

Abstract
The histone lysine methyltransferase nuclear receptor-binding SET domain protein 2 (NSD2, also known as WHSC1/MMSET) is an epigenetic modifier and is thought to play a driving role in oncogenesis. Both NSD2 overexpression and point mutations that increase its catalytic activity are associated with several human cancers. Although NSD2 is an attractive therapeutic target, no potent, selective, and bioactive small molecule inhibitors of NSD2 have been reported to date, possibly due to the challenges of developing high-throughput assays for NSD2. Here, to establish a platform for the discovery and development of selective NSD2 inhibitors, we optimized and implemented multiple assays. We performed quantitative high-throughput screening with full-length WT NSD2 and a nucleosome substrate against a diverse collection of bioactive small molecules comprising 16,251 compounds. We further interrogated 174 inhibitory compounds identified in the primary screen with orthogonal and counter assays and with activity assays based on the clinically relevant NSD2 variants E1099K and T1150A. We selected five confirmed inhibitors for follow-up, which included a radiolabeled validation assay, surface plasmon resonance studies, methyltransferase profiling, and histone methylation in cells. We found that all five NSD2 inhibitors bind the catalytic SET domain and one exhibited apparent activity in cells, validating the workflow and providing a template for identifying selective NSD2 inhibitors. In summary, we have established a robust discovery pipeline for identifying potent NSD2 inhibitors from small-molecule libraries.
Keywords: epigenetics, cancer, high-throughput screening (HTS), histone methylation, nucleosome, oncogene, small molecule, lysine methyltransferase inhibitor, MMSET, NSD2, WHSC1
Introduction
Epigenetic modifiers are widely recognized as targets for therapeutic intervention, due to their critical roles in regulating gene expression and chromatin integrity in addition to their dysregulation in a range of human pathologies. In particular, the nuclear receptor-binding SET domain (NSD)2 family of histone lysine methyltransferase enzymes, NSD1, NSD2/WHSC1/MMSET, and NSD3/WHSC1L1, have all been implicated as cancer therapeutic targets (1). The catalytic SET domain (Suppressor of variegation, Enhancer of zeste, Trithorax) of NSD family enzymes catalyzes the mono- and di-methylation of the ϵ-amine of lysine 36 of histone H3 (H3K36), utilizing the methyl donor S-adenosyl-l-methionine (SAM) (Fig. 1A). The nonoverlapping roles of the NSD family of enzymes in normal physiology are attributed to a variety of additional domains, including a PWWP (proline–tryptophan–tryptophan–proline) domain, plant homeodomain, zinc finger domains, and a high-mobility group box domain (1).
Figure 1.
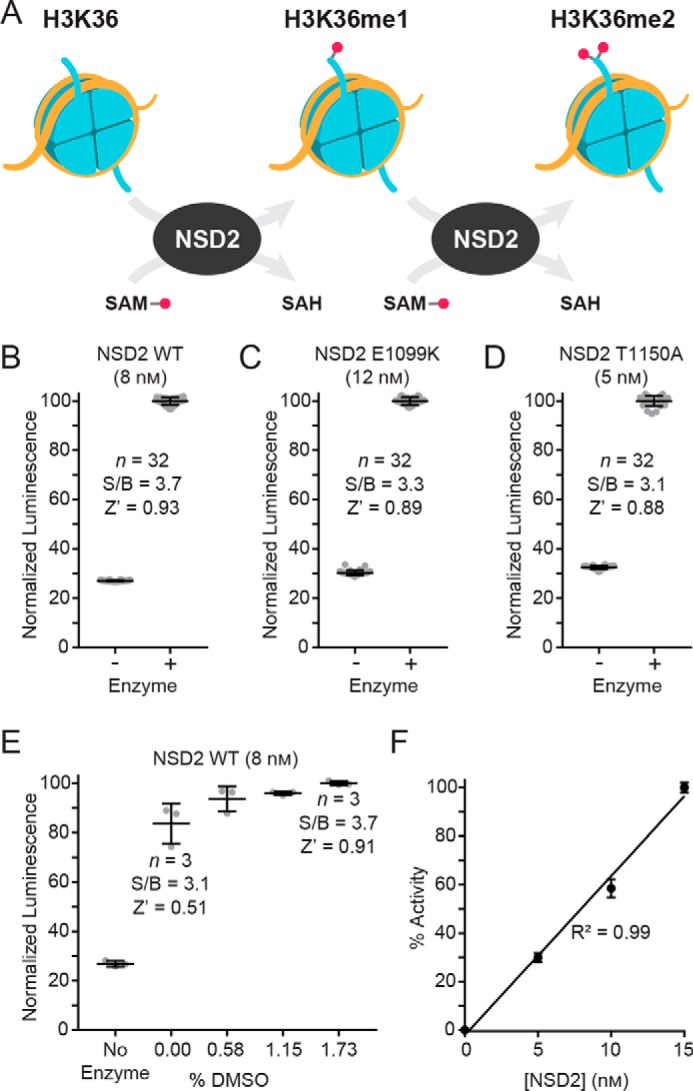
Optimization of the MTase-Glo primary assay. A, reaction scheme for the methylation of H3K36 by NSD2. B–D, optimized assay conditions for the full-length NSD2 WT (8 nm) (B), E1099K (12 nm) (C), or T1150A (5 nm) (D) enzymes with 500 nm nucleosomes, 0.58% DMSO, and 1 μm SAM. The mean normalized luminescence values ± S.D. (n = 32) are plotted for B–D. The assays are robust with S/B values >3 and Z′-factor values ∼0.9. E, titrations of DMSO demonstrated that the assay performance is not diminished by the introduction of vehicle up to 1.7% (mean ± S.D.; n = 3). F, linear correlation is observed between the WT NSD2 enzyme concentration and methyltransferase activity (mean ± S.D.; n = 8).
NSD2 has been implicated as a therapeutic target for a variety of cancers. Because the gene is located within the Wolf-Hirschhorn syndrome critical region of chromosome 4, NSD2 is also known as Wolf-Hirschhorn syndrome candidate 1 (WHSC1) (2). NSD2 was first described as a gene dysregulated by the t(4;14)(p16.3;q32.3) translocation in ∼15% of multiple myeloma (MM) cases and is called MMSET (2–4). The translocation results in a fusion transcript of NSD2 with the immunoglobulin heavy chain and increased NSD2 expression. The t(4;14) translocation can cause overexpression of both NSD2 and fibroblast growth factor receptor 3 (FGFR3) (2, 3). However, NSD2 is thought to be the primary oncogenic driver of the t(4;14)+ MM subtype because NSD2 is universally overexpressed, whereas FGFR3 is not expressed in ∼30% of MM cases (4–6). The role of NSD2 in driving t(4;14)+ MM pathogenesis was supported by knockdown of NSD2 in MM t(4;14)+ cell lines, which led to reduced growth and tumorigenesis (7–11). Conversely, transfection of t(4;14)− cells with NSD2 promotes tumorigenesis and oncogenic transformation of primary cells via elevated levels of dimethylated H3K36 (H3K36me2) (12). Numerous studies have linked increased expression of NSD2 with globally increased levels of H3K36me2 (9, 12–21). High expression of NSD2 protein has been demonstrated in many different human cancer types, including bladder, brain, gastrointestinal, lung, liver, ovary, skin, and uterus (18, 20, 22–28).
Notably, NSD2 is among the most frequently mutated genes in pediatric cancer genomes (29). The NSD2 SET domain variant, E1099K, was identified in both acute lymphoblastic leukemia tumors and cell lines with increased H3K36me2 that lack the t(4;14) translocation (21, 30). Sequence results of >1,000 pediatric cancer genomes, representing 21 different cancers, revealed the E1099K variant in 14% of t(12;21) ETV6-RUNX1 containing acute lymphoblastic leukemias (21). NSD2 is also among the most frequently mutated genes found in mantle cell lymphoma tumors, where both E1099K and T1150A variants are observed (31). The E1099K variant has also been reported in chronic lymphocytic leukemia (CLL) and lung and stomach cancers (32–35). Recombinant NSD2 E1099K showed higher in vitro activity compared with the WT enzyme (21). Ectopic expression of NSD2 E1099K induced H3K36me2 and promoted transformation, whereas knockdown of the enzyme reduced cell line proliferation and tumorigenesis (21).
Although NSD2 is an attractive therapeutic target, few small molecule inhibitors have been reported, and none demonstrate the desirable attributes of high-quality chemical probes (36). The compound LEM-06 (IC50 = 900 μm) was discovered by virtual screening against an NSD2 homology model (37). The antiparasitic drug suramin inhibits NSD2 (IC50 = 0.3–21 μm) but is a pan-inhibitor of methyltransferases (38, 39) as well as other enzymes (40). Likewise, the nonspecific histone lysine methyltransferase inhibitor chaetocin (IC50 = 3–6 μm) showed similar inhibition of NSD1–3 (39). The natural product sinefungin is a close structural analog of SAM and a modest inhibitor of NSD2 (IC50 = 26–30 μm) (41, 42). Structure–activity relationships have been reported for sinefungin analogs, the most potent of which inhibited the SET domains of NSD2 (IC50 = 1.8 μm) and SETD2 (IC50 = 0.29 μm) (41). Also, a peptide inhibitor of NSD2, PTD2 (IC50 = 3–22 μm), has been reported that was derived from the histone H4 sequence (43).
A major challenge in screening for small molecule inhibitors is that native NSD2 requires nucleosomes as a substrate (17). Interestingly, the apparent specific activity of NSD2 is higher with HeLa-derived nucleosomes compared with recombinant nucleosomes, which has been attributed to unknown modifications of the native substrate (17). Thus, native nucleosomes purified from HeLa are likely a more physiologically relevant substrate than recombinant nucleosomes. Recombinant NSD2 does not act on peptides and is thus not amenable to the commonly adapted histone-derived peptide screening platforms. Here, we report biochemical assay development and pilot-scale screening using full-length recombinant WT, E1099K, and T1150A NSD2 enzymes with native HeLa nucleosomes as a substrate. Chemical libraries were screened in a three-dose–point quantitative high-throughput screening (qHTS) format (44) with the Methyltransferase-Glo (MTase-GloTM) bioluminescence assay (45). A number of counter and orthogonal assays were also developed to characterize the hits from the primary screen. Confirmed hits were validated with a radiolabeled SAM substrate-based NSD2 activity assay and further interrogated with binding studies by surface plasmon resonance (SPR), methyltransferase profiling, and activity assessments in U-2 OS cells.
Results
Primary assay development and optimization
To identify small molecule inhibitors of the WT NSD2, the MTase-Glo assay was optimized for use as a primary assay, with whole nucleosomes as the substrate, in 1,536-well format with 4-μl reaction volumes (Fig. 1B). The MTase-Glo assay reagent measures methyltransferase activity through the coupling of the NSD2 reaction product S-adenosyl-l-homocysteine (SAH) to a bioluminescent signal (45). The assay was further optimized for the NSD2 variants E1099K (Fig. 1C) and T1150A (Fig. 1D), to enable insight into cross-inhibition of clinically-relevant NSD2 enzymes. The enzyme concentrations were optimized to allow a robust signal-to-background ratio (>3.0) in a 15-min reaction at room temperature while consuming ≤20% substrate. The three optimized assays were robust with Z′-factor values near 0.9. There was no reduction in assay performance in the presence of up to 1.7% DMSO, which represents a triple aliquot of fixed volume (23 nl) transferred via a Kalypsys pintool equipped with a 1,536-pin array (Fig. 1E) (46). A titration of WT NSD2 demonstrated a linear correlation with methyltransferase activity as expected (Fig. 1F).
qHTS for NSD2 inhibitors
The primary screen was carried out by performing qHTS against 16,251 compounds at three concentrations (115, 57.5, and 11.5 μm) in 1,536-well plates (Table 1). Commercially-available libraries screened included the LOPAC (1,280 compounds), Prestwick (1,360 compounds), MicroSource (2,000 compounds), and Tocris (1,304 compounds) collections. Additionally, four NCATS libraries were screened, including an epigenetics-focused collection (284 compounds), a natural products library (2,108 compounds), the NPACT library (5,099 compounds), and the NCATS Pharmaceutical Collection (2,816 compounds) (47). These libraries, enriched with pharmacologically active compounds, were selected to evaluate the suitability of the primary and secondary assays to identify NSD2 inhibitors. The overall quality of the primary screen data from 43 plates was high, with average values for the Z′-factor of 0.92 ± 0.02, signal-to-background (S/B) of 3.28 ± 0.31, and coefficient of variation (CV) for the DMSO control of 1.68 ± 0.49% (Fig. 2, A–C). The screen was conducted over a period of 4 months, so these data demonstrate excellent day-to-day reproducibility.
Table 1.
Optimized protocol for the NSD2 MTase-Glo UltraGlo luciferase qHTS assay

Figure 2.

Quantitative high-throughput screening and secondary screening for inhibitors of NSD2 WT and variant enzymes. A–D, statistics from the qHTS primary screen against full-length WT NSD2 with the MTase-Glo methyltransferase assay for 43 1,536-well plates. A, overall assay performance of the primary screen was robust with an average Z′-factor value of 0.92. B, average signal-to-background ratio was 3.28. C, average % CV for the DMSO control was 1.68. D, of the 16,251 compounds screened, 536 were identified as active (3.3% hit rate). E, primary and secondary assay triage process. Eight compound libraries (16,251 compounds) were screened in qHTS format at three concentrations, resulting in 536 hits. From the initial hit list, 289 compounds were selected as cherry picks and further evaluated in 11-point dose response. Of the 289 cherry picks, the activities of 48 were confirmed against NSD2 with the primary and orthogonal assays with no activity observed by the counter assays. The majority of these compounds was also active against the NSD2 variants E1099K (45 compounds) and T1150A (44 compounds).
Hit confirmation and secondary screening
Of the 16,251 compounds evaluated in the primary screen, 536 compounds were active with a maximum response ≥50%, corresponding to a hit rate of 3.3% (Fig. 2D). Promiscuity scores were calculated for each hit, and any compounds with a promiscuity score higher than 0.2 were eliminated from the cherry-picking. Next, the remaining hits were evaluated by a structural filter to eliminate the pan-assay interference compounds (PAINS) (48). Finally, 289 hit molecules were selected as cherry picks and prepared in 11-point concentration series from library stock solutions for further studies (Fig. 2E).
First, 174 of the cherry picks were confirmed as active in the primary assay (a 60% confirmation rate). Actives were defined as having concentration-response curves (CRCs) in the classes of 1.1, 1.2, 2.1, 2.2, and 3. In brief, classes 1.1 and 1.2 are the highest-confidence complete CRCs containing both upper and lower asymptotes with efficacies >80% and ≤80%, respectively. Classes 2.1 and 2.2 are incomplete CRCs having only one asymptote with efficacies >80% and ≤80%, respectively. Class 3 CRCs show activity at only the highest concentration or are poorly fit. Class 4 CRCs are inactive, having a curve-fit of insufficient efficacy or lacking a fit altogether (44). Second, an MTase-Glo counter screen was implemented without NSD2 but containing 200 nm SAH (mimicking 20% substrate conversion), which identified 37 assay interference compounds that might act by inhibiting the coupling enzymes, luciferase or the luminescent signal. Potential redox cycling by compounds was assessed with an Amplex Red assay performed in the presence of reducing agents (49), and 63 compounds were found to be active with a threshold of 3σ. To provide additional evidence for on-target activity against NSD2, the orthogonal EPIgeneous homogeneous time-resolved fluorescence (HTRF) methyltransferase assay was utilized with reaction conditions identical to the primary assay (Fig. 3). The assay measures the NSD2 reaction product SAH, which competitively displaces d2-labeled SAH that is pre-bound to anti-SAH labeled with Lumi4-Tb, resulting in a loss of FRET signal (50).
Figure 3.
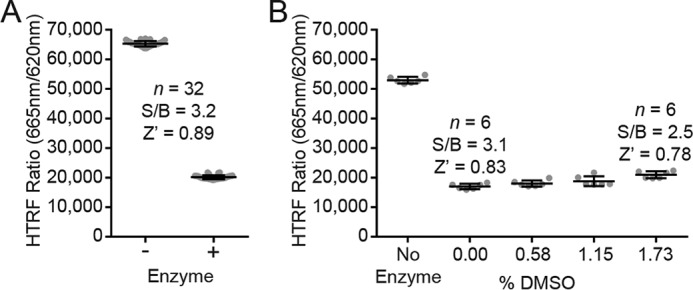
Optimized EPIgeneous HTRF methyltransferase assay with the full-length NSD2 WT enzyme (8 nm), 500 nm nucleosomes, 0.58% DMSO, and 1 μm SAM. A, mean HTRF ratio values ± S.D. for n = 32 replicates are plotted, showing robust performance with S/B values >3 and a Z′-factor value of ∼0.9. B, titrations of DMSO demonstrated that the assay performance is not diminished by the introduction of vehicle (mean ± S.D.; n = 6).
Among the 37 compounds found to be active with the MTase-Glo counter assay was a known luciferase inhibitor NCGC00183809 (Fig. 4A). The compound shares structural commonality with the firefly luciferase inhibitor PTC-124 (51, 52). As such, it was likely a false-positive hit that inhibits the UltraGlo luciferase enzyme utilized in the MTase-Glo assay. This notion was supported by the results of the orthogonal assay, which did not indicate inhibition of NSD2 activity. UltraGlo luciferase is a genetically evolved firefly luciferase containing 70 mutations to improve its robustness, thermal stability, and resistance to interference compounds (45, 53, 54), which is consistent with the 158-fold reduced potency of NCGC00183809 to UltraGlo compared with firefly luciferase (Fig. 4A). The nonspecific methyltransferase inhibitor 3-deazaneplanocin A (DZNep) showed similar activities in both the MTase-Glo primary and counter assays; however, no inhibition of NSD2 activity was observed with the orthogonal assay (Fig. 4B).
Figure 4.

Primary and secondary assays characterize hit compounds against full-length NSD2 WT and variant enzymes. A, NCGC00183809 was identified by the primary assay (IC50 = 2.2 μm; Hill slope = −1.84) and contains the PTC-124 moiety (red) (51, 52). The compound showed similar activity with the counter assay (IC50 = 1.8 μm; Hill slope = −1.85) but was substantially more potent against firefly luciferase (IC50 = 11 nm; Hill slope = −2.26). No activity against NSD2 was observed with the EPIgeneous HTRF orthogonal assay. The data of WT NSD2 MTase-Glo are shown as the mean value ± S.D. of n = 2 technical replicates. B, DZNep showed similar activities between the primary (IC50 = 1 μm; Hill slope = −1.63) and counter assays (IC50 = 0.87 μm; Hill slope = −1.95). No activity was observed with the orthogonal assay. C, confirmation of chaetocin as an NSD2 inhibitor. Chaetocin was identified as a hit from the primary screen, and the activity was confirmed by primary (IC50 = 8.5 μm; Hill slope = −1.09) and orthogonal assays (IC50 = 67 μm; Hill slope = −1.37). No activity was observed with the MTase-Glo counter assay or Amplex Red assay. Chaetocin also inhibits the NSD2 variants E1099K (IC50 = 19 μm; Hill slope = −0.93) and T1150A (IC50 = 9.6 μm; Hill slope = −1.15). Data from the MTase-Glo Counter and Amplex Red assays resulted from single experiments. All other data are shown as mean values ± S.D. of n technical replicates (n = 3 for WT NSD2 MTase-Glo and n = 2 for HTRF orthogonal, NSD2 E1099K, and NSD2 T1150A). D, primary assay showed inhibition of WT (IC50 = 0.9 μm; Hill slope = −1.4), E1099K (IC50 = 2.8 μm; Hill slope = −1.51), and T1150A NSD2 (IC50 = 2.9 μm; Hill slope = −1.44) enzymes by DA3003-1, whereas no activity was observed with the counter assay. Inhibition of WT NSD2 was further supported by the orthogonal assay. However, DA3003-1 showed activity with the Amplex Red assay (IC50 = 0.9 μm; Hill slope = 1.25) indicating redox activity.
After the orthogonal assay, 48 confirmed hits remained that were not active in the two counter screens (Fig. 2E). Of the 48 confirmed inhibitors, 45 were also active against the E1099K enzyme, and 44 showed activity against the T1150A variant. One of these hits is the nonspecific histone lysine methyltransferase inhibitor chaetocin, which was reported to inhibit NSD1, NSD2, and NSD3 (39). Chaetocin inhibited both WT and variant NSD2 enzymes without showing activity by the MTase-Glo counter assay or Amplex Red assay (Fig. 4C). The hit DA3003-1 is known to be redox-active (49, 55), and this was corroborated by the Amplex Red counter screen (Fig. 4D). Redox activity is undesirable, because it can result in nonspecific modulation of proteins, activation of cell pathways with redox-switches, and cytotoxicity (56). DA3003-1 was nevertheless selected for follow-up studies due to its submicromolar potency with the primary assay (IC50 = 0.9 μm). In addition to chaetocin and DA3003-1, three other hits were selected for further studies that have not been previously linked to NSD2 inhibition: PF-03882845, TC LPA5 4, and ABT-199 (Table 2). In anticipation of a full-fledged HTS, these five compounds were selected to validate our post-HTS workflow, which is intended to further evaluate compound activities.
Table 2.
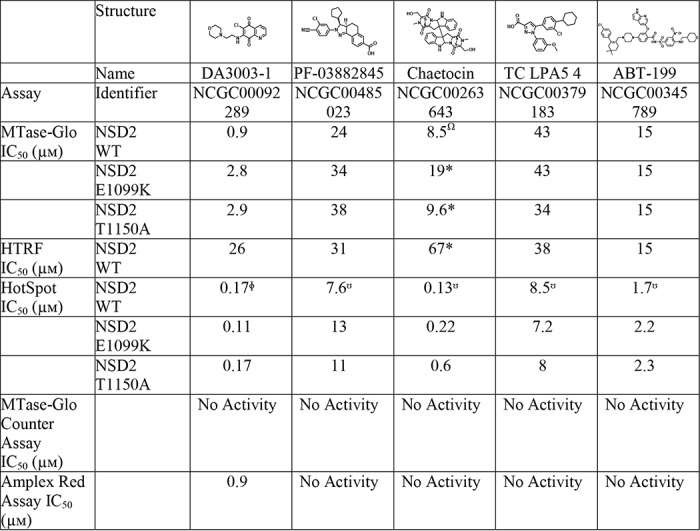
Biochemical activities of five compounds that inhibit NSD2 WT and variant enzymes
All values are derived from one experiment unless followed by a symbol, in which case the value represents a global fit to n technical replicate experiments (*, n = 2; Ω, n = 3; Φ, n = 4; ☋, n = 5).
Potency evaluation with the HotSpot radiolabel assay
Biochemical activities of the five selected hit compounds against WT and variant NSD2 enzymes were further validated by the radioisotope-based HotSpot assay (Fig. 5). The HotSpot assay incorporates [3H]SAM to assess total histone methylation by direct measurement of the filter-bound tritiated substrate, without the need for coupling enzymes or antibodies (38). Both DA3003-1 and chaetocin inhibited WT and variant NSD2 enzymes with submicromolar potencies, whereas PF-03882845, TC LPA5 4, and ABT-199 inhibited the enzymes at low-micromolar concentrations (Table 2). Although chaetocin is known to inhibit NSD2, inhibition by DA3003-1, PF-03882845, TC LPA5 4, and ABT-199 has not been reported. Notably, the HotSpot assay was consistently more sensitive than the MTase-Glo and HTRF assays for both WT and variant enzymes (Table 2). Overall, the reaction conditions were very similar. The MTase-Glo and HTRF assays utilized 8 nm NSD2, 500 nm nucleosomes, 50 mm Tris-HCl, pH 8.8, 5 mm MgCl2, 50 mm NaCl, 1 mm TCEP, 0.01% Tween; and the HotSpot assay used 10 nm NSD2, 400 nm nucleosomes, 50 mm Tris-HCl, pH 8.5, 5 mm MgCl2, 50 mm NaCl, 1 mm DTT, 0.01% Brij35. A comparison of IC50 values determined by the HotSpot assay for all five compounds from reactions containing either 1 mm TCEP or DTT did not suggest that potency differences were due to the reducing agent (data not shown). Each of the five compounds inhibited WT, E1099K, and T1150A NSD2 enzymes with similar potencies (Table 2).
Figure 5.
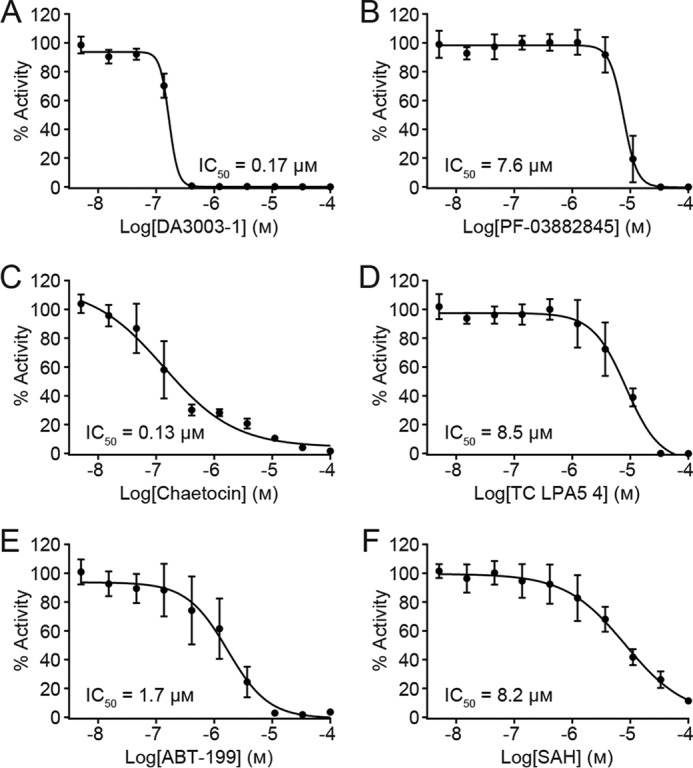
Inhibition of full-length WT NSD2 activity toward nucleosomes measured by the radiolabeled HotSpot assay with a 10-point concentration series of inhibitor. DA3003-1 potently inhibits NSD2 with an IC50 value of 0.17 μm (Hill slope = −5.57; n = 4) (A); PF-03882845 inhibits NSD2 activity with an IC50 value of 7.6 μm (Hill slope = −3.68; n = 5) (B); chaetocin is a potent inhibitor of NSD2 activity (IC50 = 0.13 μm; Hill slope = −0.71; n = 5) (C); TC LPA5 4 inhibits NSD2 with an IC50 value of 8.5 μm (Hill slope = −1.5; n = 5) (D); ABT-199 inhibits NSD2 with an IC50 value of 1.7 μm (Hill slope = −1.23; n = 5) (E). The positive control SAH inhibits NSD2 with an IC50 value of 8.2 μm (Hill slope = −0.85; n = 4) (F). Data are plotted as the mean value ± S.D. of n technical replicate experiments.
Direct binding of inhibitors to the NSD2 SET domain
To support an on-target mechanism of action for the five inhibitors, SPR was used to determine whether each inhibitor interacts with the catalytic SET domain of NSD2. As expected, the two positive controls, cofactor SAM and product SAH, both bound the NSD2 SET domain with low micromolar affinities (Table 3). With the exception of chaetocin, the inhibitors bound the NSD2 SET domain stoichiometrically, with dissociation constants (Kd) comparable with the in vitro IC50 values determined by the HotSpot assay (Table 2). Chaetocin bound the SET domain with an apparent dissociation constant of 20 nm. Additionally, the data indicated super-stoichiometric binding, which might be due to a binding ratio higher than 1:1 that is consistent with chaetocin's two disulfide moieties forming adducts with the NSD2 protein or compound aggregation (data not shown) (57). These data indicate that all five compounds might mediate inhibition of NSD2 by directly binding to the catalytic SET domain.
Table 3.
Direct binding of hit compounds to the NSD2 SET domain as measured by surface plasmon resonance, with SAH and SAM as positive controls
| Compound name | kon | koff | Kd |
|---|---|---|---|
| m−1 s−1 | s−1 | μm | |
| SAH | 2.33 × 103 | 1.19 × 10−2 | 5.1 |
| SAM | 4.06 × 103 | 1.70 × 10−2 | 4.2 |
| DA03003-1 | 3.67 × 104 | 1.34 × 10−2 | 0.37 |
| PF-03882845 | 8.55 × 103 | 3.31 × 10−2 | 3.9 |
| Chaetocin | 2.21 × 103 | 4.59 × 10−5 | 0.02 |
| TC LPA5 4 | 4.29 × 103 | 3.59 × 10−2 | 8.4 |
| ABT-199 | 4.39 × 103 | 3.64 × 10−2 | 8.3 |
Selectivity assessment by methyltransferase profiling
Methyltransferase profiling of the five NSD2 inhibitors was performed to examine the selectivity toward NSD2 compared with other methyltransferases. Activities of the inhibitors were evaluated against 35 other methyltransferases, including NSD1 and NSD3, with the HotSpot methyltransferase assay technology (Table 4). DA3003-1 was the least selective methyltransferase inhibitor and showed potent inhibition of nearly every enzyme except DOT1L (no activity) and GLP (weak inhibition). Furthermore, DA3003-1 inhibited 27 enzymes potently with submicromolar IC50 values. Both PF-03882845 and TC LPA5 4 inhibited most enzymes, albeit with weak potencies, but showed some selectivity to the PRMT5–MEP50 complex and the MLL4 complex. Chaetocin inhibited 18 methyltransferases, 12 with submicromolar potencies (including all NSD enzymes) and two with IC50 values above 75 μm. ABT-199 inhibited 23 methyltransferases, two of which have IC50 values above 65 μm. The NSDs were among the most potently inhibited enzymes, with IC50 values in the low-micromolar range.
Table 4.
Selectivity of inhibitor compounds toward WT NSD2 was assessed by profiling 37 other methyltransferases, including the E1099K and T1150A NSD2 variants, with the HotSpot methyltransferase assay
The IC50 values for all test compounds are colored as a heat map with green indicating potent inhibition and red indicating weak inhibition. No color indicates no measurable activity. All values are derived from a single experiment except for those of WT NSD2, which result from a global fit of multiple data sets as indicated in Table 2.

Inhibitor activities in a cell-based assay
To evaluate the activities of the five NSD2 inhibitors in cells, U-2 OS human osteosarcoma cells were chosen due to a relatively high expression of endogenous NSD2 protein (18). To establish a baseline for reduced NSD2 activity, siRNA was used to knock down NSD2 in U-2 OS cells (Fig. 6A). Variations in NSD2 transcript knockdown were observed among the three unique siRNA molecules, and greater reductions in NSD2 corresponded to lower levels of H3K36me2 (Fig. 6, B and C). Knockdown of NSD2 did not appear to reduce the proliferation of U-2 OS cells (Fig. 6D). Next, the cells were treated with each of the five compounds in 10-point dose response from 0.0025 to 50 μm for 96 h. NSD2 inhibition should result in reduced H3K36me2. Total histone H3 and H3K36me2 levels were measured by Western blot analysis. The positive control DZNep was tested at a concentration of 10 μm in parallel with each compound and was also evaluated in dose response (Fig. 6E). Densitometry was used to quantify both H3K36me2 and total H3, and the densities of H3K36me2 were normalized to those of H3 (Fig. 6F). The growth of U-2 OS cells over 96 h in the presence of test compounds at the same concentrations was also evaluated (Fig. 6G). The IC50 value of DZNep for reducing H3K36me2 levels in U-2 OS cells was 390 nm with a modest dose-dependent reduction in U-2 OS confluency (IC50 = 180 nm). Similar to DZNep, DA3003-1 treatment also resulted in a dose-dependent reduction in H3K36me2 (IC50 = 545 nm); however, higher drug concentrations resulted in cytotoxicity (CC50 = 270 nm) similar to chaetocin and the control bortezomib, both of which were cytotoxic at all concentrations tested. PF-03882845 reduced H3K36me2 (IC50 = 3.2 μm) over a range of concentrations that had negligible influence on growth; however, substantial cell death at concentrations above 16.7 μm was observed during the Western blot analysis experiment (Fig. 6E), so data from the two highest concentrations were excluded from the potency calculation. TC LPA5 4 did not reduce H3K36me2 over the concentrations tested, and minimal reduction in growth was observed. ABT-199 modestly reduced H3K36me2 levels at 16.7 μm; however, higher concentrations were cytotoxic.
Figure 6.

Analysis of siRNA and inhibitor activities toward U-2 OS human osteosarcoma cells. NSD2 transcript levels in U-2 OS cells were reduced by a 72-h transfection with siWHSC1 (A), siWHSC1 (B), or siWHSC1 (C) compared with siRNA control or transcript levels in cells that were nontransfected. B, Western blot analysis of the same cell samples in A indicates that the level of H3K36me2 reduction corresponds to the extent of NSD2 transcript knockdown. C, densitometry was performed with the Western blotting in B, and the H3K36me2 values were normalized to those of total H3. D, proliferation of U-2 OS cells 72 h after a 72-h transfection with the indicated siWHSC1. E, U-2 OS cells were treated with the indicated concentrations of test compounds or 10 μm of the global histone methyltransferase inhibitor DZNep, which served as a positive control. After 96 h of treatment, whole-cell lysates were subjected to Western blotting analyses. The blots were probed with anti-histone H3K36me2 and anti-histone H3 antibodies, respectively. F, densitometry was performed, and the values for H3K36me2 densities were normalized to the total H3 densities. The resulting data for DZNep (IC50 = 390 nm; Hill slope = −1.34), PF-03882845 (IC50 = 3.2 μm; Hill slope = −1.74), DA3003-1 (IC50 = 545 nm; Hill slope = −0.81), and ABT-199 were plotted and fit to a nonlinear four-parameter equation. The data for TC LPA5 4 was fit to a linear equation. G, U-2 OS cells were treated with the indicated concentrations of test compounds, and percent confluency of the cells at 96 h is shown as area under the curve. Bortezomib was included as a control for cytotoxicity.
Discussion
A number of histone lysine methyltransferases have been implicated as attractive therapeutic targets in the field of oncology, and small molecule inhibitors are in different stages of preclinical and clinical development (59, 60). Selective inhibitors of NSD2 are of major interest both to advance basic research and for therapeutic development. However, NSD2 is regarded as a challenging target (61), and no selective small molecule inhibitors of NSD2 have been reported to date. Challenges in studying NSD2 in vitro include that the target of NSD2 methylation depends on the nature of the substrate (17), and full-length NSD2 is only active against a nucleosome substrate (38), which may be cost prohibitive in many cases.
A wide variety of methyltransferase assays have been described in the literature (38, 45, 62–67); however, few optimized assays have been reported for NSD2 in plate formats to enable HTS. Recently, a radiolabeled [3H]SAM assay was optimized for the NSD2 SET domain with both histone octamer (Z′ = 0.69) and nucleosome (Z′ = 0.75) substrates in a 384-well format (39). The assay utilizing a nucleosome substrate was used to screen 1,040 compounds from the Prestwick Chemical Library at a single concentration of 25 μm. Although the reported assays show robust performance in the 384-well format, the use of radiolabeled reagents for HTS is a challenge for many laboratories due to safety regulations and disposal costs.
Herein, we report the implementation and validation of two optimized homogeneous NSD2 activity assays in the highly miniaturized 1,536-well format for the identification of small molecule inhibitors from chemical libraries. The assays were utilized to screen the full-length WT NSD2 enzyme against a nucleosome substrate in qHTS format with three concentrations of test compounds. The use of qHTS reduces both false-positive and false-negative hits common to single-point HTS and facilitates selection of actives (44). For the primary screen, we used the recently reported Methyltransferase-Glo assay reagent with a sensitive bioluminescent readout (45). A similar approach has been applied for the discovery of NSD1 inhibitors by HTS (68). After screening eight libraries, including numerous pharmacologically active collections, containing over 16,000 compounds, many hits were identified, including chaetocin, which is known to inhibit NSD2 (39). By incorporating orthogonal and counter screens, hits were prioritized for subsequent follow-up studies.
Among the confirmed active inhibitors, DA3003-1, PF-03882845, chaetocin, TC LPA5 4, and ABT-199 were selected for further characterization. In vitro potencies were determined by the HotSpot assay, which is a direct readout of the NSD2 reaction product (38). The HotSpot assay is very similar in format to the traditional gold standard radioisotope detection used in conjunction with gel electrophoresis or MS. The five compounds inhibited WT NSD2 as well as the E1099K and T1150A variant enzymes.
DA3003-1 is a cell-permeable Cdc25 phosphatase inhibitor that potently and irreversibly inhibits all Cdc25 isoforms, including Cdc25A (IC50 = 29 nm), Cdc25B2 (IC50 = 95 nm), and Cdc25C (IC50 = 89 nm) (69). It is known that DA3003-1 is capable of redox cycling (49, 55), which was verified here by the Amplex Red counter screen. In addition to potent inhibition of NSD2 activity, our data demonstrate that DA3003-1 bound the SET domain with a strong affinity (Kd = 370 nm, Table 3), which is comparable with its potency (IC50 = 170 nm, Table 2). Together, this suggests that DA3003-1 inhibits NSD2 through a direct interaction with the catalytic SET domain, although it is most likely nonspecific. Notably, DA3003-1 inhibited 27 of the 38 methyltransferase enzymes tested with submicromolar potencies and another eight with IC50 values near 1 μm. The potency of DA3003-1 in cells with respect to reducing H3K36me2 (IC50 = 545 nm) corresponds to cytotoxicity (CC50 = 270 nm).
The Pfizer compound PF-03882845 is a highly potent mineralocorticoid receptor antagonist (IC50 = 0.755 nm) (70). In comparison, it inhibited NSD2 in vitro with an ∼10,000-fold weaker potency (IC50 = 7.6 μm) and bound the SET domain (Kd = 3.9 μm) within 2-fold of the IC50 value. Profiling studies indicated that PF-03882845 inhibits many other methyltransferases with modest potencies, although it inhibits the histone arginine methyltransferase PRMT5 with a submicromolar potency. Interestingly, PF-03882845 reduced H3K36me2 in cells with a potency (3.2 μm) within 2-fold of the biochemical IC50 value. Concentrations at and above 17 μm resulted in substantial cell death in the Western blot analysis experiment, but only minimal reductions in cell growth were observed with up to 50 μm drug at 96 h in the cell growth assay.
TC LPA5 4 was first reported by Sanofi Aventis as a selective lysophosphatidic acid receptor 5 (LPA5) antagonist that inhibited LPA-mediated human platelet aggregation with an IC50 value of 2.2 μm (71). The biochemical potency of TC LPA5 4 against NSD2 (IC50 = 8.5 μm) was nearly identical to its affinity to the catalytic SET domain (Kd = 8.4 μm). The methyltransferase activity profiles of TC LPA5 4 and PF-03882845 showed striking similarities (Table 4). Both compounds inhibited WT NSD2 with an IC50 value near 8 μm. Also, both compounds were most potent against the PRMT5–MEP50 complex with nearly identical IC50 values. Furthermore, both compounds had ∼3-fold weaker potency against the MLL4 complex. Compared with the PRMT5–MEP50 complex, the potency values of PF-03882845 were at least 8-fold weaker against 31 other methyltransferases. Similarly, compared with the PRMT5–MEP50 complex, the potency value of TC LPA5 4 is at least 7-fold weaker against 28 other methyltransferases. No activity or toxicity was observed for TC LPA5 4 in U-2 OS cells.
The fungal mycotoxin chaetocin is known to inhibit NSD2 (39), so identifying it as a hit further validated our screening approach. Of the 38 methyltransferases profiled, chaetocin inhibited 12 with submicromolar potencies. Notably, the methyltransferase profiling indicated chaetocin potencies of 740 nm against SUV39H1 and 570 nm against SUV39H2, which is consistent with a previously reported value of 600 nm (72). Chaetocin was initially reported to be a specific inhibitor of the histone lysine methyltransferase SU(VAR)3-9 both in vitro and in vivo (72). However, the two disulfide bonds of chaetocin can complicate bioassay interpretation because of the potential for redox activity and covalent modification of proteins (36). Indeed, reports have indicated that the activity against histone lysine methyltransferases is due to chemical modification of the enzyme by the disulfide groups (73, 74). Superstoichiometric binding of chaetocin to the NSD2 SET domain was observed by surface plasmon resonance, which might be due to the formation of direct compound–thiol adducts. The affinity of chaetocin to the NSD2 SET domain was strong (Kd = 20 nm) with a biochemical potency about 7-fold weaker. Chaetocin was cytotoxic to U-2 OS cells at all concentrations tested.
ABT-199, also known as GDC-0199 or venetoclax, binds BCL-2 with a subnanomolar affinity (Ki <0.010 nm) and is approved by the Food and Drug Administration for the treatment of CLL (75, 76). The biochemical potency of ABT-199 against WT NSD2 (IC50 = 1.7 μm) was similar against the NSD2 variants, NSD1 and NSD3 (Table 4). The compound was most potent against the MLL1 and MLL4 complexes. The affinity of ABT-199 to the NSD2 SET domain (Kd = 8.3 μm) is nearly 5-fold weaker than the biochemical potency. At a concentration of 50 μm, ABT-199 was cytotoxic, consistent with its use in oncology, but any influences on cellular H3K36me2 levels were negligible.
DZNep is a carbocyclic analog of adenosine and a derivative of the antibiotic neplanocin-A. It was initially reported as a competitive inhibitor of SAH hydrolase at picomolar concentrations (77). DZNep has since been reported as a global histone methylation inhibitor when used at substantially higher concentrations (36, 78, 79). Although DZNep was identified as a hit from the primary screen, similar activities were observed with the MTase-Glo primary and counter assays, and no inhibition of NSD2 activity was observed with the orthogonal assay. The potency of DZNep in reducing H3K36me2 in U-2 OS cells (IC50 = 390 nm; Fig. 6F) is similar to its potency in reducing H3K27me3 in SU-DHL-6 cells (IC50 = 160 nm) when assessed by the same method of Western blot analysis (80). DZNep has been also shown to reduce H3K36me2 in SW480 cells at a concentration of 5 μm (81).
The purpose of this study was to establish an HTS discovery pipeline for NSD2 and to evaluate the workflow for identifying high-quality tool inhibitors of NSD2. The majority of the molecules screened were from pharmacologically active libraries that served to validate the primary and secondary assays. The identification of known methyltransferase inhibitors, including chaetocin and DZNep, further validated the workflow. During this pilot, many known interference compounds were identified by the secondary assays, thereby demonstrating how such bad actors behave among the various assays. Five actives selected from the primary screen were shown to bind the catalytic SET domain and inhibit NSD2 activity in vitro. Although these studies confirm inhibition of NSD2, they do not rule out inhibition by intractable mechanisms of action, such as nonspecific reactivity, redox, or aggregation. Two of the five compounds reduced H3K36me2 in U-2 OS cells, but the mechanisms are likely to be complicated and involve multiple targets. These studies provide a basis for the future discovery and development of novel selective NSD2 inhibitors by establishing a robust workflow for identifying and triaging hits from high-throughput screens.
Experimental procedures
Chemicals, reagents, and libraries
MTase-Glo (V7602) was purchased from Promega, Inc. (Madison, WI). The EPIgeneous HTRF methyltransferase assay (catalog no. 62SAHPEB) was purchased from Cisbio (Bedford, MA). Dimethyl sulfoxide (DMSO) was purchased from ThermoFisher Scientific (Pittsburgh, PA). The libraries screened include the following: Library of 1280 Pharmacologically Active Compounds (LOPAC1280; Sigma); Tocris (1,304 compounds, Tocris Bioscience, Bristol, UK); Prestwick (1,360 compounds, Prestwick Chemical, San Diego); MicroSource (2,000 compounds, MicroSource Discovery Systems, Gaylordsville, CT); NPC (the NCATS Pharmaceutical Collection (2,816 compounds) (47); NPACT (NCATS Pharmacologically Active Chemical Toolbox, 5,099 compounds); an epigenetic collection (284 compounds); and a natural products library (2,108 compounds). The reference compound DZNep was purchased from Selleckchem and dissolved with DMSO to a 10 mm stock. The U-2 OS human osteosarcoma cell line and McCoy's 5A medium were purchased from American Type Culture Collection (Manassas, VA). U-2 OS cells were grown in McCoy's 5A medium supplemented with 10% fetal bovine serum (FBS), 100 μg/ml penicillin, and 100 μg/ml streptomycin. Cultures were maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. The 12% BisTris gel, nitrocellulose membrane, Lipofectamine RNAiMAX reagent, and High-Capacity RNA-to-cDNA kit were purchased from ThermoFisher Scientific. The RNeasy mini kit and AllStars Hs Cell Death Control siRNA were obtained from Qiagen (Germantown, MD). The TaqMan Gene Expression Master Mix and TaqMan WHSC1 human (Hs00983720_m1) were obtained from Applied Biosystems (Foster City, CA). Silencer Select Negative Control No. 2 siRNA, siWHSC1 A (Assay ID 42800), siWHSC1 B (Assay ID 42732), siWHSC1 C (Assay ID 42650), and human GAPDH endogenous control (4352934E) were obtained from Life Technologies, Inc. Dimethyl histone H3 (Lys-36) rabbit mAb (2901) and histone H3 mouse mAb (3638) were purchased from Cell Signaling Technology. Anti-rabbit IgG IRDye 680RD and anti-mouse IgG IRDye 800CW secondary antibodies were purchased from LI-COR.
Enzymes and substrates
Human ASH1L (residues 2046–2330; GenBankTM accession no. NM_018489) was expressed in Escherichia coli with an N-terminal polyhistidine tag. Human DNMT1 (residues 2–1632; NM_001130823) was expressed in an insect cell/baculovirus expression system as an N-terminal GST fusion. Human DNMT3a (residues 2–912; NM_175629) was expressed in an insect cell/baculovirus expression system as an N-terminal GST fusion. Human DNMT3b (residues 2–853; NM_006892) was expressed in an insect cell/baculovirus expression system as an N-terminal GST fusion. The two proteins human DNMT3b (residues 564–853; NM_006892) containing an N-terminal polyhistidine tag and human DNMT3L (residues 160–387; NM_013369) with an N-terminal GST tag were coexpressed in an insect cell/baculovirus expression system. Human DOT1L (residues 1–416; NM_032482) was expressed in E. coli as an N-terminal GST fusion. Human recombinant EZH1 (residues 2–747; NM_001991) or EZH2 (residues 2–746; NM_001203247) was coexpressed with human recombinants AEBP2 (2–517; NM_001114176), EED (2–441; NM_003797), RbAp48 (2–425; NM_005610), and SUZ12 (2–739; NM_015355) in an insect cell/baculovirus expression system to form the five-member EZH1 or EZH2 complexes. All proteins were full-length (residue 2 through C terminus). The EED subunit incorporated an N-terminal FLAG tag, and all others included an N-terminal polyhistidine tag. Human GLP (residues 894–1298; NM_024757) and human G9a (residues 786–1210; NM_006709) were expressed as N-terminal GST fusion proteins in E. coli. Human MLL1 (residues 3745–3969; NM_005933), human WDR5 (22–334; NM_017588), RbBP5 (1–538; NM_005057), Ash2L (2–534; NM_001105214), and DPY-30 (1–99; NM_0325742) were expressed in E. coli with N-terminal polyhistidine tags, assembled as a complex (two copies of DPY-30), and stored in 20 mm Tris-HCl, pH 7.5, 300 mm NaCl, 1 mm TCEP, 10% (w/v) glycerol, and 1 mm ZnCl2. Human MLL2 (residues 5319–5537; NM_003482), human MLL3 (residues 4689–4911; NM_170606), and human MLL4 (residues 2490–2715; NM_014727) were expressed in E. coli with N-terminal polyhistidine tags, and SET1B (residues 1629–1923; NM_015048) was expressed in E. coli with an N-terminal GST tag. All four were assembled in complexes as MLL1, as described above. Human recombinant NSD1 (residues 1538–2696; NM_022455) was expressed with an N-terminal polyhistidine tag in an insect cell/baculovirus expression system. Human recombinant NSD2 (residues 2–1365; NM_001042424) was expressed with an N-terminal polyhistidine tag in an insect cell/baculovirus expression system. Human recombinant NSD2-SET domain for SPR studies (residues 934–1241; NM_001042424) was expressed with an N-terminal polyhistidine tag in E. coli. Human recombinant NSD3 (residues 1021–1322; NM_023034) was expressed in E. coli as an N-terminal GST fusion. Human PRDM9 (residues 2–414; NM_020227) was expressed in an insect cell/baculovirus expression system as an N-terminal GST fusion. Human recombinant PRMT1 (residues 2–371; NM_001536) was expressed in E. coli as an N-terminal GST fusion. Human recombinant PRMT3 (residues 2–531; NM_005788) was expressed in E. coli with an N-terminal polyhistidine tag. Human recombinant PRMT4 (residues 2–608; NM_199141) was expressed in E. coli as an N-terminal GST fusion. Human recombinant PRMT5 (residues 2–637; NM_006109) was expressed with an N-terminal FLAG tag in an insect cell/baculovirus expression system. The complex of PRMT5–MEP50 was coexpressed with PRMT5 as described above with MEP50 (residues 2–342; NM_024102) containing an N-terminal polyhistidine tag in an insect cell/baculovirus expression system. Human recombinant PRMT6 (residues 2–375; NM_018137) and PRMT7 (residues 2–692; NM_019023.2) were expressed with N-terminal polyhistidine tags in an insect cell/baculovirus expression system. Human recombinant PRMT8 (residues 61–394; NM_019854) was expressed in E. coli with N- and C-terminal polyhistidine tags. Human recombinant SET7/9 (residues 2–366; NM_030648) was expressed in E. coli as an N-terminal GST fusion with a C-terminal polyhistidine tag. Human recombinant SETD2 (residues 1434–1711; NM_014159) and SMYD1 (residues 2–490; NM_198274), both with N-terminal GST fusions, were expressed in E. coli. Human recombinant SMYD2 (residues 2–433; NM_020197) was expressed in E. coli with an N-terminal polyhistidine tag. Human recombinant SUV39H1 (residues 44–412; NM_003173) and SUV39H2 (residues 48–410; NM_001193424), both with C-terminal polyhistidine tags, were expressed in E. coli. Human recombinant SUV420H1-tv2 (transcript variant 2, residues 2–393; NM_016028) was expressed as an N-terminal GST fusion in an insect cell/baculovirus expression system. Purified nucleosomes (HMT(35–130)) were obtained from HeLa according to Schnitzler (82). Core histones, including histone 5 (HMT(35–435)), were purified from chicken erythrocytes by a modification of the method of Schnitzler (82) followed by acid extraction/dialysis (83). Human recombinant histone H2A (residues 1–130; NM_021052) and histone H3.3 (residues 1–136; NM_002107), both untagged, were expressed in E. coli. Human recombinant GST-GAR (glycine- and arginine-rich sequence from the N terminus of fibrillarin, residues 2–78; NM_001436) was expressed in E. coli as an N-terminal GST fusion. The following substrates were purchased from vendors: poly(dI-dC)(dI-dC) from Sigma; λ DNA from New England Biolabs; histone H4 from BPS Biosciences, and histone H3(1–21) peptide from AnaSpec. The following reference compounds were purchased from vendors: SAH and chaetocin from Cayman Chemicals, and LLY 507 and ryuvidine from R & D Systems.
Methyltransferase-Glo assay
MTase-Glo assays were performed by a multistep format in white solid bottom 1,536-well plates (Greiner, catalog no. 789175-F). First, 23 nl of compounds (or DMSO control) were pin-transferred into 3 μl of reaction buffer (50 mm Tris-HCl, pH 8.8, 5 mm MgCl2, 50 mm NaCl, 1 mm TCEP, and 0.01% Tween 20) containing 666.7 nm (500 nm final) nucleosomes and either 10.7 nm (8 nm final) WT, 16 nm (12 nm final) E1099K, or 6.67 nm (5 nm final) T1150A NSD2 enzyme or no enzyme (low activity control, columns 2–3). Plates were then incubated for 30 min at room temperature prior to reaction initiation with 1 μl of 4 μm (1 μm final) SAM in reaction buffer and incubated at room temperature for 15 min. Upon completion, methyltransferase conversion of SAM to SAH was then detected using a two-step detection system where 1 μl of MTase-Glo reagent was first added to each well to convert SAH to ADP for 30 min at room temperature. Secondly, 5 μl of MTase-Glo Detection Solution was added to each well and allowed to incubate for 30 min at room temperature to convert ADP to ATP, which was then measured by luminescence detection using a ViewLux uHTS Microplate Imager (PerkinElmer Life Sciences) and compared with control samples to determine relative activity.
Methyltransferase-Glo counter assay
MTase-Glo counter assay was performed with identical procedures as with the MTase-Glo primary assay but without NSD2 enzyme or nucleosomes. Instead, 200 nm SAH was added to mimic the reaction with 20% substrate conversion.
EPIgeneous HTRF methyltransferase assay
The EPIgeneous HTRF methyltransferase assay was performed using white solid bottom 1,536-well plates (Greiner, catalog no. 789175-F). First, 23 nl of compounds (or DMSO control) were pin-transferred into 3 μl of reaction buffer (50 mm Tris-HCl, pH 8.8, 5 mm MgCl2, 50 mm NaCl, 1 mm TCEP and 0.01% Tween 20) containing 666.7 nm (500 nm final) nucleosomes and either 10.7 nm (8 nm final) WT NSD2 enzyme or no enzyme (low activity control, columns 2–3). Plates were then incubated for 30 min at room temperature prior to reaction initiation with 1 μl of 4 μm (1 μm final) SAM in reaction buffer and incubated at room temperature for 15 min. After the incubation period, 0.8 μl of EPIgeneous Detection Buffer One was added to each well, followed by a 10-min incubation at room temperature. Next, anti-SAH/Lumi4-Tb solution was prepared according to the manufacturer's instructions, and 1.6 μl of the solution were added to each well. Finally, the SAH-d2 conjugate was prepared as a 32-fold dilution according to the manufacturer's instructions, and 1.6 μl of the solution was added to each well. The assay plate was allowed to incubate for 1 h at room temperature before detection of the HTRF signal using an Envision plate reader (PerkinElmer Life Sciences).
Amplex red (10-acetyl-3,7-dihydroxyphenoxazine) assay
The assay was adapted from a previously described protocol to assess redox cycling of compounds in the presence of reducing agents (49). 23 nl of compounds (or DMSO control) were pin-transferred into 2.5 μl of HBSS (ThermoFisher Scientific; containing 1.26 mm CaCl2, 0.49 mm MgCl2, 1 g/liter d-glucose) in black 1,536-well plates. Compound fluorescence was measured immediately (READ 0) using a ViewLux uHTS microplate imager (PerkinElmer Life Sciences) equipped with excitation 525/20 and emission 598/25 filters. 2.5 μl of a 2× Amplex Red solution (100 μm Amplex Red (Cayman Chemical, Ann Arbor, MI), 200 μm DTT (ThermoFisher Scientific), and 2 units/ml horseradish peroxidase (Sigma); diluted in HBSS and protected from light) were added to each well. Fluorescence was measured after a 15-min incubation at room temperature (READ 1), using ViewLux settings identical to read 0. Activity was calculated using corrected fluorescence values (READ 1 minus READ 0), which were compared with control samples (negative = vehicle; positive = 46 μm walrycin B).
HotSpot methyltransferase assay
The HotSpot radioisotope-based methyltransferase assays were performed as described previously (58, 84) with the following modifications. Standard substrate concentrations were 5 μm peptide or protein substrate, or 0.05 mg/ml for nucleosomes and core histones, and 1 μm SAM, unless otherwise mentioned. For control compound IC50 determinations, the test compounds were diluted in DMSO and then added to the enzyme/substrate mixtures in nanoliter aliquots by using an acoustic technology (Echo550; Labcyte) with a 20-min preincubation. The reaction was initiated by the addition of [3H]SAM (tritiated SAM, PerkinElmer Life Sciences) and incubated at 30 °C for 1 h. The reaction was detected by a filter-binding method. Data analysis was performed using GraphPad Prism software.
Surface plasmon resonance
Surface plasmon resonance measurements were performed using a Biacore 8K (GE Healthcare) at 25 °C. Human recombinant NSD2-SET domain was immobilized to a Serial-S CM5 Sensorchip (GE Healthcare) using the classic amine-coupling method in immobilization buffer containing 10 mm HEPES, 150 mm NaCl, 0.5 mm TCEP, and 0.05% v/v surfactant P20. Single cycle kinetic measurements were performed in running buffer containing 50 mm Tris, pH 8.8, 50 mm NaCl, 0.5 mm TCEP, 5 mm MgCl2, 0.05% surfactant P20, and 2% DMSO. Compounds were diluted in a 3-fold series with running buffer, and the DMSO concentration was carefully matched to 2%. Compound solutions were then injected over the prepared sensorchip at a flow rate of 80 μl/min for 80 s and allowed to dissociate over a period of 200–600 s. Data analysis was performed with Biacore 8K evaluation software using the 1:1 kinetic binding model.
Methyltransferase profiling
All methyltransferase profiling was performed with the HotSpot methyltransferase assay format as described above. The final DMSO concentration in the reaction was adjusted to 1% DMSO for all profiling. The reaction buffer for EZH1 and EZH2 was 50 mm Tris-HCl, pH 8.0, 0.01% Brij35, 1 mm EDTA, 1 mm DTT, and 1 mm PMSF. The reaction buffer for SET8 and PRMT5 was 50 mm Tris-HCl, pH 8.5, 0.01% Brij35, and 1 mm DTT. The reaction buffer for NSD3 was 50 mm Bicine, pH 8.5, 0.01% Brij35, and 1 mm DTT. The reaction buffer for all other HMTs was 50 mm Tris-HCl, pH 8.5, 50 mm NaCl, 5 mm MgCl2, 1 mm DTT, and 1 mm PMSF. The reaction buffer for DNMTs was 50 mm Tris-HCl, pH 7.5, 5 mm EDTA, 0.01% Brij35, 5 mm DTT, 0.1 mm PMSF, and 5% glycerol.
siRNA knockdown of NSD2
All siRNA transfections were performed using 6-well plates. Within five wells, 128 μl of siRNA (either 40 nm Silencer Select Negative control, 5 nm siWHSC1 C (Assay ID 42650), 5 nm siWHSC1 B (Assay ID 42732), 5 nm siWHSC1 A (Assay ID 42800), or 20 nm AllStars Cell Death Control) were complexed with 3.2 μl of RNAiMax transfection reagent in 1 ml of McCoy's 5A for 15 min at ambient temperature. The sixth well contained cells only without siRNA and lipid complex. One hundred thousand cells in 1 ml of McCoy's, 20% FBS were added to each well. The plates were maintained at room temperature for 45 min before incubation at 37 °C, 5% CO2. After 72 h, the cells were utilized for an NSD2 transcript analysis, a cell growth assay, and Western blot analysis of H3K36me2 and total H3 levels. For the transcript analysis, RNA was extracted using an RNeasy mini kit. Following RNA extraction, cDNA was generated with the High-Capacity RNA-to-cDNA kit. RT-PCR was conducted using a TaqMan Gene Expression Master Mix, TaqMan WHSC1 human (Hs00983720_m1), and human GAPDH endogenous control (4352934E). For the cell growth assay, 1,750 cells were plated in 40-μl volumes (McCoy's 5A + 10% FBS + 0.5× penicillin/streptomycin) in a 384-well cell carrier plate (PerkinElmer Life Sciences), and the plates were sealed with Breathe-Easy sealing membrane (Sigma). Images were captured every 4 h up to 72 h with an IncuCyte ZOOM System (Essen BioScience). For the Western blot analysis, the cells were lysed with 1× SDS sample buffer and blotted as described below.
Western blotting analyses
Test compounds were dissolved with DMSO to a 10 mm stock. U-2 OS cells were seeded in 12-well plates at a density of 0.5 × 106/well in complete culture medium and placed into the incubator at 37 °C, 5% CO2. After overnight incubation, the cells were treated with test compounds (10-point concentration series with 3-fold dilution, 0.0025–50 μm) or with reference compound DZnep (10 μm single dose) and allowed to incubate for an additional 96 h. Following the incubation with compound, culture medium was removed, and the cells were washed once with ice-cold PBS. The cells were lysed with 1× SDS sample buffer (62.5 mm Tris-HCl, pH 6.8, 2% SDS w/v, 10% glycerol, 0.01% w/v bromphenol blue, 50 mm DTT), and the lysates were sonicated three times in 3-s increments at 15 A. Cell lysate samples (14 μl) were subjected to SDS-PAGE and transferred onto nitrocellulose membranes by the iBlot dry blotting system. The membranes were blocked with 2% nonfat milk blocking buffer for 1 h and then probed with anti-histone H3 (dimethyl Lys-36) primary antibody overnight. Anti-rabbit IgG IRDye 680RD secondary antibody was used to detect the primary antibody. Then the blots were washed three times with 1× TBS buffer plus 0.01% Tween 20 and re-probed with anti-histone H3 primary antibody and anti-mouse IgG IRDye 800CW secondary antibody. The membranes were scanned with a LI-COR Odyssey Fc Imaging System. The specific bands of interest were quantified by LI-COR Image Studio Lite software.
U-2 OS cell growth assay
U-2 OS (ATCC HTB-96) cells were obtained directly from the ATCC and cultured according to the recommended culturing conditions. At cell passage 2, 1,750 cells were plated in 40-μl volumes (McCoy's 5A + 10% FBS + 0.5× penicillin/streptomycin) in a 384-well cell carrier plate (PerkinElmer Life Sciences) and incubated overnight. At 16 h, test compounds were delivered in 195-nl aliquots by pin transfer. The plates were sealed with Breathe-Easy sealing membrane (Sigma), and images were captured every 4 h up to 96 h with an IncuCyte ZOOM System (Essen BioScience).
Data analysis
Data normalization and curve fitting were performed using in-house informatics tools. Briefly, raw plate reads for each titration point were first normalized relative to the DMSO-only wells (100% activity) and no enzyme control wells (0% activity) and were then corrected by applying a plate-wise block pattern correction algorithm to remove any plate edge effects and systematic background noise. Active compounds from the primary HTS were defined as having a maximum response ≥50%. To determine compound activities from the 11-point qHTS, the concentration-response data for each sample were plotted and modeled by a four-parameter logistic fit yielding IC50 and efficacy (maximal response) values as described previously (44). The activities were designated as classes 1–4 according to the type of concentration-response curve observed. Active compounds were defined as having concentration-response curves in the classes of 1–3. The promiscuity score for each compound was defined as (number of assays in which the compound is active)/(total number of assays in which the compound was tested). A compound with a promiscuity score higher than 0.2 was considered as a “frequent hitter” to be eliminated from the follow-up studies.
Author contributions
N. P. C., S. C. K., A. S., H. M., A. J., and M. D. H. conceptualization; N. P. C., S. C. K., M. S., H. M., and A. J. data curation; N. P. C., S. C. K., M. J. H., K. Y. H., Y. W., Q. C., E. K., J. W., H. M., O. W. L., M. S., M. L.-N., and A. J. formal analysis; N. P. C., S. C. K., A. S., M. L.-N., H. M., A. J., and M. D. H. supervision; N. P. C., S. C. K., and O. W. L. validation; N. P. C., S. C. K., M. J. H., K. Y. H., Y. W., Q. C., E. K., J. W., S. C., D. M. C., K. C.-C. C., P. S., H. M., and M. L.-N. investigation; N. P. C., S. C. K., M. J. H., K. Y. H., Y. W., Q. C., E. K., J. W., S. C., D. M. C., K. C.-C. C., P. S., A. S., M. L.-N., H. M., A. J., and M. D. H. methodology; N. P. C., S. C. K., M. J. H., K. Y. H., Y. W., Q. C., E. K., J. W., H. M., and M. D. H. writing-original draft; N. P. C., K. R. B., M. S., A. S., H. M., and M. D. H. project administration; N. P. C., S. C. K., M. J. H., and M. D. H. writing-review and editing; N. P. C., M. J. H., and K. R. B. visualization.
Acknowledgment
We thank Jayme L. Dahlin for helpful discussions and critical reading of this manuscript.
This work was supported by the National Center for Advancing Translational Sciences (NCATS) Division of Pre-Clinical Innovation Intramural Program. K. Y. H., Y. W., Q. C., E. K., J. W., and H. M. are employed by Reaction Biology. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
- NSD
- nuclear receptor-binding SET domain
- qHTS
- quantitative high-throughput screening
- SAM
- S-adenosyl-l-methionine
- SPR
- surface plasmon resonance
- HTRF
- homogeneous time-resolved fluorescence
- DZNep
- 3-deazaneplanocin A
- SAH
- S-adenosyl-l-homocysteine
- MMSET
- multiple myeloma SET domain
- MM
- multiple myeloma
- CLL
- chronic lymphocytic leukemia
- CRC
- concentration-response curve
- TCEP
- tris(2-carboxyethyl)phosphine
- PMSF
- phenylmethylsulfonyl fluoride
- HBSS
- Hanks' balanced salt solution
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- Bicine
- N,N-bis(2-hydroxyethyl)glycine
- MTase-GloTM
- Methyltransferase-Glo
- S/B
- signal-to-background
- LPA
- lysophosphatidic acid
- NCATS
- National Center for Advancing Translational Sciences
- CV
- coefficient of variation.
References
- 1. Bennett R. L., Swaroop A., Troche C., and Licht J. D. (2017) The role of nuclear receptor-binding SET domain family histone lysine methyltransferases in cancer. Cold Spring Harb. Perspect. Med. 7, a026708 10.1101/cshperspect.a026708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chesi M., Nardini E., Lim R. S., Smith K. D., Kuehl W. M., and Bergsagel P. L. (1998) The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood 92, 3025–3034 [PubMed] [Google Scholar]
- 3. Stec I., Wright T. J., van Ommen G. J., de Boer P. A., van Haeringen A., Moorman A. F., Altherr M. R., and den Dunnen J. T. (1998) WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum. Mol. Genet. 7, 1071–1082 10.1093/hmg/7.7.1071 [DOI] [PubMed] [Google Scholar]
- 4. Keats J. J., Reiman T., Maxwell C. A., Taylor B. J., Larratt L. M., Mant M. J., Belch A. R., and Pilarski L. M. (2003) In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood 101, 1520–1529 10.1182/blood-2002-06-1675 [DOI] [PubMed] [Google Scholar]
- 5. Santra M., Zhan F., Tian E., Barlogie B., and Shaughnessy J. Jr. (2003) A subset of multiple myeloma harboring the t(4;14)(p16;q32) translocation lacks FGFR3 expression but maintains an IGH/MMSET fusion transcript. Blood 101, 2374–2376 10.1182/blood-2002-09-2801 [DOI] [PubMed] [Google Scholar]
- 6. Keats J. J., Maxwell C. A., Taylor B. J., Hendzel M. J., Chesi M., Bergsagel P. L., Larratt L. M., Mant M. J., Reiman T., Belch A. R., and Pilarski L. M. (2005) Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood 105, 4060–4069 10.1182/blood-2004-09-3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lauring J., Abukhdeir A. M., Konishi H., Garay J. P., Gustin J. P., Wang Q., Arceci R. J., Matsui W., and Park B. H. (2008) The multiple myeloma associated MMSET gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood 111, 856–864 10.1182/blood-2007-05-088674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brito J. L., Walker B., Jenner M., Dickens N. J., Brown N. J., Ross F. M., Avramidou A., Irving J. A., Gonzalez D., Davies F. E., and Morgan G. J. (2009) MMSET deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells. Haematologica 94, 78–86 10.3324/haematol.13426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martinez-Garcia E., Popovic R., Min D. J., Sweet S. M., Thomas P. M., Zamdborg L., Heffner A., Will C., Lamy L., Staudt L. M., Levens D. L., Kelleher N. L., and Licht J. D. (2011) The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 117, 211–220 10.1182/blood-2010-07-298349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang Z., Wu H., Chuai S., Xu F., Yan F., Englund N., Wang Z., Zhang H., Fang M., Wang Y., Gu J., Zhang M., Yang T., Zhao K., Yu Y., et al. (2013) NSD2 is recruited through its PHD domain to oncogenic gene loci to drive multiple myeloma. Cancer Res. 73, 6277–6288 10.1158/0008-5472.CAN-13-1000 [DOI] [PubMed] [Google Scholar]
- 11. Marango J., Shimoyama M., Nishio H., Meyer J. A., Min D. J., Sirulnik A., Martinez-Martinez Y., Chesi M., Bergsagel P. L., Zhou M. M., Waxman S., Leibovitch B. A., Walsh M. J., and Licht J. D. (2008) The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood 111, 3145–3154 10.1182/blood-2007-06-092122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kuo A. J., Cheung P., Chen K., Zee B. M., Kioi M., Lauring J., Xi Y., Park B. H., Shi X., Garcia B. A., Li W., and Gozani O. (2011) NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 44, 609–620 10.1016/j.molcel.2011.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zheng Y., Sweet S. M., Popovic R., Martinez-Garcia E., Tipton J. D., Thomas P. M., Licht J. D., and Kelleher N. L. (2012) Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proc. Natl. Acad. Sci. U.S.A. 109, 13549–13554 10.1073/pnas.1205707109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Popovic R., Martinez-Garcia E., Giannopoulou E. G., Zhang Q., Zhang Q., Ezponda T., Shah M. Y., Zheng Y., Will C. M., Small E. C., Hua Y., Bulic M., Jiang Y., Carrara M., Calogero R. A., et al. (2014) Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 10, e1004566 10.1371/journal.pgen.1004566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sankaran S. M., Wilkinson A. W., Elias J. E., and Gozani O. (2016) A PWWP domain of histone-lysine N-methyltransferase NSD2 binds to dimethylated Lys-36 of histone H3 and regulates NSD2 function at chromatin. J. Biol. Chem. 291, 8465–8474 10.1074/jbc.M116.720748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zheng Y., Fornelli L., Compton P. D., Sharma S., Canterbury J., Mullen C., Zabrouskov V., Fellers R. T., Thomas P. M., Licht J. D., Senko M. W., and Kelleher N. L. (2016) Unabridged analysis of human histone H3 by differential top-down mass spectrometry reveals hypermethylated proteoforms from MMSET/NSD2 overexpression. Mol. Cell. Proteomics 15, 776–790 10.1074/mcp.M115.053819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Y., Trojer P., Xu C. F., Cheung P., Kuo A., Drury W. J. 3rd., Qiao Q., Neubert T. A., Xu R. M., Gozani O., and Reinberg D. (2009) The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J. Biol. Chem. 284, 34283–34295 10.1074/jbc.M109.034462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hudlebusch H. R., Santoni-Rugiu E., Simon R., Ralfkiær E., Rossing H. H., Johansen J. V., Jørgensen M., Sauter G., and Helin K. (2011) The histone methyltransferase and putative oncoprotein MMSET is overexpressed in a large variety of human tumors. Clin. Cancer Res. 17, 2919–2933 10.1158/1078-0432.CCR-10-1302 [DOI] [PubMed] [Google Scholar]
- 19. Ezponda T., Popovic R., Shah M. Y., Martinez-Garcia E., Zheng Y., Min D. J., Will C., Neri A., Kelleher N. L., Yu J., and Licht J. D. (2013) The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene 32, 2882–2890 10.1038/onc.2012.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saloura V., Cho H. S., Kiyotani K., Alachkar H., Zuo Z., Nakakido M., Tsunoda T., Seiwert T., Lingen M., Licht J., Nakamura Y., and Hamamoto R. (2015) WHSC1 promotes oncogenesis through regulation of NIMA-related kinase-7 in squamous cell carcinoma of the head and neck. Mol. Cancer Res. 13, 293–304 10.1158/1541-7786.MCR-14-0292-T [DOI] [PubMed] [Google Scholar]
- 21. Jaffe J. D., Wang Y., Chan H. M., Zhang J., Huether R., Kryukov G. V., Bhang H. E., Taylor J. E., Hu M., Englund N. P., Yan F., Wang Z., Robert McDonald E. 3rd, Wei L., Ma J., et al. (2013) Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat. Genet. 45, 1386–1391 10.1038/ng.2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li J., Yin C., Okamoto H., Mushlin H., Balgley B. M., Lee C. S., Yuan K., Ikejiri B., Glasker S., Vortmeyer A. O., Oldfield E. H., Weil R. J., and Zhuang Z. (2008) Identification of a novel proliferation-related protein, WHSC1 4a, in human gliomas. Neuro Oncol. 10, 45–51 10.1215/15228517-2007-036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hudlebusch H. R., Skotte J., Santoni-Rugiu E., Zimling Z. G., Lees M. J., Simon R., Sauter G., Rota R., De Ioris M. A., Quarto M., Johansen J. V., Jørgensen M., Rechnitzer C., Maroun L. L., Schroder H., et al. (2011) MMSET is highly expressed and associated with aggressiveness in neuroblastoma. Cancer Res. 71, 4226–4235 10.1158/0008-5472.CAN-10-3810,10.1158/1538-7445.AM2011-4226 [DOI] [PubMed] [Google Scholar]
- 24. Xiao M., Yang S., Chen J., Ning X., Guo L., Huang K., and Sui L. (2013) Overexpression of MMSET in endometrial cancer: a clinicopathologic study. J. Surg. Oncol. 107, 428–432 10.1002/jso.23234 [DOI] [PubMed] [Google Scholar]
- 25. Yang S., Zhang Y., Meng F., Liu Y., Xia B., Xiao M., Xu Y., Ning X., Li H., and Lou G. (2013) Overexpression of multiple myeloma SET domain (MMSET) is associated with advanced tumor aggressiveness and poor prognosis in serous ovarian carcinoma. Biomarkers 18, 257–263 10.3109/1354750X.2013.773082 [DOI] [PubMed] [Google Scholar]
- 26. Okabe H., Satoh S., Kato T., Kitahara O., Yanagawa R., Yamaoka Y., Tsunoda T., Furukawa Y., and Nakamura Y. (2001) Genome-wide analysis of gene expression in human hepatocellular carcinomas using cDNA microarray: identification of genes involved in viral carcinogenesis and tumor progression. Cancer Res. 61, 2129–2137 [PubMed] [Google Scholar]
- 27. Kim J. Y., Kee H. J., Choe N. W., Kim S. M., Eom G. H., Baek H. J., Kook H., Kook H., and Seo S. B. (2008) Multiple-myeloma-related WHSC1/MMSET isoform RE-IIBP is a histone methyltransferase with transcriptional repression activity. Mol. Cell. Biol. 28, 2023–2034 10.1128/MCB.02130-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kassambara A., Klein B., and Moreaux J. (2009) MMSET is overexpressed in cancers: link with tumor aggressiveness. Biochem. Biophys. Res. Commun. 379, 840–845 10.1016/j.bbrc.2008.12.093 [DOI] [PubMed] [Google Scholar]
- 29. Huether R., Dong L., Chen X., Wu G., Parker M., Wei L., Ma J., Edmonson M. N., Hedlund E. K., Rusch M. C., Shurtleff S. A., Mulder H. L., Boggs K., Vadordaria B., Cheng J., et al. (2014) The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat. Commun. 5, 3630 10.1038/ncomms4630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oyer J. A., Huang X., Zheng Y., Shim J., Ezponda T., Carpenter Z., Allegretta M., Okot-Kotber C. I., Patel J. P., Melnick A., Levine R. L., Ferrando A., Mackerell A. D. Jr., Kelleher N. L., Licht J. D., and Popovic R. (2014) Point mutation E1099K in MMSET/NSD2 enhances its methyltransferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia 28, 198–201 10.1038/leu.2013.204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beà S., Valdés-Mas R., Navarro A., Salaverria I., Martín-Garcia D., Jares P., Giné E., Pinyol M., Royo C., Nadeu F., Conde L., Juan M., Clot G., Vizàn P., Di Croce L., et al. (2013) Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 110, 18250–18255 10.1073/pnas.1314608110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Imielinski M., Berger A. H., Hammerman P. S., Hernandez B., Pugh T. J., Hodis E., Cho J., Suh J., Capelletti M., Sivachenko A., Sougnez C., Auclair D., Lawrence M. S., Stojanov P., Cibulskis K., et al. (2012) Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120 10.1016/j.cell.2012.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fabbri G., Rasi S., Rossi D., Trifonov V., Khiabanian H., Ma J., Grunn A., Fangazio M., Capello D., Monti S., Cresta S., Gargiulo E., Forconi F., Guarini A., Arcaini L., et al. (2011) Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J. Exp. Med. 208, 1389–1401 10.1084/jem.20110921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Holmfeldt L., Wei L., Diaz-Flores E., Walsh M., Zhang J., Ding L., Payne-Turner D., Churchman M., Andersson A., Chen S. C., McCastlain K., Becksfort J., Ma J., Wu G., Patel S. N., et al. (2013) The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat. Genet. 45, 242–252 10.1038/ng.2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cancer Genome Atlas Research Network, Weinstein J. N., Collisson E. A., Mills G. B., Shaw K. R., Ozenberger B. A., Ellrott K., Shmulevich I., Sander C., and Stuart J. M. (2013) The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120 10.1038/ng.2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Arrowsmith C. H., Audia J. E., Austin C., Baell J., Bennett J., Blagg J., Bountra C., Brennan P. E., Brown P. J., Bunnage M. E., Buser-Doepner C., Campbell R. M., Carter A. J., Cohen P., Copeland R. A., et al. (2015) The promise and peril of chemical probes. Nat. Chem. Biol. 11, 536–541 10.1038/nchembio.1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. di Luccio E. (2015) Inhibition of nuclear receptor binding SET domain 2/multiple myeloma SET domain by LEM-06 implication for epigenetic cancer therapies. J. Cancer Prev. 20, 113–120 10.15430/JCP.2015.20.2.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Horiuchi K. Y., Eason M. M., Ferry J. J., Planck J. L., Walsh C. P., Smith R. F., Howitz K. T., and Ma H. (2013) Assay development for histone methyltransferases. Assay Drug Dev. Technol. 11, 227–236 10.1089/adt.2012.480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Allali-Hassani A., Kuznetsova E., Hajian T., Wu H., Dombrovski L., Li Y., Gräslund S., Arrowsmith C. H., Schapira M., and Vedadi M. (2014) A basic post-SET extension of NSDs is essential for nucleosome binding in vitro. J. Biomol. Screen. 19, 928–935 10.1177/1087057114525854 [DOI] [PubMed] [Google Scholar]
- 40. McGeary R. P., Bennett A. J., Tran Q. B., Cosgrove K. L., and Ross B. P. (2008) Suramin: clinical uses and structure-activity relationships. Mini Rev. Med. Chem. 8, 1384–1394 10.2174/138955708786369573 [DOI] [PubMed] [Google Scholar]
- 41. Tisi D., Chiarparin E., Tamanini E., Pathuri P., Coyle J. E., Hold A., Holding F. P., Amin N., Martin A. C., Rich S. J., Berdini V., Yon J., Acklam P., Burke R., Drouin L., et al. (2016) Structure of the epigenetic oncogene MMSET and inhibition by N-alkyl sinefungin derivatives. ACS Chem. Biol. 11, 3093–3105 10.1021/acschembio.6b00308 [DOI] [PubMed] [Google Scholar]
- 42. Richon V. M., Johnston D., Sneeringer C. J., Jin L., Majer C. R., Elliston K., Jerva L. F., Scott M. P., and Copeland R. A. (2011) Chemogenetic analysis of human protein methyltransferases. Chem. Biol. Drug Des. 78, 199–210 10.1111/j.1747-0285.2011.01135.x [DOI] [PubMed] [Google Scholar]
- 43. Morrison M. J., Boriack-Sjodin P. A., Swinger K. K., Wigle T. J., Sadalge D., Kuntz K. W., Scott M. P., Janzen W. P., Chesworth R., Duncan K. W., Harvey D. M., Lampe J. W., Mitchell L. H., and Copeland R. A. (2018) Identification of a peptide inhibitor for the histone methyltransferase WHSC1. PLoS ONE 13, e0197082 10.1371/journal.pone.0197082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Inglese J., Auld D. S., Jadhav A., Johnson R. L., Simeonov A., Yasgar A., Zheng W., and Austin C. P. (2006) Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. U.S.A. 103, 11473–11478 10.1073/pnas.0604348103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hsiao K., Zegzouti H., and Goueli S. A. (2016) Methyltransferase-Glo: a universal, bioluminescent and homogenous assay for monitoring all classes of methyltransferases. Epigenomics 8, 321–339 10.2217/epi.15.113 [DOI] [PubMed] [Google Scholar]
- 46. Michael S., Auld D., Klumpp C., Jadhav A., Zheng W., Thorne N., Austin C. P., Inglese J., and Simeonov A. (2008) A robotic platform for quantitative high-throughput screening. Assay Drug Dev. Technol. 6, 637–657 10.1089/adt.2008.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang R., Southall N., Wang Y., Yasgar A., Shinn P., Jadhav A., Nguyen D. T., and Austin C. P. (2011) The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci. Transl. Med. 3, 80ps16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dahlin J. L., Nissink J. W., Strasser J. M., Francis S., Higgins L., Zhou H., Zhang Z., and Walters M. A. (2015) PAINS in the assay: chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J. Med. Chem. 58, 2091–2113 10.1021/jm5019093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Johnston P. A., Soares K. M., Shinde S. N., Foster C. A., Shun T. Y., Takyi H. K., Wipf P., and Lazo J. S. (2008) Development of a 384-well colorimetric assay to quantify hydrogen peroxide generated by the redox cycling of compounds in the presence of reducing agents. Assay Drug Dev. Technol. 6, 505–518 10.1089/adt.2008.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kimos M., Burton M., Urbain D., Caudron D., Martini M., Famelart M., Gillard M., Barrow J., and Wood M. (2016) Development of an HTRF assay for the detection and characterization of inhibitors of catechol-O-methyltransferase. J. Biomol. Screen. 21, 490–495 10.1177/1087057115616793 [DOI] [PubMed] [Google Scholar]
- 51. Auld D. S., Thorne N., Maguire W. F., and Inglese J. (2009) Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc. Natl. Acad. Sci. U.S.A. 106, 3585–3590 10.1073/pnas.0813345106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Auld D. S., Lovell S., Thorne N., Lea W. A., Maloney D. J., Shen M., Rai G., Battaile K. P., Thomas C. J., Simeonov A., Hanzlik R. P., and Inglese J. (2010) Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc. Natl. Acad. Sci. U.S.A. 107, 4878–4883 10.1073/pnas.0909141107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tanega C., Shen M., Mott B. T., Thomas C. J., MacArthur R., Inglese J., and Auld D. S. (2009) Comparison of bioluminescent kinase assays using substrate depletion and product formation. Assay Drug Dev. Technol. 7, 606–614 10.1089/adt.2009.0230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Auld D. S., Southall N. T., Jadhav A., Johnson R. L., Diller D. J., Simeonov A., Austin C. P., and Inglese J. (2008) Characterization of chemical libraries for luciferase inhibitory activity. J. Med. Chem. 51, 2372–2386 10.1021/jm701302v [DOI] [PubMed] [Google Scholar]
- 55. Soares K. M., Blackmon N., Shun T. Y., Shinde S. N., Takyi H. K., Wipf P., Lazo J. S., and Johnston P. A. (2010) Profiling the NIH small molecule repository for compounds that generate H2O2 by redox cycling in reducing environments. Assay Drug Dev. Technol. 8, 152–174 10.1089/adt.2009.0247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dahlin J. L., Baell J., and Walters M. A. (2004) Assay Interference by Chemical Reactivity. in Assay Guidance Manual, NCBI, National Institutes of Health, Bethesda [Google Scholar]
- 57. Giannetti A. M., Koch B. D., and Browner M. F. (2008) Surface plasmon resonance based assay for the detection and characterization of promiscuous inhibitors. J. Med. Chem. 51, 574–580 10.1021/jm700952v [DOI] [PubMed] [Google Scholar]
- 58. Anastassiadis T., Deacon S. W., Devarajan K., Ma H., and Peterson J. R. (2011) Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1039–1045 10.1038/nbt.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McGrath J., and Trojer P. (2015) Targeting histone lysine methylation in cancer. Pharmacol. Ther. 150, 1–22 10.1016/j.pharmthera.2015.01.002 [DOI] [PubMed] [Google Scholar]
- 60. Liu Q., and Wang M. W. (2016) Histone lysine methyltransferases as anti-cancer targets for drug discovery. Acta Pharmacol. Sin. 37, 1273–1280 10.1038/aps.2016.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cain C. (2013) NSD2 momentum. SciBX: Science-Business eXchange 0;6(39) 10.1038/scibx.2013.1083 [DOI] [Google Scholar]
- 62. Luo M. (2012) Current chemical biology approaches to interrogate protein methyltransferases. ACS Chem. Biol. 7, 443–463 10.1021/cb200519y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Allali-Hassani A., Wasney G. A., Siarheyeva A., Hajian T., Arrowsmith C. H., and Vedadi M. (2012) Fluorescence-based methods for screening writers and readers of histone methyl marks. J. Biomol. Screen. 17, 71–84 10.1177/1087057111422256 [DOI] [PubMed] [Google Scholar]
- 64. Quinn A. M., Allali-Hassani A., Vedadi M., and Simeonov A. (2010) A chemiluminescence-based method for identification of histone lysine methyltransferase inhibitors. Mol. Biosyst. 6, 782–788 10.1039/b921912a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kumar M., Zielinski T., and Lowery R. G. (2015) Biochemical assay development for histone methyltransferases using a transcreener-based assay for S-Adenosylhomocysteine. Assay Drug Dev. Technol. 13, 200–209 10.1089/adt.2014.609 [DOI] [PubMed] [Google Scholar]
- 66. Chi H., Takemoto Y., Nsiama T. K., Kato T., Nishino N., Ito A., and Yoshida M. (2014) Design and synthesis of peptide-MCA substrates for a novel assay of histone methyltransferases and their inhibitors. Bioorg. Med. Chem. 22, 1268–1275 10.1016/j.bmc.2014.01.011 [DOI] [PubMed] [Google Scholar]
- 67. Ma H., Howitz K. T., Horiuchi K. Y., and Wang Y. (2015) Histone methyltransferase activity assays. in Epigenetics for Drug Discovery, pp. 267–287, Royal Society of Chemistry, London/Cambridge, UK [Google Scholar]
- 68. Drake K. M., Watson V. G., Kisielewski A., Glynn R., and Napper A. D. (2014) A sensitive luminescent assay for the histone methyltransferase NSD1 and other SAM-dependent enzymes. Assay Drug Dev. Technol. 12, 258–271 10.1089/adt.2014.583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lazo J. S., Aslan D. C., Southwick E. C., Cooley K. A., Ducruet A. P., Joo B., Vogt A., and Wipf P. (2001) Discovery and biological evaluation of a new family of potent inhibitors of the dual specificity protein phosphatase Cdc25. J. Med. Chem. 44, 4042–4049 10.1021/jm0102046 [DOI] [PubMed] [Google Scholar]
- 70. Orena S., Maurer T. S., She L., Eudy R., Bernardo V., Dash D., Loria P., Banker M. E., Tugnait M., Okerberg C. V., Qian J., and Boustany-Kari C. M. (2013) PF-03882845, a non-steroidal mineralocorticoid receptor antagonist, prevents renal injury with reduced risk of hyperkalemia in an animal model of nephropathy. Front. Pharmacol. 4, 115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kozian D. H., Evers A., Florian P., Wonerow P., Joho S., and Nazare M. (2012) Selective non-lipid modulator of LPA5 activity in human platelets. Bioorg. Med. Chem. Lett. 22, 5239–5243 10.1016/j.bmcl.2012.06.057 [DOI] [PubMed] [Google Scholar]
- 72. Greiner D., Bonaldi T., Eskeland R., Roemer E., and Imhof A. (2005) Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3–9. Nat. Chem. Biol. 1, 143–145 10.1038/nchembio721 [DOI] [PubMed] [Google Scholar]
- 73. Cherblanc F. L., Chapman K. L., Brown R., and Fuchter M. J. (2013) Chaetocin is a nonspecific inhibitor of histone lysine methyltransferases. Nat. Chem. Biol. 9, 136–137 10.1038/nchembio.1187 [DOI] [PubMed] [Google Scholar]
- 74. Cherblanc F. L., Chapman K. L., Reid J., Borg A. J., Sundriyal S., Alcazar-Fuoli L., Bignell E., Demetriades M., Schofield C. J., DiMaggio P. A. Jr., Brown R., and Fuchter M. J. (2013) On the histone lysine methyltransferase activity of fungal metabolite chaetocin. J. Med. Chem. 56, 8616–8625 10.1021/jm401063r [DOI] [PubMed] [Google Scholar]
- 75. Souers A. J., Leverson J. D., Boghaert E. R., Ackler S. L., Catron N. D., Chen J., Dayton B. D., Ding H., Enschede S. H., Fairbrother W. J., Huang D. C., Hymowitz S. G., Jin S., Khaw S. L., Kovar P. J., et al. (2013) ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208 10.1038/nm.3048 [DOI] [PubMed] [Google Scholar]
- 76. Leverson J. D., Phillips D. C., Mitten M. J., Boghaert E. R., Diaz D., Tahir S. K., Belmont L. D., Nimmer P., Xiao Y., Ma X. M., Lowes K. N., Kovar P., Chen J., Jin S., Smith M., et al. (2015) Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 7, 279ra40 10.1126/scitranslmed.aaa4642 [DOI] [PubMed] [Google Scholar]
- 77. Glazer R. I., Hartman K. D., Knode M. C., Richard M. M., Chiang P. K., Tseng C. K., and Marquez V. E. (1986) 3-Deazaneplanocin: a new and potent inhibitor of S-adenosylhomocysteine hydrolase and its effects on human promyelocytic leukemia cell line HL-60. Biochem. Biophys. Res. Commun. 135, 688–694 10.1016/0006-291X(86)90048-3 [DOI] [PubMed] [Google Scholar]
- 78. Miranda T. B., Cortez C. C., Yoo C. B., Liang G., Abe M., Kelly T. K., Marquez V. E., and Jones P. A. (2009) DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 8, 1579–1588 10.1158/1535-7163.MCT-09-0013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tan J., Yang X., Zhuang L., Jiang X., Chen W., Lee P. L., Karuturi R. K., Tan P. B., Liu E. T., and Yu Q. (2007) Pharmacologic disruption of polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 21, 1050–1063 10.1101/gad.1524107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Qian J., Lu L., Wu J., and Ma H. (2013) Development of multiple cell-based assays for the detection of histone H3 Lys27 trimethylation (H3K27me3). Assay Drug Dev. Technol. 11, 449–456 10.1089/adt.2013.515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Benoit Y. D., Laursen K. B., Witherspoon M. S., Lipkin S. M., and Gudas L. J. (2013) Inhibition of PRC2 histone methyltransferase activity increases TRAIL-mediated apoptosis sensitivity in human colon cancer cells. J. Cell. Physiol. 228, 764–772 10.1002/jcp.24224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Schnitzler G. R. (2001) Isolation of histones and nucleosome cores from mammalian cells. Curr. Protoc. Mol. Biol. 2001, Chapter 21, Unit 21.5 10.1002/0471142727.mb2105s50 [DOI] [PubMed] [Google Scholar]
- 83. Shechter D., Dormann H. L., Allis C. D., and Hake S. B. (2007) Extraction, purification and analysis of histones. Nat. Protoc. 2, 1445–1457 10.1038/nprot.2007.202 [DOI] [PubMed] [Google Scholar]
- 84. Ma H., Deacon S., and Horiuchi K. (2008) The challenge of selecting protein kinase assays for lead discovery optimization. Expert Opin. Drug Discov. 3, 607–621 10.1517/17460441.3.6.607 [DOI] [PMC free article] [PubMed] [Google Scholar]