

Abstract
A computationally guided synthetic route to a free silanide derived from tris(3‐methylindol‐2‐yl)methane ([(tmim)Si]−) through nucleophilic substitution on the SiII precursor (Idipp)SiCl2 is reported (Idipp=2,3‐dihydro‐1,3‐bis(2,6‐diisopropylphenyl)‐1H‐imidazol‐2‐ylidene). This approach circumvents the need for strained tetrahedral silanes as synthetic intermediates. Computational investigations show that the electron‐donating properties of [(tmim)Si]− are close to those of PMe3. Experimentally, the [(tmim)Si]− anion is shown to undergo clean complexation to the base metal salts CuCl and FeCl2, demonstrating the potential utility as a supporting ligand.
Keywords: metal complexes, constrained geometry, density functional theory, silicon, silyl ligand
Silicon(II) compounds are attracting attention as strongly electron‐donating ligands for transition metals, often surpassing the widely used phosphine and carbene ligands.1 In particular, a range of neutral silylenes (R2Si:)—often stabilized by Lewis base coordination—have been prepared and are finding applications as ancillary ligands in catalytic transformations.2 The anionic analogues, silanides ([R3Si]−), can be expected to be even stronger donor ligands. However, the reducing character and the ensuing high reactivity of the metal–silyl bond limits the application of silanides as supporting ligands to mostly multidentate architectures.3
While most silanides are stabilized by interaction with an alkali‐metal cation, there are a few examples of stable “naked” silanides derived from polysilanes4 and borosilanes.5 Of particular interest is the recent observation by the groups of Krempner and Breher that substituents containing remote Lewis base functionalities, such as silylethers or pyrazoles, allow isolation of zwitterionic silanides, in which the counterion is encapsulated away from the Si− center (Scheme 1).6 Generally, silanides are derived from tetrasubstituted silicon precursors, most often by reductive cleavage of an Si−X bond (X=halide, pyrazolide), by nucleophilic cleavage of Si−Si bonds with an alkoxide, or, in rare cases, by deprotonation of an Si−H bond.7
Scheme 1.
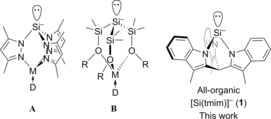
Zwitterionic silanides1e, 6b,6c, 8 and the all‐organic analogue described here. R=Me, C2H5OCH3; D=donor; M=alkali metal or transition metal.
Here we describe the synthesis of the all‐organic, free silanide [(tmim)Si]− (1, tmimH3=tris(3‐methylindol‐2‐yl)methane, Scheme 1). The Si− center is stabilized sterically and electronically by electron‐withdrawing N‐indolyl substituents incorporated in a bicyclo[2.2.2]octane structure. The coordination chemistry of 1 with FeII and CuI demonstrates the ability of silanide 1 to form stable metal complexes. The donor properties of 1 are investigated computationally and experimentally.
The tmim scaffold has been used by Barnard and Mason to prepare the stable phosphine (tmim)P,9 which is isoelectronic to 1, suggesting that the latter should not suffer from excessive strain. However, unsuccessful initial attempts to synthesize tetrahedral SiIV precursors to 1, such as (tmim)SiCl and (tmim)SiH, prompted us to perform more detailed strain‐enthalpy calculations.10 To this end, we calculated the enthalpy of the homodesmotic reactions depicted in Table 1 at the TPSS/TZVP level, which provides an estimate of the strain of the corresponding cage compound. As expected, the experimentally accessible (tmim)P displays a very small strain of +1.2 kcal mol−1. The targeted Si− analogue 1 affords a slight negative reaction enthalpy (−1.6 kcal mol−1), suggesting that it may be stable. In contrast, the strain is significantly higher for the tetrahedral silanes (ΔH R=H=13.1 kcal mol−1 and ΔH R=Cl=14.4 kcal mol−1, respectively). This difference can be understood by orbital hybridization theory. In compliance with Bent's Rule,11 compounds featuring a lone pair on the central element will use hybrid orbitals with high p character to form bonds with the nitrogen atoms, which allows smaller N−Si−N angles favored by the cage structure. These calculations motivated the search for a synthetic route to the targeted Si− compound 1 that would not involve tetrahedral SiIV intermediates.
Table 1.
Homodesmotic reactions calculated for ER=SiCl, SiH, P, Si−. Reaction enthalpies and N−E−N angles of the ER‐containing entities.
| |||
|---|---|---|---|
| ER= | ΔH/kcal mol−1 | Angle/° open | Angle/° cage |
| SiCl | 14.4 | 109.2 | 101.7 |
| SiH | 13.1 | 109.0 | 100.6 |
| P | 1.2 | 99.9 | 94.3 |
| Si− | −1.6 | 95.8 | 89.9 |
Therefore, we investigated the idea that a silanide could be synthesized by direct substitution at SiII. We turned our attention to the recently developed (Idipp)SiCl2 (Idipp=2,3‐dihydro‐1,3‐bis(2,6‐diisopropylphenyl)‐1H‐imidazol‐2‐ylidene), which has been shown to act as a convenient SiII synthon.12 A first substitution attempt with the trisodium salt (tmim)Na3 in THF afforded a complex mixture, from which silylsilylene 2 ((tmim)Si−SiCl(Idipp)) could be identified as a component in the X‐ray crystal structure (Figure 1). Due to the very low yield of this species, further analytical data could not be obtained. The observation of 2 suggests that a nucleophilic attack of 1 on a second equivalent (Idipp)SiCl2 is kinetically competitive with the reaction of tmim with (Idipp)SiCl2 to form 1. Enhancing the nucleophilicity of the [tmim]3− trianion to favor the first substitution reaction by using the tripotassium salt and one equivalent of 18‐crown‐6 gratifyingly allowed clean substitution of both chlorides of (Idipp)SiCl2 to afford [1][K(18‐c‐6)] in 55 % yield (Scheme 2).
Figure 1.
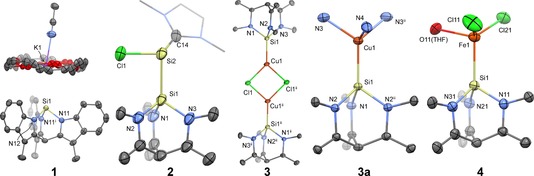
Molecular structure in the crystal of 1, 2, 3, 3 a, and 4.25 Ellipsoids are drawn at 50 % probability. For clarity, hydrogen atoms, K+(18‐c‐6) and cocrystallized solvent are omitted where necessary, and only the core atoms are plotted in 2, 3, 3 a, and 4. Symmetry codes: i=x, 1−y, z; ii=−x, −y, −z; iii=x, −y, z. The crown ether in 1 is disordered on a mirror plane. The unit cell of 4 contains two independent molecules of which one is shown (see the Supporting Information).
Scheme 2.
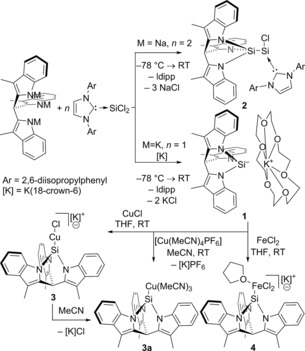
Synthesis and complexation reactions of 1.
A single set of 1H NMR resonances for [1][K(18‐c‐6)] in the aromatic region indicates threefold symmetry, consistent with a bicyclo[2.2.2]octane topology. In MeCN solution, [1][K(18‐c‐6)] exists as solvent‐separated ion pair, as evidenced by distinct diffusion coefficients for the anion and cation measured by DOSY NMR.13 Accordingly, the 29Si NMR shows a resonance at −48.1 ppm, which is in good agreement with the DFT‐calculated value of δ=−46.6 ppm for the free silanide anion (see the Supporting Information). Moreover, it is close to the observed shift for A (Scheme 1, −38.6, M=Mo)1e and significantly different from the electronically distinct B (−194.7 ppm, M=K, R=C2H5OMe).6b Crystals suitable for X‐ray diffraction were grown from an acetonitrile solution at −35 °C. The structure shows an ion pair with, at most, a weak interaction according to the long Si–K distance (3.8807(6) Å). Reported distances for contact ion pairs of Si−⋅⋅⋅K+(18‐c‐6)4a,4b, 14 range from 3.2911(16)14a to 3.9413(18) Å,14b featuring silanide 1 at the longer‐distance part of that spectrum, exceeded only by the cyclosilyl dianion reported by Fischer et al.14b The bicyclo[2.2.2]octane core possesses N−Si−N angles of 90.30(6)–91.14(4)°, consistent with a strong p character of the bonding orbitals and in agreement with the DFT‐predicted angles (see above).
The properties of 1 as a ligand were first investigated computationally. Regarding the steric properties of the ligand, the Tolman cone angle was determined to be 194.6°, which is the same as that of P(o‐tol)3 (194(6)°, see the Supporting Information).15a Additionally, the %V bur was calculated to be 40.1 % based on a M–Si distance of 2.0 Å, in between those of PCy3 and PtBu3 (37.1 and 42.4 %, resp.).15b,15c The frontier orbitals of 1 are localized on the aromatic system, with the lone pair on silicon occupying the HOMO−4 at an energy of 9.6 kcal mol−1 below the HOMO. The in silico analogue of the Tolman electronic parameter (CEP) has been determined for a range of neutral 2 e− donor ligands, including NHCs and phosphines, in [IrLCp(CO)] (Cp=cyclopentadienyl) by Gusev et al.16a (see the Supporting Information). Perrin et al. proved applicability for charged ligands.16b Typical values are (CO)=2115 cm−1 for PF3, 2049 cm−1 for PPh3, 2039 cm−1 for 1,3‐bis(methyl)‐4,5‐dihydroimidazol‐2‐ylidene, 2033 cm−1 for 1,3‐bis(methyl)imidazol‐2‐ylidene, and 2028 cm−1 for PtBu3. We also determined the CEP for a range of silanides with electron‐withdrawing groups (Table 2). The calculated vibrational frequency puts ligand 1 ( (CO)=2038 cm−1) at the weak‐donor extreme in the list of silanide ligands, just beyond Si(C2F5)3 ( (CO)=2034 cm−1) and ligand LiA ( (CO)=2024 cm−1). Remarkably, anion 1 is a weaker donor than the relatively weak σ‐donating, neutral, base‐stabilized silylenes1f, 16c (VII, VIII: (CO)=2011 and 2013 cm−1, respectively). The electronic properties of 1 can be best compared with PMe3 ( (CO)=2037 cm−1). Interestingly, the nonstrained tris(3‐methylindol‐N‐yl)silyl with (CO)=2021 cm−1 is a stronger donor than 1, illustrating the stabilizing effect of the cage structure on the silicon‐centered lone pair.
Table 2.
Computational (CO) of SiII ligands in [IrLCp(CO)].
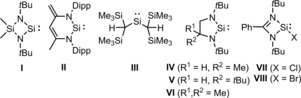
| |||
|---|---|---|---|
| L | (CO)/[cm−1] | L | (CO)/[cm−1] |
| VII | 2011 | VI | 2044 |
| VIII | 2013 | II | 2045 |
| Si(MI)3 [a] | 2021 | IV | 2045 |
| [LiSi(Pz)3] (LiA) | 2024 | III | 2046 |
| Si(C2F5)3 | 2034 | I | 2047 |
| PMe3 | 2037 | V | 2048 |
| 1 | 2038 | – | |
[a] MI=3‐methylindol‐1‐yl.
The ability of 1 to act as a ligand for transition metals was demonstrated with copper(I) and iron(II) chlorides. First, complexation of 1 to CuCl in THF produces the poorly soluble chlorocuprate 3 (Scheme 2). The 29Si NMR resonance could not be observed, likely due to the large quadrupole moment of the bound copper atom.17 The monomeric nature of chlorocuprate 3 was suggested by ESI‐MS (THF, [C28H22N3SiCuCl]−=526.0345 a.u.) and established by DOSY NMR. In contrast, the X‐ray crystal structure of 3 displays a centrosymmetric dimer with a Cu2Cl2 diamond core.1e, 18, 19, 20 The Cu−Si distance (2.1905(9) Å) is marginally shorter than that reported for the monomeric silyl chlorocuprate based on ligand A (Scheme 1, 2.197(2) Å)1e and hence the shortest reported for copper silanides, presumably because of the large Si s character of the bonding orbital.21 In acetonitrile, complex 3 dissociates to the neutral tris(acetonitrile) coppersilyl complex 3 a by elimination of [K(18‐c‐6)]Cl, as was established by comparison of 1H NMR data from an authentic sample of 3 a synthesized from [1][K(18‐c‐6)] and [Cu(MeCN)4PF6]. An X‐ray crystal structure of 3 a reveals that formal substitution of Cl for MeCN (3→3 a) causes a slight elongation of the Cu−Si bond (Δd=0.0201(11) Å).
Finally, complexation of 1 to FeIICl2 in THF afforded the anionic dichloroferrate complex 4. The 1H NMR spectrum features one set of six paramagnetically shifted resonances in addition to that of the K+(18‐c‐6) counterion, suggesting retention of the local C 3 symmetry. The solid‐state structure of 4 contains two independent molecules in the unit cell and confirms the anionic nature of the complex, displaying Si−Fe distances of 2.4482(12) and 2.4589(12) Å. Only one high‐spin FeII complex of a monodentate silylene is known, which forms at low temperatures.22 A small number of high‐spin, monodentate, silyliron complexes have been reported, by the groups of Tilley, Goff, and Tatsumi, for simple SiR3 silyls (R=H, alkyl, aryl, SiMe3),23 but 4 is the first nitrogen‐supported silyl complex of this kind.
Interestingly, the geometry around silicon is sensitive to coordination. In the ideal case, the sum of angles is 328.5° for sp3 hybridization and 270° for the nonhybridized extreme. The N−Si−N angle sum increases from 272.58(10)° in uncoordinated 1 to 278.75(14)° in 3 a, 280.8(2)° in 3, and 282.9(3)/281.8(3)° in 4, which can be explained by an increasing p character of the lone pair upon binding to a Lewis acid and a consequent decrease in the p character of the Si−N bonding orbitals. This is confirmed by Natural Bond Orbital (NBO) analysis24 of 1, 3 a, 3, and 4: the s character of the silicon Natural Hybrid Orbital (NHO), corresponding to the lone pair in 1 and forming the M−Si bond in the complexes, decreases from 62.8 % in 1 to 57.4 % for 3 a, 59.3 % for 3, and 58.9 % for 4.
In summary, nucleophilic substitution on the neutral SiII compound (Idipp)SiCl2 afforded a free silanide (1) derived from tris(3‐methylindol‐2‐yl)methane (tmimH3). This approach avoids strained tetrahedral silane intermediates, and may find further applications in silanide synthesis. The ability of 1 to form transition‐metal complexes was demonstrated: coordination to CuI afforded the dimeric silyl cuprate [Cu(Si(tmim))μ‐Cl]2 2−, featuring the shortest known silyl copper distance, and the neutral acetonitrile‐solvated analogue [Cu(Si(tmim))(NCMe)3]. Complexation to FeCl2 gave the corresponding ferrate [FeCl2(Si(tmim))(THF)]−, a rare example of a high‐spin silyliron complex. These results show that the reactive lone pair of a silanide can be efficiently stabilized by a combination of inductive effects and constrained geometry, making these structures attractive candidates as donor ligands for homogeneous catalysis. Studies in this direction are ongoing in our laboratories.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Marc‐Etienne Moret obtained an MSc degree in chemistry from the Ecole Polytechnique Fédérale de Lausanne (EPFL) in 2005, and a PhD degree from the Eidgenössische Technische Hochschule Zürich (ETHZ) with Prof. P. Chen in 2009. In 2010, he moved to the California Institute of Technology (Caltech) for postdoctoral research in the group of Prof. J. C. Peters on the activation of N2 by synthetic iron complexes. Since 2012, he is Assistant Professor in the group of Organic Chemistry and Catalysis at Utrecht University (UU). His research interests revolve around the design of novel ligands for small‐molecule activation and catalysis with first‐row transition metals.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Support with NMR spectroscopic analysis by Dr. J.T.B.H. Jastrzebski is gratefully acknowledged. We acknowledge funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement PIIF‐GA‐2012–327306 (IIF‐Marie Curie grant awarded to M.E.M.), the Dutch National Research School Combination Catalysis (NRSC‐C), and the Sectorplan Natuur‐ en Scheikunde (Tenure‐track grant at Utrecht University). This work was sponsored by NWO Exacte en Natuurwetenschappen (Physical Sciences) for the use of supercomputer facilities, with financial support from the Netherlands Organization for Scientific Research (NWO). The X‐ray diffractometer was financed by the NWO.
L. Witteman, T. Evers, M. Lutz, M.-E. Moret, Chem. Eur. J. 2018, 24, 12236.
References
- 1.
- 1a. Wang W., Inoue S., Irran E., Driess M., Angew. Chem. Int. Ed. 2012, 51, 3691–3694; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3751–3754; [Google Scholar]
- 1b. Wiberg N., Coord. Chem. Rev. 1997, 163, 217–252; [Google Scholar]
- 1c. Lerner H., Coord. Chem. Rev. 2005, 249, 781–798; [Google Scholar]
- 1d. Li J., Merkel S., Henn J., Meindl K., Döring A., Roesky H. W., Ghadwal R. S., Stalke D., Inorg. Chem. 2010, 49, 775–777; [DOI] [PubMed] [Google Scholar]
- 1e. Styra S., González-Gallardo S., Armbruster F., Oña-Burgos P., Moos E., Vonderach M., Weis P., Hampe O., Grün A., Schmitt Y., Gerhards M., Menges F., Gaffga M., Niedner-Schatteburg G., Breher F., Chem. Eur. J. 2013, 19, 8436–8446; [DOI] [PubMed] [Google Scholar]
- 1f. Benedek Z., Szilvási T., RSC Adv. 2015, 5, 5077–5086. [Google Scholar]
- 2.
- 2a. Fürstner A., Krause H., Lehmann C. W., Chem. Commun. 2001, 2372–2373; [DOI] [PubMed] [Google Scholar]
- 2b. Gallego D., Brück A., Irran E., Meier F., Kaupp M., Driess M., Hartwig J. F., J. Am. Chem. Soc. 2013, 135, 15617–15626; [DOI] [PubMed] [Google Scholar]
- 2c. Wang W., Inoue S., Enthaler S., Driess M., Angew. Chem. Int. Ed. 2012, 51, 6167–6171; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 6271–6275; [Google Scholar]
- 2d. Metsänen T. T., Gallego D., Szilvási T., Driess M., Oestreich M., Chem. Sci. 2015, 6, 7143–7149; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2e. Gallego D., Inoue S., Blom B., Driess M., Organometallics 2014, 33, 6885–6897; [Google Scholar]
- 2f. Zhang M., Liu X., Shi C., Ren C., Ding Y., Roesky H. W., Z. Anorg. Allg. Chem. 2008, 634, 1755–1758; [Google Scholar]
- 2g. Brück A., Gallego D., Wang W., Irran E., Driess M., Hartwig J. F., Angew. Chem. Int. Ed. 2012, 51, 11478–11482; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 11645–11649; [Google Scholar]
- 2h. Blom B., Enthaler S., Inoue S., Irran E., Driess M., J. Am. Chem. Soc. 2013, 135, 6703–6713. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Simon M., Breher F., Dalton Trans. 2017, 46, 7976–7997; [DOI] [PubMed] [Google Scholar]
- 3b. Okazaki M., Ohshitanai S., Iwata M., Tobita H., Ogino H., Coord. Chem. Rev. 2002, 226, 167–178. [Google Scholar]
- 4.
- 4a. Ichinohe M., Toyoshima M., Kinjo R., Sekiguchi A., J. Am. Chem. Soc. 2003, 125, 13328–13329; [DOI] [PubMed] [Google Scholar]
- 4b. Jenkins D. M., Teng W., Englich U., Stone D., Ruhlandt-Senge K., Organometallics 2001, 20, 4600–4606; [Google Scholar]
- 4c. Abersfelder K., Scheschkewitz D., J. Am. Chem. Soc. 2008, 130, 4114–4121; [DOI] [PubMed] [Google Scholar]
- 4d. Ichinohe M., Kinjo R., Sekiguchi A., Organometallics 2003, 22, 4621–4623; [Google Scholar]
- 4e. Molev G., Tumanskii B., Sheberla D., Botoshansky M., Bravo-Zhivotovskii D., Apeloig Y., J. Am. Chem. Soc. 2009, 131, 11698–11700; [DOI] [PubMed] [Google Scholar]
- 4f. Nakamoto M., Fukawa T., Lee V. Y., Sekiguchi A., J. Am. Chem. Soc. 2002, 124, 15160–15161. [DOI] [PubMed] [Google Scholar]
- 5. Scheschkewitz D., Hofmann M., Ghaffari A., Amseis P., Präsang C., Mesbah W., Geiseler G., Massa W., Berndt A., J. Organomet. Chem. 2002, 646, 262–270. [Google Scholar]
- 6.
- 6a. Hitchcock P. B., Lappert M. F., Layh M., Chem. Commun. 1998, 0, 2179–2180; [Google Scholar]
- 6b. Li H., Hope-Weeks L. J., Krempner C., Chem. Commun. 2011, 47, 4117–4119; [DOI] [PubMed] [Google Scholar]
- 6c. Armbruster F., Fernández I., Breher F., Dalton Trans. 2009, 5612; [DOI] [PubMed] [Google Scholar]
- 6d. Mashin E., Kratish Y., Kaushansky A., Bravo-Zhivotovskii D., Apeloig Y., Struct. Chem. 2017, 28, 537–544; [Google Scholar]
- 6e. Li H., Aquino A. J. A., Cordes D. B., Hase W. L., Krempner C., Chem. Sci. 2017, 8, 1316–1328; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6f. Li H., Hung-Low F., Krempner C., Organometallics 2012, 31, 7117–7124; [Google Scholar]
- 6g. Thalangamaarachchige V. D., Unruh D. K., Cordes D. B., Krempner C., Inorg. Chem. 2015, 54, 4189–4191; [DOI] [PubMed] [Google Scholar]
- 6h. Armbruster F., Augenstein T., Oña-Burgos P., Breher F., Chem. Eur. J. 2013, 19, 17899–17906. [DOI] [PubMed] [Google Scholar]
- 7. Schwarze N., Steinhauer S., Neumann B., Stammler H.-G., Hoge B., Angew. Chem. Int. Ed. 2016, 55, 16156–16160; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16390–16394. [Google Scholar]
- 8. Krempner C., Chisholm M. H., Gallucci J., Angew. Chem. Int. Ed. 2008, 47, 410–413; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 416–420. [Google Scholar]
- 9. Barnard T. S., Mason M. R., Inorg. Chem. 2001, 40, 5001–5009. [DOI] [PubMed] [Google Scholar]
- 10.Gaussian 09. D01. 2013, full reference can be found in SI.
- 11. Bent H. A., Chem. Rev. 1961, 61, 275–311. [Google Scholar]
- 12.
- 12a. Ghadwal R. S., Roesky H. W., Merkel S., Henn J., Stalke D., Angew. Chem. Int. Ed. 2009, 48, 5683–5686; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 5793–5796; [Google Scholar]
- 12b. Ghadwal R. S., Azhakar R., Roesky H. W., Acc. Chem. Res. 2013, 46, 444–456; [DOI] [PubMed] [Google Scholar]
- 12c. Geiß D., Arz M. I., Straßmann M., Schnakenburg G., Filippou A. C., Angew. Chem. Int. Ed. 2015, 54, 2739–2744; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 2777–2782; [Google Scholar]
- 12d. Al-Rafia S. M. I., McDonald R., Ferguson M. J., Rivard E., Chem. Eur. J. 2012, 18, 13810–13820; [DOI] [PubMed] [Google Scholar]
- 12e. Hadlington T. J., Abdalla J. A. B., Tirfoin R., Aldridge S., Jones C., Nagase S., Power P. P., Mountford P., Aldridge S., Chem. Commun. 2016, 52, 1717–1720. [DOI] [PubMed] [Google Scholar]
- 13. Pregosin P. S., Magn. Reson. Chem. 2017, 55, 405–413. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Klapötke T. M., Vasisht S. K., Fischer G., Mayer P., J. Organomet. Chem. 2010, 695, 667–672; [Google Scholar]
- 14b. Fischer R., Konopa T., Baumgartner J., Marschner C., Organometallics 2004, 23, 1899–1907; [Google Scholar]
- 14c. Wallner A., Hlina J., Konopa T., Wagner H., Baumgartner J., Marschner C., Flörke U., Organometallics 2010, 29, 2660–2675. [Google Scholar]
- 15.
- 15a. Tolman C. A., J. Am. Chem. Soc. 1970, 92, 2956–2965; [Google Scholar]
- 15b. Poater A., Cosenza B., Correa A., Giudice S., Ragone F., Scarano V., Cavallo L., Eur. J. Inorg. Chem. 2009, 2009, 1759–1766; [Google Scholar]
- 15c. Clavier H., Nolan S. P., Chem. Commun. 2010, 46, 841. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Gusev D. G., Organometallics 2009, 28, 763–770; [Google Scholar]
- 16b. Perrin L., Clot E., Eisenstein O., Loch J., Crabtree R. H., Inorg. Chem. 2001, 40, 5806–5811; [DOI] [PubMed] [Google Scholar]
- 16c. Meltzer A., Präsang C., Driess M., J. Am. Chem. Soc. 2009, 131, 7232–7233. [DOI] [PubMed] [Google Scholar]
- 17. Vingerhoets P., Flanagan K. T., Avgoulea M., Billowes J., Bissell M. L., Blaum K., Brown B. A., Cheal B., De Rydt M., Forest D. H., Geppert C., Honma M., Kowalska M., Krämer J., Krieger A., Mané E., Neugart R., Neyens G., Nörtershäuser W., Otsuka T., Schug M., Stroke H. H., Tungate G., Yordanov D. T., Phys. Rev. C 2010, 82, 64311. [Google Scholar]
- 18.
- 18a. Khan S., Ahirwar S. K., Pal S., Parvin N., Kathewad N., Organometallics 2015, 34, 5401–5406; [Google Scholar]
- 18b. Parvin N., Dasgupta R., Pal S., Sen S. S., Khan S., Dalton Trans. 2017, 46, 6528–6532. [DOI] [PubMed] [Google Scholar]
- 19. Farwell J. D., Hitchcock P. B., Lappert M. F., Protchenko A. V., J. Organomet. Chem. 2007, 692, 4953–4961. [Google Scholar]
- 20. Heine A., Stalke D., Angew. Chem. Int. Ed. Engl. 1993, 32, 121–122; [Google Scholar]; Angew. Chem. 1993, 105, 90–92. [Google Scholar]
- 21.
- 21a. Troadec T., Prades A., Rodriguez R., Mirgalet R., Baceiredo A., Saffon-Merceron N., Branchadell V., Kato T., Inorg. Chem. 2016, 55, 8234–8240; [DOI] [PubMed] [Google Scholar]
- 21b. Tan G., Blom B., Gallego D., Driess M., Organometallics 2014, 33, 363–369. [Google Scholar]
- 22. Hänninen M. M., Pal K., Day B. M., Pugh T., Layfield R. A., Dalton Trans. 2016, 45, 11301–11305. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Roddick D. M., Tilley T. D., Rheingold A. L., Geib S. J., J. Am. Chem. Soc. 1987, 109, 945–946; [Google Scholar]
- 23b. Kim Y. O., Goff H. M., J. Am. Chem. Soc. 1988, 110, 8706–8707; [Google Scholar]
- 23c. Heyn R. H., Tilley T. D., Inorg. Chim. Acta 2002, 341, 91–98; [Google Scholar]
- 23d. Turculet L., Feldman J. D., Tilley T. D., Organometallics 2003, 22, 4627–4629; [Google Scholar]
- 23e. Hatanaka T., Ohki Y., Tatsumi K., Eur. J. Inorg. Chem. 2013, 2013, 3966–3971. [Google Scholar]
- 24.E. D. Glendening, J, K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis, F. Weinhold, NBO 6.0, 2013.
- 25. https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/chem.201801435 1814522, 1814523, 1814524, 1814525, and 1814526, contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary