

1.
To the Editor:
Acute myeloid leukemia (AML) is a heterogeneous malignancy characterized by chromosomal aberrations and somatic mutations that identify biologically distinct subsets and guide risk stratification for therapy.1 Treatment‐associated changes in clonal architecture are common in AML, with emergence or clearance of specific sub‐clones driving sensitivity and resistance to therapy. Therefore, the molecular characterization of emerging clones may facilitate the selection of optimal targeted therapies and rational combinations.
Venetoclax, a selective BCL‐2 inhibitor, induced a complete response or complete response with incomplete blood recovery (CR/CRi) in 6/32 (19%) patients with AML who either had relapsed/refractory disease or were medically unfit for intensive chemotherapy.2 In this report, we present a comparison of genetic biomarkers observed in pre‐ and post‐treatment specimens from 29 of the 32 patients enrolled on this phase II study. Measurable reduction in bone marrow (BM) blast counts was observed in 15/29 (52%) of the patients, including CR/CRi in 6, a ≥50% reduction in BM blasts in 5, and a more modest blast reduction of <50% in 4 (Supporting Information Figure 1). The remaining patients (14/29, 48%) had no blast reduction.
We investigated the presence of somatic mutations commonly associated with AML in baseline and end‐of‐treatment samples. DNA isolated from blood and bone marrow specimens was analyzed by next‐generation sequencing using the TruSight Myeloid panel (Illumina), the FoundationOne Heme panel (Foundation Medicine), or whole exome sequencing (MD Anderson Cancer Center, Khalifa Institute). Comparison of mutations at baseline and end of treatment is shown in Figure 1A.
Figure 1.
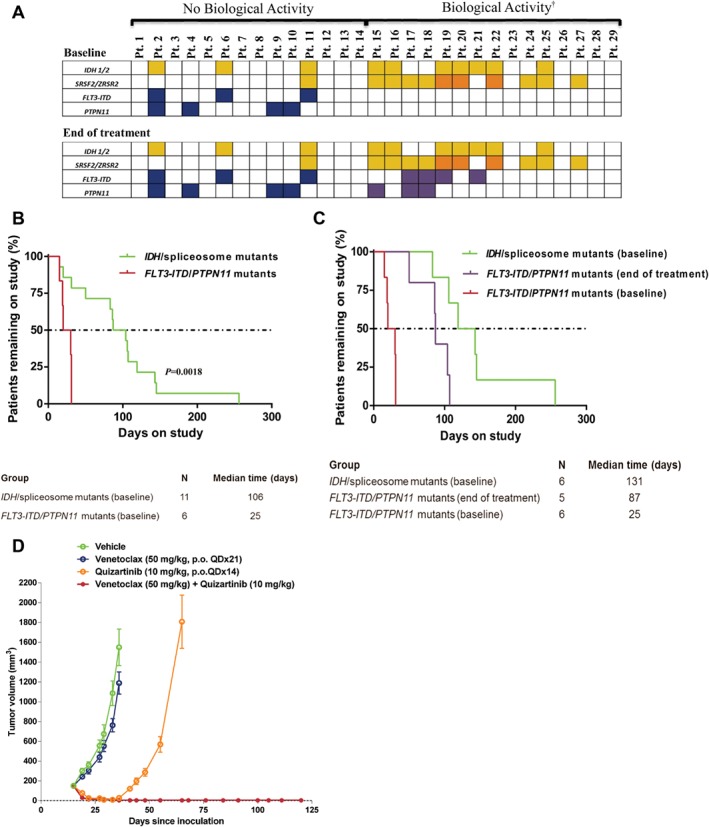
Mutations affecting response to venetoclax in AML. A, Mutations observed pre‐therapy and end of treatment with single agent venetoclax therapy. Yellow indicates IDH or SRSF2 mutations; orange indicates ZRSR2 mutations; blue indicates FLT3‐ITD or PTPN11 mutations; purple indicates newly detected FLT3‐ITD or PTPN11 mutations; blank cells indicate specific mutation was not detected, †Biological activity defined as any reduction in BM blast count while on venetoclax therapy; B, Time on study for patients with mutations associated with intrinsic sensitivity (pre‐therapy IDH/spliceosome mutants) and intrinsic resistance (pre‐therapy FLT3‐ITD/PTPN11 mutants) to venetoclax; C, Time on study for patients with mutations associated with intrinsic sensitivity, intrinsic resistance, and acquisition of mutations associated with acquired resistance (FLT3‐ITD/PTPN11 mutants) to venetoclax; D, Tumor growth inhibition by venetoclax plus quizartinib in mice xenografted with FLT3‐ITD+ MV‐4–11 cells
At baseline, 10/29 (34%) patients had mutations in isocitrate dehydrogenase 1/2 (IDH1/2) genes. Of these, 7 (70%) had a reduction in BM blasts, including 3 CR/CRi. At baseline, 11/29 (38%) patients had spliceosome mutations in SRSF2 or ZRSR2. Ten (88%) of these patients had a decrease in BM blasts, including 3 CR/CRi. Seven patients had both IDH1/2 and spliceosome mutations with BM blast reductions observed in 6 (86%). In total, 11/14 (79%) patients with mutations in IDH1/2 or SRSF2/ZRSR2 had evidence of BM blast reduction, including 4 CR/CRi, implicating these as possible markers of sensitivity to venetoclax (Figure 1A).
Among 14 patients who did not have a decrease in BM blasts on venetoclax treatment, 3 (21%) had FMS‐like tyrosine kinase‐3‐internal tandem duplication (FLT3‐ITD) and 4 (29%) had protein tyrosine phosphatase, non‐receptor type 11 (PTPN11) mutation at baseline, with 1 having both. Three patients had baseline mutations in both the IDH/spliceosome and FLT3‐ITD/PTPN11 groups and these three were the only patients who harbored IDH/spliceosome mutations and did not have BM blast reductions on venetoclax. The median time on study was 106 days (range, 50–256) for the IDH/spliceosome+ (n = 11) and 25 days (range, 15–31) for FLT3‐ITD/PTPN11 + patients (n = 6) (P = .0018, Wilcoxon) (Figure 1B). These data suggest that FLT3‐ITD or PTPN11 mutations in AML may produce intrinsic/primary resistance to venetoclax.
We also performed mutational analysis on matched end‐of‐treatment samples from 20 patients at the time of AML progression/therapy termination. The IDH1/2, SRSF2/ZRSR2, FLT3‐ITD, and PTPN11 mutations identified prior to treatment were still present at the end of therapy in all patients. Notably, in 5 IDH/spliceosome+ patients that were negative for FLT3‐ITD/PTPN11 mutations at baseline, FLT3‐ITD (n = 2), PTPN11 (n = 1), or both (n = 2) mutations were now detected in the end‐of‐treatment samples. The median time on study for these five patients was 87 days (range, 50–107) as compared to 131 days (range, 83–256) for the six patients who had IDH/spliceosome mutations at baseline and did not acquire FLT3 or PTPN11 mutations (Figure 1C). Furthermore, two patients in whom venetoclax initially induced BM blast reductions had both FLT3‐ITD and PTPN11 mutations newly detectable at the end of treatment in different subclones, based on allele frequency.
Based on our sequencing findings, we assessed the combination of venetoclax with the small‐molecule FLT3 inhibitor quizartinib in the FLT3‐ITD+ mutant xenograft model MV‐4–11 (Supporting Information Methods). In vitro, the MV‐4–11 cells were sensitive to BCL‐2 inhibition by venetoclax.3 However, similar to our clinical observations, venetoclax did not inhibit the growth of these tumors when implanted in vivo. Daily dosing of quizartinib induced tumor regressions in this model, although the tumors regrew following cessation of therapy. Strikingly, co‐treatment with venetoclax and quizartinib induced similar tumor regressions as quizartinib alone but with significantly increased durability, preventing tumor re‐emergence for up to 3 months post‐cessation of treatment (Figure 1D). These data suggest that combining venetoclax with FLT3 inhibitors could be highly effective for the treatment of FLT3‐mutated AML and may also prevent the emergence of FLT3‐mutated, venetoclax‐resistant sub‐clones in patients who do not have an already detectable FLT3 mutation.
In summary, our data suggest that SRSF2/ZRSR2 and IDH1/2 mutations may predict sensitivity to venetoclax therapy in AML. Chan et al. previously demonstrated that IDH1/2 mutations can sensitize leukemic cells to venetoclax.4 However, of the 10 IDH1/2‐mutated AML samples assessed in this trial, 7 had co‐occurring spliceosome mutations, making it difficult to determine whether only one or both of these mutations together predict for venetoclax sensitivity. Recent findings suggest that IDH2 and SRSF2 mutations cooperate to induce a lethal transplantable myeloproliferative neoplasm.5 Additionally, SRSF2 mutation is known to induce alternative splicing of genes involved in the apoptotic pathway, a possible link to venetoclax sensitivity.6 We note that FLT3‐ITD or PTPN11 mutations may confer primary and secondary resistance to venetoclax. Consistent with this, previous studies have shown that FLT3‐ITD or PTPN11 mutations can enhance the expression of anti‐apoptotic BCL‐2 relatives like BCL‐XL and MCL‐1.7, 8 When combined with venetoclax, the FLT3 inhibitor quizartinib induced more durable responses in FLT3‐ITD+ tumor‐bearing mice than either agent alone (Figure 1D). Thus, simultaneous targeting of BCL‐2 and FLT3 may be one approach to overcome primary resistance and prevent emergence of secondary resistance to venetoclax therapy in AML patients.
CONFLICT OF INTEREST
Vivian Ruvolo, Zixing Wang, and Ken Chen: nothing to disclose. Evelyn McKeegan was an employee of AbbVie, Inc. at the time of the study and may own stock. Marina Konopleva: consulted for and received research grants from AbbVie Inc. and Genentech. Naval Daver: received research grants from AbbVie Inc. and Genentech. Brenda Chyla, Kelly Doyle, Xin Huang, Andrew Souers, Joel Leverson, Jalaja Potluri, Erwin Boghaert, Anahita Bhathena, Relja Popovic: employees of AbbVie Inc. and may own stock.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting information 1
ACKNOWLEDGMENTS
Venetoclax is being developed by AbbVie and Genentech. AbbVie and Genentech provided financial support for the study and participated in the design, study conduct, analysis, and interpretation of data as well as the writing, review and approval of this Correspondence. The authors thank the patients and their families, study coordinators, and support staff. Medical writing support was provided by Namrata Bhatnagar, Ph.D., an employee of AbbVie.
Funding information AbbVie; Genentech
REFERENCES
- 1. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6(10):1106–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mali RS, Laseter EA, Doyle K, et al. FLT3‐ITD activation mediates resistance to the BCL‐2 selective antagonist, venetoclax, in FLT3‐ITD mutant AML models. Blood. 2017;130:1348. [Google Scholar]
- 4. Chan SM, Thomas D, Corces‐Zimmerman MR, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL‐2 dependence in acute myeloid leukemia. Nat Med. 2015;21(2):178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoshimi A, Lin KT, Wiseman D, et al. Spliceosomal dysfunction is a critical mediator of IDH2 mutant leukemogenesis. Blood. 2017;130:473. [Google Scholar]
- 6. Kim E, Ilagan JO, Liang Y, et al. SRSF2 mutations contribute to myelodysplasia by mutant‐specific effects on exon recognition. Cancer Cell. 2015;27(5):617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen L, Chen W, Mysliwski M, et al. Mutated Ptpn11 alters leukemic stem cell frequency and reduces the sensitivity of acute myeloid leukemia cells to Mcl1 inhibition. Leukemia. 2015;29(6):1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kasper S, Breitenbuecher F, Heidel F, et al. Targeting MCL‐1 sensitizes FLT3‐ITD‐positive leukemias to cytotoxic therapies. Blood Cancer J. 2012;2(3):e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting information 1