

Abstract
Background and objectives: This retrospective study analysed a case series of subjects with citrin deficiency, and aims to present the molecular and clinical characterization of this disease in the Hong Kong Chinese population for the first time. Patients and Methods: Data from medical records of eighteen patients with citrin deficiency (years 2006–2015) were retrieved. Demographic data, biochemical parameters, radiological results, genetic testing results, management, and clinical outcome were collected and analysed. Results: Eighteen patients with diagnosis of citrin deficiency were recruited. All 18 patients carried at least one common pathogenic variant c.852_855delTATG in SLC25A13. Prolonged jaundice (neonatal intrahepatic cholestasis caused by citrin deficiency, NICCD) was the most common presenting symptom, in conjunction with elevated plasma citrulline, threonine, alkaline phosphatase, and alpha-fetoprotein levels. The abnormal biochemical parameters including liver derangement returned to normal range in most of the cases by 6 months of age after the introduction of a lactose-free formula. There were a few cases with atypical presentations. Two subjects did not present with NICCD, and were subsequently diagnosed later in life after their siblings presented with symptoms of citrin deficiency at one month of age and subsequently received a molecular diagnosis. One patient with citrin deficiency also exhibited multiple liver hemangioendotheliomas, which subsided gradually after introduction of a lactose-free formula. Only one patient from this cohort was offered expanded metabolic screening at birth. She was not ascertained by conducted newborn screening and was diagnosed upon presentation with cholestatic jaundice by 1 month of age. Conclusion: This is the first report of the clinical and molecular characterization of a large cohort of patients with citrin deficiency in Hong Kong. The presentation of this cohort of patients expands the clinical phenotypic spectrum of NICCD. Benign liver tumors such as hemangioendotheliomas may be associated with citrin deficiency in addition to the well-known association with hepatocellular carcinoma. Citrin deficiency may manifest in later infancy period with an NICCD-like phenotype. Furthermore, this condition is not always ascertained by expanded newborn metabolic screening testing.
Keywords: Citrin deficiency, NICCD, FTTDCD, CTLN2, Hemangioendothelioma
1. Introduction
Citrin deficiency is an autosomal recessive condition that encompasses a broad phenotypic spectrum of clinical phenotypes including neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD: OMIM#605814) [1], the intermediate type failure to thrive and dyslipidemia caused by citrin deficiency (FTTDCD) [2], and the adult-onset citrullinemia type II (CTLN2: OMIM#603471) [1,3,4]. The condition is caused by biallelic pathogenic variants in SLC25A13 on chromosome 7q21.3 [5,6]. Citrin deficiency can manifest in the neonatal period or infancy with an NICCD phenotype. NICCD is characterized by neonatal or infantile onset intrahepatic cholestatic jaundice with raised plasma citrulline, arginine, ammonia, alpha-fetoprotein, and liver parenchymal cellular infiltration [7]. Most of the NICCD patients respond well with a dietary management of lactose-free formula. Very rarely, liver transplant was needed in few affected subjects [8,9]. Some of the subjects with citrin deficiency will develop an intermediate phenotype, FTTDCD beyond neonatal period [10], while some remain asymptomatic until adulthood. This intermediate phenotype is mostly associated with failure to thrive and dyslipidemia. The factors that would confer a higher risk for subjects with NICCD or FTTDCD to develop CTLN2 remain unknown for the most part. Subjects with this latter condition may present with recurrent late childhood or adult onset hyperammonemic episodes with liver dysfunction and neuropsychiatric symptoms [1,3,4]. The initial case series were reported in Japanese and other Asian populations. However, subjects have now been reported from different ethnicities [1,2,4,10,11]. Further research studies are essential to elucidate the pathophysiology underlying this diverse spectrum. This study aims to present the clinical and molecular spectrum of citrin deficiency in a cohort of Chinese patients in Hong Kong and to discuss a rare incidence of liver hemangioendothelioma in one of the subjects.
2. Materials and methods
2.1. Subjects
Medical records of paediatric patients aged 0–18 with the diagnosis of citrin deficiency from 2007 to 2017 were retrieved from the medical record system of a Joint Metabolic Clinic located at Prince of Wales Hospital, New Territories, Hong Kong Special Administrative Region. Eighteen patients were identified with a diagnosis of citrin deficiency. Demographic data including age, gender, birth weight, gestation, ethnicity, age of presentation, clinical symptoms and signs, and biochemical findings were collected. The treatment and outcome of the patients were also retrieved. Since subjects 1 and 2 in the cohort did not present with NICCD and were diagnosed late in life, their biochemical data were not used to compare with the data from the other 16 patients who presented with NICCD.
2.2. Newborn screening
Only one patient out of the 18 patients in this cohort received newborn metabolic screening testing. All the other patients did not receive newborn metabolic screening.
2.3. Molecular testing
Tiered molecular testing was conducted. It included targeted mutation analysis by Sanger sequencing for coding exons 1, 6, 7, 9, 11, 14, 16, 17 of the SLC25A13 gene. Flanking introns (20 nucleotides) were directly sequenced in both directions. Long range PCR analysis was performed to screen for the 3 kb insertion mutation in intron 16 (IVS16ins3kb) currently known as c.1750+72_1751-4dup17insNM_138459.3:2667. If tiered testing was not informative, Sanger sequencing of the remaining exons of SLC25A13 was performed.
3. Results
3.1. Patient demographics
Twelve female and six male patients with diagnosis of citrin deficiency were recruited in this study. Out of the 18 patients with citrin deficiency, two subjects did not exhibit NICCD and were ascertained after their siblings were molecularly diagnosed with citrin deficiency. Sixteen patients presented with an NICCD phenotype with onset from 1 month to 4 months of life. The median age of presentation was 48 days of life (range: 32 days to 4 months). The median birth weight was 2.65 kg (range: 2.07–3.48 kg). (Fig. 1, Table 1).
Fig. 1.

Plasma amino acid levels for 18 subjects at presentation (citrulline, threonine, threonine to serine ratio).
“ ” represents the normal cut off levels of amino acids citrulline, threonine, and threonine/serine ratio in plasma samples.
” represents the normal cut off levels of amino acids citrulline, threonine, and threonine/serine ratio in plasma samples.
“ ” represents the distribution of levels of plasma amino acid of each subjects. Blue dots represent plasma citrulline values, red dots represent plasma threonine values, and green dots represent plasma threonine/serine ratios.
” represents the distribution of levels of plasma amino acid of each subjects. Blue dots represent plasma citrulline values, red dots represent plasma threonine values, and green dots represent plasma threonine/serine ratios.
Table 1.
Biochemical characteristics of 16 citrin deficiency patients with NICCD phenotype.
| Biochemical findings | Number of positive case (%) |
|---|---|
| Anaemia (Hb < 10 g/dL) at presentation | 4 (25) |
| Prothrombin time (>12.4 s) | 12 (75) |
| Hypoglycemia (Glucose <3.9 mmol/L) | 2 (12.5) |
| Raised total bilirubin (>17 umol/L) | 16 (100) |
| Raised direct bilirubin (>5 umol/L) | 16 (100) |
| Elevated ALT (>67 IU/L) | 1 (6) |
| Elevated GGT (>95 U/L) | 13 (81) |
| Hypoalbuminaemia (<35 g/L) | 12 (75) |
| Raised AFP at presentation (at least twice the upper normal range for age) | 16 (100) |
| Raised ALP at presentation (>380 IU/L) | 13 (81) |
| Raised Triglyceride (> 1.7 mmol/L) | 1 (6) |
3.2. Dietary preference
All 18 patients have a strong preference toward protein-rich foods especially fish and egg, and dislike high carbohydrate foods such as rice, sweets, and noodles. Four patients reported nausea after intake of high carbohydrate foods.
3.3. Biochemical findings
All 16 (100%) NICCD patients presented with cholestatic jaundice, and 81% of them had significantly raised alkaline phosphatase at presentation. Sixteen subjects (100%) had markedly elevated alpha-fetoprotein (AFP) levels at presentation (Fig. 1, Table 1). The biochemical abnormalities began to improve within a month and returned to a normal range within six months after switching the formula to a soy based or lactose free formula milk in 15 out of 16 patients. The liver function of one patient (subject 12) did not improve soon after initiation of dietary modifications consisting of a lactose-free formula in conjunction with fat-soluble vitamin supplements. It took four months for the cholestatic jaundice to resolve and ten months for his liver panel to normalize. All 16 patients with NICCD had completely normal alpha-fetoprotein levels by 12 months of age.
3.4. Molecular findings
Genetic test results showed that 11 out of 18 patients were homozygous for the c.852_855delTATG (p.Met285Profs*2) pathogenic variant in SLC25A13 and the remainder (6/18) of the patients were compound heterozygous for the c.852_855delTATG pathogenic variant and another pathogenic variant in SLC25A13. The other pathogenic variants included c.640C > T (p.Gln214*), c.1638_1660dup23 (p.Ala554Glyfs*17), and c.1750+72_1751-4dup17insNM_138459.3:2667. One patient (1/18) had one pathogenic variant c.852_855delTATG, but the other identified variant, c.754 + 6 T > G (IVS7 + 6 T > G), was a variant of unknown significance (VUS). One novel likely pathogenic missense variant c.284C > A(p.Ala95Asp) was identified in compound heterozygosity with the common c.852_855delTATG pathogenic variant in one patient (subject 9) presenting with an NICCD phenotype (Table 2)). This novel variant is predicted to be highly damaging/deleterious by software SIFT and Polyphen-2.
Table 2.
Demographic data and molecular findings of 18 patients with citrin deficiency.
| Subject | Gender | Birth weight (kg) | Gestation at birth | Age at presentation | Mutation 1 | Mutation 2 |
|---|---|---|---|---|---|---|
| 1 | M | 3 | 40w | 9y | c.852_855delTATG (p.Met285Profs*2) | c.852_855delTATG (p.Met285Profs*2) |
| 2 | M | 2.6 | 37w5d | 4y | c.852_855delTATG (p.Met285Profs*2) | c.852_855delTATG (p.Met285Profs*2) |
| 3 | F | 2.9 | 38w3d | 33d | c.852_855delTATG (p.Met285Profs*2) | c.852_855delTATG (p.Met285Profs*2) |
| 4 | M | 2.85 | 39w | 35d | c.852_855delTATG (p.Met285Profs*2) | c.852_855delTATG (p.Met285Profs*2) |
| 5 | M | 3.485 | 40w | 34d | c.852_855delTATG (p.Met285Profs*2) | c.852_855delTATG (p.Met285Profs*2) |
| 6 | F | 2.35 | 39w2d | 35d | c.852_855delTATG (p.Met285Profs*2) | c.754 + 6 T > G (IVS7 + 6 T > G) |
| 7 | F | 3.19 | 39w6d | 47d | c.852_855delTATG (p.Met285Profs*2) | c.852_855delTATG (p.Met285Profs*2) |
| 8 | F | 2.07 | 32w | 4 m | c.852_855delTATG (p.Met285Profs*2) | c.1750+72_1751-4dup17insNM_138459.3:2667 |
| 9 | F | 2.8 | 38w5d | 3 m | c.852_855delTATG (p.Met285Profs*2) | c.284C > A (p.Ala95Asp) |
| 10 | F | 2.61 | 39w | 2 m | c.852_855delTATG (p.Met285Profs*2) | c.852_855delTATG (p.Met285Profs*2) |
| 11 | F | 2.2 | 37w5d | 2 m8d | c.852_855delTATG (p.Met285Profs*2) | c.1750+72_1751-4dup17insNM_138459.3:2667 |
| 12 | F | 2.65 | 38w | 32d | c.852_855delTATG (p.Met285Profs*2) | c.852_855delTATG (p.Met285Profs*2) |
| 13 | F | 2.85 | 39w | 48d | c.852_855delTATG (p.Met285Profs*2) | c.640C > T (p.Gln214*) |
| 14 | F | 2.13 | 37w5d | 84d | c.852_855delTATG (p.Met285Profs*2) | c.852_855delTATG (p.Met285Profs*2) |
| 15 | F | 2.822 | 38w | 64d | c.852_855delTATG (p.Met285Profs*2) | c.852_855delTATG (p.Met285Profs*2) |
| 16 | M | 2.6 | 38w3d | 39d | c.852_855delTATG (p.Met285Profs*2) | c.1638_1660dup23 (p.Ala554Glyfs*17) |
| 17 | F | 3.12 | 40w | 35d | c.852_855delTATG (p.Met285Profs*2) | c.852_855delTATG (p.Met285Profs*2) |
| 18 | F | 2.59 | 39w5d | 31d | c.852_855delTATG (p.Met285Profs*2) | c.1638_1660dup23 (p.Ala554Glyfs*17) |
3.5. Patient 1 and Patient 2
Patients 1 and 2 were both molecularly diagnosed later in life (9 years and 4 years of age respectively) after their siblings were diagnosed with citrin deficiency due to their NICCD presentation. They never developed intrahepatic cholestasis in their newborn period and both exhibited a peculiar dietary preference for protein-rich foods and aversion to carbohydrates at the time of their molecular confirmation of citrin deficiency. Patient 1 (sibling of patient 3) was born at term in Mainland China. His past history revealed jaundice with onset at 3 months of age due to suspected viral hepatitis. His condition gradually improved at 11 months of age. Subsequently, his symptoms resolved. He had no history of failure to thrive and dyslipidemia. Patient is currently 17 years of age. He has always demonstrated a food preference for protein-rich foods but otherwise no other symptoms. He has not exhibited a neuropsychiatric phenotype. His plasma citrulline level at diagnosis was normal but since then it has remained mildly elevated most of the time. His liver function, AFP, and lipid profile were normal at diagnosis at 9 years of age and have remained normal since then Patient 2 (sibling of patient 5) was born at full term. He had a history of failure to thrive during early infancy and his body weight was less than 3rd centile. However, he had neither cholestatic jaundice nor liver derangement at that time. Since diagnosis at age 4 years, he has always had normal plasma citrulline levels, AFP, and lipid profile. Growth parameters returned to normal range after 1 year of age. This patient has continued to display a food preference for protein-rich foods and aversion to carbohydrates.
3.6. Patient 8
Patient 8 was a preterm female infant born at 32 weeks gestation with a birth weight 2.07 kg. She presented at 4 months of life with progressive cholestatic jaundice and hepatomegaly. Alpha-fetoprotein was markedly raised with the highest level at 69901 μg/L (reference: ≤ 77 μg/L) at presentation, and elevated ALT ranged from 114 IU/L to highest level at 345 IU/L (Reference: <67 IU/L). An MRI of the liver showed multiple focal liver hemangioendotheliomas and diffuse liver parenchymal cellular infiltration (Fig. 2A). She had no other cutaneous stigmata of haemangioma. Patient was managed with the introduction of a lactose-free formula with fat soluble vitamin supplements. Subsequently, the biochemical liver derangements improved. Follow-up MRI of the liver performed at the age of two years showed a complete resolution of all the T2-hyperintense lesions (Fig. 2B). Alpha-fetoprotein levels returned to normal range at 9 months of age. However, this patient had a small body-build with low body weight. His weight did not improve after the introduction of dietary modifications and has remained at the 3rd centile all along. There is no dyslipidemia, but her plasma citrulline level remains intermittently elevated with the highest concentration of 499umol/L (reference: 3–35 umol/L). She has demonstrated a strong preference for protein rich foods and a dislike for carbohydrates including rice.
Fig. 2.
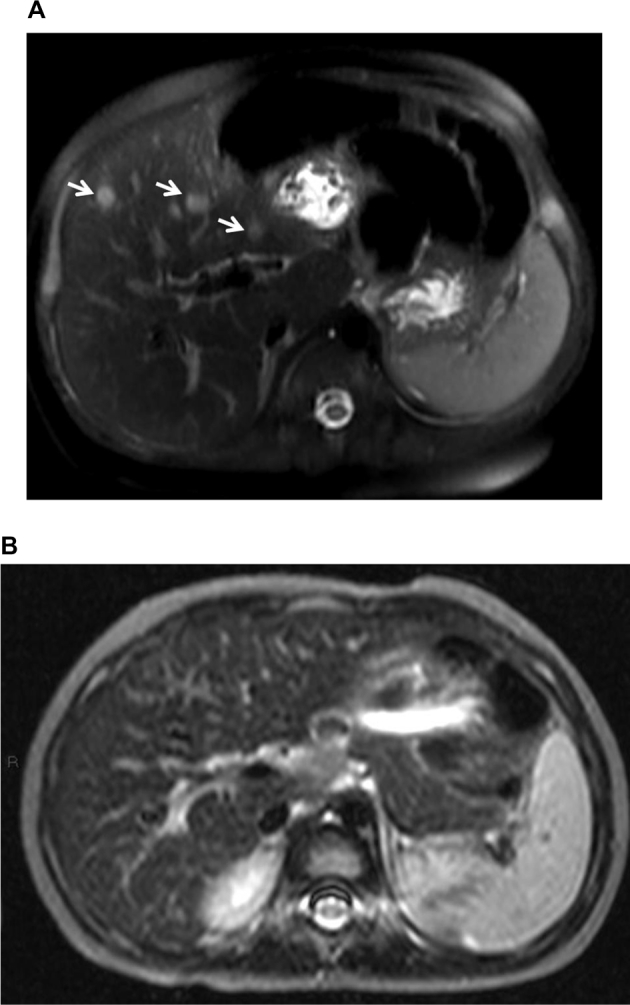
A. MRI of the liver (patient 8) at 5 months of age, a T2 -weighted fat saturated image of the liver at the level of right main portal vein shows multiple roundish hyperintense lesions (arrows) suggestive of haemangiomas. B. At age 2 years, T2 -weighted fat saturated image of the liver at the same level shows complete resolution of all hyperintense lesions.
3.7. Patient 12
Patient 12 was born at full term with birth weight 2.65 kg. He presented with prolonged cholestatic jaundice on day 32 of life. Initial radiological findings showed no gallbladder and repeated E-HIDA scan showed impaired hepatic tracer excretion. An intra-operative cholangiogram was performed to rule out the possibility of biliary atresia at 1.5 months of age. His persistent cholestatic jaundice prompted a liver biopsy that showed prominent cholestasis with giant cell hepatitis pattern and pericellular fibrosis. Dietary modification in conjunction with fat-soluble vitamin supplementation was started right after diagnosis at around 5 weeks of life. However, his liver function continued to deteriorate during the first few months with a prothrombin time up to 19.8 s (reference: 9.5–12 s) and his albumin was 26 g/L (reference: 35-53 g/L). His ALT increased from 96 IU/L to 798 IU/L (reference: <67 IU/L). The condition of patient 12 improved over 4 months of time, and the liver function including ALT, PT, and albumin levels returned to a normal baseline at around 11 months of life.
3.8. Patient 17
Patient 17 was born at term with a birth weight of 3.12 kg. She presented with prolonged neonatal jaundice at day 43 of life. Her clinical presentation was compatible with NICCD phenotype, and her lipid profile showed elevated triglycerides with level 2.4 (ref: <1.7 mmol/L).
3.9. Patient 18
Patient 18 was born at full term with a birth weight of 2.59 kg. Newborn screening was performed on day 2 of life but the level of citrulline was below the detection threshold (cut off <37umol/L). She later presented at around 1 month of life with an NICCD phenotype and was determined to have an elevated plasma citrulline of 680umol/L.
4. Discussion
Here we present for the first time the clinical and molecular spectrum of citrin deficiency in a cohort of Chinese patients from Hong Kong.
In this cohort, 11 out of 18 citrin deficiency patients were homozygous for a c.852_855delTATG (p.Met285Profs*2) pathogenic variant in SLC25A13 and the remainder 7 patients in the cohort are compound heterozygous for the c.852_855del. and with another pathogenic variant in SLC25A13. C.852_855del is the first reported pathogenic variant in SLC25A13 [1] and it is the most frequently observed among Southern Chinese patients with citrin deficiency [2,3,[12], [13], [14]]. The frequency of the c.852_855 delTATG/c.852_855 delTATG genotype in our cohort was approximately 60%. This was a slightly higher frequency than the relative frequency of 49% observed in South China in a previous study [14]. Previous haplotype studies done in patients with citrin deficiency in Mainland China have estimated that the common c.852_855delTATG mutation originated in South China around Guangxi and Yunan areas as a result of a founder effect, whereas the relative frequency of this mutation tended to decrease in North China due to migration and genetic drift [14]. The other pathogenic variants including c.640C > T, c.1638_1660dup23 (p.Ala554Glyfs*17), and IVS16ins3kb (currently known as c.1750+72_1751-4dup17insNM_138459.3:2667) have been reported in previous studies [11,13,15]. The common c.1638-1660dup 23 was found in 2 subjects (12.5%) in our cohort and the other common c.1750+72_1751-4dup17insNM_138459.3:2667 was also found in 2 subjects (12.5%). These two mutations have been found with a frequency of 8.5% and 10% in a recent publication of a large cohort of Chinese patients with citrin deficiency [14]. Compared to previous studies in Southern Chinese populations, one of the four frequent mutations (7.5%) observed in South China, c.615 + 5G > A was not found in our small cohort [[12], [13], [14]]. One novel variant c.284C > A(p.Ala95Asp) has been identified in subject 9. This variant was predicted to be highly damaging/deleterious by both SIFT and Polyphen-2. It has not been identified in ClinVar and ExAc databases. Although most of the variants observed in this condition are truncating, the presence of p.Ala95Asp variant emphasizes the emerging contribution of missense variants in this disorder [[16], [17], [18]]. Regarding the other novel variant, c.754 + 6 T > G (IVS7 + 6 T > G), it has not been found in gnomAD, ExAC, ClinVar, Dnabler, or LSDB. Although it was found in a child with citrin deficiency based on clinical and biochemical features and in compound heterozygosity with the common deletion observed in Southern Chinese patients with this condition, it would still be classified as a VUS.
Twelve subjects in this cohort had prolonged prothrombin time (Table 1). Although several patients have been reported in the literature with abnormal coagulation pattern associated with bleeding diathesis and in severe cases with disseminated intravascular coagulation and lack of response to vitamin K due to liver failure [10,19] these features were not present in the majority of our subjects whose prolonged prothrombin time normalized within a month of dietary intervention except for patient 12 whose prothrombin time returned to baseline at 11 months of age.
Two subjects with NICCD in our cohort (patient 6 and patient 17) had hypoglycemia at presentation that subsided immediately after dietary intervention (Table 1). There was no evidence of liver failure, lactic acidosis, or severe ketoacidosis in either patient. Hypoglycemia is not usually thought to be part of the observed biochemical abnormalities of citrin deficiency. However, it has been previously reported in patients with this condition [20,21] and should alert to the possibility of citrin deficiency in the setting of neonatal intrahepatic cholestasis.
Subject 8 presented in early infancy with multiple s liver hemangioendotheliomas which resolved gradually after two years of dietary treatment. This subject had a milder clinical course when compared to a previously reported case of citrin deficiency who also presented in infancy with a hemangioendothelioma, massive ascites, and progressed to end-stage liver failure requiring liver transplantation. [22]. Several case reports have usually pointed to an epidemiological connection between hepatocellular carcinoma (HCC) and citrin deficiency [[7], [8], [9]]. Moreover, liver tumors do not present in the early stages of the disease as in these two cases but are rather diagnosed in subjects with CTLN2 from 21 to 66 years of age [23]. Liver pathology at the time of diagnosis of HCC has included fibrosis, steatosis, and fibrosis. However, in other cases of HCC these findings have been absent and the HCC was thought to be secondary to non-alcoholic fatty changes in the liver [24]. The lesions associated with the liver hemangioendothelioma in this patient shrank in size, and completely resolved after the initiation and continuation of a lactose-free formula for two years. At the moment, no conclusive direct cause and effect relationship can be established for this comorbidity. However, these two cases of hemangioendothelioma suggest a possible association between citrin deficiency and this type of liver tumor, expanding the spectrum of liver tumors presenting in subjects with citrin deficiency.
The lack of symptomatic disease in the newborn period in patient 8 emphasizes that patients may not be symptomatic initially and may develop intrahepatic cholestatic jaundice in later infancy. The presence of an asymptomatic neonatal period and later development of intrahepatic cholestasis confirms the notion that there may be a later presentation of cholestatic jaundice in infancy with no evidence of NICCD. The liver derangement improved with the introduction of a lactose-free formula and fat-soluble vitamin supplement. Other patients with citrin deficiency may have an unremarkable neonatal period but may subsequently present later as was the case with an infant who died of acute liver failure at 8 months or the case of the infant who presented with failure to thrive at 7 months of age [8,10].
Following previous publications that report approximately half of all patients with citrin deficiency not being ascertained by expanded newborn screening in blood spots [25], it is evident from patient 18 that citrin deficiency could have a normal newborn metabolic screening result, as the analyte in this patient was normal in the blood spot obtained during the second day of life. In both private and public newborn metabolic screening programs in Hong Kong, there is only one screening card collected at 24–72 h of life for well full term babies. It is important to emphasize that citrin deficiency should be considered in the differential diagnosis of intrahepatic cholestasis and/or liver dysfunction even in the presence of a normal expanded newborn screening result.
Two of the 18 patients (patients 1 and 2) had no abnormal biochemical parameters suggestive of citrin deficiency (plasma citrulline levels, AFP, lipid profile, and hepatic profile), when their molecular diagnosis was established in childhood. Occasionally patient 1 has exhibited mildly elevated plasma citrulline levels. None of these patients had confirmed NICCD. Although patient 2 had transient FTT in infancy, his growth parameters have been normal since then and without dietary intervention. These findings demonstrate that there could be minimally symptomatic disease in childhood in citrin deficiency. Patients 1 and 2 were homozygous for a c.852_855del in SLC25A13. Their respective siblings with same pathogenic variants presented with a prolonged NICCD phenotype. These findings point to the fact that in citrin deficiency there could be intra-familial phenotypic heterogeneity among subjects with same molecular findings, suggesting the presence of genetic and environmental modifiers.
Both patients 1 and 2 were not completely clinically asymptomatic at the time of molecular diagnosis as they exhibited a peculiar food preference that was also shared by the other 16 subjects reported in this cohort. Reduced carbohydrate intake has been previously reported in patients with citrin deficiency [26]. Double knockout (KO) mice for mitochondrial glycerol-3-phosphate dehydrogenase and citrin have shown a suppressed intake of sucrose, glycerol, and ethanol. The results suggested the aversion behavior observed in these mice to these solutions may be mediated by hepatic metabolic perturbations, resulting in a behavioral response to increased hepatic cytosolic NADH and a decreased cellular adenine nucleotide pool [27]. It has been speculated that these findings may underlie the dietary selection manifested by patients with citrin deficiency.The presence of a food preference for protein-rich foods in conjunction with an aversion for carbohydrates should prompt clinicians to suspect citrin deficiency even if other signs and symptoms are absent at the time of evaluation. Moreover, the history of FTT in patient 2 in addition to his dietary preference should have compelled an evaluation for citrin deficiency.
Among our 18 patients, only one patient (patient 17) had raised plasma triglyceride levels, whereas the remainder of the patients had no evidence of dyslipidemia. The triglyceride was mildly raised with a level of 2.4 mmol/L (reference: <1.7 mmol/L). Subject 2 had transient FTT in infancy that spontaneously resolved without dietary treatment. A prospective natural history study should beconducted in this cohort of subjects to determine how many of them may develop FTTDCD. Moreover, long-term follow-up into adulthood of the subjects from this cohort and citrin deficiency patients into adulthood should be pursued given the risk of acute encephalopathic hyperammonemia and HCC, including in those subjects without cirrhotic livers [[4], [28]].
In conclusion, the study describes for the first time the clinical and molecular spectrum of citrin deficiency in a cohort of 18 Southern Chinese patients in Hong Kong SAR. Patients with same molecular finding may present very diversely, ranging from asymptomatic in the neonatal period to remarkable liver derangement which may last throughout infancy. Furthermore, it is essential to investigate the diagnostic possibility of citrin deficiency even in patients with normal newborn screening results. Therefore, it is important to have a high index of suspicion to diagnose this disorder. The presence of liver hemangioendothelioma in one of our subjects in conjunction with a previously reported case expands the clinical spectrum of tumors found in this condition. Moreover, the presentation of cholestatic jaundice in later infancy from this cohort should point to the fact that citrin deficiency is associated with a continuum of clinical phenotypes that may not necessarily present within a clear-cut and specific timeframe. It is important to recognize that hypoglycemia could be part of the presenting biochemical phenotype. Furthermore, the presence of a peculiar food preference for protein-rich foods should be enough to prompt clinicians to suspect citrin deficiency even if other symptoms are not present.
Conflict of interest
No conflict of interest of all parties.
Acknowledgement
We acknowledge the support of the Joint BCM-CUHK Center of Medical Genetics, The Chinese University of Hong Kong, Prince of Wales Hospital, ShaTin, New Territories, Hong Kong SAR.
Contributor Information
S.C. Chong, Email: chongsc@cuhk.edu.hk.
F. Scaglia, Email: fscaglia@bcm.edu.
References
- 1.Saheki T., Kobayashi K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD) J. Hum. Genet. 2002;47(7):333–341. doi: 10.1007/s100380200046. [DOI] [PubMed] [Google Scholar]
- 2.Song Y.Z., Guo L., Yang Y.L., Han L.S., Kobayashi K., Saheki T. Failure to thrive and dyslipidemia caused by citrin deficiency: a novel clinical phenotype. Zhongguo dang dai er ke za zhi= Chin. J. Contemp. Pediatr. 2009;11(5):328–332. [PubMed] [Google Scholar]
- 3.Fukumoto K., Sumida Y., Yoshida N., Sakai K., Kanemasa K., Itoh Y., Mitsufuji S., Kataoka K., Okanoue T. A case of adult-onset type II citrullinemia having a liver histology of nonalcoholic steatohepatitis (NASH). Nihon Shokakibyo Gakkai zasshi. Jpn. J. Gastroenterol. 2008;105(2):244–251. [PubMed] [Google Scholar]
- 4.Tsai C.W., Wu M.S., Yang C.C., Chen H.L., Hwu W.L., Wu M.Z., Liu K.L. Homozygous SLC25A13 mutation in a Taiwanese patient with adult-onset citrullinemia complicated with steatosis and hepatocellular carcinoma. J. Formos. Med. Assoc. 2006;105(10):852–856. doi: 10.1016/S0929-6646(09)60274-6. [DOI] [PubMed] [Google Scholar]
- 5.Yamaguchi N., Kobayashi K., Yasuda T., Nishi I., Iijima M., Nakagawa M., Osame M., Kondo I., Saheki T. Screening of SLC25A13 mutations in early and late onset patients with citrin deficiency and in the Japanese population: Identification of two novel mutations and establishment of multiple DNA diagnosis methods for nine mutations. Hum. Mutat. 2002;19(2):122–130. doi: 10.1002/humu.10022. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka T., Nagao M., Tsutsumi H. Application of mutation analysis for the previously uncertain cases of adult-onset type II citrullinemia (CTLN2) and their clinical profiles. Tohoku J. Exp. Med. 2002;198(2):89–97. doi: 10.1620/tjem.198.89. [DOI] [PubMed] [Google Scholar]
- 7.Soeda J., Yazaki M., Nakata T., Miwa S., Ikeda S.I., Hosoda W., Iijima M., Kobayashi K., Saheki T., Kojiro M., Miyagawa S.I. Primary liver carcinoma exhibiting dual hepatocellular-biliary epithelial differentiations associated with citrin deficiency: a case report. J. Clin. Gastroenterol. 2008;42(7):855–860. doi: 10.1097/01.mcg.0000225683.29841.9c. [DOI] [PubMed] [Google Scholar]
- 8.Zhang M.H., Gong J.Y., Wang J.S. Citrin deficiency presenting as acute liver failure in an eight-month-old infant. World J Gastroenterol: WJG. 2015;21(23):7331. doi: 10.3748/wjg.v21.i23.7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamamori A., Okano Y., Ozaki H., Fujimoto A., Kajiwara M., Fukuda K., Kobayashi K., Saheki T., Tagami Y., Yamano T. Neonatal intrahepatic cholestasis caused by citrin deficiency: severe hepatic dysfunction in an infant requiring liver transplantation. Eur. J. Pediatr. 2002;161(11):609–613. doi: 10.1007/s00431-002-1045-2. [DOI] [PubMed] [Google Scholar]
- 10.Dimmock D., Kobayashi K., Iijima M., Tabata A., Wong L.J., Saheki T., Lee B., Scaglia F. Citrin deficiency: a novel cause of failure to thrive that responds to a high-protein, low-carbohydrate diet. Pediatrics. 2007;119(3):e773–e777. doi: 10.1542/peds.2006-1950. [DOI] [PubMed] [Google Scholar]
- 11.Ko J.M., Kim G.H., Kim J.H., Kim J.Y., Choi J.H., Ushikai M., Saheki T., Kobayashi K., Yoo H.W. Six cases of citrin deficiency in Korea. Int. J. Mol. Med. 2007;20(6):809–815. [PubMed] [Google Scholar]
- 12.Song Y.-Z., Deng M., Chen F.-P., Wen F., Guo L., Cao S.-L., Gong J., Xu H., Jiang G.-Y., Zhong L., Kobayashi K., Saheki T., Wang Z.-N. Genotypic and phenotypic features of citrin deficiency: Five-year experience in a Chinese pediatric Center. Int. J. Mol. Med. 2011;28:33–40. doi: 10.3892/ijmm.2011.653. [DOI] [PubMed] [Google Scholar]
- 13.Chen R., Wang X.H., Fu H.Y., Zhang S.R., Abudouxikuer K., Saheki T., Wang J.S. Different regional distribution of SLC25A13 mutations in Chinese patients with neonatal intrahepatic cholestasis. World J Gastroenterol: WJG. 2013;19(28):4545. doi: 10.3748/wjg.v19.i28.4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin W.-X., Zeng H.-S., Zhang Z.-H., Mao M., Zheng Q.-Q., Zhao S.-T., Cheng Y., Chen F.-P., Wen W.-R., Song Y.-Z. Molecular diagnosis of pediatric patients with citrin deficiency in China: SLC25A13 mutation spectrum and the geographic distribution. Sci. Rep. 2016;11(6):29732. doi: 10.1038/srep29732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H., Shu S., Chen C., Huang Z., Wang D. Novel mutations in the SLC25A13 gene in a patient with NICCD and severe manifestations. J. Pediatr. Endocrinol. Metab. 2015;28(3–4):471–475. doi: 10.1515/jpem-2014-0278. [DOI] [PubMed] [Google Scholar]
- 16.Avdjieva-Tzavella D.M., Ivanova M.B., Todorov T.P., Todorova A.P., Panteleeva E.I., Tincheva S.S., Lazarova E.A., Kathom H.M., Yaneva P.G., Tincheva R.S. First Bulgarian case of citrin deficiency caused by one novel and one recurrent mutation in the SLC25A13 gene. Genet. Couns. 2014;25(3):271–276. [PubMed] [Google Scholar]
- 17.Zhang Z.H., Lin W.X., Deng M., Zhao S.T., Zeng H.S., Chen F.P., Song Y.Z. Clinical, molecular and functional investigation on an infant with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) PLoS One. 2014;9(2) doi: 10.1371/journal.pone.0089267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dimmock D., Maranda B., Dionisi-Vici C., Wang J., Kleppe S., Fiermonte G., Bai R., Hainline B., Hamosh A., O'Brien W.E., Scaglia F. Citrin deficiency, a perplexing global disorder. Mol. Genet. Metab. 2009;96(1):44–49. doi: 10.1016/j.ymgme.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 19.Zhao X.J., Tang X.M., Zha Q.B., Shi S.S., Song Y.Z., Xiao X.M. Prenatal diagnosis of citrin deficiency in a Chinese family with a fatal proband. Tohoku J. Exp. Med. 2011;225(4):273–276. doi: 10.1620/tjem.225.273. [DOI] [PubMed] [Google Scholar]
- 20.Hachiso M., Oda Y., Goto M., Kobayashi K., Saheki T., Ohura T., Noma S., Kitanaka S. Citrin deficiency presenting with ketotic hypoglycaemia and hepatomegaly in childhood. Eur. J. Pediatr. 2005;164(2):109–110. doi: 10.1007/s00431-004-1549-z. [DOI] [PubMed] [Google Scholar]
- 21.Ohura T., Kobayashi K., Tazawa Y., Abukawa D., Sakamoto O., Tsuchiya S., Saheki T. Clinical pictures of 75 patients with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) J. Inherit. Metab. Dis. 2007;30(2):139–144. doi: 10.1007/s10545-007-0506-1. [DOI] [PubMed] [Google Scholar]
- 22.Shigeta T., Kasahara M., Kimura T., Fukuda A., Sasaki K., Arai K., Nakagawa A., Nakagawa S., Kobayashi K., Soneda S., Kitagawa H. Liver transplantation for an infant with neonatal intrahepatic cholestasis caused by citrin deficiency using heterozygote living donor. Pediatr. Transplant. 2010 Nov;14(7):E86–E88. doi: 10.1111/j.1399-3046.2009.01172.x. [DOI] [PubMed] [Google Scholar]
- 23.Erez A., Shchelochkov O.A., Plon S.E., Scaglia F., Lee B. Insights into the pathogenesis and treatment of cancer from inborn errors of metabolism. Am. J. Hum. Genet. 2011;88(4):402–421. doi: 10.1016/j.ajhg.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hagiwara N., Sekijima Y., Takei Y.I., Ikeda S.I., Kawasaki S., Kobayashi K., Saheki T. Hepatocellular Carcinoma in a Case of Adult-onset Type II Citrullinemi. Intern. Med. 2003;42(10):978–982. doi: 10.2169/internalmedicine.42.978. [DOI] [PubMed] [Google Scholar]
- 25.Tamamori A., Fujimoto A., Okano Y., Kobayashi K., Saheki T., Tagami Y., Takei H., Shigematsu Y., Hata I., Ozaki H., Tokuhara D. Effects of citrin deficiency in the perinatal period: feasibility of newborn mass screening for citrin deficiency. Pediatr. Res. 2004;56(4):608. doi: 10.1203/01.PDR.0000139713.64264.BC. [DOI] [PubMed] [Google Scholar]
- 26.Saheki T., Kobayashi K., Terashi M., Ohura T., Yanagawa Y., Okano Y., Hattori T., Fujimoto H., Mutoh K., Kizaki Z., Inui A. Reduced carbohydrate intake in citrin-deficient subjects. J. Inherit Metab. Dis. 2008;31(3):386–394. doi: 10.1007/s10545-008-0752-x. [DOI] [PubMed] [Google Scholar]
- 27.Saheki T., Inoue K., Ono H., Fujimoto Y., Furuie S., Yamamura K.I., Kuroda E., Ushikai M., Asakawa A., Inui A., Eto K., Kadowaki T., Moriyama M., Sinasac D.S., Yamamoto T., Furukawa T., Kobayashi K. Oral aversion to dietary sugar, ethanol and glycerol correlates with alterations in specific hepatic metabolites in a mouse model of human citrin deficiency. Mole. Genet. Metab. 2017;120(4):306–316. doi: 10.1016/j.ymgme.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 28.NG Y.W., Chan A.O., Au Yeung Y.T., Lau G.T., Cheng C.W., Shek C.C., Tiu S.C. Hyperammonemic encephalopathy in an adult patient with citrin deficiency associated with a novel mutation. Hong Kong Med. J. 2011;17(5):410–413. [PubMed] [Google Scholar]