Fig. 5, A and B, and Fig. S4). In the elongation complex assembled with the scaffold 2 and 8-mer RNA plus GTP, the DNA:RNA hybrid is traceable for 7 bp (Fig. 5, C and D, and Fig. S4). The electron density map also shows a GTP bound at the i+1 site base paired with the TS DNA base (Fig. 5, C and D, and Fig. S4). GTP is not incorporated into the RNA as indicated by the presence of the triphosphate group in GTP and a 4.8 Å distance from its α phosphate to the 3′ OH group of RNA not optimal for catalysis (Fig. 5, C and D). Apparently, despite the correct binding of GTP at the i+1 site, RNAPII-binding restraints disable catalysis in crystallo. In both elongation complexes RNA primers were designed to anneal to the same DNA segment forming DNA:RNA hybrids of the same length. The observed difference in the traceable lengths of the DNA:RNA hybrids may be attributed to topological restraints of accommodation of the longer 5′ end of 12-mer RNA in the crystalline RNAPII transcription complex. We speculate that the 5′ end of 12-mer RNA is unable to efficiently thread through the RNA exit pore and becomes disordered in the complex.
Figure 4.

The crystal structure of N4 RNAPII elongation complex. A, overall view of the RNAPII elongation complex assembled on scaffold 1. The orientation of the complex is the same as of the binary complex in Fig. 1C. N4 RNAPII subunits are shown as semi-transparent surface models colored as in Fig. 1B. DNA and RNA are shown as stick models in magenta and red, correspondingly. The location of the RNA exit pore is indicated. B, the Thumb subdomain is disordered in the RNAPII elongation complex. C, organization of the DNA:RNA hybrid in the elongation complex and location of the RNA exit pore. RNAPII is shown as a surface model of light blue (gp15) and light green (gp16) colors; some regions of the enzyme in frontal projection were removed to show the catalytic cleft. DNA (magenta) and RNA (red) are shown as stick models overlaid on electron density maps.
Figure 5.

N4 RNAPII contacts with DNA and RNA in the elongation complexes. A, the overall view of the catalytic cleft in the elongation complex with scaffold 1. RNAPII is shown as a ribbon model, DNA and RNA are shown as stick models; two catalytic aspartates are shown as stick models with a modeled magnesium ion shown as a yellow sphere; residues are colored as in Fig. 1C. B, schematic representation of contacts between RNAPII and DNA and RNA residues in the elongation complex with scaffold 1. Residues are depicted using the same color/shape scheme as in Fig. 3C; traceable RNA residues are shown as red rectangles; disordered RNA residues are shown as salmon rectangles. C, the overall view of the catalytic cleft in the elongation complex with scaffold 2. RNAPII is shown as a ribbon model; DNA and RNA are shown as stick models; two catalytic aspartates are shown as stick models with a modeled magnesium ion shown as a yellow sphere; residues are colored as in Fig. 1C. GTP in the i+1 site is shown in green. D, schematic representation of contacts between RNAPII and DNA and RNA residues in the elongation complex assembled on scaffold 2. Residues are depicted using the same color/shape scheme as in Fig. 3C; traceable RNA residues are shown as red rectangles.
Discussion
The X-ray crystal structure of the DNA-bound N4 RNAPII reveals details of the architecture of a minimum RNAP of T7-like family of enzymes. The overall structure of RNAPII, particularly that of the polymerase domain, is similar to other RNAPs. The major structural difference in RNAPII concerns a substantial reduction in size of the NTD. Being significantly smaller than its counterparts in other T7-like enzymes, the NTD of N4 RNAPII, nevertheless, contains the structural analogs of the conserved promoter DNA recognition motifs such as the AT-rich recognition motif and the β-intercalating hairpin. In factor-independent T7 RNAP, mobility of the NTD enables conformational transitions from the promoter-bound nonprocessive initiation complex (where the AT-rich recognition motif and the β-intercalating hairpin are engaged in specific DNA contacts and DNA duplex melting) to the processive sequence–independent elongation complex (in which these structural elements locate far from DNA). Thus, the NTD of T7 RNAP is the major determinant of specific transcription initiation at early stages of transcription and a key contributor to the enzyme processivity during later stage of transcription. In factor-dependent RNAPs the role of NTD appears to be different. In hmtRNAP, the NTD is not capable of refolding. Instead, it serves primarily as a binding platform for transcription factors, TFAM and TFB2M for the transcription initiation and TEFM for the transcription elongation (16, 17). Only in complex with transcription factors TFAM and TFB2M, the AT-rich recognition motif and the β-intercalating hairpin of the hmtRNAP NTD participate in promoter DNA binding for positioning the transcription start site of DNA at the active site. In N4 RNAPII, the role of the NTD in promoter binding and melting appears to reduce even further; the AT-rich recognition motif and the β-intercalating hairpin locate at a short distance, unable to establish specific interactions with DNA (Fig. 2C). This finding suggests that the NTD of RNAPII serves merely as a platform for assembly with transcription factors that recruit the enzyme to premelted promoter DNA. Other evidence suggesting that RNAPII lacks capability for promoter recognition by itself is provided by its specificity loop. Although it maintains the same architecture as the one found in T7 RNAP, it lacks positively charged residues at the tip (Fig. 2B) and packs against the NTD (Fig. 1C), limiting its flexibility.
Presently, understanding of mechanisms of transcription by T7-like RNAPs is biased by availability of crystal structures obtained for enzymes with cores formed by single polypeptide chains. The overall geometry of heterodimeric N4 RNAPII shows good correlation with other members of the family (Fig. 1B). However, a heterodimeric nature of RNAPII appears to provide additional capabilities to the enzyme. First, the hinge-like organization of the dimerization site between gp15 and gp16 subunits (Fig. 4A) may be important for fast loading of TS DNA to the active site. Second, RNAPII dimerization is associated with the increased capacity of the catalytic cleft of the polymerase domain, which can accommodate a longer, up to 8 bp, DNA:RNA hybrid without structural constraints or rearrangements. Unconstrained accommodation of a growing DNA:RNA hybrid may represent an efficient mechanism optimized for rapid transition from initiation to elongation. As it has been shown for T7 RNAP, this enzyme remains bound to the promoter and translocates the active center along the template by the mechanism of DNA scrunching during early transcription initiation (5). Gradual accumulation of topological stress within the T7 RNAP initiation complex triggered by the growing DNA:RNA hybrid induces refolding of the enzyme to the elongation conformation. Transition from initiation to elongation has been shown to be a major barrier for many polymerases; this process constitutes a significant fraction of time required for transcription of an average gene and results in nonproductive reiterative cycles of RNA synthesis and abortion (28). Although additional crystallographic studies are required to address the architecture of the complete RNAPII initiation complex, heterodimeric organization of N4 RNAPII may represent the mechanism of adaptation for minimizing an energetic barrier on the way to processive transcription.
One of the evidences indicating that RNAPII evolved toward facilitated transition from transcription initiation to elongation is the finding of a preformed RNA exit pore in the promoter-bound enzyme not engaged in transcription. In both the T7-like single-subunit RNAPs and cellular multisubunit RNAPs, the RNA exit pore is formed in the course of extension of nascent transcript (15, 16, 22, 29). Funneling the 5′ end of transcript to the RNA exit pore after its separation from TS DNA greatly stabilizes the transcription complex and contributes to processive RNA synthesis. The presence of the RNA exit pore in the RNAPII prior to RNA synthesis suggests that the enzyme bypasses the need for structural rearrangements from transcription initiation to elongation stages.
We, however, note that there is one fundamental structural difference between RNAPII and hmtRNAP. Transition from initiation to elongation in hmtRNAP is accompanied by the formation of the RNA exit channel underneath the intercalating hairpin separating RNA from DNA at the upstream boundary of the DNA:RNA hybrid (15). An important role in maintaining the processivity of the hmtRNAP elongation complex plays the transcription factor TEFM that binds to the enzyme to cover the RNA exit channel turning it into a wide pore (16). In RNAPII the RNA exit pore exists prior to synthesis of an RNA transcript, and its parameters resemble that in T7 RNAP elongation complex rather than in hmtRNAP. The RNA exit pore in RNAPII supports the hypothesis that the enzyme performs all stages of transcription without undergoing major structural changes.
The structures of RNAPII also provide a clue about the most functional arrangement of domains in naturally split T7-like enzymes. There have been a number of studies reporting functional activities of T7 RNAP split into two to four fragments (splitting occurring in the NTD (residues 67 and 179) or the Fingers (residue 601)) as shown recently (27, 30). Surprisingly, such variants of T7 RNAP remained functional but showed decreased activities compared with the WT enzyme. A structural analysis shows that the observed reduced activities of split versions of T7 RNAP may be caused by their lower stabilities because of relatively small binding surfaces of the interacting polypeptides. In this regard, N4 RNAPII shows an example of a split T7-like RNAP with a flexible but stable subunit arrangement. If RNAPII were split into the NTD and the polymerase domain as in the split versions of T7 RNAP, the resulting heterodimer would be significantly less stable and, likely, prone to dissociation during transcription (Fig. S3B). The structures of RNAPII suggest that a compromise between maximum stability of the heterodimer and maintaining flexibility of the catalytic cleft may be achieved by splitting enzyme single polypeptide at the Palm subdomain.
Experimental procedures
Protein expression and purification
N4 RNAPII was prepared by co-expressing gp15 and N-terminal hexahistidine-tagged gp16 subunits in Escherichia coli strain BL21 transformed with plasmid pAD1 (25). RNAPII was purified by Ni2+-affinity chromatography and heparin column chromatography as described (18). Se-Met derivative of N4 RNAPII was expressed under the condition of suppressed methionine biosynthesis (31). The proteins were exchanged into the storage buffer (40 mm Tris-HCl, pH 8.0, 200 mm NaCl, 5 mm β-mercaptoethanol, and 0.1% Tween 20) by dialysis, concentrated to 20 mg/ml by centrifugation (Vivaspin 20 concentrator, Sartorius AG), flash-frozen in liquid nitrogen, and stored at −80 °C.
Crystallizations of the RNAPII–promoter DNA complex and elongation complex and determination of their structures by X-ray crystallography
For crystallization of the RNAPII–DNA complex, we used DNA oligonucleotide designed to self-anneal with the formation of a partially single-stranded hairpin-like template containing the consensus sequence of N4 middle promoter (Fig. 1A) (5′-CCCACCTGCAAAACGGTCTGCGAATCTCTCTGATTCGCAGACCGTTTT-3′). The RNAPII–DNA complex was formed by mixing equimolar amounts of N4 RNAPII (20 mg/ml) and DNA followed by incubation for 10 min at 22 °C. The crystals were obtained by hanging-drop vapor diffusion method at 22 °C with the crystallization solution containing 0.17 m sodium acetate, 0.085 m sodium cacodylate, pH 6.5, 15% PEG8000, 15% glycerol, and 5 μm spermine. Hexagonal crystals appeared overnight and reached their maximum dimensions of 0.2 × 0.2 × 0.1 mm in 3–4 days. Crystals were harvested from crystallization drops and directly frozen in liquid nitrogen.
Crystals of the N4 RNAPII elongation complexes were prepared by soaking RNA into the RNAPII–DNA complex crystals prepared as described above. DNA sequences used for crystallizations were 5′-AACCCACCAAAAAACGGTCTGCGAATCTCTCTGATTCGCAGACCGTTTT-3′ (EC scaffold 1) and 5′-CCCACCAAAAAAACGGTCTGCGAATCTCTCTGATTCGCAGACCGTTTT-3′ (EC scaffold 2) (Fig. 5, B and D). Crystals of RNAPII–DNA complexes were soaked in the crystallization solution supplemented by 0.5 mm RNA (RNA12, 5′-AAAAUUUGGUGG-3′ for EC1 and RNA8, 5′-UUUGGUGG-3′ for EC2) and incubated overnight at 4 °C. Crystals of RNAPII elongation complex containing the scaffold 2 were additionally soaked overnight in the crystallization solution supplemented with 1 mm GTP and 20 mm MgCl2. After soaking, crystals were harvested and directly frozen in liquid nitrogen.
The diffraction datasets for Se-Met crystals were collected at the X29 beamline of the National Synchrotron Light Source (Brookhaven National Laboratory, Upton, NY), and datasets for native crystals were collected at the F1 beamline of the Cornell High Energy Synchrotron Source (Cornell University, Ithaca, NY). The crystallographic datasets were processed using HKL2000 (32). The crystal structure of RNAPII–DNA complex was determined by selenium single-wavelength anomalous diffraction (SAD) method using the suite of programs PHENIX (33). The crystals containing Se-Met–labeled RNAPII belong to C2 space group with two RNAPII–DNA complexes per asymmetric unit, whereas the crystals containing native RNAPII belong to I4 space group with one RNAPII–DNA complex per asymmetric unit.
With the anomalous signal from Se-Met, 42 of a possible 44 selenium sites in the asymmetric unit were located and the experimental phase (figure of merit: 0.287) was calculated by using Automated Structure Solution (AutoSol) in PHENIX. Density modification by Automated Model Building (AutoBuild) in PHENIX yielded an excellent map and ∼86% of the model was built automatically. Manual model building of protein and DNA were done by Coot. The structures of binary and elongation complexes containing native RNAP were determined by the molecular replacement using Automated Molecular Replacement (Phaser-MR) in PHENIX. Final coordinates and structure factors have been deposited to the Protein Data Bank (PDB) with the accession codes listed in the Table S1.
In vitro transcription assay
N4 RNAPII and promoter DNA (same as used for crystallization of the binary complex) complex was assembled by incubating 5 μm DNA and 5 μm RNAPII in the transcription buffer (40 mm Tris-HCl, pH 7.9, 15 mm MgCl2, 5 mm β-mercaptoethanol) for 10 min at 22 °C. RNA transcriptions were initiated by adding 400 μm GTP or GTP and UTP along with 0.1 μCi of [γ-32P]GTP. The reactions were stopped after 10 min by adding an equal volume of the stop solution (90% formamide, 50 mm EDTA). The 32P-labeled RNAs were resolved by denaturing gel (20% acrylamide, 7 m urea) electrophoresis, visualized by Phosphor Imager Typhoon 9410 (GE Healthcare) and analyzed using the software Image Quant 5.1 (GE Healthcare).
Gel mobility shift assay
N4 middle promoter template (5′-CCCACCTGCAAAACGGTCTGCGAATCT CTCTGATTCGCAGACCGTTTT-3′) was labeled with 32P at the 5′ end using T4 polynucleotide kinase (New England Biolabs). 5 μm template was mixed with different amounts of N4 RNAPII in the transcription buffer (40 mm Tris-HCl, pH 7.9, 15 mm MgCl2, 5 mm β-mercaptoethanol), incubated for 10 min at 22 °C, and loaded to a nondenaturing 7% acrylamide gel. DNAs were visualized by Phosphor Imager Typhoon 9410 (GE Healthcare) and analyzed using the software Image Quant 5.1 (GE Healthcare).
Author contributions
V. M. and K. S. M. formal analysis; V. M. and K. S. M. investigation; V. M. writing-original draft; K. S. M. supervision; K. S. M. funding acquisition; K. S. M. validation; K. S. M. writing-review and editing.
Supplementary Material
Acknowledgments
We thank the staff at the NSLS and MacCHESS for support of crystallographic data collections. We thank Prof. Lucia Rothman-Denes for providing the N4 RNAPII expression vector and critical reading of the manuscript.
This work was supported by National Institutes of Health Grant GM087350 (to K. S. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4 and Tables S1 and S2.
The atomic coordinates and structure factors (codes 6DT7, 6DT8, and 6DTA) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- RNAP
- RNA polymerase
- vRNAP
- virion-encapsidated RNAP
- mtRNAP
- mitochondrial RNAP
- hmtRNAP
- human mitochondrial RNAP
- NTD
- N-terminal domain
- TS
- template-strand
- Se-Met
- selenomethionine.
References
- 1. Cermakian N., Ikeda T. M., Cedergren R., and Gray M. W. (1996) Sequences homologous to yeast mitochondrial and bacteriophage T3 and T7 RNA polymerases are widespread throughout the eukaryotic lineage. Nucleic Acids Res. 24, 648–654 10.1093/nar/24.4.648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chamberlin M., McGrath J., and Waskell L. (1970) New RNA polymerase from Escherichia coli infected with bacteriophage T7. Nature 228, 227–231 10.1038/228227a0 [DOI] [PubMed] [Google Scholar]
- 3. McAllister W. T., and Raskin C. A. (1993) The phage RNA polymerases are related to DNA polymerases and reverse transcriptases. Mol. Microbiol. 10, 1–6 10.1111/j.1365-2958.1993.tb00897.x [DOI] [PubMed] [Google Scholar]
- 4. Sousa R., Chung Y. J., Rose J. P., and Wang B. C. (1993) Crystal structure of bacteriophage T7 RNA polymerase at 3.3 Å resolution. Nature 364, 593–599 10.1038/364593a0 [DOI] [PubMed] [Google Scholar]
- 5. Cheetham G. M., Jeruzalmi D., and Steitz T. A. (1999) Structural basis for initiation of transcription from an RNA polymerase-promoter complex. Nature 399, 80–83 10.1038/19999 [DOI] [PubMed] [Google Scholar]
- 6. Cheetham G. M., and Steitz T. A. (1999) Structure of a transcribing T7 RNA polymerase initiation complex. Science 286, 2305–2309 10.1126/science.286.5448.2305 [DOI] [PubMed] [Google Scholar]
- 7. Durniak K. J., Bailey S., and Steitz T. A. (2008) The structure of a transcribing T7 RNA polymerase in transition from initiation to elongation. Science 322, 553–557 10.1126/science.1163433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tahirov T. H., Temiakov D., Anikin M., Patlan V., McAllister W. T., Vassylyev D. G., and Yokoyama S. (2002) Structure of a T7 RNA polymerase elongation complex at 2.9 Å resolution. Nature 420, 43–50 10.1038/nature01129 [DOI] [PubMed] [Google Scholar]
- 9. Yin Y. W., and Steitz T. A. (2002) Structural basis for the transition from initiation to elongation transcription in T7 RNA polymerase. Science 298, 1387–1395 10.1126/science.1077464 [DOI] [PubMed] [Google Scholar]
- 10. Jeruzalmi D., and Steitz T. A. (1998) Structure of T7 RNA polymerase complexed to the transcriptional inhibitor T7 lysozyme. EMBO J. 17, 4101–4113 10.1093/emboj/17.14.4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gleghorn M. L., Davydova E. K., Rothman-Denes L. B., and Murakami K. S. (2008) Structural basis for DNA-hairpin promoter recognition by the bacteriophage N4 virion RNA polymerase. Mol. Cell 32, 707–717 10.1016/j.molcel.2008.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Basu R. S., and Murakami K. S. (2013) Watching the bacteriophage N4 RNA polymerase transcription by time-dependent soak-trigger-freeze X-ray crystallography. J. Biol. Chem. 288, 3305–3311 10.1074/jbc.M112.387712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Murakami K. S., Davydova E. K., and Rothman-Denes L. B. (2008) X-ray crystal structure of the polymerase domain of the bacteriophage N4 virion RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 105, 5046–5051 10.1073/pnas.0712325105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ringel R., Sologub M., Morozov Y. I., Litonin D., Cramer P., and Temiakov D. (2011) Structure of human mitochondrial RNA polymerase. Nature 478, 269–273 10.1038/nature10435 [DOI] [PubMed] [Google Scholar]
- 15. Schwinghammer K., Cheung A. C., Morozov Y. I., Agaronyan K., Temiakov D., and Cramer P. (2013) Structure of human mitochondrial RNA polymerase elongation complex. Nat. Struct. Mol. Biol. 20, 1298–1303 10.1038/nsmb.2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hillen H. S., Morozov Y. I., Sarfallah A., Temiakov D., and Cramer P. (2017). Structural basis of mitochondrial transcription initiation. Cell 171, 1072–1081.e1010 10.1016/j.cell.2017.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hillen H. S., Parshin A. V., Agaronyan K., Morozov Y. I., Graber J. J., Chernev A., Schwinghammer K., Urlaub H., Anikin M., Cramer P., and Temiakov D. (2017). Mechanism of transcription anti-termination in human mitochondria. Cell 171, 1082–1093.e1013 10.1016/j.cell.2017.09.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Willis S. H., Kazmierczak K. M., Carter R. H., and Rothman-Denes L. B. (2002) N4 RNA polymerase II, a heterodimeric RNA polymerase with homology to the single-subunit family of RNA polymerases. J. Bacteriol. 184, 4952–4961 10.1128/JB.184.18.4952-4961.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kazmierczak K. M., Davydova E. K., Mustaev A. A., and Rothman-Denes L. B. (2002) The phage N4 virion RNA polymerase catalytic domain is related to single-subunit RNA polymerases. EMBO J. 21, 5815–5823 10.1093/emboj/cdf584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lenneman B. R., and Rothman-Denes L. B. (2015) Structural and biochemical investigation of bacteriophage N4-encoded RNA polymerases. Biomolecules 5, 647–667 10.3390/biom5020647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheetham G. M., and Steitz T. A. (2000) Insights into transcription: structure and function of single-subunit DNA-dependent RNA polymerases. Curr. Opin. Struct. Biol. 10, 117–123 10.1016/S0959-440X(99)00058-5 [DOI] [PubMed] [Google Scholar]
- 22. Basu R. S., Warner B. A., Molodtsov V., Pupov D., Esyunina D., Fernández-Tornero C., Kulbachinskiy A., and Murakami K. S. (2014) Structural basis of transcription initiation by bacterial RNA polymerase holoenzyme. J. Biol. Chem. 289, 24549–24559 10.1074/jbc.M114.584037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rothman-Denes L. B., and Schito G. C. (1974) Novel transcribing activities in N4-infected Escherichia coli. Virology 60, 65–72 10.1016/0042-6822(74)90366-3 [DOI] [PubMed] [Google Scholar]
- 24. vander Laan K., Falco S. C., and Rothman-Denes L. B. (1977) The program of RNA synthesis in N4-infected Escherichia coli. Virology 76, 596–601 10.1016/0042-6822(77)90241-0 [DOI] [PubMed] [Google Scholar]
- 25. Carter R. H., Demidenko A. A., Hattingh-Willis S., and Rothman-Denes L. B. (2003) Phage N4 RNA polymerase II recruitment to DNA by a single-stranded DNA-binding protein. Genes Dev. 17, 2334–2345 10.1101/gad.1121403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abravaya K., and Rothman-Denes L. B. (1989) In vitro requirements for N4 RNA polymerase II-specific initiation. J. Biol. Chem. 264, 12695–12699 [PubMed] [Google Scholar]
- 27. Shis D. L., and Bennett M. R. (2013) Library of synthetic transcriptional AND gates built with split T7 RNA polymerase mutants. Proc. Natl. Acad. Sci. U.S.A. 110, 5028–5033 10.1073/pnas.1220157110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tang G. Q., Roy R., Bandwar R. P., Ha T., and Patel S. S. (2009) Real-time observation of the transition from transcription initiation to elongation of the RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 106, 22175–22180 10.1073/pnas.0906979106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Steitz T. A. (2009) The structural changes of T7 RNA polymerase from transcription initiation to elongation. Curr. Opin. Struct. Biol. 19, 683–690 10.1016/j.sbi.2009.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Segall-Shapiro T. H., Meyer A. J., Ellington A. D., Sontag E. D., and Voigt C. A. (2014) A “resource allocator” for transcription based on a highly fragmented T7 RNA polymerase. Mol. Syst. Biol. 10, 742 10.15252/msb.20145299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Doublie S. (1997) Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 276, 523–530 10.1016/S0076-6879(97)76075-0 [DOI] [PubMed] [Google Scholar]
- 32. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 33. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.