

Abstract
A transition metal‐free N‐arylation of primary and secondary amines with diaryliodonium salts is presented. Both acyclic and cyclic amines are well tolerated, providing a large set of N‐alkyl anilines. The methodology is unprecedented among metal‐free methods in terms of amine scope, the ability to transfer both electron‐withdrawing and electron‐donating aryl groups, and efficient use of resources, as excess substrate or reagents are not required.
Keywords: anilines, arylamines, diaryliodonium salts, hypervalent iodine, metal-free reactions
Arylated amine derivatives are ubiquitous in nature, as well as in medicinal applications and materials science,1 and has led to an intense research focus on the development of efficient synthetic methodologies for this compound class. Transition metal catalyzed cross‐couplings have provided a wide range of arylamines, on an industrial scale, as intermediates towards pharmaceutically active compounds.1b, 2 More recently, C−H activation has also proven efficient for N‐arylation of amines.3
Still, the drawbacks associated with transition‐metal catalysis, including toxicity, cost, need for substrate‐dependent designer ligands, and risk of product contamination has led to an increased focus on the development of metal‐free methodologies for C−N bond formation.4 While nucleophilic aromatic substitution generally has rather narrow aryl scope,5 fluoroarenes lacking highly electron‐withdrawing substituents can be utilized in the arylation of secondary amines under strongly basic conditions at high temperature (Scheme 1 a).6 Iminomalonates have recently been introduced as electrophilic aminating reagents with arylmetal reagents. This strategy has a wide scope, although requiring extra steps for attachment and cleavage of the umpolung reagent.7
Scheme 1.
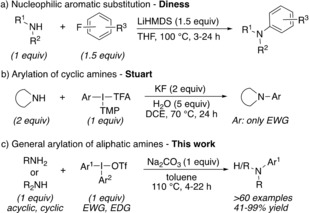
Metal‐free N‐arylation of amines. DCE=1,2‐dichloroethane, EDG=electron‐donating group, EWG=electron‐withdrawing group, HMDS=hexamethyldisilazide, TFA=trifluoroacetate, Tf=trifluoromethanesulfonyl, TMP=trimethoxyphenyl.
Other important advances in metal‐free C−N bond formation include organocatalytic radical coupling,8 reactions with arynes,9 and hypervalent iodine reagents.10 Antonchick and co‐workers have demonstrated that (diacetoxyiodo)benzene (DIB) and related iodine(III) reagents can oxidize the nitrogen atom and enable nucleophilic attack by a variety of arenes.11 Organocatalytic N‐arylations of amides and anilines, with in situ formation of iodine(III) from iodoarene precursors, have also been reported.11a, 12 While useful in many reactions, both aryne reactions and DIB oxidations are limited by the moderate regioselectivity often observed, and DIB cannot be used to arylate aliphatic amines.
Diaryliodonium salts are bench‐stable iodine(III) reagents that can be easily synthesized from iodoarenes or arenes by one‐pot reactions.13 Their efficiency in arylations has been demonstrated with a wide range of nucleophiles under metal‐free conditions.14 Arylation of certain nitrogen nucleophiles has been well established with diaryliodonium salts,15 but reactions with amines have proven difficult.16 In 2016, Stuart and co‐workers reported the first synthetically useful, intermolecular arylation of amines with diaryliodonium salts (Scheme 1 b).17 In this pioneering paper, a range of electron‐deficient aryl groups could be transferred using excess amounts of cyclic, secondary amines in halogenated solvent. The limited substrate scope and environmental aspects of this procedure inspired us to initiate an investigation towards a general methodology. Herein we present the efficient arylation of primary amines, as well as acyclic and cyclic secondary amines. The reactions are high‐yielding without excess reagents, and tolerate diaryliodonium salts with both electron‐withdrawing (EWG) and electron‐donating (EDG) substituents (Scheme 1 c).
The optimization was performed with the primary amine 1 a and the nitrophenyliodonium salts 2 in a 1:1 ratio in refluxing toluene (Table 1).18, 19 The reaction showed poor conversion in the absence of an external base, also upon increased loading of 2 a (entries 1 and 2). To the contrary, excess amine proved efficient, with the second equivalent of amine likely acting as base (entry 3). To comply with our aim to develop an efficient arylation procedure, the use of excess substrate was highly unattractive. We hence investigated the addition of bases, such as triethylamine, which indeed improved the yield of the product 3 a (entry 4). Several inorganic bases could also be employed, including K3PO4 and carbonates,18 and sodium carbonate pleasingly delivered 3 a in 81 % yield (entry 5). The counterion of the diaryliodonium salts 2 proved important, as reactions with the iodonium bromide 2 b were severely retarded, whereas the tetrafluoroborate 2 c and tosylate 2 d were more compatible (entries 6–8). The reaction was also evaluated with the trimethoxyphenyl (TMP) salts 2 e and 2 f, as TMP has recently proven to be an efficient “dummy group” in arylations with unsymmetric iodonium salts.20 Indeed, 2 e and 2 f could both be employed under our reaction conditions, albeit with moderate results (entries 9 and 10). To the contrary, only decomposition was observed when 1 a was reacted with 2 f using Stuart's recently reported procedure for cyclic amines,17 illustrating the large difference between these protocols.
Table 1.
Selected results from the optimization of reaction conditions.[a]
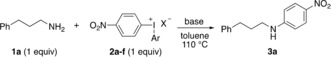
| Entry | 2 (X) | Ar | Base (equiv) | t [h] | Yield [%][b] |
|---|---|---|---|---|---|
| 1 | 2 a (OTf) | Ph | – | 1 | 35 |
| 2[c] | 2 a (OTf) | Ph | – | 2 | 25 |
| 3[d] | 2 a (OTf) | Ph | – | 2 | 86 |
| 4 | 2 a (OTf) | Ph | Et3N (1.0) | 2 | 74 |
| 5 | 2 a (OTf) | Ph | Na2CO3 (1.0) | 4 | 81 |
| 6 | 2 b (Br) | Ph | Na2CO3 (1.0) | 4 | 4 |
| 7 | 2 c (BF4) | Ph | Na2CO3 (1.0) | 4 | 54 |
| 8 | 2 d (OTs) | Ph | Na2CO3 (1.0) | 4 | 77 |
| 9 | 2 e (OTs) | TMP | Na2CO3 (1.0) | 4 | 62 |
| 10 | 2 f (TFA) | TMP | Na2CO3 (1.0) | 4 | 43 |
[a] 2 (0.1 mmol), base and 1 a (0.1 mmol) in anhydrous, degassed toluene (0.5 mL) under Ar. [b] Yield of the isolated product. [c] 2 a (2 equiv). [d] 1 a (2 equiv). Ts=para‐toluenesulfonyl.
The substrate scope was investigated under the reaction conditions in entry 5 of Table 1, that is, without excess substrate or reagents. To our satisfaction, a variety of primary amines, cyclic and acyclic secondary amines, proved suitable for the transformation. As previous protocols were incompatible with primary amines,16, 17 the scope investigation was focused on this substrate class. Nitrophenylation of primary amines with 2 a could be performed within 4 hours, as depicted in Scheme 2 a. Amines with various hydrocarbon chains were efficiently monoarylated to provide the arylamines 3 a–e in 79–91 % yield. Cycloalkyl substituents were also tolerated, with improved reactivity for larger ring sizes (3 f–h). Even the highly sterically hindered adamantyl amine could be arylated to provide 3 i in 60 % yield within a 24 hour reaction time. Furthermore, substrates containing a variety of functional groups, including methoxy, bromide, pyridyl, and silicate proved suitable (3 j–m). Importantly, the arylation of (R)‐1‐phenylethylamine proceeded without erosion of the ee value (3 n). Aromatic amines can also be employed, as demonstrated by the transformation of aniline into the diarylamine 3 o in 91 % yield.
Scheme 2.
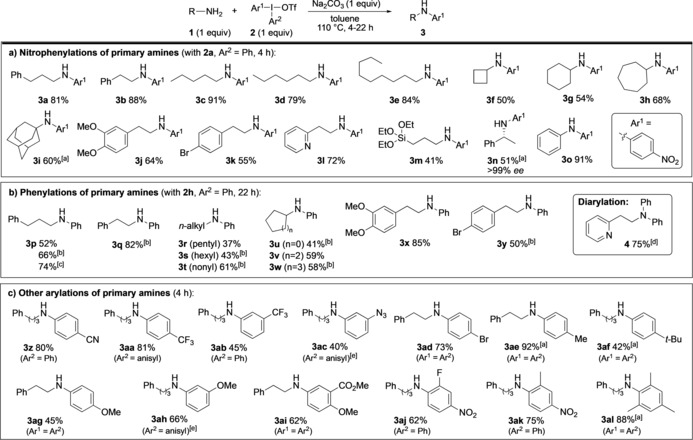
Arylation of primary amines. [a] Reaction time 22–24 h. [b] Amine 1 (1.5 equiv). [c] Amine 1 (2 equiv). [d] Yield based on 2 h. [e] OTs anion.
Diaryliodonium salts lacking EWG groups are generally more difficult to apply in metal‐free arylations, because of decreased reactivity for ligand coupling and increased propensity to form byproducts by either aryne or radical pathways.14, 21 We were thus keen to investigate more challenging arylations, and started by examining reactions with diphenyliodonium triflate (2 h). Pleasingly, amines with acyclic and cyclic substituents could be phenylated to the products 3 p–w in moderate to high yields upon increasing the reaction time to 22 hours (Scheme 2 b). Functional groups like methoxy and bromide were well tolerated, and delivered the arylamines 3 x (85 %) and 3 y (50 %), respectively. Formation of minor amounts of separable, diarylated byproduct was observed with some substrates, and could be avoided by using the reagent 2 h as the limiting reagent.18 As illustrated with 3 p (footnotes [b,c]), this change increased the yield of the isolated product at the expense of a less atom efficient reaction. Surprisingly, 2‐(2‐pyridyl)ethylamine underwent selective diarylation under standard reaction conditions, and furnished the diarylamine 4 in 75 % yield without any sign of monoarylated product.18
The scope investigation was continued using a range of symmetric and unsymmetric diaryliodonium salts (Scheme 2 c). As expected, electron‐deficient aryl groups were easily transferred, providing products with cyano or CF3 substituents (3 z–ab) within 4 hours. An azido‐substituted iodonium salt could be utilized to reach the azidophenylated product 3 ac, which is well suited for further transformations. Importantly, also electron‐donating diaryliodonium salts could be employed. Alkyl‐substituted aryl groups were easily transferred, and 3 ae was obtained in excellent yield. Even highly electron‐rich iodonium salts proved compatible, as demonstrated by transfer of methoxy‐substituted aryl groups to yield the amines 3 ag–ai. Steric hindrance was well tolerated also in the aryl groups, as illustrated by the densely functionalized arylamines 3 aj–al.
Arylation of secondary, cyclic amines was expected to be more facile than primary amines, as such amines were previously the only suitable substrates.17 Indeed, cyclic amines proved highly reactive under our standard reaction conditions without excess reagents (Scheme 3 a). Piperidines, morpholine, and thiomorpholine provided the products 5 a–d in good to excellent yields with 2 a. While N‐phenylpiperazine proved less suitable (5 e), pyrrolidine and tetrahydroisoquinoline were efficiently arylated, delivering 5 f and 5 g, respectively, in excellent yields. The preferential arylation of secondary, cyclic amines over primary was illustrated by a competition experiment that delivered 5 c and 3 a in 4:1 ratio (see Scheme S5 in the Supporting Information).
Scheme 3.
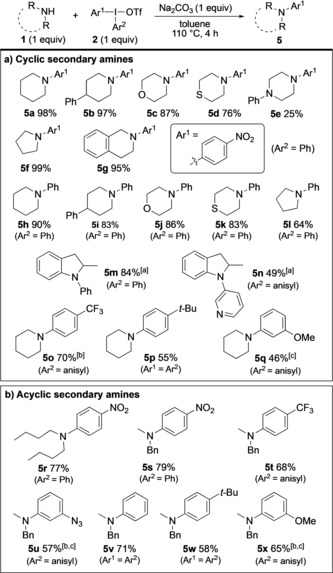
Arylation of secondary amines. [a] Reaction time 22 h. [b] Inseparable from Ar2I. [c] OTs anion.
A selection of cyclic amines was subsequently phenylated with Ph2IOTf (2 h) in high yields (Scheme 3 a), including piperidines (5 h, 5 i) and morpholine (5 j, 86%). The latter compares very well to the literature,17 where 5 j was only obtained in 55 %, despite a large excess of morpholine. Thiomorpholine, pyrrolidine, and 2‐methylindoline were also suitable substrates (5 k–m). Arylation with other iodonium salts was accomplished next, thus providing the pyridyl‐decorated indoline 5 n, as well as piperidines 5 o–q with EWG‐ and EDG‐substituted aryl moieties.
The arylation of acyclic secondary amines was initially investigated with one symmetric and one unsymmetric dialkylamine, providing products 5 r and 5 s, respectively, in similar, good yields (Scheme 3 b). N‐Methyl benzylamine was then treated with a range of symmetric and unsymmetric iodonium salts. To our satisfaction, both EWG‐ and EDG‐substituted aryl groups could be transferred, providing the products 5 t–x. In all cases, reactions with unsymmetric diaryliodonium salts proceeded with high chemoselectivity using the indicated dummy groups. This approach facilitates the synthesis of the iodonium salts, the recovery of the resulting iodoarene in large scale applications, and the atom efficiency when transferring complex aryl groups. Separation of the formed Ar2I from the product proved challenging in a few cases, and we are currently investigating other dummy groups for those substrates.
Control reactions with the radical scavenger 1,1‐diphenylethylene (DPE) and the aryne trap furan were conducted to understand the mechanism, and found to not alter the reaction outcome.18 Furthermore, all products were formed in a regiospecific manner, and we hence suggest that the arylation proceeds by the commonly accepted ligand coupling mechanism depicted in Scheme 4. The amine reacts with the diaryliodonium salt to give the charged T‐shaped intermediate A, which is then deprotonated in the presence of base (or excess amine) to give the intermediate B. Subsequent ligand coupling provides the arylated product and ArI. Alternatively, the four‐coordinated intermediate C, where two amines are coordinated to the iodine, could be a more reactive intermediate for ligand coupling. Such intermediates were recently reported in a mechanistic study on O‐arylation.21
Scheme 4.
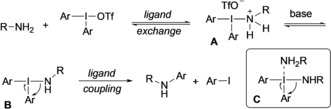
Suggested arylation mechanism.
In conclusion, we have developed an efficient, metal‐free N‐arylation of aliphatic amines with diaryliodonium salts under mild reaction conditions without excess reagents. The methodology is applicable to primary amines, as well as secondary cyclic and acyclic amines, containing a variety of functional groups. Both electron‐deficient and electron‐donating aryl groups, as well as heteroaryl groups can be transferred. Steric hindrance is well tolerated in both coupling partners, and no erosion of ee values was observed. Extension of the methodology to amino acid derivatives, as well as mechanistic studies are currently ongoing, and will be reported in due course.
Experimental Section
General procedure for the synthesis of the arylamines 3–5: The diaryliodonium salt 2 (0.1 mmol) and Na2CO3 (0.1 mmol, 1.0 equiv) were added to an oven‐dried microwave vial, which was sealed with a microwave vial cap. The vial was kept in vacuum for 15 minutes through a needle and then flushed with argon. This procedure was repeated for 3–4 times. The amine 1 (0.1 mmol, 1.0 equiv) was added by syringe followed by anhydrous and degassed toluene (0.5 mL), and the mixture was stirred at 110 °C for the indicated time. After completion of the reaction, it was brought down to RT and subjected to purification by silica gel column chromatography with a n‐pentane/EtOAc or n‐pentane/Et2O gradient to obtain the arylamines 3–5.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Olle Engkvist Byggmästare Foundation (2014/645) and the Swedish Research Council (2015‐04404) are kindly acknowledged for financial support.
N. Purkait, G. Kervefors, E. Linde, B. Olofsson, Angew. Chem. Int. Ed. 2018, 57, 11427.
References
- 1.
- 1a. Hili R., Yudin A. K., Nat. Chem. Biol. 2006, 2, 284; [DOI] [PubMed] [Google Scholar]
- 1b. Schlummer B., Scholz U., Adv. Synth. Catal. 2004, 346, 1599; [Google Scholar]
- 1c. Wang C., Dong H., Hu W., Liu Y., Zhu D., Chem. Rev. 2012, 112, 2208; [DOI] [PubMed] [Google Scholar]
- 1d. Roughley S. D., Jordan A. M., J. Med. Chem. 2011, 54, 3451. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Muci A. R., Buchwald S. L., in Cross-Coupling Reactions: A Practical Guide (Ed.: N. Miyaura), Springer, Berlin, Heidelberg, 2002, pp. 131; [Google Scholar]
- 2b. Evano G., Blanchard N., Toumi M., Chem. Rev. 2008, 108, 3054. [DOI] [PubMed] [Google Scholar]
- 3. Jiao J., Murakami K., Itami K., ACS Catal. 2016, 6, 610. [Google Scholar]
- 4.
- 4a. Garrett C. E., Prasad K., Adv. Synth. Catal. 2004, 346, 889; [Google Scholar]
- 4b. Sun C.-L., Shi Z.-J., Chem. Rev. 2014, 114, 9219. [DOI] [PubMed] [Google Scholar]
- 5. Caron S., Ghosh A., in Practical Synthetic Organic Chemistry, Wiley, Hoboken, 2011, pp. 237. [Google Scholar]
- 6. Borch Jacobsen C., Meldal M., Diness F., Chem. Eur. J. 2017, 23, 846. [DOI] [PubMed] [Google Scholar]
- 7. Kattamuri P. V., Yin J., Siriwongsup S., Kwon D.-H., Ess D. H., Li Q., Li G., Yousufuddin M., Richardson P. F., Sutton S. C., Kürti L., J. Am. Chem. Soc. 2017, 139, 11184. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Louillat-Habermeyer M.-L., Jin R., Patureau F. W., Angew. Chem. Int. Ed. 2015, 54, 4102; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4175; [Google Scholar]
- 8b. Martínez C., Bosnidou A. E., Allmendinger S., Muñiz K., Chem. Eur. J. 2016, 22, 9929. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Bhunia A., Yetra S. R., Biju A. T., Chem. Soc. Rev. 2012, 41, 3140; [DOI] [PubMed] [Google Scholar]
- 9b. Liu Z., Larock R. C., Org. Lett. 2003, 5, 4673; [DOI] [PubMed] [Google Scholar]
- 9c. Pintori D. G., Greaney M. F., Org. Lett. 2010, 12, 168; [DOI] [PubMed] [Google Scholar]
- 9d. Cano R., Ramón D. J., Yus M., J. Org. Chem. 2011, 76, 654; [DOI] [PubMed] [Google Scholar]
- 9e. Pal K. B., Mahanti M., Nilsson U. J., Org. Lett. 2018, 20, 616. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Yoshimura A., Zhdankin V. V., Chem. Rev. 2016, 116, 3328; [DOI] [PubMed] [Google Scholar]
- 10b. Muñiz K., Top. Curr. Chem. 2016, 373, 105; [DOI] [PubMed] [Google Scholar]
- 10c. Murarka S., Antonchick A. P., Top. Curr. Chem. 2016, 373, 75. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Antonchick A. P., Samanta R., Kulikov K., Lategahn J., Angew. Chem. Int. Ed. 2011, 50, 8605; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8764; similar reactions were simultaneously published by Chang and DeBoef: [Google Scholar]
- 11b. Kim H. J., Kim J., Cho S. H., Chang S., J. Am. Chem. Soc. 2011, 133, 16382; [DOI] [PubMed] [Google Scholar]
- 11c. Kantak A. A., Potavathri S., Barham R. A., Romano K. M., DeBoef B., J. Am. Chem. Soc. 2011, 133, 19960; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11d. Samanta R., Antonchick A. P., Synlett 2012, 23, 809; [Google Scholar]
- 11e. Manna S., Matcha K., Antonchick A. P., Angew. Chem. Int. Ed. 2014, 53, 8163; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8302. [Google Scholar]
- 12.
- 12a. Manna S., Serebrennikova P. O., Utepova I. A., Antonchick A. P., Chupakhin O. N., Org. Lett. 2015, 17, 4588; [DOI] [PubMed] [Google Scholar]
- 12b. Lucchetti N., Scalone M., Fantasia S., Muñiz K., Adv. Synth. Catal. 2016, 358, 2093. [Google Scholar]
- 13.
- 13a. Hossain M. D., Kitamura T., Tetrahedron 2006, 62, 6955; [Google Scholar]
- 13b. Bielawski M., Zhu M., Olofsson B., Adv. Synth. Catal. 2007, 349, 2610; [Google Scholar]
- 13c. Zhu M., Jalalian N., Olofsson B., Synlett 2008, 592; [Google Scholar]
- 13d. Bielawski M., Aili D., Olofsson B., J. Org. Chem. 2008, 73, 4602; [DOI] [PubMed] [Google Scholar]
- 13e. Seidl T. L., Sundalam S. K., McCullough B., Stuart D. R., J. Org. Chem. 2016, 81, 1998; [DOI] [PubMed] [Google Scholar]
- 13f. Carreras V., Sandtorv A. H., Stuart D. R., J. Org. Chem. 2017, 82, 1279; [DOI] [PubMed] [Google Scholar]
- 13g. Lindstedt E., Reitti M., Olofsson B., J. Org. Chem. 2017, 82, 11909. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Merritt E. A., Olofsson B., Angew. Chem. Int. Ed. 2009, 48, 9052; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9214; [Google Scholar]
- 14b. Olofsson B., Top. Curr. Chem. 2016, 373, 135; [Google Scholar]
- 14c. Aradi K., Tóth B. L., Tolnai G. L., Novák Z., Synlett 2016, 27, 1456. [Google Scholar]
- 15.Recent examples of N-arylation:
- 15a. Gonda Z., Novák Z., Chem. Eur. J. 2015, 21, 16801 (pyrazoles); [DOI] [PubMed] [Google Scholar]
- 15b. Tinnis F., Stridfeldt E., Lundberg H., Adolfsson H., Olofsson B., Org. Lett. 2015, 17, 2688 (amides); [DOI] [PubMed] [Google Scholar]
- 15c. Reitti M., Villo P., Olofsson B., Angew. Chem. Int. Ed. 2016, 55, 8928; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9074 (nitrite, azide); [Google Scholar]
- 15d. Lucchetti N., Scalone M., Fantasia S., Muñiz K., Angew. Chem. Int. Ed. 2016, 55, 13335; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13529 (phthalimides). [Google Scholar]
- 16.Amine arylations:
- 16a. Beringer F. M., Brierley A., Drexler M., Gindler E. M., Lumpkin C. C., J. Am. Chem. Soc. 1953, 75, 2708 (nitrophenylation of methylamine (5 equiv)); [Google Scholar]
- 16b. Carroll M. A., Wood R. A., Tetrahedron 2007, 63, 11349 (anilines); [Google Scholar]
- 16c. Li J., Liu L., RSC Adv. 2012, 2, 10485; [Google Scholar]
- 16d. Riedmueller S., Nachtsheim B. J., Synlett 2015, 26, 651 (indolines). [Google Scholar]
- 17. Sandtorv A. H., Stuart D. R., Angew. Chem. Int. Ed. 2016, 55, 15812; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16044. [Google Scholar]
- 18.See the Supporting Information for details.
- 19.Unlike many metal-free arylations with iodonium salts, the use of anhydrous, degassed solvent proved important.
- 20.
- 20a. Malmgren J., Santoro S., Jalalian N., Himo F., Olofsson B., Chem. Eur. J. 2013, 19, 10334; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Stuart D. R., Chem. Eur. J. 2017, 23, 15852. [DOI] [PubMed] [Google Scholar]
- 21. Stridfeldt E., Lindstedt E., Reitti M., Blid J., Norrby P.-O., Olofsson B., Chem. Eur. J. 2017, 23, 13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary