

Abstract
The availability of a receptor for theranostic pretargeting approaches was assessed by use of a new click‐chemistry‐based deactivatable fluorescence‐quenching concept. The efficacy was evaluated in a cell‐based model system featuring both membranous (available) and internalized (unavailable) receptor fractions of the clinically relevant receptor chemokine receptor 4 (CXCR4). Proof of concept was achieved with a deactivatable tracer consisting of a CXCR4‐specific peptide functionalized with a Cy5 dye bearing a chemoselective azide handle (N3‐Cy5‐AcTZ14011). Treatment with a Cy7 quencher dye (Cy7‐DBCO) resulted in optically silent Cy7‐[click]‐Cy5‐AcTZ14011. In situ, a >90 % FRET‐based reduction of the signal intensity of N3‐Cy5‐AcTZ14011 [K D=(222.4±25.2) nm] was seen within minutes after quencher addition. In cells, discrimination between the membranous and the internalized receptor fraction could be achieved through quantitative assessment of quenching/internalization kinetics. Similar evaluation of an activatable tracer variant based on the same targeting moiety (Cy5‐S‐S‐Cy3‐AcTZ14011) was unsuccessful in vitro. As such, using the described deactivatable approach to screen membrane receptors and their applicability in receptor‐(pre‐)targeted theranostics can become straightforward.
Keywords: click chemistry, deactivatable, fluorescence, pretargeting, theranostics
Introduction
Pretargeting is a theranostic strategy that has been used to enhance the specific target‐to‐background ratios of (radioisotope‐based) receptor‐mediated imaging and therapeutic agents.1 In this concept, an (unlabeled) pretargeting vector with a high receptor‐targeting affinity is administered and then allowed to circulate until an optimal target‐to‐background ratio is obtained (e.g., a few days for antibodies vs. hours for peptides). Initial targeting is followed by the administration of a secondary vector that has favorable clearance kinetics, but strongly interacts with the initial (receptor‐bound) pretargeting vector. By this approach, effective labeling with minimal side effects can be achieved.1c Recent developments in orthogonal conjugation techniques, often referred to as “click‐chemistry”,2 allow for the formation of a covalent bond between the secondary vector and the initial receptor‐targeting agent.3 Although there is a clear preference for copper‐free “click‐chemistry”, there is still debate about which alkyne system(s) would be most reactive/suitable for in vitro and/or in vivo applications. Common examples are dibenzocyclooctyne (DBCO), bicyclo[6.1.0]nonyne (BCN), difluorocyclooctyne (DIFO), and trans‐cyclooctene (TCO) moieties.2, 4
An important requirement both for direct targeting and for the secondary binding step in a pretargeting concept is the accessibility of the initial pretargeting vector to the extracellular matrix. Receptor internalization that occurs through endogenous endocytosis within the timeframe between administration of the first and the secondary agent can limit the efficiency of this interaction.1b, 5 For that reason, quantification of internalization kinetics is of importance. Internalization itself can be assessed by using directly targeting radiotracers,6 but unfortunately these assays can be cumbersome and time‐consuming. Here the use of fluorescence instead of radioactivity might prove to be more advantageous, because a direct in vitro read‐out and quantification of the receptors exposed to the extracellular matrix can be achieved by using the original pretargeting vector without the need for further manipulation of the cells. Furthermore, fluorescence provides the opportunity to identify the location of the target receptors in living cells: on the membrane, for example, or in the cytoplasm.7
A new deactivatable sensor technique based on the reasoning that dye–dye FRET interactions can be used either to quantify or to silence the fluorescence intensity (Scheme 1) was developed. In contrast to “activatable” approaches for the assessment of receptor internalization,8 this new approach does not require additional enzymatic activation steps (e.g., S−S bond cleavage) for emission of a fluorescence signal at a specific location in the cell (Scheme 1). In the deactivatable setup described here, a non‐cell‐penetrating Cy7 quencher dye decorated with a chemoselective DBCO handle is directed towards a Cy5‐azide‐labeled pretargeting peptide that specifically targets a receptor for which the membranous localization and internalization kinetics can be controlled during in vitro evaluation.9 Chemoselective conjugation between the quencher and the labeled peptide was used to drive deactivation of the (exposed) membranous Cy5‐related fluorescence (Scheme 1 A and C). To highlight the differences between the working mechanisms of the new deactivatable approach and an activatable approach, an activatable tracer variant based on the same targeting moiety was used as a reference (Scheme 1 B and D).
Scheme 1.
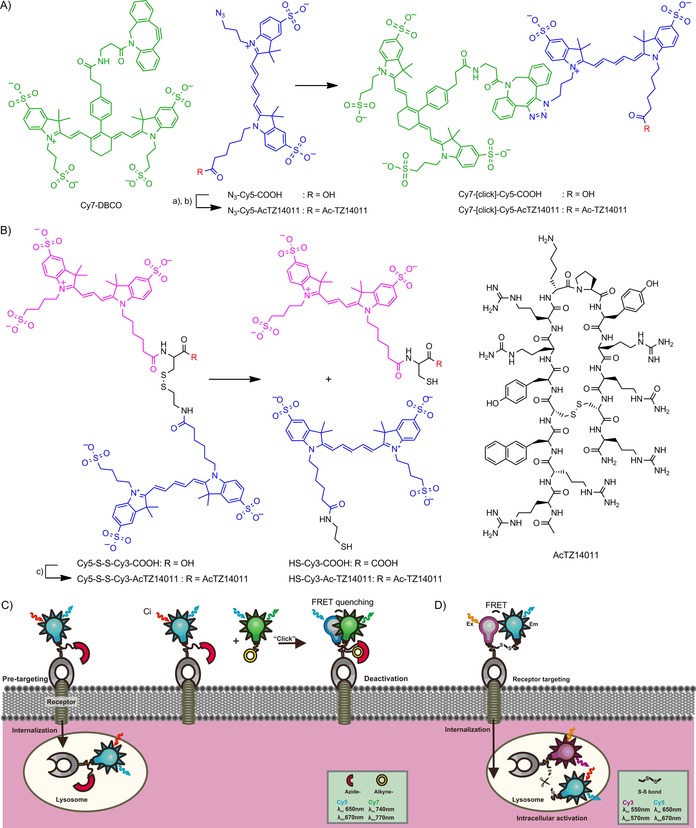
Deactivatable and activatable targeting concepts. A) Structure of the pretargeting agent N3‐Cy5‐AcTZ14011 and the reaction between it and the quencher dye Cy7‐DBCO. a) (PfpO)2O, NMM, DMF; b) Ac‐TZ14011, DMSO/buffer pH 7.4; c) PyBOP, NMM, DMSO. B) Structures of the activatable imaging label Cy5‐S‐S‐Cy3‐COOH and of the receptor‐targeted agent Cy5‐S‐S‐Cy3‐AcTZ14011 and the reaction with reducing agents. The structure of AcTZ14011 is provided in the insert on the right. Schematic overview of the deactivatable concept with C) membrane targeting in the first pretargeting step (targeting moiety in gray, azide in red, and Cy5 in blue), followed by internalization, and Ci) FRET‐based deactivation after addition of the second vector (quencher dye, alkyne in yellow and Cy7 in green). D) Schematic overview of the direct targeting‐based activatable concept with S−S bond cleavage after internalization (Cy3 in purple and Cy5 in blue).
Results and Discussion
Synthesis
The synthesis of N3‐Cy5‐COOH was performed as shown in Scheme 2. The phthalimide‐protected amine‐containing sulfonated indolenine building block 1 was used. With this building block, together with a carboxylic‐acid‐functionalized indolenine, the asymmetric phthalimide‐ and acid‐functionalized Cy5 dye 2 was synthesized by a known solid‐phase method.10 Notably, the synthesized product was of sufficient purity for further use in subsequent chemical steps. After phthalimide removal, the amine functionality of the amine‐ and acid‐functionalized dye 3 was converted into an azide moiety by using the imidazole‐1‐sulfonyl azide diazotransfer reagent,11 yielding the azide‐ and acid‐functionalized N3‐Cy5‐COOH.
Scheme 2.
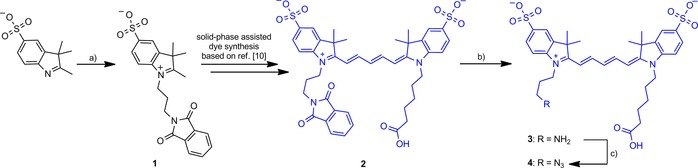
Synthesis of N3‐Cy5‐COOH. a) N‐(3‐Bromopropyl)phthalimide in p‐dichlorobenzene; b) MeNH2 in EtOH/H2O; c) Imidazole‐1‐sulfonyl azide⋅HCl, NMM, CuSO4 (cat.), MeCN/H2O.
Selective receptor targeting was induced by use of the Ac‐TZ14011 peptide. This peptide, which selectively targets chemokine receptor 4 (CXCR4), combines high receptor affinity with high (in vivo) stability and allows easy functionalization through the presence of a single amine group (d‐Lys8).12
Conjugation of N3‐Cy5‐COOH to the Ac‐TZ14011 peptide by using the coupling reagent bis(pentafluorophenyl)carbonate yielded N3‐Cy5‐AcTZ14011. The activatable reference imaging label Cy5‐S‐S‐Cy3‐COOH was synthesized by using a strategy similar to that described by Yang et al.8c (see the Supporting Information for a more detailed description of the synthesis of Cy5‐S‐S‐Cy3‐COOH and Cy5‐S‐S‐Cy3‐AcTZ14011).
Photophysical and (de)activation properties
Prior to assessment of tracer kinetics, the photophysical properties of the new clickable dye were evaluated. An absorption maximum of 647 nm and an associated fluorescence emission maximum of 665 nm (Figure 1 A), as well as a molar extinction coefficient of 2.5×105 m −1 cm−1 in PBS were found.13 The quantum yield of N3‐Cy5‐COOH was 26 %, which is nearly identical to the quantum yield of the Cy5.18 dye that was used as a reference (27 %).14
Figure 1.
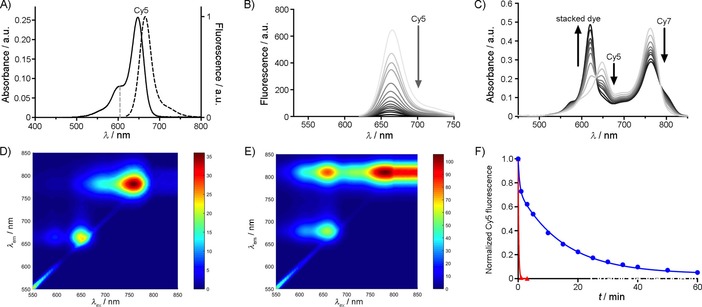
Photophysical properties of N3‐Cy5‐COOH. A) Absorption (—) and emission spectrum (⋅⋅⋅⋅) of N3‐Cy5‐COOH. B) Fluorescence spectrum over time after addition of two equivalents of the Cy7‐DBCO quencher to N3‐Cy5‐COOH until a plateau was reached at 60 min (5 min intervals between measurements). C) Repeated measurement over the course of 60 min of the absorbance spectrum of N3‐Cy5‐COOH after mixing with Cy7‐DBCO (5 min intervals between measurements). 2D fluorescence intensity graphs of D) N3‐Cy5‐COOH prior to addition of Cy7‐DBCO, and E) the Cy7‐[click]‐Cy5‐AcTZ14011 complex at 60 min after addition of Cy7‐DBCO to N3‐Cy5‐COOH. F) Normalized changes in fluorescence intensity over time (60 min) after addition of Cy7‐DBCO to N3‐Cy5‐COOH (•) and N3‐Cy5‐Ac‐TZ14011 (▴).
In situ, the deactivation properties (Figure 1 B–F) were initially analyzed with the unconjugated label N3‐Cy5‐COOH. This helped to avoid solubility issues and charge‐mediated intermolecular interactions that could affect the characterization. Deactivation efficiency and kinetics were assessed after addition of Cy7‐DBCO under click conditions. Addition of Cy7‐DBCO resulted in a decrease in fluorescence of N3‐Cy5‐COOH (Figure 1 B), consistently with the formation of Cy7‐ [click]‐Cy5‐COOH. Formation of the conjugate was accompanied by a slight visual color change and quenching of the Cy5 emission. Consistently with FRET between Cy5 and Cy7, a 90 % decrease in Cy5‐related fluorescence was observed within 90 min, with a fast decrease in signal already visible after 2–5 minutes (Figure 1 C).
Unexpectedly, no FRET‐induced Cy7 fluorescence upon excitation at 620 nm could be observed for Cy7‐[click]‐Cy5‐COOH [phosphate‐buffered saline (PBS), Figure 1 B and C]. The absorption spectrum of Cy7‐[click]‐Cy5‐COOH revealed a blue‐shifted absorbance peak upon formation of the deactivatable construct that was different from the absorbance peaks of N3‐Cy5‐COOH and Cy7‐DBCO alone (PBS, Figure 1 C). Combined with the Cy7‐fluorescence quenching, this peak was indicative of the occurrence of π‐stacking.13 To confirm this assumption, the same absorption spectrum was also determined in a solvent that disfavors stacking (DMSO). This resulted in disappearance of the blue‐shifted absorption peak (Figure 1 D) and the appearance of FRET‐induced Cy7 fluorescence (Figure 1 E).
Hence, π‐stacking does not only occur between identical cyanine dye molecules but can also occur between different cyanine dyes.
A significant difference in fluorescence intensity upon quenching of N3‐Cy5‐AcTZ14011 and N3‐Cy5‐CooH with the Cy7‐DBCO quencher was observed. The fluorescence quenching kinetics (Figure 1 F) were at least an order of magnitude faster in the peptide conjugate, resulting in near‐instantaneous fluorescence quenching for the conjugated peptide. Additionally, absorbance spectroscopy revealed significantly broader peaks, a signature of π‐stacking (see Figure S1 in the Supporting Information).
The photophysical properties of the activatable reference tracer are described in Figure S4. For this tracer, a FRET efficiency of 55 % was measured, and addition of dithiothreitol (DTT), a strong thiol‐based reductant, resulted in a 45 % increase in signal intensity for Cy3 and a concomitant decrease in the Cy5‐related signal (Figure S4 B and C, 140 min). This confirmed abolition of the FRET effect as a result of cleavage of the disulfide bond (Figure S4 D and E). In line with literature findings, no indications of π‐stacking between the sulfonated Cy3 and Cy5 dyes was observed.13 DTT reduction of the complete activatable tracer Cy5‐S‐S‐Cy3‐AcTZ14011 induced a tenfold increase in Cy3‐fluorescence, similarly to the case of Cy5‐S‐S‐Cy3‐COOH (Figure S4 C), thus indicating a limited effect of the peptide on the FRET label.
Receptor affinity
Evaluation of new fluorescence‐based sensor techniques in clinically relevant receptor‐based model systems should increase the validity of the outcome, and thus their applicability. The typical example of a suitable model receptor used here is CXCR4, a receptor that has been linked to increased aggressiveness and invasiveness in numerous cancer types.15 The validity of this receptor as a theranostic target is underlined by its overexpression in, for example, breast cancer, but also in Ewing sarcoma and multiple melanoma.16 For the latter, theranostic applications based on CXCR4 have recently become available in the form of Pentixather.16a The suitability of CXCR4 as a mechanistic model in the assessment of a new sensor technique for evaluation of receptor availability in pretargeting approaches is further exemplified by its versatile kinetics: internalization of this membrane‐bound receptor occurs upon ligand binding at 37 °C, whereas when cultured at 4 °C this receptor is confined to the cell membrane,9 allowing assessment of different forms of receptor availability and kinetics.
The affinity of N3‐Cy5‐AcTZ14011 for the CXCR4 receptor was assessed in vitro, by using CXCR4‐overexpressing MDAMB231 X4 cells.17 For N3‐Cy5‐AcTZ14011 an almost ideal saturation binding curve was obtained. From these results, a K D of (222.4±25.2) nm could be deducted (Figure S2 A), which is similar to those previously reported for other fluorescent Ac‐TZ14011‐analogues.7a, 16b, 18 The affinity of an activatable variant (Cy5‐S‐S‐Cy3‐AcTZ14011) was shown to be significantly less (K D>500 nm, Figure S2 B). This large difference in affinity can be attributed to the difference in size between the imaging labels in the activatable tracer variant and N3‐Cy5‐AcTZ14011, it having previously been shown that the size of the imaging label can negatively affect overall tracer properties.19
Deactivation in vitro
Fluorescence confocal microscopy was used to monitor both the membrane binding and the internalization of N3‐Cy5‐AcTZ14011 conjugates (Figure 2). Pretargeting with N3‐Cy5‐AcTZ14011 at 4 °C (conditions that support membranous localization of the receptor) resulted in a homogeneous staining of the cell membrane (Figure 2 A i, with Cy5‐fluorescence in red). After addition of Cy7‐DBCO and subsequent formation of Cy7‐[click]‐Cy5‐AcTZ14011 (Scheme 1), a fast decrease in membranous signal intensity was seen in the first 5 minutes [(31.4±5.2) %]. Over the course of 30 minutes a total decrease of (79.4±2.8) % was seen (Figure 2 A ii). During this period the intrinsic CXCR4‐linked green fluorescent protein (GFP) signal, and thus the amount of CXCR4 present on the membrane, was not altered (Figure S3). No decrease in Cy5‐related fluorescence was seen when Cy7‐DBCO was added to cells that were prestained with a “non‐click” reference tracer (Cy5‐Ac‐TZ14011,20 Figure 2 B, in blue), thus confirming the bioorthogonal click reaction in Cy7‐[click]‐Cy5‐AcTZ14011.
Figure 2.
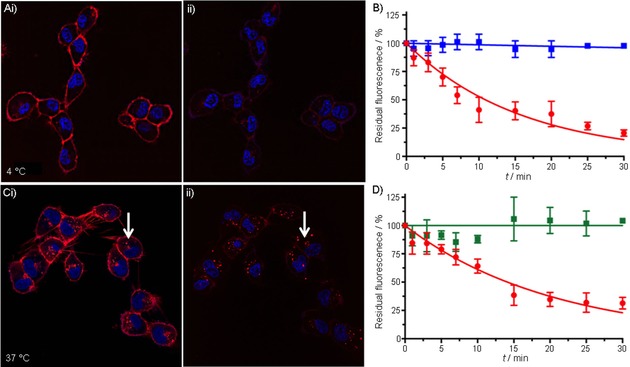
Confocal‐imaging‐based assessment of membrane availability and internalization kinetics. A) Confocal images of CXCR4‐expressing MDAMB231 X4 cells i: after 1 h incubation with N3‐Cy5‐AcTZ14011 at 4 °C, and ii: 30 min after subsequent addition of Cy7‐DBCO. B) Quantified membrane‐related Cy5‐fluorescence of N3‐Cy5‐AcTZ14011 (•) or non‐azide‐containing Cy5‐AcTZ14011 (▪) over the course of 30 min after addition of Cy7‐DBCO. C) Confocal images of MDAMB231 X4 cells i: after 1 h incubation with N3‐Cy5‐AcTZ14011 at 37 °C, and ii: 30 min after subsequent addition of Cy7‐DBCO (arrows indicate internalized tracer⋅receptor complex located in lysosomes in the cytoplasm). D) Quantified membrane‐related (•) and internalized (▪) N3‐Cy5‐AcTZ14011 fluorescence over the course of 30 min after addition of Cy7‐DBCO. For confocal images: Cy5‐signal in red, nuclear staining in blue.
Incubation at 37 °C (conditions that support receptor internalization) resulted in internalization of N3‐Cy5‐AcTZ14011 (Figure 2 C i). Again, rapid quenching of the membrane‐related fluorescence signal was observed after addition of Cy7‐DBCO [(21.0±4.1) % after 5 and (68.4±5.2) % after 30 min, Figure 2 C ii, Cy5 in red], whereas the Cy5 fluorescence intensity of the internalized fraction remained unchanged (Figure 2 Ci i and D). Thus, this approach not only allowed evaluation of the availability of receptor–tracer complexes at the membrane for a secondary functionalization step, but also allowed quantitative assessment of the internalized fraction of N3‐Cy5‐AcTZ14011 (Figure 2 D). With this setup, receptors with fast internalization kinetics could be deemed less suitable for pretargeting approaches or could be used to synchronize application of the second vector to a timeframe in which the availability of the receptor and initial vector is high enough.
Recently Knorr and colleagues showed a similar but reversed concept, in which a bioorthogonal handle was used to “turn on” rather than decrease fluorescence.21 Although this could be an interesting approach for evaluation of membrane receptor expression, this approach does not allow assessment of receptor kinetics because fluorescence was decoupled from the non‐internalizing second‐step vector, leaving the internalized fraction unstained. It could, in theory, be visualized by using an activatable approach. Unfortunately, though, fluorescence confocal experiments with the activatable agent Cy5‐S‐S‐Cy3‐AcTZ14011 were unsuccessful (Figure S5). Although addition of the tracer to the cells resulted in some membrane staining, this staining did not decrease over time, and no intracellular increase in Cy3 emission (indicating internalization and subsequent cleavage of the S−S bond) was seen. This observation is in contradiction with previous reports that show the ability of targeted activatable tracers to become fluorescent after endosomal processing.22 Because it had been shown that S−S bond cleavage was feasible in situ (Figure S4), the absence of an intracellular signal can most likely be attributed to a lack of internalization of the agent as a result of the chemical composition (e.g., relative size, charge distribution, or lipophilicity) of the imaging label. Perhaps in the future the two approaches could complement each other. This would, however, require a different design for the activatable component.
By demonstrating that a fluorescence‐based deactivatable “click‐chemistry” approach allows superior mapping of the internalization kinetics of CXCR4 under different conditions, we have generated a diagnostic approach that can be used to assess the validity of a plurality of membrane receptor targets for pretargeting applications. The incorporation of fluorescence in pretargeting approaches has, however, been described previously.7b, 23 We have used the DBCO moiety as a model alkyne system that proved to be sufficiently reactive in the presented study setup. As such, it is highly likely that the same setup would be equally, or even more, effective when using alkyne systems with faster reaction kinetics, such as BCN.4a Obviously the presented deactivatable concept is also likely to serve as an assay for pretargeting mechanisms that are not based on “click‐chemistry” but work through molecular recognition (e.g., streptavidin/biotin or bifunctional antibodies).
Conclusion
The presented click‐chemistry concept allowed quantitative assessment of membrane‐bound CXCR4 receptors available for pretargeting. By demonstrating that receptor availability varied at different incubation temperatures, our findings underline how the presented deactivatable approach might be used as a future tool for the selection of membrane receptors and their compatibility with (in vivo) pretargeting theranostic approaches.
Experimental Section
General: All chemicals and solvents were obtained from commercial sources and used without further purification. HPLC was performed with a Waters HPLC system, a 1525EF pump, and a 2489 UV/VIS detector. For preparative HPLC a Dr. Maisch, GmbH (Ammerbuch‐Entringen, Germany) Reprosil‐Pur 120 C18‐AQ 10 μm (250×20 mm) column was used (12 mL min−1), whereas for semipreparative HPLC a Dr. Maisch, GmbH Reprosil‐Pur C18‐AQ 10 μm (250×10 mm) column was used (5 mL min−1). Analytical HPLC was performed with a Dr. Maisch, GmbH Reprosil‐Pur C18‐AQ 5 μm (250 4.6 mm) or a Dr. Maisch, GmbH Reprosil‐Pur C18‐AQ 5 μm (250×10 mm) column with a gradient of trifluoroacetic acid (TFA, 0.1 %) in H2O/CH3CN 95:5 to TFA (0.1 %) in H2O/CH3CN 5:95 over 40 minutes (1 mL min−1) being employed. Mass spectrometry was performed with a Bruker microflex MALDI‐TOF instrument, α‐cyano‐4‐hydroxycinnamic acid as matrix, and granuliberin R as internal standard. UPLC/MS was performed with a Waters Acquity UPLC‐MS system, an Acquity UPLC photodiode array detector, and a SQ Detector mass spectrometer. Here a flow rate of 0.5 mL min−1 was used [Waters BEH C18 130 Å 1.7 μm (100×2.1 mm) column]. NMR spectra of the new dye and of phthalimidopropyl‐sulfoindolenine were obtained with a Bruker AV 400 or 500 spectrometer (400 MHz 1H NMR or 500 MHz 1H NMR, respectively) and the chemical shifts were measured relative to tetramethylsilane (TMS). Absorption spectra were recorded with an Ultrospec 3000 spectrometer (Amersham, Pharmacia Biotech) and subtraction of a solvent blank. Fluorescence measurements were performed with a PerkinElmer LS 55 fluorescence spectrometer equipped with a red‐sensitive photomultiplier tube (PMT). The fluorescence properties of dyes were determined according to published procedures.13 Chloromethyl polystyrene resin (1 % DVB, 200–400 mesh, 1.6–1.8 mmol g−1) was obtained from TCI chemicals. Cy7‐DBCO quencher was purchased from Jena Bioscience, Germany.
3‐Phthalimidopropyl trimethylindolenine (1): A mixture of potassium trimethylsulfoindolenine (1.5 g, 5.4 mmol), N‐(3‐bromopropyl)phthalimide (4.5 g, 16.8 mmol, 3 equiv), and tetrabutylammonium iodide (0.27 mmol, 0.05 equiv) in 1,2‐dichlorobenzene (15 mL) was heated to 100 °C for 18 h, followed by 3 h at 150 °C. The crude indolenine slurry was subsequently precipitated in Et2O (250 mL). The majority of the supernatant was removed, and from the remaining suspension the precipitate was collected by centrifugation (560 g) and washed twice with Et2O. Initial purification of the crude building block was performed by silica column chromatography (MeOH/CH2Cl2 10→20 %). Further purification of this material by silica column chromatography (MeOH/EtOAc 25 %) and precipitation from Et2O yielded the title compound as a pale yellow solid (0.8 g, 1.8 mmol, 34 % yield). R f=0.38 (MeOH/EtOAc 25 %); 1H NMR (400 MHz, CD3OD): δ=7.93–7.73 (m, 4 H; Phth‐Ar‐H), 7.65–7.59 (m, 1 H; Ar‐H), 7.56 (d, J=1.7 Hz, 1 H; Ar‐H), 6.64 (d, J=8.3 Hz, 1 H; Ar‐H), 3.74 (t, J=7.4 Hz, 2 H; N‐CH2‐), 3.67 (t, J=7.1 Hz, 2 H; N‐CH2‐), 2.10–2.97 (m, 2 H; CH2‐CH 2‐CH2), 1.34 ppm (s, 6 H; C‐(CH3)2); HRMS (MALDI‐TOF): m/z calcd for C22H23N2O5S: 427.13 [M+H]+; found: 426.65. The characteristic peak of the indolenine 2‐methyl moiety was not observed, presumably due to proton exchange with the deuterated methanol.
Synthesis of Cy5‐(SO3)phthalimidyl‐(SO3)COOH dye 2: The synthesis of Cy5‐(SO3)phthalimidyl‐(SO3)COOH was based on a slightly adapted variant of the procedure previously described by Lopalco et al.10 In brief, carboxypentyl indolenine (940 mg, 2 mmol, 2 equiv) and malonaldehyde dianilide hydrochloride (540 mg, 2.2 mmol, 2.2 equiv) were dissolved in AcOH/Ac2O (1:1, v/v, 15 mL) and subsequently heated to 120 °C for 2 h. After having cooled to room temperature, the now dark‐colored mixture was precipitated in Et2O (300 mL) and the hemicyanine was collected as a brown precipitate. This was then washed once with Et2O and twice with EtOAc. The crude hemicyanine was dissolved in DMF (50 mL) and added to previously prepared Merrifield‐resin‐bound 4‐aminophenol (750 mg resin, 1 mmol amine moieties, 1 equiv) in a polypropylene vessel (75 mL) with frit and mixed with N2 bubbling for 1 h. The resin was repeatedly washed with DMF (50 mL) and CH2Cl2 (50 mL) until all the brown coloration was eluted. Next, 1 (210 mg, 0.5 mmol, 0.5 equiv) and pyridine/Ac2O (3:1, v/v, 12 mL) were added to the washed resin and the suspension was mixed for 2 h. The resulting deep blue filtrate was separated from the beads by filtration, and the beads were washed once more with DMF. The collected filtrate fractions were combined and precipitated from Et2O (300 mL) to afford the crude dye as a dark blue solid (250 mg) that was used directly in the next reaction step. A small amount was purified by preparative HPLC for analysis. Analytical HPLC t R=29.9 min. 1H NMR (400 MHz, [D6]DMSO): δ=8.30–8.43 (m, 2 H; cyanine bridge CH‐CH‐CH‐CH‐CH‐CH), 7.92–7.79 (m, 6 H; 4×phthalimide C−H+aryl), 7.59–7.67 (m, 2 H; aryl), 7.33–7.41 (m, 2 H; aryl), 6.48 (t, J=12.4 Hz, 1 H; CH‐CH‐CH‐CH‐CH‐CH), 6.24–6.38 (m, 2 H; cyanine bridge CH‐CH‐CH‐CH‐CH‐CH), 4.22 (br t, 2 H; N‐CH 2‐CH2‐CH2‐NPhth), 4.12 (br t, J=6.8 Hz, 2 H; N‐CH 2‐CH2‐CH2‐CH2‐CH2‐COOH), 3.71 (t, J=7.2 Hz, 2 H; N‐CH2‐CH2‐CH 2‐NPhth), 2.21 (t, J=7.2 Hz, 2 H; N‐CH2‐CH2‐CH2‐CH2‐CH 2‐COOH), 2.00–2.10 (m, 2 H; N‐CH2‐CH 2‐CH2‐NPhth), 1.71 (2×s+m, 14 H; 4×indolenine CH3+N‐CH2‐CH 2‐CH2‐CH2‐CH2‐COOH), 1.62–1.48 (m, 2 H; N‐CH2‐CH2‐CH2‐CH 2‐CH2‐COOH), 1.33–1.43 ppm (m, 2 H; N‐CH2‐CH2‐CH 2‐CH2‐CH2‐COOH); HRMS (MALDI‐TOF): m/z calcd for C42H46N3O10S2: 816.26 [M+H]+; found: 816.00.
Synthesis of Cy5‐(SO3)amine‐(SO3)COOH dye 3: Compound 2 (100 mg) was dissolved in a methylamine solution in EtOH (33 %, 8 mL), to which water (1 mL) was added. The solution was allowed to stir overnight, yielding a golden brown solution. Excess methylamine was removed by N2 bubbling, followed by rotary evaporation. The now blue residue was dissolved in MeOH and precipitated in Et2O. The blue precipitate was collected by precipitation and was purified by preparative HPLC. After MS analysis, the fractions containing product were pooled and lyophilized to yield the title compound as a blue solid (25 mg, 36 μmol, 18 % yield over two steps from the dye building blocks). Analytical HPLC t R=24.6 min. 1H NMR (500 MHz, [D6]DMSO): δ=8.34–8.46 (m, 2 H; cyanine bridge CH‐CH‐CH‐CH‐CH‐CH), 7.85 (2×d J=11.4 Hz, 2 H; aryl), 7.67–7.74 (br m, 2 H; NH2), 7.63–7.67 (m, 2 H; aryl), 7.33–7.41 (m, 2 H; aryl), 6.57 (t, J=12.3 Hz, 1 H; cyanine bridge CH‐CH‐CH‐CH‐CH‐CH), 6.26–6.41 (2×d, J=13.8 Hz, 2×1 H; cyanine bridge CH‐CH‐CH‐CH‐CH‐CH), 4.11–4.21 (m, 4 H; N‐CH 2‐CH2‐CH2‐NH3+ N‐CH 2‐CH2‐CH2‐CH2‐CH2‐COOH), 2.86–2.95 (m, 2 H; N‐CH2‐CH2‐CH 2‐NH2), 2.21 (t, J=7.2 Hz, 2 H; N‐CH2‐CH2‐CH2‐CH2‐CH 2‐COOH), 1.90–2.04 (m, 2 H; N‐CH2‐CH 2‐CH2‐NH2), 1.71 (2×s+m, 14 H; 4×indolenine CH3+ N‐CH2‐CH 2‐CH2‐CH2‐CH2‐COOH), 1.50–1.60 (m, 2 H; N‐CH2‐CH2‐CH2‐CH 2‐CH2‐COOH) 1.32–1.42 ppm (m, 2 H; N‐CH2‐CH2‐CH 2‐CH2‐CH2‐COOH); HRMS (MALDI‐TOF): m/z calcd for C34H44N3O8S2: 686.26 [M+H]+; found: 686.43.
Synthesis of N3‐Cy5‐COOH (4): Compound 3 (25.0 mg, 36 μmol) was dissolved in a mixture of H2O (3 mL) and MeCN (1 mL) and the pH was adjusted to approximately 8 with 4‐methylmorpholine (NMM) . A catalytic amount of CuSO4 (1 mg) was added, followed by imidazole‐1‐sulfonyl azide⋅HCl11 (15 mg, 72 μmol, 2 equiv). The reaction mixture was stirred for 2 h, after which a second portion of imidazole‐1‐sulfonyl azide⋅HCl (5.0 mg, 24 μmol, 0.7 equiv) was added and stirring was continued for 30 minutes. The reaction mixture was then purified by preparative HPLC after addition of KCl (20 mg) and subsequently lyophilized to yield the title product as a blue solid (9.0 mg, 12 μmol, 33 % yield). Analytical HPLC t R=29.1 min. 1H NMR (400 MHz, [D6]DMSO, with added ethylene carbonate (4 mm) as internal standard13): δ=8.30–8.45 (m, 2 H; cyanine bridge CH‐CH‐CH‐CH‐CH‐ CH), 7.83 (2×d, J=9.1 Hz, 2 H; aryl), 7.62–7.67 (m, 2 H; aryl), 7.30–7.38 (m, 2 H; aryl), 6.60 (t, J=12.3 Hz, 1 H; cyanine bridge CH‐CH‐CH‐CH‐CH‐CH), 6.26–6.42 (2×d, J=13.7 Hz, 2×1 H; cyanine bridge CH‐CH‐CH‐CH‐CH‐CH), 4.05–4.20 (m, 4 H; N‐CH 2‐CH2‐CH2‐N3+ N‐CH 2‐CH2‐CH2‐CH2‐CH2‐COOH), 3.49 (under water peak, 2 H; N‐CH2‐CH2‐CH 2‐N3), 2.21 (t, J=7.2 Hz, 2 H; N‐CH2‐CH2‐CH2‐CH2‐CH 2‐COOH), 1.90–2.05 (m, 2 H; N‐CH2‐CH 2‐CH2‐N3), 1.71 (2×s+m, 14 H; 4×indolenine CH3+N‐CH2‐CH 2‐CH2‐CH2‐CH2‐COOH), 1.50–1.60 (m, 2 H; N‐CH2‐CH2‐CH2‐CH 2‐CH2‐COOH), 1.32–1.43 ppm (m, 2 H; N‐CH2‐CH2‐CH 2‐CH2‐CH2‐COOH); HRMS (MALDI‐TOF): m/z calcd for C34H44N3O8S2: 712.25 [M+H]+; found: 712.89.
Synthesis of N3‐Cy5‐AcTZ14011 (5): Compound 4 (3.6 mg, 4.8 μmol) was dissolved in dry DMF (250 μL), after which NMM (5 μL), followed by bis(pentafluorophenyl)carbonate (4 mg, 10 μmol), were added in order to form the active ester. This mixture was stirred for 1 h. The crude activated dye was obtained after precipitation in tert‐butyl methyl ether (MTBE, 10 mL) and was subsequently spun down in a centrifuge. The pellet was washed with EtOAc (10 mL) and was used directly for coupling to the targeting peptide without further purification. A stock solution of this dye‐Pfp ester was made in DMSO (20 mg mL−1); of this, 50 μL (1 μmol) was added to Ac‐TZ14011 (1 μmol, 5 mg). This was then diluted in a mixture of phosphate buffer (pH 8.0, 50 mm, 1.5 mL) and acetonitrile (400 μL) and stirred for 16 h. The reaction mixture was then acidified with a drop of TFA, purified by semipreparative HPLC (100 min gradient), and lyophilized to yield the title compound as a blue solid (0.7 mg, 250 nmol, 5 % yield). Purity was >93 % as assayed by UPLC/MS at 220 nm. Analytical HPLC t R=28.5 min. HRMS (ESI): m/z calcd for C126H185N40O26S4: 934.11 [M+3 H]3+; found: 934.26; m/z calcd for C128H186F3N40O28S4: 972.11 [M+3 H+TFA]3+; found: 972.44; m/z calcd for C126H186N40O26S4: 700.83 [M+4 H]4+; found: 701.50; m/z calcd for C130H186F6N40O30S4: 1514.65 [M+2 H+2 TFA]2+; found: 1514.80.
Photophysical properties of N3‐Cy5‐COOH and N3‐Cy5‐AcTZ14011 with Cy7‐DBCO quencher: Cy7‐DBCO in DMSO (800 μm, 7.5 μL) was added to a solution of N3‐Cy5‐COOH in PBS (100 μm, 30 μL), and the resulting mixture was gently shaken for 60 minutes. The resulting solution was diluted 100‐fold in DMSO or PBS and a 2D fluorescence heat map was generated and compared with those of freshly prepared mixtures of N3‐Cy5‐COOH (1 μm in PBS or DMSO) and Cy7‐DBCO (800 μm in DMSO, 7.5 μL). Absorbance spectra of the four samples were also measured. The quenching rates were also investigated by fluorescence measurements with solutions of N3‐Cy5‐COOH or N3‐Cy5‐AcTZ14011 in PBS (1 μm, 3 mL) after addition of Cy7‐DBCO in DMSO (800 μm, 7.5 μL, 2 equiv). Fluorescence and absorbance measurements were performed at 5 min intervals.
Cells: MDAMB213 X4 breast cancer cells of human origin, in which overexpression of the CXCR4 receptor was acquired after transfection with a GFP‐tagged version of the human CXCR4 gene, were kindly provided by Dr. Gary D. Luker (University of Michigan, Ann Arbor, US).17 Cells were cultured under standard culture conditions in Gibco's minimum essential medium (MEM) enriched with fetal bovine serum (10 %) and penicillin/streptomycin (Life Technologies, Inc.)
Flow cytometry: Cells were trypsinized and aliquoted (3×105 cells per sample). For saturation binding experiments, different concentrations ranging between 0 and 2000 nm in PBS (50 μL) were added. Cells were incubated with either of the tracers for one hour at 4 °C. Next, the cells were washed twice with BSA in PBS (0.1 %, 300 μL) and resuspended in BSA in PBS (0.1 %, 300 μL). Fluorescence was measured with a FACSCanto II instrument (BD Biosciences) in the APC‐A channel (λ ex 645 nm, λ em 660 nm). Cells were gated on forward and side scatter and intrinsic GFP expression. For each sample, 10 000 gated cells were analyzed, and all experiments were performed in triplicate. The normalized geometric means were fitted with equations in the GraphPad Prism 5 software. The K D values were calculated by using the “Binding—Saturation, One site—Total nonlinear regression” equation according to previously described procedures.5, 7a, 18
Fluorescence confocal imaging: Cells were seeded on glass‐bottomed culture dishes (MatTek corporation) and placed in the incubator overnight. Cells were then incubated with tracer (1 μm) for 1 h either at 4 °C (for assessment of membrane binding) or at 37 °C (for assessment of internalization). After incubation, the cells were washed three times with PBS. Confocal microscopy was performed on viable cells with an SP8WL fluorescence confocal microscope (Leica). Prior to imaging, the nuclei of the cells were stained with Hoechst 33342 (1 mg mL−1 stock, ThermoFisher). Sequential scanning settings were used to visualize all fluorescent features: Hoechst (nuclear staining; λ ex 405 nm, λ em 425–475 nm), intrinsic CXCR4‐coupled GFP (λ ex 488 nm, λ em 525–575 nm), and Cy5 (λ ex 633 nm, λ em 650–700 nm). The Cy7‐DBCO quencher in DMSO stock (800 μm, 1 μL per sample) was added immediately after images had been taken with pretargeting tracer N3‐Cy5‐AcTZ14011 (set at t=0′). For evaluation of staining over time, images were obtained over a period of 6 min at room temperature, with a 2 min time interval for the first 15 min and a 5 min time interval for the remaining 45 min. Experiments under identical conditions with use of the non‐deactivatable reference tracers Cy5‐AcTZ14011 and Cy5‐S‐S‐Cy3‐AcTZ14011 (Supporting Information) were used as controls.
To quantify the signal intensity within the confocal images, pixel intensities were measured with the aid of Fiji software.24 Regions of interest were assigned around the whole cell and within the cell (minimum of four cells per image). To assess the membrane signal, the internal signal was then subtracted from the total signal intensity in the cell. The signal intensity at t=0 was set at 100 %, after which the difference over time was determined relative to t=0. Experiments were performed in triplicate.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was financially supported by a Netherlands Organization for Scientific Research STW‐VIDI grant (grant no. STW BGT11272) and a NWO‐TOP grant (GO 854.10.012), a European Research Council under the European Union′s Seventh Framework Program (FP7/2007–2013) grant (no. 2012–306890), and the 2015–2016 Post‐Doctoral Molecular Imaging Scholar Program Grant granted by the Society of Nuclear Medicine and Molecular Imaging (SNMMI) and the Education and Research Foundation for Nuclear Medicine and Molecular Imaging.
S. van der Wal, C. M. de Korne, L. G. L. Sand, D. M. van Willigen, P. C. W. Hogendoorn, K. Szuhai, F. W. B. van Leeuwen, T. Buckle, ChemBioChem 2018, 19, 1758.
References
- 1.
- 1a. Boerman O. C., van Schaijk F. G., Oyen W. J., Corstens F. H., J. Nucl. Med. 2003, 44, 400–411; [PubMed] [Google Scholar]
- 1b. Goldenberg D. M., Chang C. H., Rossi E. A., McBride W. J., Sharkey R. M., Theranostics 2012, 2, 523–540; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Goldenberg D. M., Sharkey R. M., Oncogene 2007, 26, 3734–3744. [DOI] [PubMed] [Google Scholar]
- 2. Debets M. F., van Hest J. C., Rutjes F. P., Org. Biomol. Chem. 2013, 11, 6439–6455. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Jewett J. C., Bertozzi C. R., Chem. Soc. Rev. 2010, 39, 1272–1279; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Rossin R., Robillard M. S., Curr. Opin. Chem. Biol. 2014, 21, 161–169. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Murrey H. E., Judkins J. C., Am Ende C. W., Ballard T. E., Fang Y., Riccardi K., Di L., Guilmette E. R., Schwartz J. W., Fox J. M., Johnson D. S., J. Am. Chem. Soc. 2015, 137, 11461–11475; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Notni J., Wester H. J., Chem. Eur. J. 2016, 22, 11500–11508. [DOI] [PubMed] [Google Scholar]
- 5. Buckle T., Kuil J., van den Berg N. S., Bunschoten A., Lamb H. J., Yuan H., Josephson L., Jonkers J., Borowsky A. D., van Leeuwen F. W., PLoS One 2013, 8, e48324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Liu G., Dou S., Yin D., Squires S., Liu X., Wang Y., Rusckowski M., Hnatowich D. J., Cancer Biother. Radiopharm. 2007, 22, 33–39; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Sharkey R. M., van Rij C. M., Karacay H., Rossi E. A., Frielink C., Regino C., Cardillo T. M., McBride W. J., Chang C. H., Boerman O. C., Goldenberg D. M., J. Nucl. Med. 2012, 53, 1625–1632. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Buckle T., van der Wal S., van Malderen S. J., Muller L., Kuil J., van Unen V., Peters R. J., van Bemmel M. E., McDonnell L. A., Velders A. H., Koning F., Vanhaeke F., van Leeuwen F. W., Theranostics 2017, 7, 624–633; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Lutje S., Rijpkema M., Goldenberg D. M., van Rij C. M., Sharkey R. M., McBride W. J., Franssen G. M., Frielink C., Helfrich W., Oyen W. J., Boerman O. C., Cancer Res. 2014, 74, 6216–6223. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Lee S., Xie J., Chen X., Curr. Top. Med. Chem. 2010, 10, 1135–1144; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Rood M. T., Oikonomou M., Buckle T., Raspe M., Urano Y., Jalink K., Velders A. H., van Leeuwen F. W., Chem. Commun. (Camb.) 2014, 50, 9733–9736; [DOI] [PubMed] [Google Scholar]
- 8c. Yang J., Chen H., Vlahov I. R., Cheng J. X., Low P. S., Proc. Natl. Acad. Sci. USA 2006, 103, 13872–13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van den Berg N. S., Buckle T., Kuil J., Wesseling J., van Leeuwen F. W., Clin. Transl. Oncol. 2011, 4, 234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lopalco M., Koini E. N., Cho J. K., Bradley M., Org. Biomol. Chem. 2009, 7, 856–859. [DOI] [PubMed] [Google Scholar]
- 11. Goddard-Borger E. D., Stick R. V., Org. Lett. 2007, 9, 3797–3800. [DOI] [PubMed] [Google Scholar]
- 12. Hanaoka H., Mukai T., Tamamura H., Mori T., Ishino S., Ogawa K., Iida Y., Doi R., Fujii N., Saji H., Nucl. Med. Biol. 2006, 33, 489–494. [DOI] [PubMed] [Google Scholar]
- 13. van der Wal K. J. S., Valentijn A. R. P. M., van Leeuwen F. W. B., Dyes Pigm. 2016, 132, 7–19. [Google Scholar]
- 14. Mujumdar R. B., Ernst L. A., Mujumdar S. R., Lewis C. J., Waggoner A. S., Bioconjugate Chem. 1993, 4, 105–111. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Müller A., Homey B., Soto H., Ge N., Catron D., Buchanan M. E., McClanahan T., Murphy E., Yuan W., Wagner S. N., Barrera J. L., Mohar A., Verastegui E., Zlotnik A., Nature 2001, 410, 50–56; [DOI] [PubMed] [Google Scholar]
- 15b. Teicher B. A., Fricker S. P., Clin. Cancer Res. 2010, 16, 2927–2931; [DOI] [PubMed] [Google Scholar]
- 15c. Peled A., Tavor S., Theranostics 2013, 3, 34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Herrmann K., Schottelius M., Lapa C., Osl T., Poschenrieder A., Hanscheid H., Luckerath K., Schreder M., Bluemel C., Knott M., Keller U., Schirbel A., Samnick S., Lassmann M., Kropf S., Buck A. K., Einsele H., Wester H. J., Knop S., J. Nucl. Med. 2016, 57, 248–251; [DOI] [PubMed] [Google Scholar]
- 16b. Kuil J., Buckle T., van Leeuwen F. W., Chem. Soc. Rev. 2012, 41, 5239–5261; [DOI] [PubMed] [Google Scholar]
- 16c. Sand L. G., Scotlandi K., Berghuis D., Snaar-Jagalska B. E., Picci P., Schmidt T., Szuhai K., Hogendoorn P. C., Eur. J. Cancer 2015, 51, 2624–2633. [DOI] [PubMed] [Google Scholar]
- 17. Luker K., Gupta M., Luker G., Biotechniques 2009, 47, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuil J., Buckle T., Yuan H., van den Berg N. S., Oishi S., Fujii N., Josephson L., van Leeuwen F. W., Bioconjugate Chem. 2011, 22, 859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bunschoten A., van Willigen D. M., Buckle T., van den Berg N. S., Welling M. M., Spa S. J., Wester H. J., van Leeuwen F. W., Bioconjugate Chem. 2016, 27, 1253–1258. [DOI] [PubMed] [Google Scholar]
- 20. Bunschoten A., Buckle T., Kuil J., Luker G. D., Luker K. E., Nieweg O. E., van Leeuwen F. W., Biomaterials 2012, 33, 867–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Knorr G., Kozma E., Schaart J. M., Nemeth K., Torok G., Kele P., Bioconjugate Chem. 2018, 29, 1312–1318. [DOI] [PubMed] [Google Scholar]
- 22. Kobayashi H., Choyke P. L., Acc. Chem. Res. 2011, 44, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cohen R., Stammes M. A., de Roos I. H., Stigter-van Walsum M., Visser G. W., van Dongen G. A., EJNMMI Res. 2011, 1, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Nat. Methods 2012, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary