

Abstract
Oxygen formation through water oxidation catalysis is a key reaction in the context of fuel generation from renewable energies. The number of homogeneous catalysts that catalyze water oxidation at high rate with low overpotential is limited. Ruthenium complexes can be particularly active, especially if they facilitate a dinuclear pathway for oxygen bond formation step. A supramolecular encapsulation strategy is reported that involves preorganization of dilute solutions (10−5 m) of ruthenium complexes to yield high local catalyst concentrations (up to 0.54 m). The preorganization strategy enhances the water oxidation rate by two‐orders of magnitude to 125 s−1, as it facilitates the diffusion‐controlled rate‐limiting dinuclear coupling step. Moreover, it modulates reaction rates, enabling comprehensive elucidation of electrocatalytic reaction mechanisms.
Keywords: electrochemical catalysis, ruthenium complexes, supramolecular chemistry, water oxidation
Renewable fuel generation is of crucial importance for the energy transition required for a sustainable society.1 In that context, water splitting is considered a “holy grail” for the production of hydrogen as a useful fuel.1, 2 As water oxidation involves two water molecules and multiple proton/electron transfers steps (2 H2O→O2+4 H++4 e−), it is a mechanistically complex half‐reaction.2 Finding suitable catalysts that engender rapid catalysis at low overpotentials is indeed challenging.3 In the past decades, molecular water oxidation catalysts (WOCs) based on various transition metals (Ru, Ir, Cu, Fe, and Ni) have been reported.2, 3, 4 These catalysts operate via the water nucleophilic attack (WNA) mechanism, or coupling of two metal–oxyl radicals (I2M).4c, 5 A survey of the WOCs reported to date reveals those that proceed via the I2M mechanism are usually capable of reaching high rates at lower overpotential,2, 6 although the highest rate has been reported for WOCs that follow WNA.7 The I2M mechanism requires sufficient concentration of the radical‐oxo intermediate to allow the coupling step to proceed in a binuclear fashion.5 Ruthenium‐based WOCs are excellent in terms of activity, overpotential, and stability.3b In particular, Ru(bda)Het2 type complexes (bda=2,2′‐bipyridine‐6,6′‐dicarboxylate, Het=aromatic N‐heterocycles) reported by Sun and co‐workers demonstrate exceptional performance.8 The complex catalyzes water oxidation at low overpotentials and a turnover frequency comparable to the natural photosystem II (100–400 s−1) through the I2M mechanism has been reported when driven by a chemical oxidant (CeIV).8b,8c, 9 The ligand effect is subtle, an analogous complex Ru(phenda)(pic)2 (phenda=[1,10] phenanthroline‐2,9‐dicarboxylic acid, pic=4‐picoline) based on the rigid phenathroline ligand catalyzes water oxidation via the WNA mechanism (Scheme 1), and higher overpotentials are required to drive the reaction.6, 10 Also, anchoring of Ru(bda)Het2 type catalysts on glassy carbon or indium tin oxide surfaces leads to site isolation of the complexes, prohibiting catalysis via the I2M mechanism, and the same complex catalyzes water oxidation via WNA with lower reaction rates.11
Scheme 1.
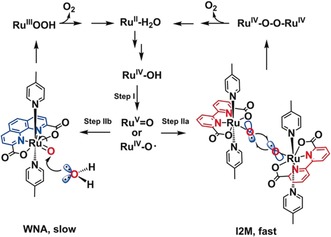
Mechanistic pathways for water oxidation with the final O−O formation via nucleophilic attack of water to the ruthenium–oxo species (WNA, blue; that is, Ru(phenda)(pic)2 10) or via interaction of two metal–oxo moieties (I2M, red; that is, Ru(bda)(pic)2 8b,8c).
Ru(bda)Het2 complexes lose their excellent performance under strongly diluted conditions as the rate‐determining step (RDS) of diffusion‐limited collision of two metal–oxyl radicals becomes slow (Scheme 1, Step IIa), which has been demonstrated by Concepcion and co‐workers.12 Furthermore, the electrochemical reaction mechanism becomes unintelligible under dilute conditions. Meyer and co‐workers have demonstrated that the Ru(bda)Het2 catalyst is first‐order in catalyst with kinetic isotope effects (KIE) around 2 implying a WNA mechanism.13 However, they could not exclude that the catalytic process proceeds via an I2M mechanism with a rate determining proton coupled electron transfer (PCET) step to form the ruthenium–oxo intermediate (Scheme 1, Step I), followed by a fast O−O coupling (Scheme 1, Step IIa).13
In parallel to water oxidation catalysis development, the field of supramolecular catalysis has progressed significantly, providing new ways to position catalysts in confined spaces.14 In this context we generated M12L24 nanospheres (M=corners Pt2+ or Pd2+, L=bispyridyl building block), initially developed by Fujita and co‐workers,15 endohedrally functionalized with guanidinium groups to which sulfonate‐bearing catalysts can be strongly bound by complementary hydrogen bonds and carboxylate functionalized substrates can be preorganized to these catalysts resulting in a 40‐fold rate enhancement for the AuI‐catalyzed cyclization of acetylenic acid.16 We anticipate that these supramolecular nanospheres are ideal to preorganize Ru(bda)Het2 type WOCs, as these should strongly benefit from high local concentration if they follow the preferred I2M mechanism. Herein, we report the application of the guanidinium functionalized M12L24 nanospheres (Figure 1 a,b) as nanoconcentrators for the supramolecular encapsulation of a homogenous WOC Ru(bda)(PySO3TBA)2 (Figure 1 c, PySO3 −=pyridine‐3‐sulfonate, TBA=tetra(n‐butyl)ammonium) by strong binding (>105 m −1)16 between the guanidinium and sulfonate moieties. Encapsulation of 12 Ru(bda)(PySO3 −)2 catalysts within the nanosphere enhanced the solution concentration of 2.5×10−5 m to a local concentration of 0.54 m, resulting in a two orders of magnitude increase in the rate of electrochemical water oxidation (0.93–125 s−1).
Figure 1.
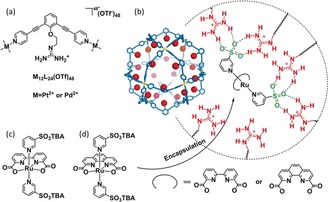
a) Molecular structures of the assembled guanidinium‐functionalized spheres. b) 3D representation of the assembled guanidinium nanosphere and catalyst encapsulation process by interactions between the guanidinium and sulfonate groups. c) Molecular structure of Ru(bda)(PySO3TBA)2. d) Molecular structure of Ru(phenda)(PySO3TBA)2.
The guanidinum functionalized building blocks, the corresponding nanospheres,16 and the ruthenium complexes9, 17 were synthesized as previously reported. Endohedral binding of Ru(bda)(PySO3 −)2 inside the nanosphere (up to 12 equiv) was confirmed by 1H NMR (Supporting Information, Figure S3).16 The addition of more than 12 equiv of guest (ratio sulfonate/guanidinium=1) results in precipitation. Diffusion‐ordered spectroscopy (DOSY) of nanosphere demonstrates a clear single band at log D=−9.6 m2 s−1, consistent with previously reported M12L24 nanospheres of similar size.15, 16 Signals originating from bound Ru(bda)(PySO3TBA)2 guests display the same log D values as the nanosphere (Supporting Information, Figure S4d), in contrast to the free Ru(bda)(PySO3TBA)2 complex (logD=−8.8 m2 s−1), supporting quantitative encapsulation. Catalyst encapsulation is further confirmed by cold‐spray‐ionization time‐of‐flight mass spectrometry (CSI‐ToF‐MS), where peaks attributed to the nanosphere with encapsulated catalyst are detected (Supporting Information, Figures S5–S12 and Table S1).
To gain more detailed insight into catalyst distribution and the effect of local catalyst concentration on catalyst mobility within the sphere, classical molecular dynamics simulations were performed (Supporting Information, Section S3). Up to 12 catalysts they dominantly reside at close distance to the guanidinium for optimal hydrogen bonding. When more than 12 catalysts are present, catalyst population at the edges significantly increases (Supporting Information, Figure S15), which experimentally manifests as precipitation from solution (see above). When the nanosphere is loaded with up to 12 ruthenium catalysts, the diffusion within the sphere is only slightly affected (Supporting Information, Figure S17), so we may safely assume that catalyst mobility in the cage is sufficient to facilitate dinuclear reaction pathways, which is important for the I2M mechanism.
Having established that up to 12 Ru(bda)(PySO3TBA)2 complexes can be bound at the endohedral site of the nanosphere, the effect of such preorganization on the electrocatalytic water oxidation performance was evaluated. Owing to the low solubility of the nanosphere in water, acetonitrile with 10 % deionized water was used as solvent with 0.1 m TBAPF6 as electrolyte. The stability of the M12L24 nanospheres in the presence of 10 % D2O and 0.1 m TBAPF6 was confirmed by 1H NMR and 1H DOSY NMR measurements (Supporting Information, Figure S18), consistent with reported data.18 Catalysis at the metal center of Ru(bda)(PySO3TBA)2 under standard conditions proceeds through the steps: RuII−OH2→RuIII−OH→RuIV−OH→RuV=O (or RuIV−O.) based on the Pourbaix diagram (Supporting Information, Figure S19). The Ru(bda)(PySO3TBA)2 exhibits higher catalytic currents than Ru(bda)(pic)2 with similar onset potential (Supporting Information, Figure S20).
The platinum nanosphere (Pt12L24) was chosen over the palladium analogue for the catalyst preorganization experiments as it usually exhibits superior stability.16 The cyclic voltammetry (CV) measurements on a solution containing 0.02 mm platinum sphere displays voltammograms that overlap with the blank, even after successive scans (Supporting Information, Figure S21). The peak currents (ip) of RuIII/RuII (Supporting Information, Figure S22) vary linearly with the square root of scan rates ( ) for the mononuclear catalyst as well as that bound in the nanosphere (Supporting Information, Figure S23), showing that the redox events take place in solution, not on adsorbed species. Interestingly, calculation of D using Randles–Sevcik equation revealed that the diffusion of the complex is about 5.4 times lower when inside the nanosphere (D=5.28×10−7 vs. 2.87×10−6 cm2 s−1; Supporting Information, Section S4.3), in line with the differences observed in DOSY NMR experiments (D=2.51×10−6 vs. 1.41×10−5 cm2 s−1). This demonstrates that Ru(bda)(PySO3 −)2 remains encapsulated by the nanosphere under electrochemical conditions.
The catalytic waves obtained for the Ru(bda)(PySO3TBA)2 that is bound in the nanopshere (6 equiv catalyst per nanosphere) shows a four‐fold increase in catalytic current compared to the one free in solution, demonstrating a higher reaction rate as a result of catalyst preorganization (Supporting Information, Figure S22). When the nanosphere was mixed with Ru(bda)(pic)2 lacking the sulfonate groups that drive encapsulation, no increase in catalytic current was observed (Supporting Information, Figure S24). Furthermore, the catalytic current of the free Ru(bda)(PySO3TBA)2 was evaluated in the presence of guanidinium‐binding sites (BuGd; Supporting Information, Figure S25), giving similar catalytic currents, showing that this hydrogen‐bonding binding alone does not lead to enhanced catalytic rates. The catalytic current varies linearly with the catalyst concentration (Supporting Information, Figure S26) for Ru(bda)(PySO3TBA)2 bound in the nanosphere (constant Ru(bda)(PySO3TBA)2/nanosphere ratio), indicating that also the encapsulated catalyst behaves as a soluble catalyst. To confirm the stability of the nanosphere system, electrolysis was conducted at 1.3 V. The current declined by about 10 % during 8000 s electrolysis, which is similar to the decline in absence of nanosphere, and throughout the experiment the current is about 3 times higher than in absence of nanosphere (Supporting Information, Figure S27a). The 1H NMR and 1H DOSY NMR spectra after electrolysis are similar to that taken before the experiment (Supporting Information, Figure S27b,c). To confirm oxygen was formed, the Faradaic efficiency was determined to be 96.3 % using a rotating ring‐disk electrode in standard conditions (Supporting Information, Figure S29).19
To demonstrate the effect of high local concentration on water oxidation catalysis, the dependence of catalytic current and rate (k cat) on the amount of catalyst per nanosphere was determined. The overall concentration of Ru(bda)(PySO3TBA)2 was kept constant (2.5×10−5 m), whereas the number of Ru(bda)(PySO3 −)2 per nanosphere was varied from 1 to 12, which translates to estimated local catalyst concentration from 0.04 to 0.54 M.20 The current densities increase considerably with the increased local catalyst concentration (Supporting Information, Figure S30), and the highest current density was achieved by the encapsulation of 12 Ru(bda)(PySO3 −)2 on average per nanosphere. The kcat was determined by combining Randles–Sevcik equation with kinetics plots (i cat/i p vs. ) (Supporting Information, Section S4.3). The clear increase of k cat was achieved by just changing the local catalyst concentration within the nanosphere (Figure 2 a), with a maximum rate of 125 s−1 which is more than 130 times higher than that observed for the non‐encapsulated system (0.93 s−1). The large enhancements in k cat suggest that this reaction proceeds via the I2M mechanism where the RDS of O−O coupling (Scheme 1, Step IIa) is promoted by catalyst preorganization in the nanosphere. Even for an average of one catalyst per nanosphere the calculated k cat is still higher than in absence of the nanosphere. These statistical mixtures contain enough nanospheres with at least two catalysts to facilitate the reaction to proceed via the favorable I2M mechanism. To further confirm this, experiments were performed in which excess nanosphere was used, indeed leading to lower catalytic currents (Supporting Information, Figure S35). When the ratio of catalyst to nanosphere was 1:8, the catalytic current and thus the rate dropped to 20 % (0.18 s−1) compared to that of the molecular catalyst in absence of nanosphere. Under these conditions, site isolation of the Ru(bda)(PySO3 −)2 in the nanosphere prevents the I2M mechanism to occur.
Figure 2.
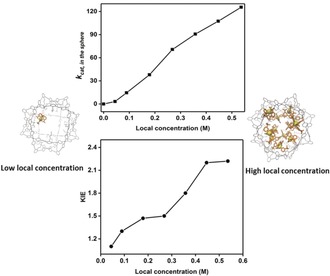
a) The dependence of k cat on the local catalyst concentration. b) The kinetic isotope effect (KIE, H2O/D2O) observed for experiments with the same Ru(bda)(PySO3TBA)2 concentration, but at various local concentration of catalyst in the nanosphere.
To further find evidence for the mechanism by which Ru(bda)(PySO3TBA)2 converts water into oxygen when located in the nanosphere, we performed KIE studies (Supporting Information, Equation (5)) and looked more carefully at the reaction rate as function of the local catalyst concentration. In the range of local catalyst concentration between 0 and 0.27 m, a second order in local catalyst is found based on a linear relationship between and local catalyst concentration (Supporting Information, Figure S36). This suggests that in this concentration regime the catalyst operates via the I2M reaction mechanism with the O−O coupling as the RDS. In line with this, a low secondary isotope effect (KIE<1.5) is observed (Figure 2 b).21 When the local catalyst concentration is higher than 0.27 m, the reaction becomes first order with respect to local catalyst concentration (Figure 2 a), while the KIE increases in this window (1.5–2.2; Figure 2 b). These observations show that by increasing the local catalyst concentration up to 0.27 m the rate‐limiting dinuclear radical coupling is facilitated (Scheme 1, Step IIa).12 At higher local catalyst concentrations, the O−O coupling becomes so fast that it is no longer rate‐limiting, and the preceding step, the oxidation of RuIV−OH (Scheme 1, Step I) via PCET to form a ruthenium–oxo species becomes RDS.12 This is the first system that confirms that water oxidation catalysis of Ru(bda)(Het)2 type complexes also proceed via the I2M mechanism during homogenous electrochemical experiments and by changing the local concentration the RDS can be switched from the radical coupling step to the preceding proton coupled oxidation step.
We evaluated the effect of the local catalyst concentration on the reaction rate using Ru(phenda)(PySO3TBA)2 (Figure 1 d) as a prototypical catalyst that catalyzes water oxidation exclusively via a WNA mechanism.10 Therefore, for this system the reaction rate should not significantly change upon increasing local catalyst concentration. In Figure 3 the relative reaction rates of Ru(phenda)(PySO3 −)2 at various local catalyst concentrations are plotted and compared to the data observed for Ru(bda)(PySO3 −)2. This clearly shows that the local Ru(phenda)(PySO3 −)2 catalyst concentration has no effect on the reaction rate, whereas for Ru(bda)(PySO3 −)2 a huge effect is observed.
Figure 3.
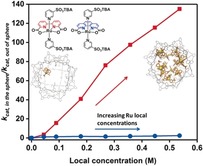
Comparison of the reaction rate improvement observed for Ru(bda)(PySO3TBA)2 (red) and Ru(phenda)(PySO3TBA)2 (blue) by changing the local catalyst concentration.
In line with the catalytic results, the diffusion calculated from simulation of the Ru(bda)(PySO3 −)2 within the cage environment indicates sufficient freedom within the nanosphere to facilitate the dinuclear radical coupling step. As such, the overall reaction proceeds at higher rates when local catalyst concentrations are high. The situation is anticipated to be different if the local concentration of Ru(bda)(PySO3 −)2 is the same, but the total concentration of ruthenium complex is increased by adding Ru(phenda)(PySO3 −)2, as in this case the lower mobility is not compensated by an average shorter distance. Indeed, when 10 equiv Ru(phenda)(PySO3 −)2 were co‐encapsulated with 2 equiv of Ru(bda)(PySO3 −)2 in the nanosphere, the catalytic rate decreased by around 80 % (Supporting Information, Figure S37a), but appeared still higher than the rate obtained for the free catalyst. Clearly, it is more difficult for the oxo intermediate to find the proper partner for the radical coupling pathway when the nanosphere is packed with Ru(phenda)(PySO3 −)2. Along these lines, in a similar experiment with 6 equiv of Ru(bda)(PySO3 −)2, the difference between the absence and presence of 6 equiv of Ru(phenda)(PySO3 −)2 is a reduction of the rate by a mere 16 % (Supporting Information, Figure S37b).
In conclusion, water oxidation catalysis is of pivotal importance to progress the field of artificial photosynthesis. The best ruthenium catalyst reported so far operates at low overpotentials and high rates, and a key element in the reaction pathway involves the final oxygen forming step by a dinuclear radical‐oxo coupling step. We have demonstrated that self‐assembled nanospheres bearing guanidinium binding sites can strongly bind sulfonate‐functionalized ruthenium catalysts. The binding of multiple catalysts (up to 12) results in extremely high local catalyst concentrations (0.54 m), which facilitates this rate determining dinuclear coupling step. Compared to the homogeneous system, the reaction rate is enhanced by two‐orders of magnitude by preorganization of the ruthenium catalysts. Moreover, we demonstrate that at very high local concentrations the reaction rate is no longer dependent the radical coupling step, effectively eliminating diffusion limitations for this particular step at very low overall catalyst concentration. Importantly, solubility limitations and other practical issues would not allow electrocatalysis to be performed in this concentration regime, and as such this supramolecular strategy is an important new method to study electrocatalysis.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support was provided by the University of Amsterdam and by the European Research Council (ERC Advanced Grant). 339 786‐NAT_CAT.
F. Yu, D. Poole, S. Mathew, N. Yan, J. Hessels, N. Orth, I. Ivanović-Burmazović, J. N. H. Reek, Angew. Chem. Int. Ed. 2018, 57, 11247.
References
- 1. Lewis N. S., Nocera D. G., Proc. Natl. Acad. Sci. USA 2006, 103, 15729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kärkäs M. D., Verho O., Johnston E. V., Åkermark B., Chem. Rev. 2014, 114, 11863. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Sala X., Romero I., Rodríguez M., Escriche L., Llobet A., Angew. Chem. Int. Ed. 2009, 48, 2842; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 2882; [Google Scholar]
- 3b. Concepcion J. J., Jurss J. W., Brennaman M. K., Hoertz P. G., Patrocinio A. O. T., Murakami Iha N. Y., Templeton J. L., Meyer T. J., Acc. Chem. Res. 2009, 42, 1954. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Blakemore J. D., Crabtree R. H., Brudvig G. W., Chem. Rev. 2015, 115, 12974; [DOI] [PubMed] [Google Scholar]
- 4b. Duan L., Tong L., Xu Y., Sun L., Energy Environ. Sci. 2011, 4, 3296; [Google Scholar]
- 4c. Garrido-Barros P., Gimbert-Suriñach C., Matheu R., Sala X., Llobet A., Chem. Soc. Rev. 2017, 46, 6088; [DOI] [PubMed] [Google Scholar]
- 4d. Hetterscheid D. G. H., Reek J. N. H., Angew. Chem. Int. Ed. 2012, 51, 9740; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9878; [Google Scholar]
- 4e. Meyer T. J., Sheridan M. V., Sherman B. D., Chem. Soc. Rev. 2017, 46, 6148. [DOI] [PubMed] [Google Scholar]
- 5. Shaffer D. W., Xie Y., Concepcion J. J., Chem. Soc. Rev. 2017, 46, 6170. [DOI] [PubMed] [Google Scholar]
- 6. Hessels J., Detz R., Koper M., Reek J., Chem. Eur. J. 2017, 23, 16413. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Matheu R., Ertem M. Z., Benet-Buchholz J., Coronado E., Batista V. S., Sala X., Llobet A., J. Am. Chem. Soc. 2015, 137, 10786; [DOI] [PubMed] [Google Scholar]
- 7b. Schulze M., Kunz V., Frischmann P. D., Würthner F., Nat. Chem. 2016, 8, 576. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Duan L., Fischer A., Xu Y., Sun L., J. Am. Chem. Soc. 2009, 131, 10397; [DOI] [PubMed] [Google Scholar]
- 8b. Duan L., Bozoglian F., Mandal S., Stewart B., Privalov T., Llobet A., Sun L., Nat. Chem. 2012, 4, 418; [DOI] [PubMed] [Google Scholar]
- 8c. Duan L., Wang L., Li F., Li F., Sun L., Acc. Chem. Res. 2015, 48, 2084. [DOI] [PubMed] [Google Scholar]
- 9. Duan L., Moyses C., Ahlquist M. S. G., Sun L., Proc. Natl. Acad. Sci. USA 2012, 109, 15584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tong L., Duan L., Xu Y., Privalov T., Sun L., Angew. Chem. Int. Ed. 2011, 50, 445; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 465. [Google Scholar]
- 11.
- 11a. Li T. T., Zhao W. L., Chen Y., Li F. M., Wang C. J., Tian Y. H., Fu W. F., Chem. Eur. J. 2014, 20, 13957; [DOI] [PubMed] [Google Scholar]
- 11b. Matheu R., Francàs L., Chernev P., Ertem M. Z., Batista V., Haumann M., Sala X., Llobet A., ACS Catal. 2015, 5, 3422. [Google Scholar]
- 12. Shaffer D. W., Xie Y., Szalda D. J., Concepcion J. J., Inorg. Chem. 2016, 55, 12024. [DOI] [PubMed] [Google Scholar]
- 13. Song N., Concepcion J. J., Binstead R. A., Rudd J. A., Vannucci A. K., Dares C. J., Coggins M. K., Meyer T. J., Proc. Natl. Acad. Sci. USA 2015, 112, 4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Leenders S. H. A. M., Gramage-Doria R., de Bruin B., Reek J. N. H., Chem. Soc. Rev. 2015, 44, 433; [DOI] [PubMed] [Google Scholar]
- 14b. Brown C. J., Toste F. D., Bergman R. G., Raymond K. N., Chem. Rev. 2015, 115, 3012; [DOI] [PubMed] [Google Scholar]
- 14c. Vriezema D. M., Aragonès M. C., Elemans J. A. A. W., Cornelissen J. J. L. M., Rowan A. E., Nolte R. J. M., Chem. Rev. 2005, 105, 1445; [DOI] [PubMed] [Google Scholar]
- 14d. Hooley R. J., Rebek J., Chem. Biol. 2009, 16, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sato S., Iida J., Suzuki K., Kawano M., Ozeki T., Fujita M., Science 2006, 313, 1273. [DOI] [PubMed] [Google Scholar]
- 16. Wang Q. Q., Gonell S., Leenders S. H. A. M., Dürr M., Ivanovic-Burmazovic I., Reek J. N. H., Nat. Chem. 2016, 8, 225. [DOI] [PubMed] [Google Scholar]
- 17. Duan L., Wang L., Inge A. K., Fischer A., Zou X., Sun L., Inorg. Chem. 2013, 52, 7844. [DOI] [PubMed] [Google Scholar]
- 18. Suzuki K., Takao K., Sato S., Fujita M., J. Am. Chem. Soc. 2010, 132, 2544. [DOI] [PubMed] [Google Scholar]
- 19. Ashford D. L., Sherman B. D., Binstead R. A., Templeton J. L., Meyer T. J., Angew. Chem. Int. Ed. 2015, 54, 4778; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4860. [Google Scholar]
- 20. Gramage-Doria R., Hessels J., Leenders S. H. A. M., Tröppner O., Dürr M., Ivanović-Burmazović I., Reek J. N. H., Angew. Chem. Int. Ed. 2014, 53, 13380; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13598. [Google Scholar]
- 21. Chen Z., Concepcion J. J., Hu X., Yang W., Hoertz P. G., Meyer T. J., Proc. Natl. Acad. Sci. USA 2010, 107, 7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary