

Abstract
Methanobrevibacter and Methanosphaera species represent some of the most prevalent methanogenic archaea in the gastrointestinal tract of animals and humans and play an important role in this environment. The aim of this study was to identify genomic features that are shared or specific for members of each genus with a special emphasis of the analysis on the assimilation of nitrogen and acetate and the utilization of methanol and ethanol for methanogenesis. Here, draft genome sequences of Methanobrevibacter thaueri strain DSM 11995T, Methanobrevibacter woesei strain DSM 11979T, and Methanosphaera cuniculi strain 4103T are reported and compared to those of 16 other Methanobrevibacter and Methanosphaera genomes, including genomes of the 13 currently available types of strains of the two genera. The comparative genome analyses indicate that among other genes, the absence of molybdopterin cofactor biosynthesis is conserved in Methanosphaera species but reveals also that the three species share a core set of more than 300 genes that distinguishes the genus Methanosphaera from the genus Methanobrevibacter. Multilocus sequence analysis shows that the genus Methanobrevibacter can be subdivided into clades, potentially new genera, which may display characteristic specific metabolic features. These features include not only the potential ability of nitrogen fixation and acetate assimilation in a clade comprised of Methanobrevibacter species from the termite gut and Methanobrevibacter arboriphilus strains but also the potential capability to utilize ethanol and methanol in a clade comprising Methanobrevibacter wolinii strain DSM 11976T, Mbb. sp. AbM4, and Mbb. boviskoreani strain DSM 25824T.
1. Introduction
The microbial ecology of the intestinal tract of humans and animals has been subject to extensive research in recent years, and it is becoming increasingly evident that commensal intestinal microbes have a strong impact on host physiology and well-being. One microbial group, the methanogenic archaea (methanogens), has been of particular interest in this regard. Methanogens have not only been implicated in greenhouse gas emission from livestock animals [1, 2] but several studies have also linked these microorganisms to specific bodyweight phenotypes in humans [3] and to differences in feed conversion efficiency in ruminants [4, 5].
The majority of the rumen and intestinal methanogens appear to belong to two of the seven known orders, the Methanobacteriales [6] and the recently described Methanomassiliicoccales [7]. Two genera of the order Methanobacteriales, Methanobrevibacter and Methanosphaera, comprise a large proportion of intestinal and rumen methanogenic archaea [8, 9]. Species of the genus Methanobrevibacter have been isolated from not only a wide range of different environments, including invertebrate and vertebrate guts, but also non-host-associated environments. The majority of the members of the genus Methanobrevibacter grow primarily hydrogenotrophically [10], using CO2 and H2 as substrates. A few strains and/or species have been shown by cultivation-independent methods to be the predominant in specific intestinal environments, such as Mbb. smithii from the human gut [8] or Mbb. ruminantium, and Mbb. gottschalkii from the rumen of sheep and cows [11]. The former three species have gained considerable attention in the analysis of the human and rumen microbiome, but there are also Mbb. isolates that are less well investigated by cultivation-independent studies, for example, Mbb. thaueri strain DSM 11995T and Mbb. woesei strain DSM 11976T . Mbb. thaueri strain DSM 11995T was isolated from cow feces while Mbb. woesei strain DSM 11976T had been isolated from goose feces [12]. The 16S rRNA gene of either of the two species shares the highest sequence identity to Mbb. smithii strain DSM 861T, Mbb. gottschalkii strain DSM 11977T, and Mbb. millerae strain DSM 16643T [12–15]. Both species have been detected in a few cultivation-independent studies on fecal and rumen samples (for Mbb. thaueri, see [16, 17] and for Mbb. woesei, see [18–20]), but besides their original description, there is very little information on the physiology and ecology of these two species.
The genus Methanosphaera is less well characterized than the genus Methanobrevibacter, and only few species have been isolated or detected by cultivation-independent methods. Two species, Msp. stadtmanae DSM 3091T and Msp. cuniculi DSM 4103T, are currently the only formally described Methanosphaera-type species [21, 22]. The first genome-sequenced Methanosphaera species, Msp. stadtmanae DSM3091T, was isolated from human feces and has been shown to be restricted to growth on methanol and hydrogen under in vitro conditions [22]. This substrate restriction can to some extent be considered to be a typical trait of the genus Methanosphaera, but its genetic basis remained poorly understood until the genome of Msp. stadtmanae strain DSM 3091T was analyzed [23]. Comparative genome analysis revealed that most genes for biosynthesis of the molybdopterin cofactor (Moco), the cofactor of formylmethanofuran dehydrogenase (fmd), are absent from the genome of this methanogen. This explained why Msp. stadtmanae strain DSM3091T is not able to grow by disproportionation of methanol nor of CO2 and hydrogen as substrates. Moreover did the analysis reveal that the genome encodes four homologues of each subunit of the methanol : coenzyme-M methyltransferase (mtaABC) [23]. The MtaABC proteins form the key enzyme complex for the utilization of methanol and had before primarily been detected in species of the order Methanosarcinales.
The second genome of a genus Methanosphaera representative, Methanosphaera sp. WGK6, has only recently become available. This methanogen's genome sequence is to large extent nearly identical to that of Msp. stadtmanae DSM 3091T, but some of the few genomic differences result in phenotypic differences between the two species. This regards mainly two genes, putative alcohol (walC) and aldehyde (walD) dehydrogenases [24]. These two genes were apparently acquired via horizontal gene transfer and seem to enable Msp. sp. WGK6 to utilize ethanol as substrate for methanogenesis (in addition to its ability to grow on methanol and hydrogen) [24]. Homologues of walC and walD genes have also been detected in genomes of other methanogens but have not been investigated systematically.
Here, draft genomes of Mbb. thaueri strain DSM 11995T, Mbb. woesei strain DSM 11979T, and Msp. cuniculi strain DSM 4103T (a draft genome of M. cuniculi DSM 4103T of similar quality was also recently published by Gilmore et al. [25]) are presented and compared to those of all other currently available Methanosphaera- and Methanobrevibacter-type strain genomes. The comparative analyses aim at investigating and identifying some of the distinctive features of the Methanobrevibacter and Methanosphaera genomes that allow differentiating the two closely related genera. The major emphasis was set on key genes that may have large impact on the overall physiology of the analyzed species, such as those involved in substrate utilization and nitrogen and acetate assimilation.
2. Materials and Methods
2.1. Cultivation of Microorganism and DNA Extraction
Mbb. thaueri DSM 11995T, Mbb. woesei DSM 11979T, and Msp. cuniculi DSM 4103T were obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ), Braunschweig, Germany. Genomic DNA was ordered by the DSMZ (Braunschweig) or was isolated using the MasterPure complete DNA purification kit (Epicentre, Madison, WI, USA).
2.2. Genome Sequencing
Extracted DNA was used to generate Illumina-shotgun libraries (Nextera_XT) according to the manufacturer's protocol (Illumina, San Diego, CA, USA). Sequencing was conducted using a MiSeq and Miseq Reagent kit v3 (2 × 300 bp paired end) as recommended by the manufacturer (Illumina). Trimmomatic 0.32 [26] was used to filter low-quality reads and for clipping of adapter contaminations. The assembly was performed with the SPAdes genome assembler software 3.11 [27]. Coverages were determined using QualiMap version 2.1 [27–29], and automatic annotation was performed using the software tool PROKKA [30], and data analysis was partly performed using the IMG/ER system (Integrated Microbial Genomes & Microbiomes) [31]. The quality and the completeness of the draft genomes have been validated with CheckM [32].
2.3. Multilocus Sequence Analysis
For multilocus sequence analysis (MLSA), total protein sequences from 19 genomes were extracted from the corresponding GenBank files using cds_extractor.pl v0.6 [33] and used for downstream analysis with an in-house pipeline at the Goettingen Genomics Laboratory [34]. In detail, proteinortho version 5 (default specification: blast = blastp v2.2.24, E value = 1e − 10, alg.-conn. = 0.1, coverage = 0.5, percent_identity = 50, adaptive_similarity = 0.95, inc_pairs = 1, inc_singles = 1, selfblast = 1, and unambiguous = 0) [35] was used to generate clusters of orthologue groups, inparalogues were removed, and MUSCLE [36] used to align the remaining sequences and poorly aligned positions were automatically filtered from the alignments using Gblocks [37]. A maximum-likelihood tree from 574 orthologues was inferred with 500 bootstraps with RAxML [38]. A phylogenetic tree was inferred with neighbour joining and 500 bootstraps. PO_2_MLSA.py is available at GitHub (https://github.com/jvollme). Visualization was done using Proteinortho results and DNAPlotter [39]. The Venn diagram of the different clades was plotted using po2group_stats (v0.1.1) [33].
2.3.1. Analysis of 16S rRNA Genes
Aligned 16S rRNA gene sequences were selected from the ARB-compatible rumen and intestinal methanogen database (RIM-DB) [14, 40]. Aligned sequences were exported in PHYLIP format to construct phylogenetic trees using all available base positions. Maximum-likelihood phylogenetic trees based on aligned archaeal 16S rRNA gene sequences were generated using RAxML version 7.3.0 [38]. Unless stated otherwise, the parameters “-m GTRGAMMA -# 500 -f a -× 2 -p 2” were used.
2.4. Nucleotide Sequence Accession Number
The annotated genomes of Mbb. thaueri DSM 11995T and Mbb. woesei DSM 11979T have been deposited at DDBJ/EMBL/GenBank under the accession MZGS00000000 and MZGU00000000, respectively. The versions described in this paper are versions MZGS01000000 and MZGU01000000, respectively. The annotated genome of Msp. cuniculi DSM 4103T has been deposited at DDBJ/EMBL/GenBank under the accession LWMS00000000. The version described in this paper is version LWMS01000000.
3. Results
3.1. General Genome Features
The Methanosphaera cuniculi strain DSM 4103T draft genome has been assembled into 48 contigs, with a N50 of 111,976 bp. The GC content of the draft genome is 28%, which is similar to the G + C content of the other two published Methanosphaera genome sequences. The genomes of Mbb. thaueri strain DSM 11995T and Mbb. woesei strain DSM 11979T were assembled into 39 contigs and 10 contigs with N50 of 178.217 bp and 240.239 bp, respectively. Plasmids were not detected from the assembled contigs in either of the two draft genomes. General features of the of Mbb. thaueri DSM 11995T, Mbb. woesei strain DSM 11979T, and Methanosphaera cuniculi strain DSM 4103T genomes and comparison with other Methanobrevibacter and Methanosphaera strains are shown in Table 1, and results of CheckM analysis (including all genomes used in the analysis) are shown in Table S1. Circular representation of the three genomes is shown in supporting Figures S1 and S2.
Table 1.
General features of Methanobrevibacter and Methanosphaera genomes.
| Size (bp) | GC content (%) | Coding percentage (%) | CDS (pseudo) | Genes (pseudo) | rRNA | tRNA | Accession | Contigs/scaffolds | CRISPR region | Original strain ID | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Methanobrevibacter filiformis DSM 11501T | 2,606,143 | 26.99 | 69.94 | 1933 | 1965 | 3 | 29 | LWMT00000000 | 237 | 6 | RFM-3 |
| Methanobrevibacter curvatus DSM 11111T | 2,414,608 | 25.72 | 70.65 | 1969 | 2004 | 4 | 31 | LWMV00000000 | 187 | 5 | RFM-2 |
| Methanobrevibacter cuticularis DSM 11139T | 2,608,702 | 26.79 | 68.12 | 2061 | 2094 | 3 | 30 | LWMW00000000 | 120 | 6 | RFM-1 |
| Methanobrevibacter arboriphilus DSM 1125T | 2,445,031 | 25.44 | 74.96 | 1963 | 2005 | 5 | 37 | JXMW00000000 | 40 | 9 | DH1 |
| Methanobrevibacter arboriphilus ANOR1 | 2,221,072 | 25.53 | 74.02 | 1993 | 2038 | 7 | 35 | CBVX000000000 | 5 | 3 | ANOR1 |
| Methanobrevibacter ruminantium DSM 1093T | 2,937,203 | 32.64 | 78.12 | 2217 | 2283 (5) | 8 | 53 | CP001719 | 1 | 4 | M1 |
| Methanobrevibacter olleyae DSM 16632T | 2,122,444 | 26.87 | 76.85 | 1813 | 1854 | 4 | 33 | FOTL00000000 | 49 | 4 | KM1H5-1P |
| Methanobrevibacter millerae DSM 16643T | 2,725,667 | 36.54 | 89.32 | 2383 | 2467 | 4 | 77 | CP011266 | 1 | 1 | ZA-10 |
| Methanobrevibacter thaueri DSM 11995T | 2,243,115 | 36.87 | 87.52 | 2138 | 2171 | 2 | 31 | MZGS00000000 | 39 | 4 | CW |
| Methanobrevibacter gottschalkii DSM 11977T | 1,879,371 | 30.02 | 88.01 | 1845 | 1889 | 7 | 34 | FOAK00000000 | 19 | NA | HO |
| Methanobrevibacter oralis DSM 7256T | 2,110,861 | 27.73 | 84.49 | 2036 | 1994 | 9 | 32 | LWMU00000000 | 99 | 2 | ZR |
| Methanobrevibacter smithii DSM 861T | 1,853,160 | 31.03 | 90.31 | 1795 | 1841 | 8 | 36 | CP000678 | 1 | 1 | PS |
| Methanobrevibacter woesei DSM 11979T | 1,543,150 | 29.90 | 89.61 | 1581 | 1614 | 2 | 31 | MZGU00000000 | 10 | 2 | GS |
| Methanobrevibacter wolinii DSM 11976T | 2,041,814 | 24.21 | 75.94 | 1700 | 1747 | 8 | 36 | JHWX00000000 | 32 | 2 | SH |
| Methanobrevibacter boviskoreani DSM 25824T | 2,045,801 | 28.98 | 78.00 | 1756 | 1799 | 4 | 36 | BAGX00000000 | 54 | 2 | JH1 |
| Methanobrevibacter sp. AbM4 | 1,998,189 | 29.04 | 76.7 | 1671 | 1716 | 49 | 36 | CP004050 | 1 | 6 | N.A. |
| Methanosphaera sp. WGK6 | 1,729,155 | 27.70 | 78.05 | 1456 | 1616 (114) | 3 | 42 | JRWK00000000 | 37 | 1 | N.A. |
| Methanosphaera stadtmanae DSM 3091T | 1,767,403 | 27.63 | 84.10 | 1534 | 1592 (2) | 12 | 42 | CP000102 | 1 | 4 | MCB-3 |
| Methanosphaera cuniculi DSM 4103T | 1,881,497 | 28.08 | 81.36 | 1585 | 1629 | 5 | 39 | LWMS00000000 | 48 | 0 | 1R-7 |
3.2. Comparative Analysis of Methanobrevibacter Genomes
Sixteen different Methanobrevibacter and three Methanosphaera genomes were included for the comparative analyses. The Methanobrevibacter strains had been isolated from a range of different intestinal environments, for example, bovine and ovine rumen [12, 41], human gut [42], and termite hindguts [43, 44], but include also the genome of the non-host-associated Mbb. arboriphilus strain DSM 1125T [45, 46]. Results of the single- and multilocus analyses are shown in Figure 1. Both analyses revealed clear separation between the two genera.
Figure 1.

Single- and multilocus sequence analysis of Methanobrevibacter and Methanosphaera species. A maximum likelihood tree (left) of 16 Methanobrevibacter and three Methanosphaera genomes was inferred with 500 bootstraps with RAxML and visualized with Dendroscope. Phylogeny of Methanobrevibacter and Methanosphaera based on the 16S rRNA gene is shown on the right. The tree was resampled 500 times, and only bootstrap values ≥ 70% are shown. The 16S rRNA tree was rooted with five Methanobacterium sequences. The scale bar indicates 0.10 inferred nucleotide substitutions per position. Red-colored dots indicate the presence of mtaABC genes in the species/clade, green-colored dots indicate the presence of walB and walC gene homologues (potential utilization of ethanol), blue-colored dots indicate the presence of nitrogenase genes, and black-colored dots indicate the presence of carbon monoxide/acetyl CO-DH genes (see also Table S3 for details).
The MLSA also supports distinct clades within the genus Methanobrevibacter (Figure 1). There is limited association of the observed clades with a specific host origin, for example, some species from the rumen form distinct clades while others cluster with Methanobrevibacter species isolated from other hosts. A BLAST-based analysis of each clade identifies genes specific for clades and genes that are shared between them (Figure 2). Among the clade-specific genes of clade 1 are catalase, nitrogenase, and also a tRNA-specific endonuclease (VapC), but most other clade-specific genes of clade 1 and the other clades are without functional annotation and await further characterization.
Figure 2.
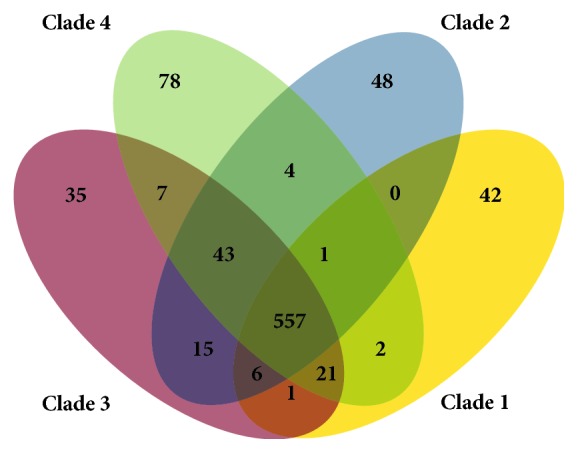
Pan/core genome analysis of four different Methanobrevibacter clades. Venn diagram showing the numbers of orthologous genes (OGs) in the core, dispensable, and specific genome of compared strains. Ortholog detection was done with the Proteinortho software (blastp) with a similarity cut-off of 50% and an E value of 1e − 10. The total numbers of genes and paralogs are depicted under the corresponding species name. Open-reading frames that were classified as pseudogenes were not included in this analysis. See also Table S3 for details on shared genes.
It was also investigated whether certain metabolic traits could be associated with specific clades as pointed out in Figure 1. This concerns mainly the metabolism of alcohols and acetate assimilation. Carbon monoxide/acetyl-CoA-synthetase complex (CODH-ACS) is—in addition to other enzymes—important for acetate assimilation, and its presence is one prerequisite for autotrophic growth. CODH-ACS is found in non-host-associated and autotrophic methanogens, for example, Methanothermobacter species [47–49], and is detected in four out of five clade 1 species, but gene homologues of the enzyme appear to be absent from the genomes of methanogens in clades 2–4.
The potential utilization of alcohols, specifically methanol and ethanol, is also less clearly distributed among the 19 genomes (and the four different clades) and there are currently no known Methanobrevibacter species that are restricted to growth on H2 and methanol/ethanol [10]. Genes for methanol utilization of the Methanosphaera/Methanosarcina type (mtaABC) are distributed broadly within the genus Methanobrevibacter as outlined in Figure 1 and have also already been reported for some strains [46, 50]. Physiological characterizations of the strains that harbor mtaABC genes (Mbb. arboriphilus strains DSM 1125T and ANOR1, Mbb. smithii strain DSM 861T, Mbb. wolinii strain DSM 11976T) is still outstanding to confirm that these species are capable of growth on methanol/methanol-hydrogen.
The utilization of ethanol by Methanobrevibacter species has only recently gained additional attention and it has been shown that Mbb. sp. AbM4 is capable of growth in the absence of hydrogen but in presence of methanol/ethanol [51, 52]. Details regarding the exact metabolism remain speculative, but the walC and walD genes recently identified in Msp. WGK6 [24] are also present in Mbb. sp. AbM4 and in the closely related Mbb. boviskoreani strain DSM 25824T, Mbb. olleyae strain DSM 16632T, and Mbb. wolinii strain DSM11976T conferring this metabolic trait potentially to the entire clade. Mbb. olleyae strain DSM 16632T is currently the only Mbb. species outside this clade that is harboring the walCD genes.
Only few additional genome sequences of Methanobrevibacter isolates other than for Mbb. smithii are currently available, but the clade-distinguishing features appear to be conserved in these isolates as well (see Table S2 for general genome features and Table S3 for shared genes).
3.3. Shared and Distinctive Genome Features in the Genomes of Methanosphaera Species
The currently known Methanosphaera species are characterized by their limited ability to use substrates for methanogenesis, for example, Msp. stadtmanae and Msp. cuniculi are able to utilize only methanol and hydrogen for growth, Msp. sp. WKG6 can also utilize ethanol, but none of the species is capable of utilizing hydrogen and CO2 or formate like organisms from closely related genera. The analysis of the Msp. stadtmanae genome helped explaining some of the distinctive physiological features of this methanogen, but comparative genome analysis with other closely related Methanosphaera and Methanobrevibacter species was not possible at that time due to the lack of sequenced genomes. After more than a decade, several Methanosphaera and Methanobrevibacter genomes have become available [24, 41, 46, 50, 53–58] and comparative analyses allow determining whether certain traits are species specific or shared by members of either or both genera. Comparative genome analysis reveals that some of the observations made for the Msp. stadtmanae genome appear to be consistent for all three available Methanosphaera genomes. Overall, more than 1000 genes are shared among all three Methanosphaera species (Figure 3), but the comparison with Methanobrevibacter genomes shows that several key genes are missing. Notably, this concerns the lack of genes for molybdopterin cofactor biosynthesis, energy-converting hydrogenase A (ehaA-Q), formate dehydrogenase (fdh), and methyl-coenzyme M reductase isoenzyme 1 mcrABCG (only genes encoding isoenzyme II are present (mrtABDG)). Despite the presence of formylmethanofuran dehydrogenase genes, it is not possible for Methanosphaera species to produce a functional enzyme due to the absence of the molybdopterin cofactor. The absence of functional formylmethanofuran dehydrogenase leads to the inability of these methanogens to grow hydrogenotrophically or to disproportionate methanol. The absence of Moco biosynthesis also seems to be specific to Methanosphaera species as all of the analyzed Methanobrevibacter genomes are capable of synthesizing this cofactor.
Figure 3.
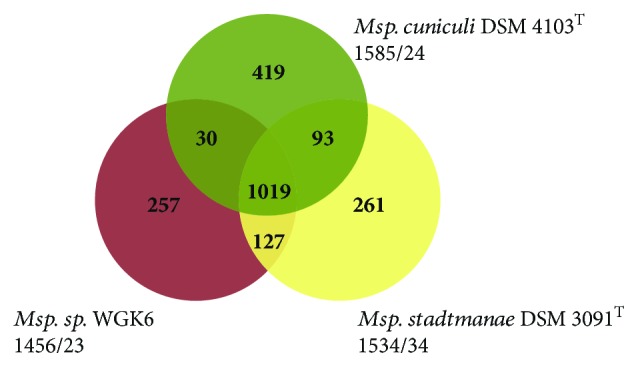
Pan/core genome analysis of three different Methanosphaera species. Venn diagram showing the numbers of orthologous genes (OGs) in the core, dispensable, and specific genome of compared strains. Ortholog detection was done with the Proteinortho software (blastp) with a similarity cut-off of 50% and an E value of 1e − 10. The total numbers of genes and paralogs are depicted under the corresponding species name. Open-reading frames that were classified as pseudo genes were not included in this analysis. See also Table S3 for details on shared genes.
One of the noteworthy differences between the known Methanosphaera species is the capability of Msp. WGK6 to utilize ethanol as an electron donor for methanogenesis. The recently reported alcohol dehydrogenase and aldehyde dehydrogenase (NL43_02835, NL43_02830) in the genome of Methanosphaera sp. WGK6 were not detected in the Msp. stadtmanae genome (as previously reported by Hoedt et al., 2016) or the Msp. cuniculi genome [25], indicating that this may be a trait of Msp. WGK6 that is shared with other less-related methanogens.
4. Discussion
The presented comparative genome analysis reveals insights into the genome content of different Methanobrevibacter species and how it compares to that of closely related Methanosphaera species. The analysis suggests that phenotypic variation among Methanobrevibacter strains may be larger than previously assumed. The comparative analysis indicates the presence of genes for methanol (in four Mbb. genomes) and ethanol utilization (in four Mbb. genomes) in the sixteen analyzed Methanobrevibacter genomes, with the Mbb. wolinii genome harboring both walCD and mtaABC gene homologues. More physiological analysis will be required to determine if the detected genes are conferring the ability to utilize ethanol/methanol to all these strains. The ability of Methanobrevibacter strains to utilize alcohols may not have been investigated in an in-depth manner previously as this trait is more typically associated with other orders of methanogens, but some studies suggest that the genes may in some cases also not be functional or may have unknown different functions, for example, growth of Mbb. smithii on methanol was not detected despite the presence of mtaABC genes (growth on methanol but not methanol and hydrogen was tested) [59]. It is noteworthy that all three sequenced strains of the Mbb. wolinii clade harbor the genes for ethanol utilization, which could point to a specific ecological role of the species in this clade; however, there are only few studies that have detected significant numbers of either of these three species in a natural environment [5, 60] making it currently difficult to determine potential cooccurrences or specific syntrophic interactions with other microorganisms.
The presence of genes that could contribute to autotrophic growth, for example, nitrogenase and CODH-ACS in clade 1 containing Mbb. species from termites and in Mbb. arboriphilus strain, has been reported previously [46], and these genes appear to be absent from species clades 2 to 4. It can only be speculated why Mbb. species of clade 1 did not undergo the same loss of these key genes like their counterparts in the clades that have primarily been isolated from the vertebrate intestinal tract. However, auxotrophy of some Mbb. species may be the result of a close symbiotic interaction with other microorganisms (and potentially the host) that provide favorable growth conditions as well as ammonium and acetate for the methanogen. It is also noteworthy that Mbb. curvatus in the “autotrophic clade” has nitrogenase genes, but apparently, no CODH complex genes and may therefore require externally supplied acetate. This could represent an intermediate stage between the potentially autotrophic strains and the other clades and/or may be an adaptation to a specific niche.
In addition to differences between species, there is currently only little information on strain diversity within a species but studies indicate that there may be considerable differences as shown for Mbb. smithii and Mbb. arboriphilus [46, 61]. However, undertaking pan-genome approaches, such as the one for Mbb. smithii by Hansen et al. [61], requires substantial cultivation efforts as there are only few strains available for each of the described methanogen species. As development of sequencing technologies continues to advance, it may also become feasible to obtain high-quality closed genomes from low amounts of starting DNA or through metagenomic approaches. Having such genomes will allow greater certainty in determining the presence and absence of specific genome features and will also enable detection of small genomic differences between strains that may go unnoticed in draft genomes.
Lastly, the results of our analyses corroborate the hypothesis that the absence of molydopterin cofactor biosynthesis is a characteristic trait shared by members of the genus Methanosphaera, while all sequenced Methanobrevibacter genomes seem to encode genes for Moco biosynthesis. However, it also needs to be considered that Methanosphaera species encode the genes for formylmethanofuran dehydrogenase and other genes required for hydrogenotrophic or methylotrophic methanogenesis [62, 63]. The presence of these genes may indicate that Methanosphaera species may still be able to utilize the aforementioned methanogenic pathways, if molybdopterin cofactor is present and taken up by the methanogen (as suggested by Fricke et al. 2006). Metatranscriptional profiling could be used to determine, if fmd and other genes for hydrogenotrophic growth are expressed at all in Methanosphaera or specifically in the absence and presence of Moco or Moco-synthesizing microorganisms. It needs to be emphasized that the analyses of this study are based on the currently available three genomes of the three Methanosphaera species that have been isolated from three different host species [23–25]. It provides strong evidence that the physiology will be similar for other Msp. species in other environments, but it can also not be ruled out that other Msp. species exist that harbor genes for Moco biosynthesis and that grow hydrogenotrophically or methylotrophically. The ability of Msp. sp. WGK6 to utilize ethanol does indicate that more phenotypic variation exists among Methanosphaera species and that further isolation and characterization of new Msp. species is necessary.
5. Conclusion
The presented study includes genomes of all currently available types of strain (and other selected isolate) of Methanobrevibacter and Methanosphaera genomes and allows comprehensive insights into the genera Methanobrevibacter and Methanosphaera. The analyses reveal that distinct clades within the genus Methanobrevibacter exist and that the lack of molybdopterin cofactor biosynthesis may be a specific trait for the genus Methanosphaera. Additional isolates and further physiological and genomic analyses will be required to determine if division of the genus Methanobrevibacter in more than one genus could be justified. The primary use of the type of strains (and other isolates) for the analysis in this study does warrant access of the scientific community to most of the analyzed Methanobrevibacter and Methanosphaera isolates and will facilitate testing of the predicted physiological phenotypes.
Acknowledgments
This work was funded by Temasek Life Sciences Laboratory. The authors thank Melanie Heinemann for the technical support.
Data Availability
The data used to the support the findings of this study are availabe in the supplemental information or -as in the case of the genomes- are downloadable using the accession numbers provided in Section 2.4.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Supplementary Materials
Figure S1: circular display of the Mbb. thaueri strain DSM 11995T and Mbb. woesei strain DSM 11979T genomes and comparison with other Methanobrevibacter genomes. The Mbb. thaueri strain DSM 11995T and the Mbb. woesei strain DSM 11979T genomes are shown in panels A and B, respectively. The genes encoded by the leading and the lagging strand of the respective species are shown in circles 1 and 2, and rRNA (pink) and tRNA (green) encoding genes are shown in circle 3. The presence of orthologous genes (red: high similarity; orange: medium similarity; yellow: low similarity; see color code below) is indicated for the other Methanobrevibacter genomes (circles 4 to 19) in comparison to that of the respective Methanobrevibacter genome. The two innermost plots represent the GC content and the GC skew (circle 20 and 21). Color code according to E values of the blastp analysis performed using Proteinortho4.26. Gray: 1e − 20 to 1; light yellow: 1e − 21 to 1e − 50; gold: 1e − 51 to 1e − 90; light orange: 1e − 91 to 1e − 100; orange: 1e − 101 to 1e − 120; red: >1e − 120.
Figure S2: circular display of the Msp. cuniculi strain DSM 4103T genome and comparison with the genomes of Msp. stadtmanae strain DSM 3091T and Msp. sp. WGK6. The genes encoded by the leading and the lagging strand of Msp. cuniculi strain DSM 4103T are shown in circles 1 and 2, and rRNA (pink) and tRNA (green) encoding genes are shown in circle 3. The presence of orthologous genes (red: high similarity; orange: medium similarity; yellow: low similarity; see color code below) is indicated for the genomes of Msp. stadtmanae strain DSM 3091T and Msp. sp. WGK6 (circles 4 and 5 in comparison to the Msp. cuniculi strain DSM 4103T genome). The two innermost plots represent the GC content and the GC skew (circles 6 and 7). Color code according to E values of the blastp analysis performed using Proteinortho4.26. Gray: 1e − 20 to 1; light yellow: 1e − 21 to 1e − 50; gold: 1e − 51 to 1e − 90; light orange: 1e − 91 to 1e − 100; orange: 1e − 101 to 1e − 120; red: >1e − 120.
Table S1: CheckM analysis to determine the completeness of genomes. Table S1A shows results from analysis of genomes from the main manuscript while Table S1B lists the results for genomes mainly mentioned in the supporting information. Table S2: general features of additional Methanobrevibacter genomes. General features of genomes mainly mentioned in the supporting information. Table S3: table of shared and unshared Methanobrevibacter and Methanosphaera genes. Highlighted in colors corresponding to those of Figure 1 are genes for CODH-ACS complex, nitrogenase, methanol, and ethanol utilization. Annotations were taken from the Mbb. arboriphilus DSM1125T genome.
References
- 1.Kittelmann S., Pinares-Patiño C. S., Seedorf H., et al. Two different bacterial community types are linked with the low-methane emission trait in sheep. PLoS One. 2014;9(7, article e103171) doi: 10.1371/journal.pone.0103171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi W., Moon C. D., Leahy S. C., et al. Methane yield phenotypes linked to differential gene expression in the sheep rumen microbiome. Genome Research. 2014;24(9):1517–1525. doi: 10.1101/gr.168245.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodrich J. K., Waters J. L., Poole A. C., et al. Human genetics shape the gut microbiome. Cell. 2014;159(4):789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carberry C. A., Waters S. M., Kenny D. A., Creevey C. J. Rumen methanogenic genotypes differ in abundance according to host residual feed intake phenotype and diet type. Applied and Environmental Microbiology. 2014;80(2):586–594. doi: 10.1128/AEM.03131-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou M., Hernandez-Sanabria E., Guan L. L. Assessment of the microbial ecology of ruminal methanogens in cattle with different feed efficiencies. Applied and Environmental Microbiology. 2009;75(20):6524–6533. doi: 10.1128/AEM.02815-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonin A. S., Boone D. R. Dworkin M., Falkow S., Rosenberg E., Schleifer K. H., Stackebrandt E. The Prokaryotes. 3rd. Vol. 3. Springer; 2006. The Order Methanobacteriales; pp. 231–243. [DOI] [Google Scholar]
- 7.Iino T., Tamaki H., Tamazawa S., et al. Candidatus Methanogranum caenicola: a novel methanogen from the anaerobic digested sludge, and proposal of Methanomassiliicoccaceae fam. nov. and Methanomassiliicoccales ord. nov., for a methanogenic lineage of the class Thermoplasmata. Microbes and Environments. 2013;28(2):244–250. doi: 10.1264/jsme2.ME12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eckburg P. B., Bik E. M., Bernstein C. N., et al. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henderson G., Cox F., Ganesh S., et al. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Scientific Reports. 2015;5(1) doi: 10.1038/srep14567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller T. L. Methanobrevibacter, Bergey’s manual of systematics of archaea and bacteria. The Archaea and Deeply Branching and Phototrophic Bacteria. 2001;1:218–226. [Google Scholar]
- 11.Seedorf H., Kittelmann S., Janssen P. H. Few highly abundant operational taxonomic units dominate within rumen methanogenic archaeal species in New Zealand sheep and cattle. Applied and Environmental Microbiology. 2015;81(3):986–995. doi: 10.1128/AEM.03018-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller T. L., Lin C. Description of Methanobrevibacter gottschalkii sp. nov., Methanobrevibacter thaueri sp. nov., Methanobrevibacter woesei sp. nov. and Methanobrevibacter wolinii sp. nov. International Journal of Systematic and Evolutionary Microbiology. 2002;52:819–822. doi: 10.1099/00207713-52-3-819. [DOI] [PubMed] [Google Scholar]
- 13.Janssen P. H., Kirs M. Structure of the archaeal community of the rumen. Applied and Environmental Microbiology. 2008;74(12):3619–3625. doi: 10.1128/AEM.02812-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seedorf H., Kittelmann S., Henderson G., Janssen P. H. RIM-DB: a taxonomic framework for community structure analysis of methanogenic archaea from the rumen and other intestinal environments. PeerJ. 2014;2, article e494 doi: 10.7717/peerj.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.St-Pierre B., Wright A.-D. G. Molecular analysis of methanogenic archaea in the forestomach of the alpaca (Vicugna pacos) BMC Microbiology. 2012;12(1):p. 1. doi: 10.1186/1471-2180-12-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishaq S. L., Sundset M. A., Crouse J., Wright A.-D. G. High-throughput DNA sequencing of the moose rumen from different geographical locations reveals a core ruminal methanogenic archaeal diversity and a differential ciliate protozoal diversity. Microbial Genomics. 2015;1(4, article e000034) doi: 10.1099/mgen.0.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salgado-Flores A., Hagen L. H., Ishaq S. L., et al. Rumen and cecum microbiomes in reindeer (Rangifer tarandus tarandus) are changed in response to a lichen diet and may affect enteric methane emissions. PLoS One. 2016;11(5, article e0155213) doi: 10.1371/journal.pone.0155213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Just N., Lecours P. B., Marcoux-Voiselle M., et al. Archaeal characterization of bioaerosols from cage-housed and floor-housed poultry operations. Canadian Journal of Microbiology. 2013;59(1):46–50. doi: 10.1139/cjm-2012-0305. [DOI] [PubMed] [Google Scholar]
- 19.Li F., Henderson G., Sun X., Cox F., Janssen P. H., Guan L. L. Taxonomic assessment of rumen microbiota using total RNA and targeted amplicon sequencing approaches. Frontiers in Microbiology. 2016;7 doi: 10.3389/fmicb.2016.00987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saengkerdsub S., Anderson R. C., Wilkinson H. H., Kim W. K., Nisbet D. J., Ricke S. C. Identification and quantification of methanogenic archaea in adult chicken ceca. Applied and Environmental Microbiology. 2006;73(1):353–356. doi: 10.1128/AEM.01931-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biavati B., Vasta M., Ferry J. G. Isolation and characterization of Methanosphaera cuniculi sp. nov. Applied and Environmental Microbiology. 1988;54(3):768–771. doi: 10.1128/aem.54.3.768-771.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller T. L., Wolin M. J. Methanosphaera stadtmaniae gen. nov., sp. nov.: a species that forms methane by reducing methanol with hydrogen. Archives of Microbiology. 1985;141(2):116–122. doi: 10.1007/BF00423270. [DOI] [PubMed] [Google Scholar]
- 23.Fricke W. F., Seedorf H., Henne A., et al. The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. Journal of Bacteriology. 2005;188(2):642–658. doi: 10.1128/JB.188.2.642-658.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoedt E. C., Cuív P. Ó., Evans P. N., et al. Differences down-under: alcohol-fueled methanogenesis by archaea present in Australian macropodids. The ISME Journal. 2016;10(10):2376–2388. doi: 10.1038/ismej.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilmore S. P., Henske J. K., Sexton J. A., et al. Genomic analysis of methanogenic archaea reveals a shift towards energy conservation. BMC Genomics. 2017;18(1):p. 639. doi: 10.1186/s12864-017-4036-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bolger A. M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bankevich A., Nurk S., Antipov D., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okonechnikov K., Conesa A., García-Alcalde F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 2016;32(2):292–294. doi: 10.1093/bioinformatics/btv566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.García-Alcalde F., Okonechnikov K., Carbonell J., et al. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics. 2012;28(20):2678–2679. doi: 10.1093/bioinformatics/bts503. [DOI] [PubMed] [Google Scholar]
- 30.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 31.Markowitz V. M., Mavromatis K., Ivanova N. N., Chen I.-M. A., Chu K., Kyrpides N. C. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25(17):2271–2278. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- 32.Parks D. H., Imelfort M., Skennerton C. T., Hugenholtz P., Tyson G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Research. 2015;25(7):1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leimbach A. Bovine E. coli mastitis comparative genomics edition. Bac-genomics-scripts. 2016 [Google Scholar]
- 34.Haack F. S., Poehlein A., Kröger C., et al. Molecular keys to the Janthinobacterium and Duganella spp. interaction with the plant pathogen Fusarium graminearum. Frontiers in Microbiology. 2016;7(1668) doi: 10.3389/fmicb.2016.01668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lechner M., Findeiß S., Steiner L., Marz M., Stadler P. F., Prohaska S. J. Proteinortho: detection of (co-) orthologs in large-scale analysis. BMC Bioinformatics. 2011;12(1):p. 124. doi: 10.1186/1471-2105-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edgar R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution. 2000;17(4):540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 38.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 39.Carver T., Thomson N., Bleasby A., Berriman M., Parkhill J. DNAPlotter: circular and linear interactive genome visualization. Bioinformatics. 2009;25(1):119–120. doi: 10.1093/bioinformatics/btn578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ludwig W., Strunk O., Westram R., et al. ARB: a software environment for sequence data. Nucleic Acids Research. 2004;32(4):1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee J.-H., Rhee M.-S., Kumar S., et al. Genome sequence of Methanobrevibacter sp. strain JH1, isolated from rumen of Korean native cattle. Genome Announcements. 2013;1(1):e00002–e00013. doi: 10.1128/genomeA.00002-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller T. L., Wolin M. J. Enumeration of Methanobrevibacter smithii in human feces. Archives of Microbiology. 1982;131(1):14–18. doi: 10.1007/BF00451492. [DOI] [PubMed] [Google Scholar]
- 43.Leadbetter J. R., Breznak J. A. Physiological ecology of Methanobrevibacter cuticularis sp. nov. and Methanobrevibacter curvatus sp. nov., isolated from the hindgut of the termite Reticulitermes flavipes. Applied and Environmental Microbiology. 1996;62(10):3620–3631. doi: 10.1128/aem.62.10.3620-3631.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leadbetter J. R., Crosby L. D., Breznak J. A. Methanobrevibacter filiformis sp. nov., a filamentous methanogen from termite hindguts. Archives of Microbiology. 1998;169(4):287–292. doi: 10.1007/s002030050574. [DOI] [PubMed] [Google Scholar]
- 45.Zeikus J. G., Henning D. L. Methanobacterium arbophilicum sp. nov. an obligate anaerobe isolated from wetwood of living trees. Antonie Van Leeuwenhoek. 1975;41(1):543–552. doi: 10.1007/BF02565096. [DOI] [PubMed] [Google Scholar]
- 46.Poehlein A., Daniel R., Seedorf H. The draft genome of the non-host-associated methanobrevibacter arboriphilus strain dh1 encodes a large repertoire of adhesin-like proteins. Archaea. 2017;2017:9. doi: 10.1155/2017/4097425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaster A.-K., Goenrich M., Seedorf H., et al. More than 200 genes required for methane formation from H2 and CO2 and energy conservation are present in Methanothermobacter marburgensis and Methanothermobacter thermautotrophicus. Archaea. 2011;2011:23. doi: 10.1155/2011/973848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liesegang H., Kaster A. K., Wiezer A., et al. Complete genome sequence of Methanothermobacter marburgensis, a methanoarchaeon model organism. Journal of Bacteriology. 2010;192(21):5850–5851. doi: 10.1128/JB.00844-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith D. R., Doucette-Stamm L. A., Deloughery C., et al. Complete genome sequence of Methanobacterium thermoautotrophicum deltaH: functional analysis and comparative genomics. Journal of Bacteriology. 1997;179(22):7135–7155. doi: 10.1128/jb.179.22.7135-7155.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Samuel B. S., Hansen E. E., Manchester J. K., et al. Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proceedings of the National Academy of Sciences. 2007;104(25):10643–10648. doi: 10.1073/pnas.0704189104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leahy S. C., Kelly W. J., Ronimus R. S., Wedlock N., Altermann E., Attwood G. T. Genome sequencing of rumen bacteria and archaea and its application to methane mitigation strategies. Animal. 2013;7(s2):235–243. doi: 10.1017/S1751731113000700. [DOI] [PubMed] [Google Scholar]
- 52.Weimar M. R., Cheung J., Dey D., et al. Development of multi-well plate methods using pure cultures of methanogens to identify new inhibitors for suppressing ruminant methane emissions. Applied and Environmental Microbiology. 2017;83(15):e00396–e00317. doi: 10.1128/aem.00396-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leahy S. C., Kelly W. J., Li D., et al. The complete genome sequence of Methanobrevibacter sp. AbM4. Standards in Genomic Sciences. 2013;8(2):215–227. doi: 10.4056/sigs.3977691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khelaifia S., Garibal M., Robert C., Raoult D., Drancourt M. Draft genome sequence of a human-associated isolate of Methanobrevibacter arboriphilicus, the lowest-G+C-content archaeon. Genome Announcements. 2014;2(1):e01181–e01113. doi: 10.1128/genomea.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kelly W. J., Li D., Lambie S. C., et al. Draft genome sequence of the rumen methanogen Methanobrevibacter olleyae YLM1. Genome Announcements. 2016;4(2):e00232–e00216. doi: 10.1128/genomeA.00232-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poehlein A., Seedorf H. Draft genome sequences of Methanobrevibacter curvatus DSM11111, Methanobrevibacter cuticularis DSM11139, Methanobrevibacter filiformis DSM11501, and Methanobrevibacter oralis DSM7256. Genome Announcements. 2016;4(3):e00617–e00616. doi: 10.1128/genomea.00617-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khelaifia S., Garibal M., Robert C., Raoult D., Drancourt M. Draft genome sequencing of Methanobrevibacter oralis strain JMR01, isolated from the human intestinal microbiota. Genome Announcements. 2014;2(1):e00073–e00014. doi: 10.1128/genomea.00073-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leahy S. C., Kelly W. J., Altermann E., et al. The genome sequence of the rumen methanogen Methanobrevibacter ruminantium reveals new possibilities for controlling ruminant methane emissions. PLoS One. 2010;5(1, article e8926) doi: 10.1371/journal.pone.0008926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miller T. L., Wolin M. J., Conway de Macario E., Macario A. J. Isolation of Methanobrevibacter smithii from human feces. Applied and Environmental Microbiology. 1982;43(1):227–232. doi: 10.1128/aem.43.1.227-232.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Federici S., Miragoli F., Pisacane V., Rebecchi A., Morelli L., Callegari M. L. Archaeal microbiota population in piglet feces shifts in response to weaning: Methanobrevibacter smithii is replaced with Methanobrevibacter boviskoreani. FEMS Microbiology Letters. 2015;362(10) doi: 10.1093/femsle/fnv064. [DOI] [PubMed] [Google Scholar]
- 61.Hansen E. E., Lozupone C. A., Rey F. E., et al. Pan-genome of the dominant human gut-associated archaeon, Methanobrevibacter smithii, studied in twins. Proceedings of the National Academy of Sciences. 2011;108(Supplement_1):4599–4606. doi: 10.1073/pnas.1000071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thauer R. K. Biochemistry of methanogenesis: a tribute to Marjory Stephenson: 1998 Marjory Stephenson Prize lecture. Microbiology. 1998;144(9):2377–2406. doi: 10.1099/00221287-144-9-2377. [DOI] [PubMed] [Google Scholar]
- 63.Thauer R. K., Kaster A.-K., Seedorf H., Buckel W., Hedderich R. Methanogenic archaea: ecologically relevant differences in energy conservation. Nature Reviews Microbiology. 2008;6(8):579–591. doi: 10.1038/nrmicro1931. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: circular display of the Mbb. thaueri strain DSM 11995T and Mbb. woesei strain DSM 11979T genomes and comparison with other Methanobrevibacter genomes. The Mbb. thaueri strain DSM 11995T and the Mbb. woesei strain DSM 11979T genomes are shown in panels A and B, respectively. The genes encoded by the leading and the lagging strand of the respective species are shown in circles 1 and 2, and rRNA (pink) and tRNA (green) encoding genes are shown in circle 3. The presence of orthologous genes (red: high similarity; orange: medium similarity; yellow: low similarity; see color code below) is indicated for the other Methanobrevibacter genomes (circles 4 to 19) in comparison to that of the respective Methanobrevibacter genome. The two innermost plots represent the GC content and the GC skew (circle 20 and 21). Color code according to E values of the blastp analysis performed using Proteinortho4.26. Gray: 1e − 20 to 1; light yellow: 1e − 21 to 1e − 50; gold: 1e − 51 to 1e − 90; light orange: 1e − 91 to 1e − 100; orange: 1e − 101 to 1e − 120; red: >1e − 120.
Figure S2: circular display of the Msp. cuniculi strain DSM 4103T genome and comparison with the genomes of Msp. stadtmanae strain DSM 3091T and Msp. sp. WGK6. The genes encoded by the leading and the lagging strand of Msp. cuniculi strain DSM 4103T are shown in circles 1 and 2, and rRNA (pink) and tRNA (green) encoding genes are shown in circle 3. The presence of orthologous genes (red: high similarity; orange: medium similarity; yellow: low similarity; see color code below) is indicated for the genomes of Msp. stadtmanae strain DSM 3091T and Msp. sp. WGK6 (circles 4 and 5 in comparison to the Msp. cuniculi strain DSM 4103T genome). The two innermost plots represent the GC content and the GC skew (circles 6 and 7). Color code according to E values of the blastp analysis performed using Proteinortho4.26. Gray: 1e − 20 to 1; light yellow: 1e − 21 to 1e − 50; gold: 1e − 51 to 1e − 90; light orange: 1e − 91 to 1e − 100; orange: 1e − 101 to 1e − 120; red: >1e − 120.
Table S1: CheckM analysis to determine the completeness of genomes. Table S1A shows results from analysis of genomes from the main manuscript while Table S1B lists the results for genomes mainly mentioned in the supporting information. Table S2: general features of additional Methanobrevibacter genomes. General features of genomes mainly mentioned in the supporting information. Table S3: table of shared and unshared Methanobrevibacter and Methanosphaera genes. Highlighted in colors corresponding to those of Figure 1 are genes for CODH-ACS complex, nitrogenase, methanol, and ethanol utilization. Annotations were taken from the Mbb. arboriphilus DSM1125T genome.
Data Availability Statement
The data used to the support the findings of this study are availabe in the supplemental information or -as in the case of the genomes- are downloadable using the accession numbers provided in Section 2.4.