

Summary
Patients with chronic lymphocytic leukaemia/small lymphocytic lymphoma (CLL/SLL) with deletion 17p [del(17p)] have poor outcomes with chemoimmunotherapy. Ibrutinib is indicated for the treatment of CLL/SLL, including del(17p) CLL/SLL, and allows for treatment without chemotherapy. This integrated analysis was performed to evaluate outcomes in 230 patients with relapsed/refractory del(17p) CLL/SLL from three ibrutinib studies. With a median of 2 prior therapies (range, 1–12), 18% and 79% of evaluable patients had del(11q) or unmutated IGHV, respectively. With a median follow‐up of 28 months, overall response rate was 85% and estimated 30‐month progression‐free and overall survival rates were 57% [95% confidence interval (CI) 50–64] and 69% (95% CI 61–75), respectively. Patients with normal lactate dehydrogenase or no bulky disease had the most favourable survival outcomes. Sustained haematological improvements in haemoglobin, platelet count and absolute neutrophil count occurred in 61%, 67% and 70% of patients with baseline cytopenias, respectively. New onset severe cytopenias and infections decreased in frequency over time. Progression‐free and overall survival with ibrutinib surpass those of other therapies for patients with del(17p) CLL/SLL. These results provide further evidence of the robust clinical activity of ibrutinib in difficult‐to‐treat CLL/SLL populations.
Keywords: 17p deletion, BTK inhibitor, chronic lymphocytic leukaemia, ibrutinib
Deletion of chromosome 17p [del(17p)] distinguishes a subgroup of patients with chronic lymphocytic leukaemia (CLL) and an exceptionally poor prognosis (Hillmen et al, 2007; Hallek et al, 2010; Zenz et al, 2010, 2012; Badoux et al, 2011). Although uncommon at the time of diagnosis (generally <5%) (Juliusson et al, 1990), the percentage of patients with CLL whose disease harbours del(17p) increases to as much as 10% when therapy is first indicated and can be identified in >40% of multiply relapsed and/or heavily pre‐treated patients (Zenz et al, 2012). Patients with CLL and del(17p) have shorter times to treatment and inferior responses to treatment than other subgroups of CLL patients (Tam & Stilgenbauer, 2015). Del(17p) is a cytogenetic abnormality resulting in loss of TP53 function, leading to resistance to chemotherapy and increased risk of Richter transformation (RT) (Hallek et al, 2010; Badoux et al, 2011; Tam & Stilgenbauer, 2015). Because median survival for patients with del(17p) CLL has typically been ≤3 years from initiation of therapy, younger, fit patients (with an otherwise long life expectancy) with del(17p) CLL have often been referred for allogeneic stem cell transplantation (Hallek et al, 2010; Zenz et al, 2010, 2012; Tam & Stilgenbauer, 2015). Until recently, safe and highly effective therapies for most patients with del(17p) have been limited, representing an important unmet medical need (O'Brien et al, 2016a).
Ibrutinib, a first‐in‐class, once‐daily inhibitor of Bruton tyrosine kinase, is approved by the US Food and Drug Administration and European Medicines Agency for the treatment of patients with CLL/small lymphocytic lymphoma (SLL), including CLL/SLL with del(17p), and allows for treatment without chemotherapy. In vitro, pharmacological inhibition of B‐cell receptor signalling with ibrutinib results in disruption of key prosurvival signals that induce apoptosis of CLL cells by a TP53‐independent mechanism (Amin et al, 2016). Objective responses are observed in most ibrutinib‐treated CLL patients, and responses appear independent of high‐risk genetic features, including del(17p) (Byrd et al, 2013). In a phase 1b/2 study of patients with CLL, 81% of 36 patients with del(17p) [relapsed/refractory (R/R) in 34 patients] achieved an objective response after ibrutinib treatment (Byrd et al, 2015). The median progression‐free survival (PFS) among the subgroup of patients with R/R del(17p) CLL was approximately 28 months (Byrd et al, 2015), which was substantially longer than the median PFS of approximately 11 months observed with either chemoimmunotherapy with FCR (fludarabine, cyclophosphamide and rituximab) (Fischer et al, 2016) or alemtuzumab (Hillmen et al, 2007) in first‐line patients. Consequently, ibrutinib was granted breakthrough therapy designation for del(17p) CLL and was given the first genotypic‐specific indication for the treatment of CLL patients, del(17p) CLL, in the United States in July 2014, and a similar indication in Europe in October 2014.
Since the initial study, many more patients with del(17p) CLL have been treated with ibrutinib, including in real‐world settings (Mato et al, 2016, UK CLL Forum 2016; Winqvist et al, 2016). To provide a more robust description of outcomes in the prospective trials in this important high‐risk patient population, we conducted a combined analysis of 243 patients with R/R del(17p) CLL who received treatment with single‐agent ibrutinib (420 mg) in 1 of 3 prospective clinical studies (Byrd et al, 2013, 2014, 2015; O'Brien et al, 2014, 2016a). This analysis characterizes disease‐specific and overall survival (OS) outcomes among these patients, and describes the results of exploratory analyses conducted to better identify factors associated with variation in survival of patients with this aggressive subtype of CLL.
Methods
Patient population
Data from patients with R/R del(17p) CLL enrolled on 1 of 3 ibrutinib clinical trials were included. Study designs for the individual trials are described in Table SI. PCYC‐1102 (NCT01105247) (Byrd et al, 2013; O'Brien et al, 2014), with long‐term follow‐up data from the extension study PCYC‐1103 (NCT01109069) (Byrd et al, 2015), was a multi‐institutional, phase 1b/2 study of single‐agent ibrutinib in patients with R/R or treatment‐naïve CLL. This analysis only included patients with R/R CLL from PCYC‐1102/1103. PCYC‐1112 (NCT01578707; RESONATE) was a multi‐institutional, phase 3 study of ibrutinib versus ofatumumab in patients with R/R CLL (Byrd et al, 2014). PCYC‐1117 (NCT01744691; RESONATE‐17) was a multi‐institutional, phase 2 study of single‐agent ibrutinib in patients with R/R CLL or SLL with del(17p) (O'Brien et al, 2016a). In all studies, patients initially received ibrutinib 420 mg once daily on a continuous basis until progressive disease (PD) or unacceptable toxicity. In PCYC‐1102/1103 a minority of patients initially received ibrutinib 840 mg once daily and these patients were not included in this analysis.
An institutional review board approved each protocol at the respective sites. All study procedures were conducted according to the principles of the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice guidelines.
The 17p deletion was assessed at study entry by fluorescence in situ hybridization (FISH) in peripheral blood according to assay‐specific definitions to determine positivity for del(17p). FISH assays were performed locally for patients enrolled on PCYC‐1112, but a central laboratory confirmed del(17p) status in studies PCYC‐1117 and PCYC‐1102/1103. TP53 mutation analysis was not performed for enrolment in these trials. Peripheral blood samples from patients enrolled on PCYC‐1102/1103 were further assessed for CpG mitogen‐stimulated complex karyotype according to methods previously described (O'Brien et al, 2014). Twenty metaphases were completely analysed whenever possible. Karyotypes were described according to the International System for Human Cytogenetic Nomenclature 2009 (Shaffer et al, 2009). A complex karyotype was defined as a karyotype with ≥3 unrelated abnormalities.
Endpoints
Overall response rate (ORR) was defined as the proportion of patients achieving complete response (CR), CR with incomplete bone marrow recovery (CRi), nodular partial response (nPR), partial response (PR), or PR with lymphocytosis (PR‐L), per the International Workshop on CLL (iwCLL) 2008 criteria modified for treatment‐related lymphocytosis (Hallek et al, 2008, 2012). Efficacy endpoints were based on investigator assessments. Other efficacy endpoints included duration of response (DOR) for responders, PFS, OS and rate of sustained haematological improvement. Sustained haematological improvement was defined as an increase in cytopenias that was sustained continuously for ≥56 days without transfusion or growth factors, as measured by the following: an increase in the platelet count or absolute neutrophil count (ANC) from baseline of ≥50% or, for haemoglobin, an increase from baseline of ≥20 g/l. Adverse events (AEs) were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0 (https://www.eortc.be/services/doc/ctc/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf), with the exception of haematological toxicities, which were graded per iwCLL 2008 criteria (Hallek et al, 2008).
Statistical analysis
Descriptive statistics, including means, standard deviations and medians for continuous variables, and proportions for discrete variables, were used to summarize the findings in each of the defined cohorts. Analyses used the all‐treated population with 420 mg of ibrutinib (≥1 dose) in PCYC‐1102/1103 excluding first‐line patients, the intent‐to‐treat population randomized to ibrutinib in PCYC‐1112, and the all‐treated population in PCYC‐1117. The ORR with a 95% confidence interval (CI) was calculated based on exact binomial distribution. The Kaplan–Meier method was used for time‐to‐event analyses. No imputation of missing values was performed. A multivariate analysis examining outcomes in the entire cohort used age (≥65 vs. <65 years), sex (male vs. female), race (white vs. other), prior therapy (≥2 vs. <2), and with versus without each of the following potential risk factor as covariates: advanced Rai stage (III/IV), bulky disease (≥5 cm), del(11q), del(13q), unmutated IGHV, trisomy 12, β2‐microglobulin (>3·5 mg/l), and lactate dehydrogenase (LDH, >250 u/l). In the multivariate analysis, P ≤ 0·05 was considered significant. The cumulative incidence function of PD due to RT and PD due to non‐RT were estimated using nonparametric maximum likelihood methods by treating the 3 subtypes of PFS events (RT, non‐RT and death) as competing risks. All continuous covariates were dichotomized by clinically meaningful cut‐off points.
Results
The baseline characteristics of the 230 patients included in the analysis are summarized in Table 1. Median age was 65 years (range, 30–89 years), with 86 patients (37%) aged ≥70 years. Most patients had advanced Rai stage disease (63%), Eastern Cooperative Oncology Group performance status ≥1 (65%) and bulky disease ≥5 cm (53%). Almost half of the patients (47%) had ≥3 prior lines of therapy.
Table 1.
Baseline patient characteristics
| Characteristic | N = 230 |
|---|---|
| Median age, years (range) | 65 (30–89) |
| ≥70 years, n (%) | 86 (37) |
| Male, n (%) | 150 (65) |
| ECOG performance status, n (%) | |
| 0 | 80 (35) |
| 1 | 149 (65) |
| 2 | 1 (<1) |
| Rai stage III–IV, n (%) | 145 (63) |
| Bulky disease ≥5 cm, n (%) | 123 (53) |
| β2M>3·5 mg/l, n/N (%) | 174/220 (79) |
| Unmutated IGHV, n/N (%) | 149/188 (79) |
| Del(11q), n/N (%)a | 40/226 (18) |
| Del(13q), n/N (%)a | 142/216 (66) |
| Median prior lines of therapy (range) | 2 (1–12) |
| ≥3 prior lines of therapy, n (%) | 108 (47) |
| Type of prior therapies, n (%) | |
| Alkylating agent | 198 (86) |
| Anti‐CD20 | 203 (88) |
| Anti‐CD20 chemoimmunotherapy | 186 (81) |
| Alemtuzumab | 61 (27) |
| Idelalisib | 4 (2) |
| Stem cell transplant | |
| Autologous | 1 (<1) |
| Allogeneic | 2 (1) |
β2M, β2‐microglobulin; ECOG, Eastern Cooperative Oncology Group.
Cytogenetic abnormalities may be co‐occurring.
At the time of analysis, median follow‐up for all patients was 28 months (range, 0·3–59 months): 41 months for PCYC‐1102/03, 31 months for PCYC‐1112 and 28 months for PCYC‐1117. One hundred and eight patients (47%) remained on ibrutinib treatment. All patients in this analysis had received ≥1 dose of ibrutinib. Figure 1 shows the best overall response according to the individual studies. The investigator‐assessed best ORR for all patients was 85% (10% CR/CRi, 3% nPR, 67% PR and 6% PR‐L). With the exception of advanced age (ORR 92% vs. 79% for patients aged <65 and ≥65 years, respectively), ORR was similar regardless of all potential risk factors examined in a multivariate logistic regression model. Median DOR for the responders was 30·7 months (range, 0·03–57·1 months). For patients who achieved CR/CRi (n = 22), an estimated 79% maintained response at 30 months compared with 55% of patients who achieved nPR/PR/PR‐L (n = 174). Sustained haematological improvement was observed in 33 of 47 patients (70%) with baseline ANC ≤ 1·5 × 109/l, in 70 of 114 (61%) with baseline haemoglobin ≤110 g/l, and in 68 of 102 (67%) with baseline platelet count ≤100 × 109/l.
Figure 1.
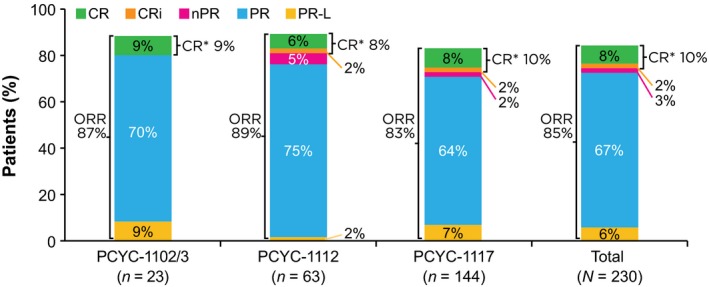
Overall response rates (ORR) as assessed by investigator. Rates are reported for each clinical trial and for the entire study population. CR, complete response; CRi, CR with incomplete bone marrow recovery; PR, partial response; PR‐L, PR with lymphocytosis; nPR, nodular PR. *CR = CR + CRi.
Estimated PFS at 12, 24 and 30 months was 80% (95% CI, 75–85), 65% (95% CI, 58–71) and 57% (95% CI, 50–64), respectively (Fig 2A). The estimated PFS at 30 months for patients with del(17p) in PCYC‐1102/1103, PCYC‐1112 and PCYC‐1117 was 58%, 55% and 57%, respectively. Overall, 74 patients had PD (32%), including 24 (10%) with RT; the cumulative incidence for RT and for progressive CLL was similar during the first year (Figure S1); however, the RT cases occurred predominantly earlier than the CLL progressions. Median time to PD was 182 days for patients with RT, whereas median time to progression for CLL without transformation was longer, at 569 days. Estimated median OS was 59·3 months (range, 0·3–59·3 months), and estimated OS at 12, 24 and 30 months was 85% (95% CI, 80–89), 77% (95% CI, 71–82) and 69% (95% CI, 61–75), respectively (Fig 2B).
Figure 2.
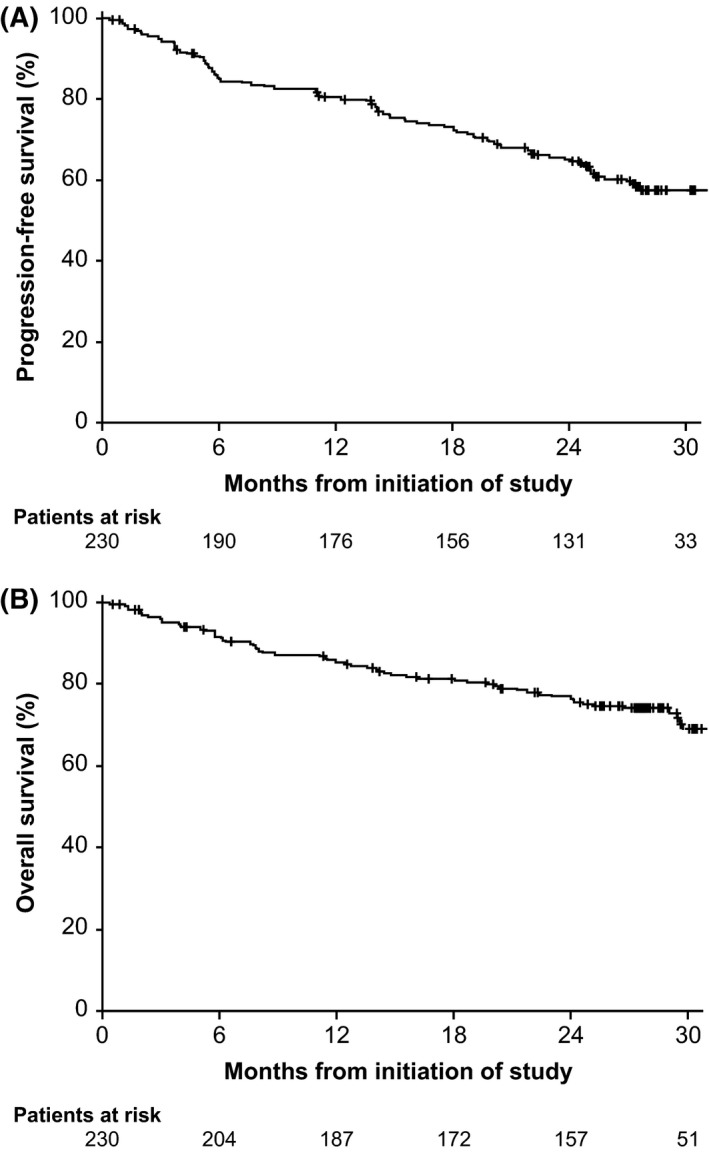
Survival outcomes. (A) Progression‐free survival and (B) overall survival at a median follow‐up of 28 months.
In exploratory analyses for baseline risk factors associated with survival outcomes in the entire population, LDH, number of prior lines of therapy and bulky disease were associated with differences in PFS outcomes, and LDH, bulky disease and β2‐microglobulin were associated with differences in OS outcomes by univariate analysis (Figure S2). In a multivariate Cox regression model, LDH and bulky disease were associated with differences in PFS and OS (P < 0·05).
Because results of previous studies have suggested that complex karyotype is significantly associated with ibrutinib treatment failure (Maddocks et al, 2015), we further examined outcomes among the 21 of 23 del(17p) patients with R/R CLL treated with 420 mg of ibrutinib enrolled on PCYC‐1102/1103 for whom data with stimulated karyotype were available. For patients with (n = 14; 67%) and without (n = 7; 33%) complex karyotype, differences in baseline characteristics included ≥3 prior therapies (86% vs. 43%), del(13q) (36% vs. 86%), high β2‐microglobulin (50% vs. 14%), advanced Rai stage (64% vs. 29%) and baseline cytopenias (79% vs. 43%). The ORR was the same for patients with and without complex karyotype, at 86% for both. Estimated median PFS for patients with complex karyotype was 26 months (95% CI, 18–43) vs. 52 months (95% CI, 1·2–not estimable) for those without complex karyotype, and OS was similarly decreased for the patients with complex karyotype [32 months (95% CI, 19–57) vs. not reached (95% CI, 1·2–not estimable)] (Fig 3).
Figure 3.
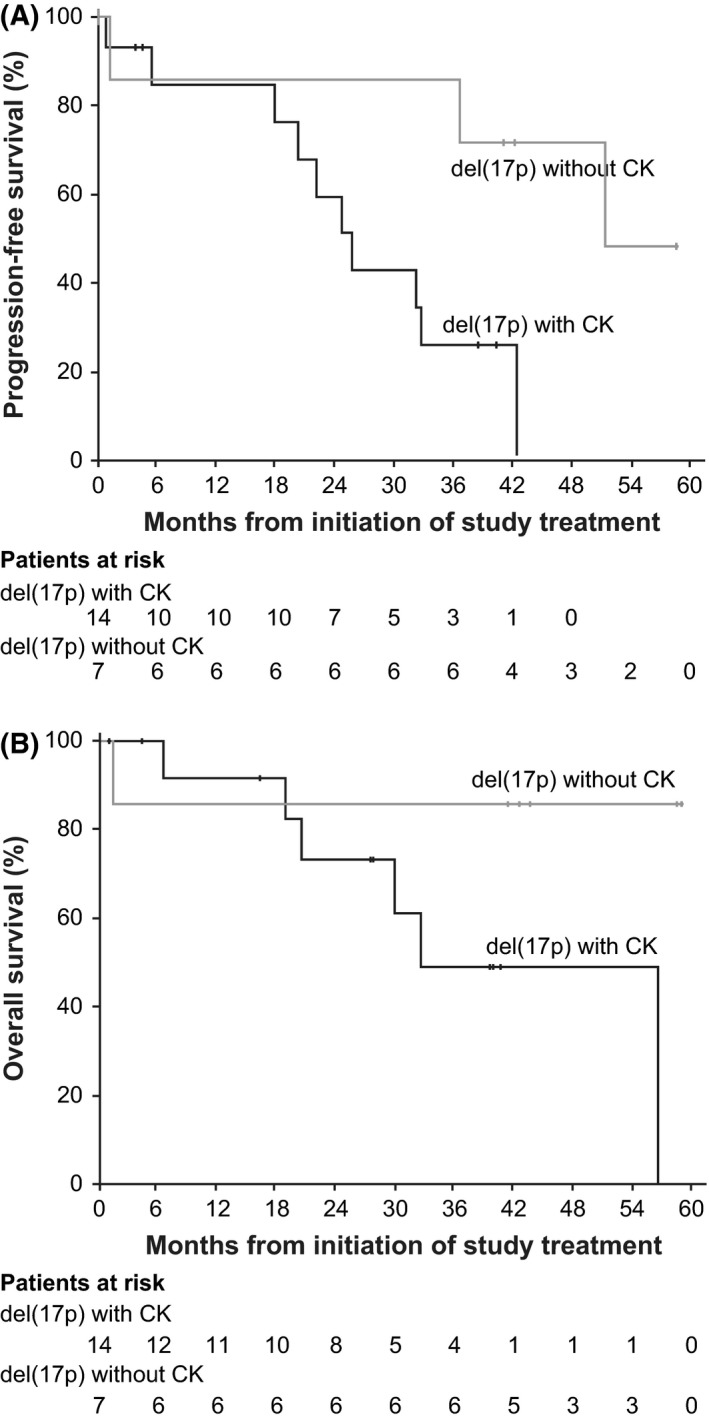
Survival outcomes for patients with (n = 14) or without (n = 7) complex karyotype in the PCYC‐1102/1103 Study. (A) Progression‐free survival and (B) overall survival. del(17p), deletion of chromosome 17p; CK, complex karyotype.
Twenty‐one patients (9%) experienced an AE leading to a dose reduction, with 15 patients experiencing these AEs during the first year of treatment. Treatment discontinuation primarily due to AEs occurred in 34 patients (15%), with over half (n = 20) occurring during the first year of treatment. Grade ≥3 AEs occurring in ≥5% of patients included neutropenia, pneumonia, hypertension, thrombocytopenia, anaemia and urinary tract infection (Table 2). The cumulative rate of grade ≥3 infections was 33% (77/230 patients); frequency of events decreased over time, as did the frequency of other AEs of clinical interest, including cytopenias (Fig 4).
Table 2.
Grade ≥3 treatment‐emergent adverse events in ≥5% of patients (median time on treatment 25 months)
| Adverse event, n (%) | Grade 3 | Grade 4 | Grade 5 | Cumulative grade ≥3 |
|---|---|---|---|---|
| Neutropenia | 12 (5) | 32 (14) | 0 (0) | 44 (19) |
| Pneumonia | 22 (10) | 2 (1) | 6 (3) | 30 (13) |
| Hypertension | 26 (11) | 0 (0) | 0 (0) | 26 (11) |
| Thrombocytopenia | 10 (4) | 10 (4) | 0 (0) | 20 (9) |
| Anaemia | 17 (7) | 2 (1) | 0 (0) | 19 (8) |
| Urinary tract infection | 12 (5) | 0 (0) | 0 (0) | 12 (5) |
Figure 4.
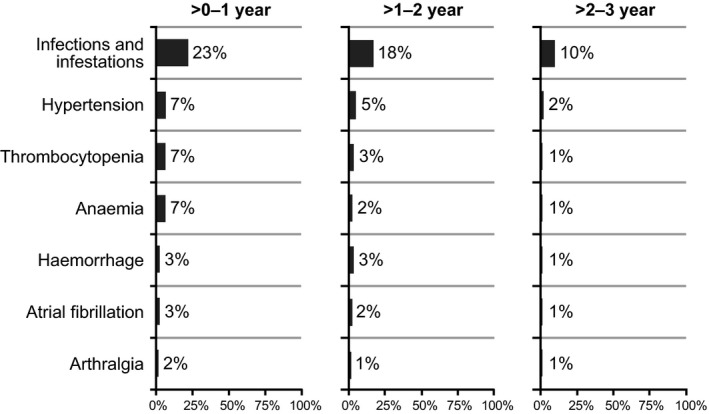
Onset of grade ≥3 adverse events of clinical interest over time.
Discussion
This analysis represents the largest summarized experience, to date, of patients with R/R del(17p) CLL receiving 420 mg of ibrutinib treatment daily within the context of prospective clinical trials. Estimated 30‐month PFS and OS were 57% and 69%, respectively. The survival data reported here not only confirm estimates from earlier reports, but also compare well with reports from other investigator‐initiated prospective and real‐world studies with ibrutinib in similar patient groups with high‐risk CLL. In a phase 2 study of single‐agent ibrutinib, investigators reported an estimated 24‐month PFS rate of 82% among patients with CLL and TP53 abnormalities, of whom 35 of 51 had previously untreated disease (Farooqui et al, 2015). Similarly, in a study combining ibrutinib and rituximab for patients with high‐risk CLL, a subgroup of mostly R/R patients with del(17p) or TP53 mutation had an 18‐month PFS rate that was 72% (Burger et al, 2014). Long‐term results from this study (median follow‐up, 47 months) showed a median PFS of 32 months for this high‐risk patient subgroup (Jain et al, 2016). In a real‐world study of ibrutinib‐treated patients enrolled in a compassionate‐use programme, the estimated 10‐month PFS and OS rates were 71% and 78%, respectively, for 50 patients with del(17p) (Winqvist et al, 2016). In a real‐world study of ibrutinib‐treated patients in a named patient scheme, the estimated 12‐month discontinuation‐free progression and OS rates were 71% and 84%, respectively for 90 patients with del(17p) (UK CLL Forum 2016). Together, the results of these clinical trials and real‐world analyses support the conclusion that ibrutinib is among the most active agents yet developed for the treatment of del(17p) CLL.
The findings reported here are for patients with R/R del(17p) CLL. Results of recent analyses suggest that ibrutinib may be more effective when used earlier in the course of disease. In an analysis of phase 3 data, O'Brien et al (2016b) reported that the 24‐month PFS was 89% for patients receiving ibrutinib after only 1 prior therapy, 80% for patients receiving 2 prior lines of therapy and 69% for patients receiving ≥3 prior lines of therapy. These findings are substantiated in the del(17p) CLL population reported in the present analysis, which demonstrated a 30‐month PFS of 65% for patients with 1 prior line of therapy and 54% for patients with ≥2 prior lines of therapy. Additionally, in a phase 2 study, the rate of CR with ibrutinib was higher in treatment‐naïve versus R/R patients with CLL/SLL and quality of responses improved over time with continued treatment (3‐year follow‐up) (Byrd et al, 2015). These results support the finding reported herein that patients with del(17p) CLL who achieved a CR/CRi (10%, n = 22) appeared more likely to be progression‐free (79%) at 30 months after initial response versus patients who achieved nPR/PR/PR‐L (55%).
Although ibrutinib undoubtedly represents a significant advance in the treatment of patients with CLL and del(17p), it nevertheless appears that many R/R patients with del(17p) will ultimately relapse with ibrutinib therapy (Byrd et al, 2015; Winqvist et al, 2016). There may be a plateau in the rate of RT in patients, suggesting that the risk of developing RT is biologically determined. Factors associated with disease progression among ibrutinib‐treated patients with del(17p) CLL are as yet poorly understood. In the present evaluation of patients with del(17p), by multivariate analysis, high LDH and bulky disease were associated with shorter PFS and OS. Of note, IGHV mutation status was not a significant factor associated with outcomes with ibrutinib therapy in this high‐risk patient population. Additionally, in patients with del(17p) in PCYC‐1102/1103, cytogenetic complexity present at treatment initiation was associated with shorter PFS and OS despite similar rates of initial response; this analysis was limited by baseline imbalances and small sample size. Woyach et al (2014) described characteristic resistance mutations arising during ibrutinib treatment among patients with CLL progression, and a subsequent report detailing outcomes from a large single‐institution cohort of ibrutinib‐treated CLL patients found that the presence of a complex karyotype rather than del(17p) was associated with a 4‐fold increase in the risk for discontinuation due to disease progression during ibrutinib treatment (Maddocks et al, 2015). Findings from subsequent studies also suggest that cytogenetic complexity is a prognostic marker of poorer outcomes in the presence or absence of del(17p) (Thompson et al, 2015; Yu et al, 2017).
Complex karyotype disease has been characterized as a highly mutagenic state (Ouillette et al, 2010), and given our evolving understanding of the molecular events underlying ibrutinib resistance, there is a plausible biological explanation for the observation reported here. Unfortunately, conventional karyotype analysis often proves unsuccessful in CLL and is typically not performed in routine clinical practice (Rogers & Jones, 2014). CpG mitogen‐stimulated methods increase the sensitivity to detect karyotype abnormalities in CLL (Thompson et al, 2015), and our data provide a strong rationale for considering more routine application of this method, particularly among patients with del(17p). The extent of prior exposure to cytotoxic chemotherapy is associated with clonal evolution to genetically high‐risk disease, including del(17p) and cytogenetic complexity (Brejcha et al, 2014; Malcikova et al, 2015), further suggesting that precision therapeutics should be used earlier in the course of disease. Likewise, the likelihood that patients in this high‐risk group with R/R disease may develop resistance to a single‐agent targeted therapy provides a compelling rationale for combination treatment approaches (Woyach et al, 2014). Although combinations with anti‐CD20 monoclonal antibodies and chemoimmunotherapy appear unlikely to significantly improve outcomes in patients with del(17p), including CR rates, over those seen with ibrutinib alone, combinations with other novel agents appear more promising (Sharman et al, 2017). Based on a strong preclinical rationale (Cervantes‐Gomez et al, 2015), ibrutinib is also being studied in combination with venetoclax in patients with CLL with and without del(17p) [NCT02756897 (MD Anderson Cancer Center); NCT02910583 (PCYC‐1142‐CA, CAPTiVATE); NCT02758665 (German CLL Study Group)].
Our findings reinforce earlier observations regarding the safety of ibrutinib treatment in this high‐risk patient population. Ibrutinib was generally well tolerated during long‐term administration, with 58% of patients being treated for longer than 2 years and 47% continuing on ibrutinib treatment at the time of data analysis. Treatment‐limiting AEs (discontinuations or dose reductions) typically occurred early (≤1 year). In general, fewer grade ≥3 AEs occurred after >1 year of ibrutinib therapy (Byrd et al, 2015). Findings from the observational study exploring the aetiology of ibrutinib discontinuation found that each addition of prior therapy increased the likelihood of ibrutinib discontinuation for non‐relapse reasons by nearly 10% (Maddocks et al, 2015). Mature follow‐up data from ongoing studies in previously untreated patients with CLL will be necessary to corroborate this finding, prospectively. Potential limitations of the present study include the retrospective nature of the analysis and that complex karyotype information was used from only 1 study, as karyotyping was not analysed by a central laboratory for the other studies.
The results of the present analysis serve to further support the conclusion that ibrutinib is among the most effective treatments for patients with del(17p) CLL, with 30‐month survival estimates that surpass other available therapies. Further, ibrutinib in this population has a favourable safety profile comparable to that of ibrutinib‐treated patients without del(17p). When administered to heavily pre‐treated patients with del(17p), which represents the majority of patients assessed in our analysis, there remains a clear risk for treatment failure with ibrutinib therapy over time. Cytogenetic complexity continues to emerge as an important marker associated with treatment failure in this group, but continued study will be necessary to understand both the clinical and molecular features driving disease progression and to select patients for studies with ibrutinib in combination with other novel agents.
Authorship contributions
JJ, ADC, DFJ, SMO, and FC developed the concept and design of the paper; JJ, AM, SC, JCB, RRF, PH, AO, CT, SS, WGW, NH, KE, FC, CZ, and SMO collected and assembled the data; JJ, KE, FC, CZ, ADC, DFJ, SMO, and NH analysed and interpreted the data; all authors wrote and gave final approval of the manuscript.
Disclosure of conflicts of interest
JJ: consultancy/advisory role and research funding from Pharmacyclics LLC, an AbbVie Company, AbbVie, and Janssen; AM: consultancy/advisory role with Celgene, Gilead, AbbVie, Janssen, and Pharmacyclics LLC, an AbbVie Company, travel accommodations from TG Therapeutics, research funding from TG Therapeutics, Acerta, DTRM, Gilead, AbbVie, and Janssen; SC: consultancy/advisory role for Janssen, Pharmacyclics LLC, an AbbVie Company, research funding from AbbVie and Pharmacyclics LLC, an AbbVie Company; JCB: research funding from Genentech, Acerta, and Pharmacyclics LLC, an AbbVie Company; RF: honoraria, consultancy/advisory role, speakers' bureau for Pharmacyclics LLC, an AbbVie Company; PH: honoraria, consultancy/advisory role, and research funding from Roche, GSK, Janssen, Gilead, AbbVie, honoraria and research funding from Novartis, Pharmacyclics LLC, an AbbVie Company, research funding from Celgene; AO: honoraria from Gilead and Janssen, research funding from GSK, Janssen, Pharmacyclics LLC, an AbbVie Company, and Gilead; CT: honoraria, consultancy/advisory role, and research funding from Janssen; SS: honoraria and consultancy/advisory role with AbbVie, Amgen, Boehringer‐Ingelheim, Celgene, Genentech, Genzyme, Gilead, GSK, Janssen, Mundipharma, Novartis, Pharmacyclics LLC, an AbbVie Company, and Hoffmann La‐Roche, speakers' bureau and travel accommodations from AbbVie, Janssen, and Pharmacyclics LLC, an AbbVie Company, research funding from AbbVie, Amgen, Boehringer‐Ingelheim, Celgene, Genentech, Genzyme, Gilead, GSK, Janssen, Mundipharma, Novartis, Pharmacyclics LLC, an AbbVie Company, and Hoffman La‐Roche; WGW: honoraria from Sanofi, Genentech/Roche, Pharmacyclics LLC, an AbbVie Company, Celgene, Gilead, GSK/Novartis, Genzyme, Merck, AbbVie, and Emergent, consulting/advisory role for GSK/Novartis, AbbVie, Genentech, Pharmacyclics LLC, an AbbVie Company, Gilead, Emergent, Sanofi, Celgene, Genzyme, and Merck, research funding from GSK/Novartis, AbbVie, Genentech, Karyopharm, Pharmacyclics, Acerta, Gilead, Janssen, Emergent, Juno, and Kite; KE: employment and patents/royalties/other intellectual property from Pharmacyclics LLC, an AbbVie Company, equity ownership with AbbVie; FC: employment and leadership with Pharmacyclics LLC, an AbbVie Company, equity ownership with AbbVie; CZ, ADC, DFJ: employment with Pharmacyclics, LLC, an AbbVie Company, equity ownership with AbbVie; SOB: consultancy/advisory role with Amgen, Astellas, Celgene GSK, Janssen, Aptose Biosciences, Vaniam Group, AbbVie, Sunesis, Alexion, Gilead, Pharmacyclics LLC, an AbbVie Company, TG Therapeutics, and Pfizer, research funding from ProNai, Regeneron, Acerta, Gilead Pharmacyclics, TG Therapeutics, and Pfizer, NH: no relevant conflicts of interest to disclose.
Supporting information
Table SI. Patients with del(17p) included in the current analysis by clinical study.
Figure S1. Cumulative incidence of Richter transformation and other (non‐Richter) progression.
Figure S2. Progression‐free survival and overall survival outcomes by baseline characteristics (univariate analysis) (A) lactate dehydrogenase, (B) prior therapies, (C) bulky disease, and (D) β2‐microglobulin.
Acknowledgements
We thank the patients who participated in these studies, their supporters, and the investigators and clinical research staff from the study centres. This manuscript was developed with editorial support from Stacey Rose, PhD, and funded by Pharmacyclics LLC, an AbbVie Company.
Presented in part at the 21st Annual Meeting of the European Haematology Association, Copenhagen, Denmark, June 9‐12, 2016.
References
- Amin, N. , Balasubramanian, S. , Saiya‐Cork, K. , Shedden, K. , Hu, N. & Malek, S.N. (2016) Cell‐intrinsic determinants of ibrutinib‐induced apoptosis in chronic lymphocytic leukemia. Clinical Cancer Research, 23, 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badoux, X.C. , Keating, M.J. , Wang, X. , O'Brien, S.M. , Ferrajoli, A. , Faderl, S. , Burger, J. , Koller, C. , Lerner, S. , Kantarjian, H. & Wierda, W.G. (2011) Cyclophosphamide, fludarabine, alemtuzumab, and rituximab as salvage therapy for heavily pretreated patients with chronic lymphocytic leukemia. Blood, 118, 2085–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejcha, M. , Stoklasova, M. , Brychtova, Y. , Panovska, A. , Stepanovska, K. , Vankova, G. , Plevova, K. , Oltova, A. , Horka, K. , Pospisilova, S. , Mayer, J. & Doubek, M. (2014) Clonal evolution in chronic lymphocytic leukemia detected by fluorescence in situ hybridization and conventional cytogenetics after stimulation with CpG oligonucleotides and interleukin‐2: a prospective analysis. Leukemia Research, 38, 170–175. [DOI] [PubMed] [Google Scholar]
- Burger, J.A. , Keating, M.J. , Wierda, W.G. , Hartmann, E. , Hoellenriegel, J. , Rosin, N.Y. , de Weerdt, I. , Jeyakumar, G. , Ferrajoli, A. , Cardenas‐Turanzas, M. , Lerner, S. , Jorgensen, J.L. , Nogueras‐Gonzalez, G.M. , Zacharian, G. , Huang, X. , Kantarjian, H. , Garg, N. , Rosenwald, A. & O'Brien, S. (2014) Safety and activity of ibrutinib plus rituximab for patients with high‐risk chronic lymphocytic leukaemia: a single‐arm, phase 2 study. The Lancet Oncology, 15, 1090–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd, J.C. , Furman, R.R. , Coutre, S.E. , Flinn, I.W. , Burger, J.A. , Blum, K.A. , Grant, B. , Sharman, J.P. , Coleman, M. , Wierda, W.G. , Jones, J.A. , Zhao, W. , Heerema, N.A. , Johnson, A.J. , Sukbuntherng, J. , Chang, B.Y. , Clow, F. , Hedrick, E. , Buggy, J.J. , James, D.F. & O'Brien, S. (2013) Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. New England Journal of Medicine, 369, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd, J.C. , Brown, J.R. , O'Brien, S. , Barrientos, J.C. , Kay, N.E. , Reddy, N.M. , Coutre, S. , Tam, C.S. , Mulligan, S.P. , Jaeger, U. , Devereux, S. , Barr, P.M. , Furman, R.R. , Kipps, T.J. , Cymbalista, F. , Pocock, C. , Thornton, P. , Caligaris‐Cappio, F. , Robak, T. , Delgado, J. , Schuster, S.J. , Montillo, M. , Schuh, A. , de Vos, S. , Gill, D. , Bloor, A. , Dearden, C. , Moreno, C. , Jones, J.J. , Chu, A.D. , Fardis, M. , McGreivy, J. , Clow, F. , James, D.F. & Hillmen, P. (2014) Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. New England Journal of Medicine, 371, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd, J.C. , Furman, R.R. , Coutre, S.E. , Burger, J.A. , Blum, K.A. , Coleman, M. , Wierda, W.G. , Jones, J.A. , Zhao, W. , Heerema, N.A. , Johnson, A.J. , Shaw, Y. , Bilotti, E. , Zhou, C. , James, D.F. & O'Brien, S. (2015) Three‐year follow‐up of treatment‐naive and previously treated patients with CLL and SLL receiving single‐agent ibrutinib. Blood, 125, 2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes‐Gomez, F. , Lamothe, B. , Woyach, J. , Wierda, W. , Keating, M. , Balakrishnan, K. & Gandhi, V. (2015) Pharmacological and protein profiling suggests venetoclax (ABT‐199) as optimal partner with ibrutinib in chronic lymphocytic leukemia. Clinical Cancer Research, 21, 3705–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqui, M.Z. , Valdez, J. , Martyr, S. , Aue, G. , Saba, N. , Niemann, C.U. , Herman, S.E. , Tian, X. , Marti, G. , Soto, S. , Hughes, T.E. , Jones, J. , Lipsky, A. , Pittaluga, S. , Stetler‐Stevenson, M. , Yuan, C. , Lee, Y.S. , Pedersen, L.B. , Geisler, C.H. , Calvo, K.R. , Arthur, D.C. , Maric, I. , Childs, R. , Young, N.S. & Wiestner, A. (2015) Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single‐arm trial. The Lancet. Oncology, 16, 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, K. , Bahlo, J. , Fink, A.M. , Goede, V. , Herling, C.D. , Cramer, P. , Langerbeins, P. , von Tresckow, J. , Engelke, A. , Maurer, C. , Kovacs, G. , Herling, M. , Tausch, E. , Kreuzer, K.A. , Eichhorst, B. , Bottcher, S. , Seymour, J.F. , Ghia, P. , Marlton, P. , Kneba, M. , Wendtner, C.M. , Dohner, H. , Stilgenbauer, S. & Hallek, M. (2016) Long‐term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood, 127, 208–215. [DOI] [PubMed] [Google Scholar]
- Hallek, M. , Cheson, B.D. , Catovsky, D. , Caligaris‐Cappio, F. , Dighiero, G. , Dohner, H. , Hillmen, P. , Keating, M.J. , Montserrat, E. , Rai, K.R. & Kipps, T.J. ; International Workshop on Chronic Lymphocytic Leukemia . (2008) Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute‐Working Group 1996 guidelines. Blood, 111, 5446–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallek, M. , Fischer, K. , Fingerle‐Rowson, G. , Fink, A.M. , Busch, R. , Mayer, J. , Hensel, M. , Hopfinger, G. , Hess, G. , von Grunhagen, U. , Bergmann, M. , Catalano, J. , Zinzani, P.L. , Caligaris‐Cappio, F. , Seymour, J.F. , Berrebi, A. , Jager, U. , Cazin, B. , Trneny, M. , Westermann, A. , Wendtner, C.M. , Eichhorst, B.F. , Staib, P. , Buhler, A. , Winkler, D. , Zenz, T. , Bottcher, S. , Ritgen, M. , Mendila, M. , Kneba, M. , Dohner, H. & Stilgenbauer, S. , International Group of Investigators; German Chronic Lymphocytic Leukaemia Study Group (2010) Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open‐label, phase 3 trial. Lancet, 376, 1164–1174. [DOI] [PubMed] [Google Scholar]
- Hallek, M. , Cheson, B.D. , Catovsky, D. , Caligaris‐Cappio, F. , Dighiero, G. , Döhner, H. , Hillmen, P. , Keating, M.J. , Montserrat, E. , Rai, K.R. & Kipps, T.J. (2012) Response assessments in chronic lymphocytic leukemia treated with novel agents causing an increase in peripheral blood lymphocytes [e‐letter, comment on Halleck et al 2008, Blood, 111 5446‐5456]. Blood, 111 Available at: http://www.bloodjournal.org/content/111/12/5446/tab-e-letters. [Google Scholar]
- Hillmen, P. , Skotnicki, A.B. , Robak, T. , Jaksic, B. , Dmoszynska, A. , Wu, J. , Sirard, C. & Mayer, J. (2007) Alemtuzumab compared with chlorambucil as first‐line therapy for chronic lymphocytic leukemia. Journal of Clinical Oncology, 25, 5616–5623. [DOI] [PubMed] [Google Scholar]
- Jain, P. , Keating, M.J. , Wierda, W. , Campbell, M. , Thompson, P.A. , Ferrajoli, A. , Estrov, Z. , Kantarjian, H. , O'Brien, S. & Burger, J. (2016) Long term follow up of treatment with ibrutinib and rituximab in patients with high‐risk chronic lymphocytic leukemia. Clinical Cancer Research, 23, 2154–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliusson, G. , Oscier, D.G. , Fitchett, M. , Ross, F.M. , Stockdill, G. , Mackie, M.J. , Parker, A.C. , Castoldi, G.L. , Guneo, A. , Knuutila, S. , Elonen, E. & Gahrton, G. (1990) Prognostic subgroups in B‐cell chronic lymphocytic leukemia defined by specific chromosomal abnormalities. New England Journal of Medicine, 323, 720–724. [DOI] [PubMed] [Google Scholar]
- Maddocks, K.J. , Ruppert, A.S. , Lozanski, G. , Heerema, N.A. , Zhao, W. , Abruzzo, L. , Lozanski, A. , Davis, M. , Gordon, A. , Smith, L.L. , Mantel, R. , Jones, J.A. , Flynn, J.M. , Jaglowski, S.M. , Andritsos, L.A. , Awan, F. , Blum, K.A. , Grever, M.R. , Johnson, A.J. , Byrd, J.C. & Woyach, J.A. (2015) Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncology, 1, 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcikova, J. , Stano‐Kozubik, K. , Tichy, B. , Kantorova, B. , Pavlova, S. , Tom, N. , Radova, L. , Smardova, J. , Pardy, F. , Doubek, M. , Brychtova, Y. , Mraz, M. , Plevova, K. , Diviskova, E. , Oltova, A. , Mayer, J. , Pospisilova, S. & Trbusek, M. (2015) Detailed analysis of therapy‐driven clonal evolution of TP53 mutations in chronic lymphocytic leukemia. Leukemia, 29, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mato, A. , Lamanna, N. , Ujjani, C.S. , Brander, D.M. , Hill, B.T. , Howlett, C. , Skarbnik, A.P. , Cheson, B.D. , Zent, C. , Pu, J.J. , Kiselev, P. , Bachow, S.H. , Winter, A.M. , Cruz, A.‐L. , Claxton, D.F. , Daniel, C. , Isaack, K. , Kennard, K.H. , Timlin, C. , Yacur, M. , Fanning, M. , Strelec, L.E. , Landsburg, D.J. , Nasta, S.D. , Schuster, S.J. , Porter, D.L. , Nabhan, C. & Barr, P.M. (2016) Toxicities and outcomes of ibrutinib‐treated patients in the United States: large retrospective analysis of 621 real world patients. Blood, 128, 3222. [Google Scholar]
- O'Brien, S. , Furman, R.R. , Coutre, S.E. , Sharman, J.P. , Burger, J.A. , Blum, K.A. , Grant, B. , Richards, D.A. , Coleman, M. , Wierda, W.G. , Jones, J.A. , Zhao, W. , Heerema, N.A. , Johnson, A.J. , Izumi, R. , Hamdy, A. , Chang, B.Y. , Graef, T. , Clow, F. , Buggy, J.J. , James, D.F. & Byrd, J.C. (2014) Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open‐label, multicentre, phase 1b/2 trial. The Lancet Oncology, 15, 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien, S. , Jones, J.A. , Coutre, S.E. , Mato, A.R. , Hillmen, P. , Tam, C. , Osterborg, A. , Siddiqi, T. , Thirman, M.J. , Furman, R.R. , Ilhan, O. , Keating, M.J. , Call, T.G. , Brown, J.R. , Stevens‐Brogan, M. , Li, Y. , Clow, F. , James, D.F. , Chu, A.D. , Hallek, M. & Stilgenbauer, S. (2016a) Ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia with 17p deletion (RESONATE‐17): a phase 2, open‐label, multicentre study. The Lancet Oncology, 17, 1409–1418. [DOI] [PubMed] [Google Scholar]
- O'Brien, S.M. , Byrd, J.C. , Hillmen, P. , Coutre, S. , Brown, J.R. , Barr, P.M. , Barrientos, J.C. , Devereux, S. , Robak, T. , Reddy, N.M. , Kipps, T.J. , Tedeschi, A. , Cymbalista, F. , Ghia, P. , Chang, S. , Ninomoto, J. , James, D.F. & Burger, J.A. (2016b) Outcomes with ibrutinib by line of therapy in patients with CLL: analyses from phase III data. Journal of Clinical Oncology, 34, 7520. [Google Scholar]
- Ouillette, P. , Fossum, S. , Parkin, B. , Ding, L. , Bockenstedt, P. , Al‐Zoubi, A. , Shedden, K. & Malek, S.N. (2010) Aggressive chronic lymphocytic leukemia with elevated genomic complexity is associated with multiple gene defects in the response to DNA double‐strand breaks. Clinical Cancer Research, 16, 835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, K. & Jones, J. (2014) Evidence‐based management of newly diagnosed chronic lymphocytic leukemia. Journal of Clinical Outcomes Management, 21, 177–188. [Google Scholar]
- Shaffer, L.G. , Slovak, M.L. & Campbell, L.J. (2009) An International System for Human Cytogenetic Nomenclature. S. Karger AG, Basel, Switzerland. [Google Scholar]
- Sharman, J.P. , Farber, C.M. , Mahadevan, D. , Schreeder, M.T. , Brooks, H.D. , Kolibaba, K.S. , Fanning, S. , Klein, L. , Greenwald, D.R. , Sportelli, P. , Miskin, H.P. , Weiss, M.S. & Burke, J.M. (2017) Ublituximab (TG‐1101), a novel glycoengineered anti‐CD20 antibody, in combination with ibrutinib is safe and highly active in patients with relapsed and/or refractory chronic lymphocytic leukaemia: results of a phase 2 trial. British Journal of Haematology, 176, 412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam, C.S. & Stilgenbauer, S. (2015) How best to manage patients with chronic lymphocytic leukemia with 17p deletion and/or TP53 mutation? Leukemia & Lymphoma, 56, 587–593. [DOI] [PubMed] [Google Scholar]
- Thompson, P.A. , O'Brien, S.M. , Wierda, W.G. , Ferrajoli, A. , Stingo, F. , Smith, S.C. , Burger, J.A. , Estrov, Z. , Jain, N. , Kantarjian, H.M. & Keating, M.J. (2015) Complex karyotype is a stronger predictor than del(17p) for an inferior outcome in relapsed or refractory chronic lymphocytic leukemia patients treated with ibrutinib‐based regimens. Cancer, 121, 3612–3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UK CLL Forum . (2016) Ibrutinib for relapsed/refractory CLL: a UK and Ireland analysis of outcomes in 315 patients. Haematologica, 101, 1563–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winqvist, M. , Asklid, A. , Andersson, P.O. , Karlsson, K. , Karlsson, C. , Lauri, B. , Lundin, J. , Mattsson, M. , Norin, S. , Standstedt, A. , Hansson, L. & Osterborg, A. (2016) Real‐world results of ibrutinib in patients with relapsed or refractory chronic lymphocytic leukemia: data from 95 consecutive patients treated in a compassionate use program. A study from the Swedish Chronic Lymphocytic Leukemia Group. Haematologica, 101, 1573–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woyach, J.A. , Furman, R.R. , Liu, T.M. , Ozer, H.G. , Zapatka, M. , Ruppert, A.S. , Xue, L. , Li, D.H. , Steggerda, S.M. , Versele, M. , Dave, S.S. , Zhang, J. , Yilmaz, A.S. , Jaglowski, S.M. , Blum, K.A. , Lozanski, A. , Lozanski, G. , James, D.F. , Barrientos, J.C. , Lichter, P. , Stilgenbauer, S. , Buggy, J.J. , Chang, B.Y. , Johnson, A.J. & Byrd, J.C. (2014) Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. New England Journal of Medicine, 370, 2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, L. , Kim, H. , Kasar, S.N. , Benien, P. , Du, W. , Hoang, K. , Aw, A. , Tesar, B. , Improgo, R. , Fernandes, S. , Radhakrishnan, S. , Klitgaard, J.L. , Lee, C. , Getz, G. , Setlur, S.R. & Brown, J.R. (2017) Survival of del17p CLL depends on genomic complexity and somatic mutation. Clinical Cancer Research, 23, 735–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenz, T. , Eichhorst, B. , Busch, R. , Denzel, T. , Habe, S. , Winkler, D. , Buhler, A. , Edelmann, J. , Bergmann, M. , Hopfinger, G. , Hensel, M. , Hallek, M. , Dohner, H. & Stilgenbauer, S. (2010) TP53 mutation and survival in chronic lymphocytic leukemia. Journal of Clinical Oncology, 28, 4473–4479. [DOI] [PubMed] [Google Scholar]
- Zenz, T. , Gribben, J.G. , Hallek, M. , Dohner, H. , Keating, M.J. & Stilgenbauer, S. (2012) Risk categories and refractory CLL in the era of chemoimmunotherapy. Blood, 119, 4101–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table SI. Patients with del(17p) included in the current analysis by clinical study.
Figure S1. Cumulative incidence of Richter transformation and other (non‐Richter) progression.
Figure S2. Progression‐free survival and overall survival outcomes by baseline characteristics (univariate analysis) (A) lactate dehydrogenase, (B) prior therapies, (C) bulky disease, and (D) β2‐microglobulin.