

Summary
Phosphatidylserine (PS) exposure increases as red cells age, and is an important signal for the removal of senescent cells from the circulation. PS exposure is elevated in red cells from sickle cell anaemia (SCA) patients and is thought to enhance haemolysis and vaso‐occlusion. Although precise conditions leading to its externalisation are unclear, high intracellular Ca2+ has been implicated. Red cells from SCA patients are also exposed to an increased oxidative challenge, and we postulated that this stimulates PS exposure, through increased Ca2+ levels. We tested four different ways of generating oxidative stress: hypoxanthine and xanthine oxidase, phenazine methosulphate, nitrite and tert‐butyl hydroperoxide, together with thiol modification with N‐ethylmaleimide (NEM), dithiothreitol and hypochlorous acid (HOCl), in red cells permeabilised to Ca2+ using bromo‐A23187. Unexpectedly, our findings showed that the four oxidants significantly reduced Ca2+‐induced PS exposure (by 40–60%) with no appreciable effect on Ca2+ affinity. By contrast, NEM markedly increased PS exposure (by about 400%) and slightly but significantly increased the affinity for Ca2+. Dithiothreitol modestly reduced PS exposure (by 25%) and HOCl had no effect. These findings emphasise the importance of thiol modification for PS exposure in sickle cells but suggest that increased oxidant stress alone is not important.
Keywords: sickle cell anaemia, oxidants, thiols, calcium, phosphatidylserine exposure
Sickle cell anaemia (SCA) is one of the commonest severe inherited disorders affecting millions of people worldwide, including some 12 000–15 000 in the UK (Piel et al, 2013). Although the aetiology has been known for over 60 years, the condition remains refractory to effective therapy, which is largely supportive. Whilst hydroxycarbamide has been identified as useful for patients with more severe symptoms, its use is not without complications and more effective treatments are keenly sought. Lack of a detailed understanding of the pathogenesis of SCA represents a major impediment to progress. Occlusion of small blood vessels, tissue ischaemia and organ damage, as well as the chronic anaemia, are important features of the condition. Through its pro‐thrombotic and pro‐phagocytic activities, red cell phosphatidylserine (PS) may participate in vascular occlusion and destruction of red cells (Hebbel, 1991; Boas et al, 1998; Setty et al, 2002; Steffen et al, 2011). This paper concerns the mechanism of red cell PS exposure and the role of oxidative damage in sickle cell disease. Understanding this aspect has clear clinical implications, with increased understanding of triggers for the the vaso‐occlusive and haemolytic complications in SCA. In addition, if the mechanisms leading to PS exposure are clarified, findings may suggest potential chemotherapeutic measures to reduce PS exposure and ameliorate some of the clinical complications of SCA.
Phosphatidylserine is usually confined to the inner leaflet of the lipid bilayer in red cell membranes (Kuypers, 1998; Haest, 2003). By analogy to apoptosis in nucleated cells, the process of PS exposure in red cells and their subsequent removal from the circulation by macrophages, has been termed eryptosis (Lang et al, 2006). In SCA, a high, but variable proportion of red cells shows PS exposure (Tait & Gibson, 1994; Kuypers et al, 1996; de Jong et al, 2001; Weiss et al, 2012), which further increases upon deoxygenation, haemoglobin polymerisation and the sickling shape change (Lubin et al, 1981; Blumenfeld et al, 1991; Weiss et al, 2012; Cytlak et al, 2013). This red cell lipid scrambling may therefore participate in the pathogenesis of SCA, and contribute to both the chronic anaemia and the acute ischaemic episodes characteristic of the condition (Rees et al, 2010), and is thus central to the clinical progression of the disease.
In mature red cells, two transport processes predominate in the maintenance of PS asymmetry: an ATP‐dependent aminophospholipid translocase (or “flippase”), which moves PS from the outer to the inner leaflet of the membrane, and a Ca2+‐dependent scrambling process (or “scramblase”), which moves PS in either direction (Haest, 2003; Bevers & Williamson, 2010). Rapid PS exposure in mature red cells requires activation of the scramblase together with inhibition of the flippase (Haest, 2003; Barber et al, 2009). Elevation of intracellular Ca2+ can elicit both these processes (Williamson et al, 1992; Basse et al, 1996; Woon et al, 1999; Haest, 2003; Bevers & Williamson, 2010). Flippase inhibition is generally considered to be more sensitive to Ca2+ (Bitbol et al, 1987; Basse et al, 1996; Kamp et al, 2001; Bevers & Williamson, 2010) although more recent work suggests that, in fact, lipid scrambling is activated at similar Ca2+ concentrations to flippase inhibition (Weiss et al, 2012; Cytlak et al, 2013). Nevertheless, our understanding of the conditions and mechanisms of PS exposure in red cells from SCA patients remains incomplete.
Other mechanisms may also participate in PS exposure. Thus, recently, the expulsion of membrane‐bound organelles by autophagy during maturation of erythrocyte precursors has been shown to result in punctuate externalised PS localised to discrete patches (Mankelow et al, 2015). Furthermore, PS may be expelled as microvesicles. The involvement of these particles in the pathogenesis of SCA and other conditions has been widely postulated (Piccin et al, 2007, 2015a, 2017; Nebor et al, 2014).
Red cells from patients with SCA contain the abnormal haemoglobin, HbS, which is able to polymerise upon deoxygenation and thereby cause cell sickling. The subsequent alterations in red cell morphology and rheology have critical deleterious consequences for red cell function. Sickle red cells also have an abnormally high cation permeability involving three main transport processes (Lew & Bookchin, 2005): the KCl cotransporter, the Ca2+‐activated K+ channel (or Gárdos channel) and an ill‐defined cation conductance (sometimes termed Psickle – Lew et al, 1997). These transporters combine to mediate solute loss with water following osmotically. The consequent elevation of red cell HbS concentration, [HbS], has the effect of markedly reducing the lag time to HbS polymerisation following deoxygenation (Eaton & Hofrichter, 1987). Psickle is activated by deoxygenation, HbS polymerisation and red cell shape change (Mohandas et al, 1986). Psickle‐mediated Ca2+ entry has an established role in activation of the Gárdos channel and may therefore increase dehydration (Rhoda et al, 1990; Lew et al, 1997). The rise in intracellular Ca2+, [Ca2+], may also play a role in lipid scrambling. Previous findings are consistent with deoxygenation‐induced Ca2+ entry, predominantly via Psickle, contributing to elevation of intracellular Ca2+ levels and leading to increased PS exposure (Weiss et al, 2012; Cytlak et al, 2013). Low micromolar levels of Ca2+ are required (Weiss et al, 2012; Cytlak et al, 2013), which although representing a higher Ca2+ affinity for scrambling than hitherto thought, still represent relatively high levels, even for deoxygenated sickle cells (Etzion et al, 1993).
Oxidative damage has long been associated with PS exposure in red cells (Jain & Shohet, 1984; Jain & Williams, 1985; Kuypers, 1998; Lang et al, 2002, 2012). This is particularly significant in diseases such as SCA in which vascular oxidative stress is increased (Rice‐Evans et al, 1986) with accumulations of highly reactive oxygen species (ROS) including the superoxide anion, hydrogen peroxide and the hydroxyl radical (Sies, 1997; Chirico & Pialoux, 2012; Voskou et al, 2015). Myeloperoxidase released from activated neutrophils may also add to oxidant challenge in SCA, through production of hypochlorous acid (HOCl) from hydrogen peroxide (Vissers et al, 1994; Mutze et al, 2003; Zhang et al, 2013). Thiol oxidation has also been associated with both inhibition of the flippase and stimulation of the scramblase (Morrot et al, 1989; Connor & Schroit, 1990; Devaux & Zachowski, 1994; Martin & Jesty, 1995; de Jong & Kuypers, 2006). In addition, the normal antioxidant capacity of the red cell is compromised in SCA patients, with low availability of antioxidant enzymes, reduced levels of glutathione (GSH) and also of non‐enzymatic antioxidants, such as vitamins C and E (Silva et al, 2013). Oxidative damage has also been associated with deleterious effects on red cell cation balance. It causes inhibition of the plasma membrane Ca2+ pump (Shalev et al, 1981; Zaidi et al, 2003), which maintains intracellular Ca2+ concentrations at low levels, perhaps via interaction with calmodulin (Gao et al, 2001), and also has marked effects on red cell potassium permeability (Gibson & Muzyamba, 2003a,2003b). A randomized controlled trial has also recently shown that l‐glutamine, believed to act predominantly as an antioxidant, reduced the frequency of acute pain in SCA (Niihara et al, 2016). Taken together, these observations are indicative of an important role of oxidative stress in pathogenesis of SCA.
We therefore postulated that oxidative challenge in SCA patients may increase red cell PS scrambling and alter the Ca2+ affinity of the process. We investigated the interaction between Ca2+ and oxidant challenge in red cells from SCA patients. Oxidative stress was provided by extracellular and intracellular superoxide anion generating systems, nitrite and tert‐butyl hydroperoxide. The effect of thiol modifications were also investigated using N‐ethylmaleimide (NEM), dithiothreitol (DTT) and HOCl.
Materials and methods
Sample collection and handling
The study was approved by the National Research Ethics Committee (reference 16/LO/1309). All research was conducted with ethical approval and in accordance with the Helsinki Declaration of 1975, as revised in 2008. Consented blood samples were taken from children homozygous for SCA (HbSS), into EDTA. The study involved 37 patients (23 males) with an average age of 11·3 ± 3·8 years. Nineteen patients were receiving hydroxycarbamide therapy and eighteen were not. Mean % HbF levels and reticulocyte counts were 10·2 ± 5·1 and 13·2 ± 5·1, respectively (all means ± standard deviation [SD], n = 37).
Chemicals, solutions, red cell preparation and full methology
The following is a brief description of the methods used. Full details of chemicals used, solutions, red cell preparation and methology are given in the Appendix S1.
Oxidant challenge and thiol modifications
Extracellular superoxide anion (SOA) and hydrogen peroxide were generated by incubation with mixtures of hypoxanthine (HO; 2 mmol/l) and xanthine oxidase (XO) at concentrations up to 0·1 U/ml, a manoeuvre that is well established to provide an oxidative challenge to red cells (Baskurt et al, 1998; Rogers et al, 2009). Using data from pilot experiments, most work was carried out with a [XO] of 0·015 U/ml. Phenazine methosulphate (PMS, 0·01–0·4 mmol/l) was used to generate intracellular superoxide anion. Nitrite (NO2) (to generate methaemoglobin) and tert‐butyl hydroperoxide (tBHP) (to generate peroxyl and alkoxyl derivatives) were used at concentrations of 1–20 mmol/l and 0·05–1·0 mmol/l, respectively. HOCl was prepared immediately before experiments and used at a final concentration of 0·001–1 mmol/l. Red cells were incubated with oxidants for 30 min at 37°C prior to measuring PS. The thiol modifiers NEM and DTT were used at concentrations of 1 mmol/l and 0·25 mmol/l and red cells were pre‐incubated for 30 min at 37°C, followed by 30 min at 37°C without or with oxidants. DTT was excluded from the second incubation step to prevent red cell lysis.
Measurement of red cell oxidative stress and red cell morphology
To measure intracellular oxidative load, red cells were first loaded with CM‐H2DCF‐DA (100 μmol/l) in the dark (for 30 min at 37°C) and washed twice. On permeation into red cells, this fluorophore is hydrolysed to the non‐fluorescent di‐hydro compound which, in the presence of reactive oxygen species (ROS), is oxidized to highly fluorescent CM‐H2dichlorofluorescein (CM‐H2‐DCF). CM‐H2DCF fluorescence was measured in the FL1 channel of a BD Accuri C6 flow cytometer (Becton Dickinson, Oxford, UK). Median FL1 fluorescence was used as an indication of the magnitude of intracellular oxidative load. For each fluorescence measurement, 10 000 events were gated. To examine red cell morphology, cells were fixed through addition of glutaraldehyde (0·3%) and examined under light microscopy, typically examining several hundred cells.
Measurement of externalised PS using fluorescent lactadherin fluorescein isothiocynate (LA‐FITC)
Accessible PS was labelled with the fluorescent PS marker, LA‐FITC. Usually this marker can only gain access to exposed PS on the outer leaflet of the membrane bilayer, unless membrane integrity has been compromised. To promote lipid scrambling, red cells were incubated with the Ca2+ ionophore bromo‐A23187 (6 μmol/l) to permeabilise red cells to Ca2+ at various [Ca2+]os clamped using EDTA (2 mmol/l). LA‐FITC was detected in the FL1 channel of a BD Accuri C6 flow cytometer using logarithmic gain (as for CM‐H2‐DCF fluorescence). For each measurement, 10 000 events were gated. XO, PMS and tBHP all showed various degrees of autofluorescence in unlabelled red cells in the absence of any fluorophore. It was therefore critical to choose appropriate concentrations of these oxidants that elevated intracellular ROS levels whilst keeping autofluorescence to manageable values. In all cases, compensation for the fluorescent overspill was carefully set using oxidant‐treated red cells, unlabelled with LA‐FITC. NO2 showed no significant autofluorescence in the FL1 channel.
Measurement of externalised PS using a prothrombinase assay
Phosphatidylserine exposure was also assessed by generating thrombin using a prothrombinase assay, following the method of Bevers et al (1982). Factor Va and Factor Xa (0·5 and 0·25 U/ml final concentration, respectively) were added, and after 2 min thrombin formation was initiated by adding prothrombin (1·1 μmol/l final concentration). After 2 and 5 min, red cell aliquots were removed and thrombin formation stopped by the addition of EDTA. Thrombin levels were then measured by adding the chromogenic substrate ThrombinChrom (0·125 mmol/l final concentration) with absorbance measured at 415 nm in an iMark microplate reader (Bio‐Rad, Hemel Hempstead, UK). In intact red cells, this thrombin assay measures only externalised PS. Additional aliquots of red cells were also lysed hypotonically through the addition of water, after which the prothrombinase assay was repeated. In lysed red cells, thrombin formation will be initiated by PS on both the inner leaflet of the membrane as well as externalised PS and thus gives a measure of total red cell PS. The assay will therefore indicate whether PS has been lost from the red cells, for example as microvesicles. Although more complicated to carry out, this prothrombinase assay has the advantage of being immune to any problems with autofluorescence or setting of positive gates in the flow cytometer.
Measurement of red cell membrane integrity
This assay used a fluorescently labelled immunoglobulin against Hb (Alexa Fluor 647 measured in FL4 of FACS) to indicate membrane integrity. Immunoglobin (molecular weight 15 kDa) cannot cross the red cell membrane unless its permeability barrier has been compromised, therefore labelling was taken as an indication of loss of membrane integrity. If this was the case, LA‐FITC would also be expected to gain access to the red cell interior (as well as the exterior) and therefore label PS in both the inner, as well as the outer, bilayer of the membrane. Positive LA‐FITC red cells would therefore not be restricted to those with only externalised PS. Red cells were treated with tBHP (0–1 mmol/l) in high potassium HEPES‐buffered saline (HK‐HBS; pH 7·4) for 30 min at 37°C), washed and resuspended. Red cells were then labelled for anti‐Hb, PS and ROS, separately or in paired assays. Loss of CM‐H2DCF signal or increase in PS signal concomitant with increased anti‐Hb signal would indicate that cell integrity was compromised. Four aliquots were prepared: (i) CM‐H2DCF‐labelled only, (ii) LA‐FITC‐labelled only, (iii) dual labelled with CM‐H2DCF and Alexa Fluor 647 conjugated anti‐Hb subunit α immunoglobulin (1:100 dilution) and (iv) dual labelled with LA‐FITC and Alexa Fluor 647 conjugated anti‐Hb subunit α immunoglobulin (1:100 dilution). Red cells were pelleted, washed once, resuspended and kept on ice in the dark until flow cytometry analysis. Median CM‐H2DCF and LA‐FITC fluorescence were detected as described above. Median fluorescence of Alexa Fluor 647 conjugated anti‐Hb was detected in the FL4 channel of a BD Accuri C6 flow cytometer.
Statistics
Results are presented as means ± SD or standard error of the mean (SEM) for blood samples from n different individuals. Where appropriate, comparisons were made using 2‐tailed Student's t‐tests and P < 0·05 was considered as significant.
Results
The effect of oxidant stress using xanthine oxidase/hypoxanthine mixtures, PMS and nitrite
Initially, three oxidants were selected to represent a range of different oxidant challenges: XO/HO (2 mmol/l) mixtures to generate extracellular ROS including SOA and hydrogen peroxide (Baskurt et al, 1998; Rogers et al, 2009), PMS to generate SOA intracellularly (Nishikimi et al, 1972; Maridonneau et al, 1983) and NO2 which promotes formation of methaemoglobin (metHb) (Muzyamba et al, 2000).
Control experiments were carried out to establish the appropriate concentrations of oxidants for use in subsequent work using the fluorophore CM‐H2DCF to measure intracellular red cell redox state. With XO/HO mixtures, CM‐H2DCF fluorescence increased with increasing enzyme activity until it reached a plateau at 0·025 units XO/ml (Fig 1A). CM‐H2DCF fluorescence also showed a similar response for PMS and NO2, with plateaux fluorescence (arbitrary units) of 78 690 ± 1423 (n = 4) achieved at 0·1 mmol/l for PMS and of 89 325 ± 4876 (n = 5) at 10 mmol/l for NO2, and concentrations of about 0·05 mmol/l for PMS and 6 mmol/l for NO2 required half maximal CM‐H2DCF fluorescence. The levels of oxidants therefore chosen for further work were 0·015 U/ml, 0·1 mmol/l and 10 mmol/l for XO, PMS and NO2, respectively.
Figure 1.
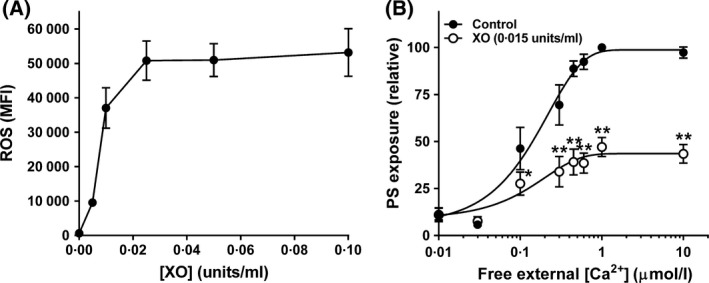
Effect of xanthine oxidase and hypoxanthine (XO/HO) mixtures on accumulation of reactive oxygen species (ROS) and phosphatidylserine (PS) exposure in red cells from patients with sickle cell anaemia (SCA). (A) Red cells were pre‐loaded with CM‐H2DCF‐DA (100 μmol/l) to measure ROS levels [as median fluorescence intensity (MFI)] or treated with the same final concentration of dimethyl sulphoxide [DMSO] (control) before incubation with hypoxanthine (HO, 2 mmol/l) and xanthine oxidase (XO, 0–0·1 U/ml) mixtures for 30 min at 37°C (n = 2). (B) Red cells were permeabilised to Ca2+ with the ionophore bromo‐A23187 (6 μmol/l), and incubated in different free [Ca2+]os, maintained using Ca2+ / 2 mmol/l EGTA mixtures, at 0·5% haematocrit for 30 min at 37°C in the absence (filled circles) or presence (open circles) of HO (2 mmol/l)/XO (0·015 U/ml) mixtures after which externalised PS was labelled with LA‐FITC, n = 8. PS exposure was normalised to that of control red cells at 1 μmol/l free [Ca2+]o (31·1 ± 3·9% of total red cells). Symbols represent means ± SEM for red cells from n different individuals. *P < 0·05; **P < 0·005.
The effect of XO/HO, PMS and NO2 on PS exposure using fluorescein isothiocyanate‐conjugated lactadherin (LA‐FITC)
Phosphatidylserine was first measured by flow cytometry using LA‐FITC. If the red cell membrane remains intact, LA‐FITC will only have access to externalised PS. Red cells were loaded intracellularly with Ca2+ using the ionophore bromo‐A23187 (6 μmol/l) and extracellular [Ca2+], [Ca2+]o, from 0 to 10 μmol/l, clamped using appropriate Ca2+ / EGTA (2 mmol/l) mixtures. Under these conditions, because of the Donnan ratio, free intracellular [Ca2+], [Ca2+]i is about double that of free extracellular values (Flatman, 1980; Muzyamba et al, 2006). Previous experiments have shown that PS exposure in Ca2+ permeabilised red cells occurs when free [Ca2+]o is increased above about 0·1 μmol/l (Weiss et al, 2012; Cytlak et al, 2013). Similar findings were found in the present work with half maximal PS exposure observed at a free [Ca2+]o of 0·34 ± 0·02 μmol/l (n = 22). When red cells were simultaneously exposed to any of the first three oxidants (XO/HO mixtures, PMS or NO2), PS exposure was unaffected in the absence of Ca2+ loading but significantly reduced in its presence – to 47 ± 5% (n = 8, P < 0·005) of controls for XO/HO mixtures, 43 ± 6% (n = 7, P < 0·005) for PMS and 64 ± 7% (n = 9, P < 0·05) for NO2 (all means ± SEM). This effect is shown for XO/HO mixtures in Fig 1B. There were no obvious differences in the response of red cells from SCA patients treated with hydroxycarbamide, with PS exposure being reduced in the presence of oxidants by about 50% whether or not patients were treated with this drug. The effect of oxidants was also similar in red cells from normal (HbAA) individuals. For example, PMS reduced PS exposure to 43 ± 18% (n = 3) of control values in HbAA red cells.
The effect of XO/HO, PMS and NO2 on PS exposure using prothrombinase assays
The decrease in PS exposure in the presence of these oxidants was unexpected as oxidants are generally thought to increase PS exposure in a number of cell types, including red cells from normal individuals and SCA patients (Cimen, 2008; Mohanty et al, 2014; Voskou et al, 2015). A possible explanation lies in the ability of red cells to shed PS into the incubation media either as the free lipid or in microvesicles (Piccin et al, 2015b), which may reduce the remaining PS present on the outside of the red cell membrane despite an increase in scrambling. To test for this possibility and to gain estimates of total red cell PS levels (i.e. that present in both bilayers of the membrane) and thereby exclude possible differential PS shedding, some of the above experiments were repeated using a prothrombinase assay to quantitate for PS. Prothrombinase assays also have the advantage that they exclude any potential artefact through the ability of many oxidants to cause autofluorescence interfering with flow cytometry measurements.
Intact red cells (i.e. in which prothrombinase has access only to externalised PS) were loaded with Ca2+ (using bromo‐A23187 as above). PS levels using the prothrombinase assay gave the same Ca2+ dependence as that for LA‐FITC‐labelled PS (Fig 2A), with half maximal activity achieved at a free [Ca2+]o of 0·6 ± 0·1 μmol/l. Treatment with XO/HO mixtures or PMS again inhibited PS levels by about 50% in the prothrombinase assay (Fig 2A for XO/HO mixtures over a full Ca2+ titration; Fig 2B for both XO/HO mixtures and PMS at two free [Ca2+]os of 0·1 and 1·0 μmol/l). These levels of inhibition were similar level to that observed in flow cytometry experiments using LA‐FITC to label exposed PS.
Figure 2.
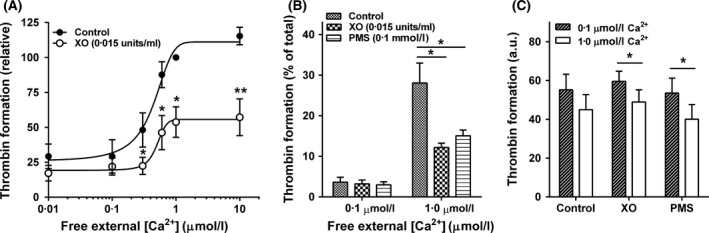
Prothrombinase activity in intact (A and B) or hypotonically lysed (C) red cells from patients with sickle cell anaemia. Red cells were treated as in Fig 1B with thrombin formation used as a measure of accessible phosphatidylserine (PS). (A) Thrombin formation in the absence (filled circles) or presence (open circles) of HO (2 mmol/l)/XO (0·015 U/ml) mixtures over a range of free extra cellular [Ca2+] ([Ca2+]o; 0·1–10 μmol/l), with thrombin formation per min normalised to that of control red cells at 1 μmol/l free [Ca2+]o. (B) Thrombin formation in the absence (control) or presence of HO (2 mmol/l)/XO (0·015 U/ml) mixtures or PMS (0·1 mmol/l), given as a percentage relative to the total value in lysed red cells at two different free [Ca2+]os (0·1 and 1 μmol/l), indicative of prothrombin activity due to externalised PS present on only the outer bilayer of the red cell membrane. (C) Total thrombin formation at 0·1 and 1 μmol/l free [Ca2+]o in the absence and presence of HO (2 mmol/l)/XO (0·015 U/ml) mixtures or PMS (0·1 mmol/l) as measured by thrombin formation (in arbitrary units, AU) of hypotonically lysed red cells to give prothrombin activity of total PS present on both the inner and outer bilayers of the red cell membrane. Symbols and histograms represent means ± SEM for red cells from 4 to 5 different individuals. *P < 0·05; **P < 0·005.
Phosphatidylserine levels were also measured in hypotonically lysed red cells using the prothrombinase assay. Hypotonic lysis allows access of the assay to PS in both bilayers of the red cell membrane and is thus a measure of total red cell PS. Under these conditions, total thrombin formation did not vary at either 0·1 or 1 μmol/l free [Ca2+]o when cells were compared in the absence or presence of XO/HO mixtures or PMS (Fig 2C). There was a loss of about 20% of total PS when free [Ca2+]o was raised from 0·1 to 1 μmol/l (Fig 2C), consistent with Ca2+‐induced PS shedding, but this reduction was similar in extent in the absence or presence of either oxidant.
From these observations, it is unlikely that the observed reduction in PS measurements in the presence of oxidants was due to an overall loss of PS through oxidant‐induced shedding into the extracellular media, excluding red cells as a possible source of PS‐containing microvesicles in response to oxidant challenge (Piccin et al, 2007; Piccin et al, 2015a) although they may participate in Ca2+‐induced PS loss.
The effect of tBHP on PS exposure
A fourth oxidant, tBHP, generates peroxyl and alkoxyl derivatives (Davies, 1989) and has previously been reported to increase PS exposure in red cells from SCA patients – using a concentration of up to 1 mmol/l in a 2‐h incubation (Lang et al, 2002). This oxidant behaved differently to the previous three. CM‐H2DCF fluorescence, indicative of oxidative load, increased with the concentration of tBHP until 0·3 mmol/l, but thereafter, at higher concentrations, fluorescence decreased markedly such that, at 0·75 mmol/l tBHP levels had fallen almost to control values (Fig 3A). With [tBHP]s of ≥0·5 mmol/l, when tested with LA‐FITC, the percentage of red cells positive for LA‐FITC fluorescence did indeed increase (from 1% to 86 ± 2% at 1 mmol/l tBHP) to include most of the red cell population (Fig 3B).
Figure 3.
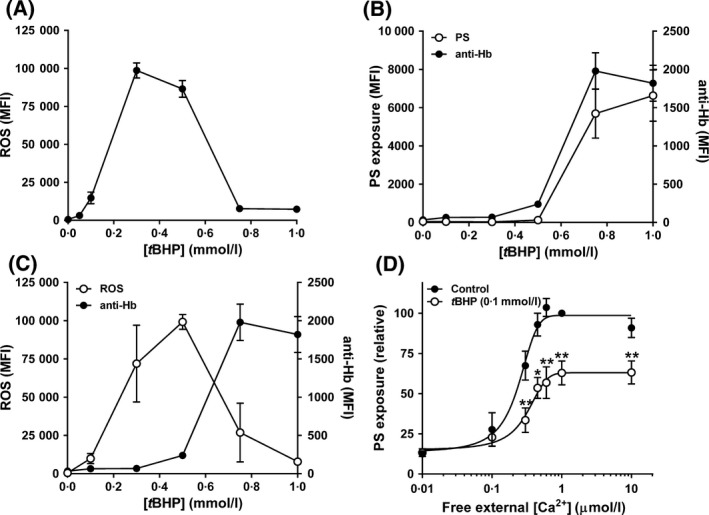
Effect of tert‐butyl hydroperoxide (tBHP) on accumulation of reactive oxygen species (ROS), phosphatidylserine (PS) exposure and membrane integrity in red cells from patients with sickle cell anaemia. (A) Red cells were pre‐loaded with CM‐H2DCF‐DA (100 μmol/l) to measure ROS levels or treated with the same final concentration of dimethyl sulphoxide [DMSO] (control) before incubation with tBHP (0–1 mmol/l) at 0·5% haematocrit for 30 min at 37°C in HK‐HBS (n = 4). (B) Red cells were double labelled with CM‐H2DCF to measure ROS levels (left ordinate) and with Alexa Fluor 647 anti‐Hb α chain (labelled anti‐Hb; right ordinate) to correlate ROS levels with labelling of intracellular haemoglobin (n = 3). (C) Red cells were double labelled with LA‐FITC to measure accessible PS and (left ordinate) and with Alexa Fluor 647 anti‐Hb α chain (labelled anti‐Hb; right ordinate) to correlate PS labelling with that of intracellular haemoglobin (n = 3). (D) Red cells were permeabilised to Ca2+ as in Fig 1B at 0·5% haematocrit for 30 min at 37°C in the absence (filled circles) or presence (open circles) of tBHP (0·1 mmol/l) after which externalised PS was labelled with LA‐FITC. PS exposure was normalised to that of control red cells at 1 μmol/l free [Ca2+]o (29·3 ± 3·4% of total red cells, n = 7). Symbols represent means ± SEM for red cells from n different individuals. *P < 0·05; **P < 0·005.
At high [tBHP] red cells become markedly leaky to cations (Ney et al, 1990). We postulated, therefore, that concentrations of tBHP higher than about 0·3 mmol/l may cause disintegrity of the red cell membrane and thereby account for the fall in CM‐H2DCF fluorescence. To test this hypothesis, cells were incubated with Alexa Fluor 647‐labelled anti‐Hb α chain immunoglobulin as well as with CM‐H2DCF‐AM and LA‐FITC. At higher levels of tBHP, CM‐H2DCF fluorescence again declined in an increasingly large population of red cells whilst the same red cells simultaneously became positive for Alexa Fluor 647 fluorescence, indicative of entry of anti‐Hb α chain immunoglobulin into the red cells (Fig 3C). At these higher [tBHP], PS positive red cells were also positive for anti‐Hb fluorescence (Fig 3C). That a large immunoglobulin (15 kDa) could gain access to the inside of the red cell implies the presence of substantial holes in the membrane that could result in the loss of red cell contents or of CM‐H2DCF itself, accounting for the fall in CM‐H2DCF fluorescence whilst access of LA‐FITC to the interior of the red cell would label PS remaining on the inside of the membrane as well as externalised PS. When tBHP concentrations were used at which red cells became positive for CM‐H2DCF fluorescence whilst remaining negative for Alexa Fluor 647 fluorescence, exposed PS labelled with LA‐FITC decreased in the presence of tBHP to 63 ± 8% (Fig 3D; means ± SEM, n = 7, P < 0·005) of controls, to a similar extent to that observed for XO/HO mixtures, PMS and NO2.
The presence of tBHP also increased cell granularity, as measured by higher side scatter (SSC) values. Compared to controls, SSC was already increased significantly at 0·3 mmol/l tBHP (159 ± 10%, P < 0·001, n = 6), reaching 211 ± 16% of control levels at 1 mmol/l tBHP (P < 0·001, n = 6). FSC also increased but changes were more variable, with a small increase to 112 ± 3% of control levels at 1 mmol/l tBHP (P < 0·005, n = 6). By contrast, SSC and FSC varied by less than 5% when cells were exposed to either PMS or NO2. Although changes were more pronounced when using mixtures of HO and higher concentrations of XO (0·1 U/ml), they were not significant (FSC: 108 ± 4%, SSC: 116 ± 11%). Notwithstanding, when observed under light microscopy (20× magnification), there was no obvious difference in morphology between untreated red cells and those exposed to any oxidant, including tBHP (up to 1 mmol/l).
The effect of sulphydryl‐modifying compounds: NEM, DTT and HOCl
Exposure of red cells to the sulphydryl oxidising reagent NEM (1 mmol/l) has been shown to stimulate PS exposure in human and mouse red cells through inhibiting the flippase and activating the scramblase (Connor & Schroit, 1990; Martin & Jesty, 1995; Kamp et al, 2001; de Jong & Kuypers, 2006). This compound was tested here in Ca2+‐clamped red cells from SCA patients. PS exposure was increased markedly in the presence of NEM (1 mmol/l), by about 400%, with the Ca2+ affinity of scramblase increasing from a 50% maximal response (EC50) value of 0·4 ± 0·07 to 0·26 ± 0·03 μmol/l free [Ca2+]o (n = 6, P < 0·03) (Fig 4A). Interestingly, the CMH2DCF signal in the presence of 1 mmol/l NEM increased by only 2‐fold compared to that in untreated red cells, to a much lower level of that obtained with the other oxidant, showing that this thiol reagent had little effect on intracellular ROS levels. In the presence of DTT (0·25 mmol/l), which protects thiol groups from oxidation, PS exposure was reduced by about 25% (Fig 4B), with no change in Ca2+ dependence of the scrambling process.
Figure 4.
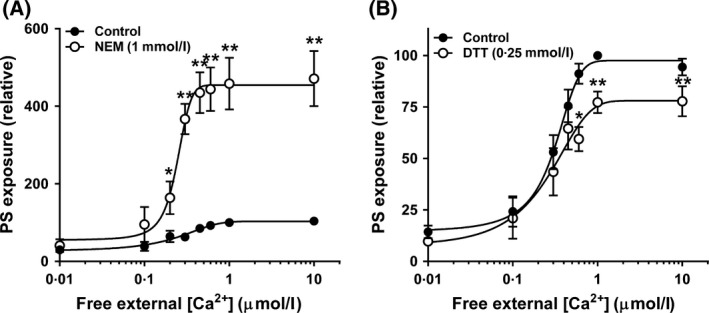
Effect of thiol modification on phosphatidylserine (PS) exposure of red cells from sickle cell anaemia patients. Red cells were pre‐incubated at 4% haematocrit in the absence (solid circles) or presence (open circles) of N‐ethylmaleimide (NEM) and dithiothreitol (DTT), then permeabilised to Ca2+ as in Fig 1B for 30 min at 37°C, after which accessible PS was labelled with LA‐FITC. (A) Effect of NEM (1 mmol/l) on PS exposure (n = 8); (B) Effect of DTT (0·25 mmol/l) on PS exposure (n = 10). PS exposure was normalised to that of control red cells (in the absence of thiol modifiers) at 1 μmol/l free [Ca2+]o (NEM control: 21·2 ± 4·7%, DTT control: 24·3 ± 4·6% of total red cells). Symbols represent means ± SEM for red cells from n different individuals. *P < 0·05; **P < 0·005.
The final oxidative manoeuvre tested was exposure HOCl. This oxidant is produced by myeloperoxidase, released from activated neutrophils, which are important mediators of vascular inflammation in SCA. Like NEM, HOCl may also react with membrane thiols. At higher concentrations of HOCl, the red cell oxidant load measured using CMH2DCF achieved a similar level to that obtained with other oxidants although no plateau was observed (Fig 5A). The effect of HOCl on PS exposure measured using LA‐FITC was tested at concentrations up to 500 μmol/l, however, no effect was observed when compared with untreated red cells (Fig 5B).
Figure 5.
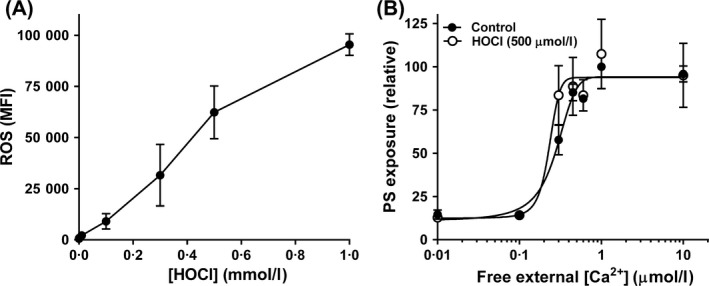
Effect of hypochlorous acid (HOCl) on phosphatidylserine (PS) exposure of red cells from sickle cell anaemia patients. (A) Red cells were pre‐loaded with CM‐H2DCF‐DA (100 μmol/l) or treated with the same final [DMSO] before incubation with HOCl (500 μmol/l) at 0·5% haematocrit for 30 min at 37°C in HK‐HBS (n = 3). (B) Red cells were permeabilised to Ca2+ as in Fig 1B at 0·5% haematocrit for 30 min at 37°C in the absence (full circles) or presence (open circles) of HOCl (500 μmol/l) after which accessible PS was labelled with LA‐FITC (n = 6). PS exposure was normalised to that of control red cells at 1 μmol/l free [Ca2+]o (32 ± 5·4% of total red cells). Symbols represent means ± SEM for red cells from n different individuals.
Discussion
Surprisingly, the present findings show that a number of oxidants – xanthine oxidase/hypoxanthine (XO/HO) mixture, PMS, NO2 and tBHP – inhibited Ca2+‐induced PS exposure in red cells from patients with SCA, with little effect on its Ca2+ dependence. In contrast, thiol oxidation with NEM markedly stimulated lipid scrambling with increase in Ca2+ affinity, whilst protection of reduced thiols with DTT inhibited PS exposure. HOCl, however, was without effect.
Red cell PS exposure is observed in numerous pathological conditions, including SCA, in which a significant, but variable (2–10%), percentage of circulating red cells are positive for externalised PS (Wood et al, 1996; Kuypers, 1998; de Jong et al, 2001; Dasgupta & Thiagarajan, 2005; Cytlak et al, 2013). SCA is associated with increased oxidative stress, neutrophil leucocytosis and vascular endothelial dysfunction, all of which are also associated with increased PS exposure (Jain, 1985; Hebbel, 1991; Kuypers, 1998; Mutze et al, 2003; Banerjee & Kuypers, 2004; Zhang et al, 2013). Recent reviews have reinforced the view that ROS are actively involved in lipid scrambling in SCA (Cimen, 2008; Mohanty et al, 2014; Voskou et al, 2015), with limited experimental evidence supporting this assumption. Aside from thiol modification with NEM (de Jong & Kuypers, 2006), however, definitive evidence for stimulation of PS exposure by oxidant challenge is scant.
We used four different oxidant treatments (XO/HO mixtures, PMS, NO2 and tBHP) to investigate their effects on PS exposure in red cells from SCA patients. These particular oxidants were chosen because of the qualitative differences in oxidative challenge that they present to the red cell. XO/HO mixtures generate SOA and hydrogen peroxide extracellularly (Baskurt et al, 1998; Rogers et al, 2009), whilst PMS generates SOA intracellularly (Nishikimi et al, 1972; Maridonneau et al, 1983); NO2 oxidises Hb to metHb (Muzyamba et al, 2000); tBHP increases generation of peroxyl and alkoxyl free radicals (Davies, 1989); whilst HOCl produced by myeloperoxidase released from neutorphils may also oxidise membrane thiols (Vissers et al, 1994; Gorudko et al, 2016). Red cell PS exposure is most reliably stimulated by elevation of intracellular Ca2+ (Bevers & Williamson, 2010) whilst a number of oxidants have previously been shown to increase red cell cation permeability (Gibson & Muzyamba, 2003a,2003b). We therefore hypothesised that oxidants may act synergistically with intracellular Ca2+ to increase PS exposure and alter its Ca2+ affinity, thereby accounting for the raised percentage of PS‐positive red cells in SCA patients. Under the experimental conditions used, however, instead of stimulating PS exposure, we found that these oxidants produced significant inhibitory effects on PS exposure (by about 40–60%) with no apparent change in Ca2+ dependence. These findings are summarised in Fig 6.
Figure 6.
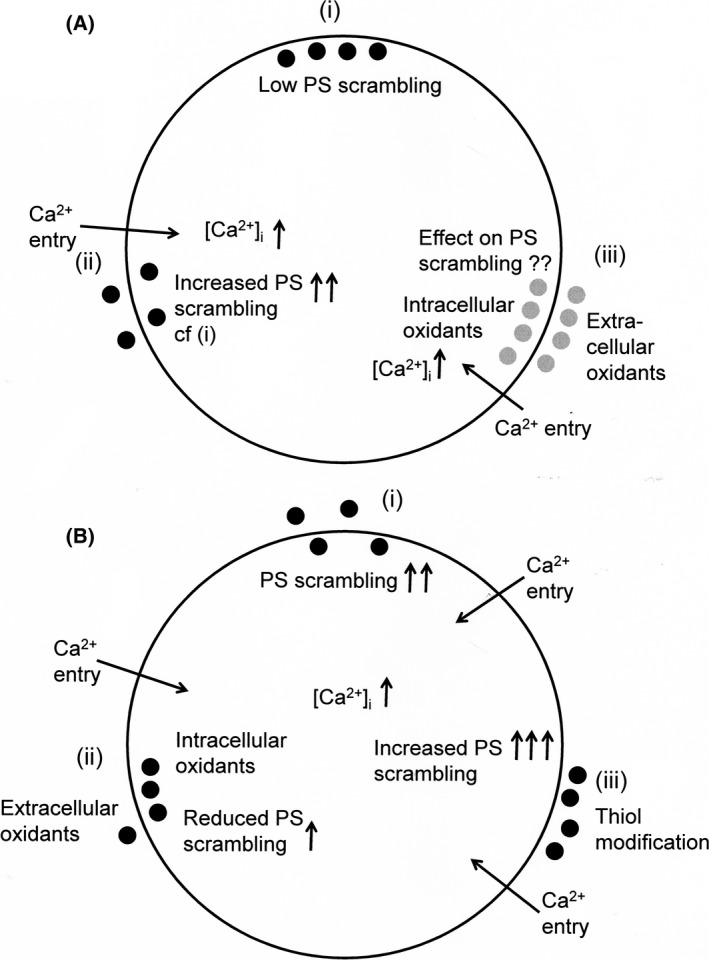
Schematic digram of stimuli affecting phosphatidylserine (PS) distribution in red cells from patients with sickle cell anaemia. (A) (i) PS is usually confined to the inner leaflet of the lipid bilayer of red cells including sickle cells through high activity of the flippase and low activity of the scramblase, as externalisation is prothrombotic and increases phagocytosis; (ii) elevation of intracellular Ca2+ ([Ca2+]i) via the deoxygenation‐induced cation conductance (or Psickle) or via ionophore promotes PS exposure increasing the possibility of microvascular occlusion; (iii) the effect of oxidants either from within the sickle cell or from the circulation is uncertain. (B) (i) As before, entry of Ca2+ increases PS exposure; (ii) most oxidants (xanthine oxidase/hypoxanthine mixtures, nitrite, phenazine methosulphate) actually reduced Ca2+‐induced PS exposure by about 50% and would reduce thrombosis; (iii) the exception was thiol oxidation which markedly increased externalisation of PS. ●, known PS distribution;  , PS distribution unknown.
, PS distribution unknown.
Oxidants are also associated with PS shedding and microvesiculation (Piccin et al, 2007, 2015b; Freikman et al, 2008; Alaarg et al, 2013) and it was possible that reduced PS levels on the outer bilayer of the red cell membrane could be due to its loss into the incubation media, notwithstanding an increase in lipid scrambling. PS may be lost as microvesicles, which have been proposed as a biomarker for SCA severity (Piccin et al, 2015a). Total thrombin formation in permeabilised red cells (using hypotonic lysis) were similar in controls and after oxidant challenge, however, which makes it unlikely that large reductions in labelled PS on the outer bilayer (up to 65%) could result from PS shedding. Microvesicles may, however, be formed in response to Ca2+‐induced PS scrambling. The mechanisms by which oxidants decrease PS exposure are unclear, although membrane lipid and protein damage, for example by peroxidation, may specifically inhibit the scrambling protein transporter. Conversely, lipid peroxidation may reduce the ability of the scramblase to translocate PS through the lipid bilayer.
The one manoeuvre which did increase PS exposure markedly was incubation with NEM to cause thiol oxidation (Fig 6). Thiol reactions are known affect the activities of the flippase and scramblase (Connor & Schroit, 1990; Martin & Jesty, 1995; Kamp et al, 2001). NEM has previously been shown to increase scrambling in normal human and mice red cells (Kamp et al, 2001; de Jong & Kuypers, 2006) and our experiments confirm this effect in red cells from SCA patients. An important difference with previous work, however, is that we observe marked effects on PS exposure at two orders of magnitude lower Ca2+ concentrations. PS exposure began with elevation of red cell Ca2+ concentration to about 100–200 nmol/l. In the opposite way, reduction of thiols by DTT reduced scrambling, as seen previously for pyridyldithioethylamine (de Jong & Kuypers, 2006). Alterations in Ca2+ affinity in our hands were modest but there was a slight increase in the presence of NEM, in contrast to previous work (de Jong & Kuypers, 2006) (cf. de Jong et al, 1997) but note the difference in haematocrit). The action of DTT in vitro suggests that circulating sickle cells are already deprived of reduced thiols (Kamp et al, 2001). Indeed, sickle cells are reported to contain lower levels of membrane thiols (Rank et al, 1985; Rice‐Evans et al, 1986). HOCl also reacts with membrane thiols. However, although giving a similar level of oxidant challenge to the red cell, as assessed using CMH2DCF, HOCl had no effect on Ca2+‐induced PS exposure. The lack of effect of HOCl suggests that NEM is able to gain access to key targets which are unavailable to HOCl. Nevertheless, it is likely that thiol modification accounts for the greater percentage of circulating sickle cells positive for PS.
Finally, red cells from SCA patients have been previously described to show increased PS exposure on incubation with tBHP (Lang et al, 2002). The concentrations used in this previous report were up to 1 mmol/l, however, with an incubation time of 2 h, after which all red cells were positive for PS (labelled using FITC‐ annexin V). Our findings that red cells treated with similar concentrations of tBHP become positive to fluorescently‐labelled antibodies to Hb α chain within 30 min suggest that membrane disintegrity occurs at these higher concentrations and that the PS label can gain access to cytoplasmic PS as well as externalised PS (Fig 7). This hypothesis is supported by previous observations that large cation leaks are induced in red cells at higher [tBHP] (Ney et al, 1990).
Figure 7.
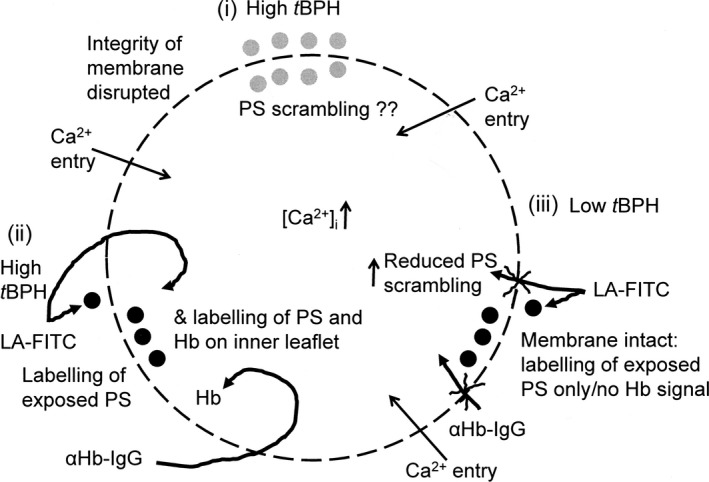
Schematic diagram of the effect of tert‐butyl hydroperoxide (tBHP) on red cells from patients with sickle cell anaemia. (i) tBHP is thought to increase phosphatidylserine (PS) exposure but relatively high concentrations have been used, which may disrupt the integrity of the membrane; (ii) in the presence of high tBHP more PS was labelled, but the ability of anti‐Hb IgG to label intracellular Hb suggests that this was because loss of membrane integrity allowed labelling of both inner leaflet PS as well as externalised PS; (iii) by contrast, with lower tBHP concentrations that do not allow entry of anti‐Hb IgG, PS exposure was reduced by 50%, as for the other oxidants (in Fig 1b). ●, known PS distribution;  , PS distribution unknown.
, PS distribution unknown.
In conclusion, the experiments described in the present work show that a number of different oxidants (XO/HO mixtures, PMS, NO2 and tBHP) reduced Ca2+‐induced PS exposure in red cells from SCA patients. Thiol oxidation by NEM was confirmed to increase PS exposure, whilst thiol reduction using DTT decreased it, consistent with the postulate that thiol oxidation in vivo contributes to increased numbers of PS‐positive red cells in SCA patients. These findings are particularly relevant to the ability of oxidative stress in the circulation in vivo in SCA patients to participate in thrombus formation, vascular occlusion and tissue ischaemia via mechanisms involving red cell PS, and demonstrate the complexity of the pathophysiology of sickle cell disease. This study gives important new information on the complex role of oxidative stress in the pathophysiology of sickle cell disease and suggests that the effects may vary depending on the precise nature of the oxidants and their physiological context.
Conflicts of interest
There are no conflicts of interest.
Author contributions
Most experiments were carried out by AH with some assistance from AN and BL; DCR and JSG designed the study; JNB and DCR acquired samples; AH and AN analysed the data; JSG, DCR, JNB and AH prepared the manuscript.
Supporting information
Appendix S1. Materials and methods.
Acknowledgments
We thank the British Heart Foundation for generous financial support (grant number 31966).
References
- Alaarg, A. , Schiffelers, R.M. , van Solinge, W.W. & van Wijk, R. (2013) Red blood cell vesiculation in hereditary hemolytic anemia. Frontiers in Physiology, 4, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee, T. & Kuypers, F.A. (2004) Reactive oxygen species and phosphatidylserine externalization in murine sickle red cells. British Journal of Haematology, 124, 391–402. [DOI] [PubMed] [Google Scholar]
- Barber, L.A. , Palascak, M.B. , Joiner, C.H. & Franco, R.S. (2009) Aminophospholipid translocase and phospholipid scramblase activities in sickle erythrocyte subpopulations. British Journal of Haematology, 146, 447–455. [DOI] [PubMed] [Google Scholar]
- Baskurt, O.K. , Temiz, A. & Meiselman, H.J. (1998) Effect of superoxide anions on red blood cell rheologic properties. Free Radical Biology and Medicine, 24, 102–110. [DOI] [PubMed] [Google Scholar]
- Basse, F. , Stout, J.G. , Sims, P.J. & Wiedmer, T. (1996) Isolation of an erythrocyte membrane protein that mediates Ca2+‐dependent transbilayer movement of phospholipid. Journal of Biological Chemistry, 271, 17205–17210. [DOI] [PubMed] [Google Scholar]
- Bevers, E.M. & Williamson, P.L. (2010) Phospholipid scrambling: an update. FEBS Letters, 584, 2724–2730. [DOI] [PubMed] [Google Scholar]
- Bevers, E.M. , Comfuriius, P. , Van Rijn, J.L.M.L. , Hemker, H.C. & Zwaal, R.F.A. (1982) Generation of prothrombin‐converting activity and the exposure of phosphatidylserine at the outer surface of platelets. European Journal of Biochemistry, 122, 429–436. [DOI] [PubMed] [Google Scholar]
- Bitbol, M. , Fellmann, P. , Zachowski, A. & Devaux, P.F. (1987) Ion regulation of phosphatidylserine and phosphatidylethanolamine outside‐inside translocation in human erythrocytes. Biochimica et Biophysica Acta, 904, 268–282. [DOI] [PubMed] [Google Scholar]
- Blumenfeld, N. , Zachowski, A. , Galacteros, F. , Beuzard, Y. & Devaux, P.F. (1991) Transmembrane mobility of phospholipids in sickle erythrcoytes: effect of deoxygenation on diffusion and asymmetry. Blood, 77, 849–854. [PubMed] [Google Scholar]
- Boas, F.E. , Forman, L. & Beutler, J.A. (1998) Phosphatidylserine exposure and red cell viability in red cell ageing and in hemolytic anemia. Proceedings of the National Academy of Sciences of the United States of America, 95, 3077–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirico, E.N. & Pialoux, V. (2012) Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB Life, 64, 72–80. [DOI] [PubMed] [Google Scholar]
- Cimen, M.Y.B. (2008) Free radical metabolism in human erythrocytes. Clinica Chimica Acta, 390, 1–11. [DOI] [PubMed] [Google Scholar]
- Connor, J. & Schroit, A.J. (1990) Aminophospholipid translocation in erythrocytes: evidence for the involvement of a specific transporter and an endofacial protein. Biochemistry, 29, 37–43. [DOI] [PubMed] [Google Scholar]
- Cytlak, U.M. , Hannemann, A. , Rees, D.C. & Gibson, J.S. (2013) Identification of the Ca2+ entry pathway involved in deoxygenation‐induced phosphatidylserine exposure in red blood cells from patients with sickle cell disease. Pflügers Archiv – European Journal of Physiology, 465, 1651–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta, S.K. & Thiagarajan, P. (2005) The role of lactadherin in the phagocytosis of phosphatidylserine‐expressing sickle red blood cells by macrophages. Haematologica, 90, 1267–1268. [PubMed] [Google Scholar]
- Davies, M.J. (1989) Detection of peroxyl and alkoxyl radicals produced by reaction of hydroperoxides with rat liver microsomal fractions. Biochemical Journal, 257, 603–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux, P.F. & Zachowski, A. (1994) Maintenance and consequences of membrane phospholipid asymmetry. Chemistry and Physics of Lipids, 73, 107–120. [Google Scholar]
- Eaton, J.W. & Hofrichter, J. (1987) Hemoglobin S gelation and sickle cell disease. Blood, 70, 1245–1266. [PubMed] [Google Scholar]
- Etzion, Z. , Tiffert, T. , Bookchin, R.M. & Lew, V.L. (1993) Effects of deoxygenation on active and passive Ca2+ transport and on the cytoplasmic Ca2+ levels of sickle cell anemia red cells. Journal of Clinical Investigation, 92, 2489–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatman, P.W. (1980) The effect of buffer composition and deoxygenation on the concentration of ionized magnesium inside human red blood cells. Journal of Physiology, 300, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freikman, I. , Amer, J. , Cohen, J.S. , Ringel, I. & Fibach, E. (2008) Oxidative stress causes membrane phospholipid rearrangement and shedding from RBC membranes – an NMR study. Biochimica et Biophysica Acta, 1778, 2388–2394. [DOI] [PubMed] [Google Scholar]
- Gao, J. , Yao, Y. & Squier, T.C. (2001) Oxidatively modified calmodulibn binds to the plasma membrane Ca‐ATPase in a nonproductive and conformationally disordered complex. Biophyiscal Journal, 80, 1791–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, J.S. & Muzyamba, M.C. (2003a) The effect of 1‐chloro‐2,4‐dinitrobenzene on K+ transport in normal and sickle human red blood cells. Journal of Physiology, 547, 903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, J.S. & Muzyamba, M.C. (2003b) Effect of phenazine methosulphate on K+ transport in human red cells. Cellular Physiology and Biochemistry, 13, 329–336. [DOI] [PubMed] [Google Scholar]
- Gorudko, I.V. , Sokolov, A.V. , Shamova, E.V. , Grigorieva, D.V. , Mironova, E.V. , Kudryavtsev, I.V. , Gusev, S.A. , Gusev, A.A. , Chekanov, A.V. , Vasilyev, V.B. , Cherenkevich, S.N. , Panasenko, O.M. & Timoshenko, A.V. (2016) Binding of human myeloperoxidase to red blood cells: Molecular targets and biophysical conequences at the plasma membrane level. Archives of Biochemistry and Biophysics, 591, 87–97. [DOI] [PubMed] [Google Scholar]
- Haest, C.W.M. (2003) Distribution and movement of membrane lipids In: Red Cell Membrane Transport in Health and Disease (eds by Bernhardt I. & Ellory J.C.), pp. 1–25. Springer Verlag, Berlin. [Google Scholar]
- Hebbel, R.P. (1991) Beyond hemoglobin polymerization: the red blood cell membrane and sickle cell disease pathophysiology. Blood, 77, 214–237. [PubMed] [Google Scholar]
- Jain, S.K. (1985) In vivo externalization of phosphatidylserine and phosphatidylethanolamine in the membrane bilayer and hypercoagubility by the lipid peroxidation of erythrocytes in rats. Journal of Clinical Investigation, 76, 281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, S.K. & Shohet, S.B. (1984) A novel phospholipid in irreversibly sickled cells: evidence for in vivo peroxidative membrane damage in sickle cell disease. Blood, 63, 362–367. [PubMed] [Google Scholar]
- Jain, S.K. & Williams, D.M. (1985) Reduced levels of plasma ascorbic acid (vitamin C) in sickle cell disease patients: its possible role in the oxidant damage to sickle cells in vivo. Clinica Chimica Acta, 149, 257–261. [DOI] [PubMed] [Google Scholar]
- de Jong, K. & Kuypers, F.A. (2006) Sulphydryl modifications alter scramblase activity in murine sickle cell disease. British Journal of Haematology, 133, 427–432. [DOI] [PubMed] [Google Scholar]
- de Jong, K. , Geldwerth, D. & Kuypers, F.A. (1997) Oxidative damage does not alter membrane phospholipid asymmetry in human erythrocytes. Biochemistry, 36, 6768–6776. [DOI] [PubMed] [Google Scholar]
- de Jong, K. , Larkin, S.K. , Styles, L.A. , Bookchin, R.M. & Kuypers, F.A. (2001) Characterization of the phosphatidylserine‐exposing subpopulations of sickle cells. Blood, 98, 860–867. [DOI] [PubMed] [Google Scholar]
- Kamp, D. , Sieberg, T. & Haest, C.W.M. (2001) Inhibition and stimulation of phospholipid scrambling activity. Consequences for lipid asymmetry, echinocytosis, and microvesiculation of erythrocytes. Biochemistry, 40, 9438–9446. [DOI] [PubMed] [Google Scholar]
- Kuypers, F.A. (1998) Phospholipid asymmetry in health and disease. Current Opinions in Hematology, 5, 122–131. [DOI] [PubMed] [Google Scholar]
- Kuypers, F.A. , Lewis, R.A. , Hua, M. , Schott, M.A. , Discher, D. , Ernst, J.D. & Lubin, B.H. (1996) Detection of altered membrane phospholipid asymmetry in subpopulations of human red blood cells using fluorescently labeled Annexin V. Blood, 87, 1179–1187. [PubMed] [Google Scholar]
- Lang, K.S. , Roll, B. , Myssina, S. , Schittenhelm, M. , Scheel‐Walter, H.‐G. , Kanz, L. , Fritz, J. , Lang, F. , Huber, S.M. & Wieder, T. (2002) Enhanced apoptosis in sickle cell anemia, thalassemia and glucose‐6‐phosphate dehydrogenase deficiency. Cellular Physiology and Biochemistry, 12, 365–372. [DOI] [PubMed] [Google Scholar]
- Lang, F. , Lang, K.S. , Lang, P.A. , Huber, S.M. & Wieder, T. (2006) Mechanisms and significance of eryptosis. Antioxidants and Redox Signalling, 8, 1183–1192. [DOI] [PubMed] [Google Scholar]
- Lang, E. , Qadri, S.M. & Lang, F. (2012) Killing me softly – suicidal erythrocyte death. International Journal of Biochemistry and Cell Biology, 44, 1236–1243. [DOI] [PubMed] [Google Scholar]
- Lew, V.L. & Bookchin, R.M. (2005) Ion transport pathology in the mechanism of sickle cell dehydration. Physiological Reviews, 85, 179–200. [DOI] [PubMed] [Google Scholar]
- Lew, V.L. , Ortiz, O.E. & Bookchin, R.M. (1997) Stochastic nature and red cell population distribution of the sickling‐induced Ca2+ permeability. Journal of Clinical Investigation, 99, 2727–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin, B. , Chiu, D. , Bastacky, J. , Roelofsen, B. & Van Deenen, L.L.M. (1981) Abnormalities in membrane phospholipid organization in sickled erythrocytes. Journal of Clinical Investigation, 67, 1643–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankelow, T.J. , Griffiths, R.E. , Trompeter, S. , Flatt, J.F. , Cogan, N.M. , Massey, E.J. & Anstee, D.J. (2015) Autophagic vesicles on mature human reticulocytes explain phosphatidylserine‐positive red cells in sickle cell disease. Blood, 126, 1831–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maridonneau, I. , Braquet, P. & Garay, R.P. (1983) Na+ and K+ transport damage induced by oxygen free radicals in human red cell membranes. Journal of Biological Chemistry, 258, 3107–3113. [PubMed] [Google Scholar]
- Martin, D.W. & Jesty, J. (1995) Calcium stimulation of procoagulant activity in human erythrocytes. Journal of Biological Chemistry, 270, 10468–10474. [DOI] [PubMed] [Google Scholar]
- Mohandas, N. , Rossi, M.E. & Clark, M.R. (1986) Association between morphologic distortion of sickle cells and deoxygenation‐induced cation permeability increases. Blood, 68, 450–454. [PubMed] [Google Scholar]
- Mohanty, J.G. , Nagababu, E. & Rifkind, J.M. (2014) Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Frontiers in Physiology, 5, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrot, G. , Herve, P. , Zachowski, A. , Fellmann, P. & Devaux, P.F. (1989) Aminophospholipid translocase of human erythrocytes: phospholipid substrate specificity and effect of cholesterol. Biochemistry, 28, 3456–3462. [DOI] [PubMed] [Google Scholar]
- Mutze, S. , Hebling, U. , Stremmel, W. , Wang, J. , Arnhold, J. , Pantopoulos, K. & Mueller, S. (2003) Myeloperoxidase‐derived hypochlorous acid antagonizes the oxidative stress‐mediated activation of iron regulatory protein 1. Journal of Biological Chemistry, 279, 40542–40549. [DOI] [PubMed] [Google Scholar]
- Muzyamba, M.C. , Speake, P.F. & Gibson, J.S. (2000) Oxidants and regulation of KCl cotransport in equine red blood cells. American Journal of Physiology, 279, C981–C989. [DOI] [PubMed] [Google Scholar]
- Muzyamba, M.C. , Campbell, E.H. & Gibson, J.S. (2006) Effect of intracellular magnesium and oxygen tension on K+‐Cl‐ cotransport in normal and sickle human red cells. Cellular Physiology and Biochemistry, 17, 121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebor, D. , Bowers, A. , Connes, P. , Hardy‐Dessources, M.‐D. , Knight‐Madden, J. , Cumming, V. , Reid, M. & Romana, M. (2014) Plasma concentration of platelet‐derived microparticles is related to painful vaso‐occlusive phenotype severity in sickle cell anemia. PLoS ONE, 9, e87243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ney, P.A. , Christopher, M.M. & Hebbel, R.P. (1990) Synergistic effects of oxidation and deformation on erythrocyte monovalent cation leak. Blood, 75, 1192–1198. [PubMed] [Google Scholar]
- Niihara, Y. , Viswanathan, K. , Miller, S.T. , Guillaume, E. , Blackwood, M. , Razon, R. , Tran, L. & Stark, C. (2016) Phase 3 study of L‐glutamine therapy in sickle cell anemia and sickle β0‐thalassemia subgroup analyses show consistent clinical improvement. Blood, 128, 1318.27609540 [Google Scholar]
- Nishikimi, M. , Rao, N.A. & Yagi, K. (1972) Reactivity of D‐amino acid oxidase with artificial electron acceptors. Biochemical and Biophysical Research Communications, 46, 849–854.4400444 [Google Scholar]
- Piccin, A. , Murphy, W.G. & Smith, O.P. (2007) Circulating microparticles: pathophysiology and clinical implications. Blood Reviews, 21, 157–171. [DOI] [PubMed] [Google Scholar]
- Piccin, A. , Murphy, C. , Eakins, E. , Kunde, J. , Corvetta, D. , Di Pierro, A. , Negri, G. , Guido, M. , Sainati, L. , McMahon, C. , Smith, O.P. & Murphy, W. (2015a) Circulating microparticles, protein C, free protein S and endothelial vascular markers in children with sickle cell anaemia. Journal of Extracellular Vesicles, 4, 28414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccin, A. , Van Schilfgaarde, M. & Smith, O. (2015b) The importance of studying red blood cells microparticles. Blood Transfusion, 13, 712–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccin, A. , Sartori, M.T. , Bisogno, G. , Van Schilfgaarde, M. , Saggiorato, G. , Pierro, A.M.D. , Corvetta, D. , Marcheselli, L. , Andrea, M. , Gastl, G. & Cesaro, S. (2017) New insights into sinusoidal obstruction syndrome. Internal Medicine Journal, 47, 1173–1183. [DOI] [PubMed] [Google Scholar]
- Piel, F.B. , Patil, A.P. , Howes, R.E. , Nyangiri, O.A. , Gething, P.W. , Dewi, M. , Temperley, W.H. , Williams, T.N. , Weatherall, D.J. & Hay, S.I. (2013) Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model‐based map and population estimates. The Lancet, 381, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank, B.H. , Carisson, J. & Hebbel, R.P. (1985) Abnormal redox status of membrane‐protein thiols in sickle erythrocytes. Journal of Clinical Investigation, 75, 1531–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees, D.C. , Williams, T.N. & Gladwin, M.T. (2010) Sickle‐cell disease. Lancet, 376, 2018–2031. [DOI] [PubMed] [Google Scholar]
- Rhoda, M.D. , Apovo, M. , Beuzard, Y. & Giraud, F. (1990) Ca2+ permeability in deoxygenated sickle cells. Blood, 75, 2453–2458. [PubMed] [Google Scholar]
- Rice‐Evans, C. , Omorphos, S.C. & Baysal, E. (1986) Sickle cell membranes and oxidative damage. Biochemical Journal, 237, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, S.C. , Said, A. , Corcuera, D. , McLaughlin, D. , Kell, P. & Doctor, A. (2009) Hypoxia limits antioxidant capacity in red blood cells by altering glycolytic pathway dominance. FASEB Journal, 23, 3159–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setty, B.N. , Kulkarini, S. & Stuart, M.J. (2002) Role of erythrocyte phosphatidylserine in sickle red cell‐endothelial adhesion. Blood, 99, 1564–1571. [DOI] [PubMed] [Google Scholar]
- Shalev, O. , Leida, M.N. , Hebbel, R.P. , Jacob, H.S. & Eaton, J.W. (1981) Abnormal erythrocyte calcium homeostasis in oxidant‐induced haemolytic disease. Blood, 58, 1232–1235. [PubMed] [Google Scholar]
- Sies, H. (1997) Oxidative stress: oxidants and antioxidants. Experimental Physiology, 82, 291–295. [DOI] [PubMed] [Google Scholar]
- Silva, D.G.H. , Belini, E. Jr , de Almeida, E.A. & Bonini‐Domingos, C.R. (2013) Oxidative stress in sickle cell disease: an overview of erythrocyte redox metabolism and current antioxidant therapeutic strategies. Free Radical Biology and Medicine, 65, 1101–1109. [DOI] [PubMed] [Google Scholar]
- Steffen, P. , Jung, A. , Nguyen, D.B. , Muller, T. , Bernhardt, I. , Kaestner, L. & Wagner, C. (2011) Stimulation of human red blood cells leads to Ca2+‐mediated intracellular adhesion. Cell Calcium, 50, 54–61. [DOI] [PubMed] [Google Scholar]
- Tait, J.F. & Gibson, D. (1994) Measurement of membrane phospholipid asymmetry in normal and sickle‐cell erythrocytes by means of annexin V binding. Journal of Laboratory and Clinical Medicine, 123, 741–748. [PubMed] [Google Scholar]
- Vissers, M.C.M. , Stern, A. , Kuypers, F. , van den Berg, J. & Winterbourn, C.C. (1994) Membrane changes associated with lysis of red blood cells by hypochlorous acid. Free Radical Biology and Medicine, 16, 703–712. [DOI] [PubMed] [Google Scholar]
- Voskou, S. , Aslan, M. , Fanis, P. , Phylactides, M. & Kleanthous, M. (2015) Oxidative stress in beta‐thalassaemia and sickle cell disease. Redox Biology, 6, 226–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, E. , Cytlak, U.M. , Rees, D.C. , Osei, A. & Gibson, J.S. (2012) Deoxygenation‐induced and Ca2+‐dependent phosphatidylserine externalisation in red blood cells from normal individuals and sickle cell patients. Cell Calcium, 51, 51–56. [DOI] [PubMed] [Google Scholar]
- Williamson, P. , Kulik, A. , Zachowksi, A. , Schlegel, R.A. & Devaux, P.F. (1992) Ca2+ induces transbilayer redistribution of all major phospholipids in human erythrocytes. Biochemistry, 31, 6355–6360. [DOI] [PubMed] [Google Scholar]
- Wood, B.L. , Gibson, D.F. & Tait, J.F. (1996) Increased erythrocyte phosphatidylserine exposure in sickle cell disease: flow‐cytometric measurement and clinical associations. Blood, 88, 1873–1880. [PubMed] [Google Scholar]
- Woon, L.A. , Holland, J.W. , Kable, E.P.W. & Roufogalis, B.D. (1999) Ca2+ sensitivity of phospholipid scrambling in human red cell ghosts. Cell Calcium, 25, 313–320. [DOI] [PubMed] [Google Scholar]
- Zaidi, A. , Barron, L. , Sharov, V.S. , Schoneich, C. , Michaelis, E.K. & Michaelis, M.L. (2003) Oxidative inactivation of purified plasma membrane Ca2+‐ATPase by hydrogen peroxide and protection by calmodulin. Biochemistry, 42, 12001–12010. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Xu, H. , Weihrauch, D. , Jones, D.W. , Jing, X. , Shi, Y. , Gourlay, D. , Oldham, K.T. , Hillery, C.A. & Pritchard, K.A. Jr (2013) Inhibition of myeloperoxidase decreases vascular oxidative stress and increases vasodilation in sickle cell disease mice. Journal of Lipid Research, 54, 3009–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Materials and methods.