

Abstract
Maintenance of cellular proteostasis relies on efficient clearance of defective gene products. For misfolded secretory proteins, this involves dislocation from the endoplasmic reticulum (ER) into the cytosol followed by proteasomal degradation. However, polypeptide aggregation prevents cytosolic dislocation and instead activates ill‐defined lysosomal catabolic pathways. Here, we describe an ER‐to‐lysosome‐associated degradation pathway (ERLAD) for proteasome‐resistant polymers of alpha1‐antitrypsin Z (ATZ). ERLAD involves the ER‐chaperone calnexin (CNX) and the engagement of the LC3 lipidation machinery by the ER‐resident ER‐phagy receptor FAM134B, echoing the initiation of starvation‐induced, receptor‐mediated ER‐phagy. However, in striking contrast to ER‐phagy, ATZ polymer delivery from the ER lumen to LAMP1/RAB7‐positive endolysosomes for clearance does not require ER capture within autophagosomes. Rather, it relies on vesicular transport where single‐membrane, ER‐derived, ATZ‐containing vesicles release their luminal content within endolysosomes upon membrane:membrane fusion events mediated by the ER‐resident SNARE STX17 and the endolysosomal SNARE VAMP8. These results may help explain the lack of benefits of pharmacologic macroautophagy enhancement that has been reported for some luminal aggregopathies.
Keywords: endolysosomes, ER‐phagy, ER‐to‐lysosome‐associated degradation, LC3 lipidation, proteasome‐resistant aggregates
Subject Categories: Autophagy & Cell Death; Membrane & Intracellular Transport; Post-translational Modifications, Proteolysis & Proteomics
Introduction
About 40% of the eukaryotic cell's proteome is synthesized in the ER (Uhlen et al, 2015). Protein folding is rather inefficient (Schubert et al, 2000; Vabulas & Hartl, 2005) and maintenance of cellular proteostasis, i.e., the capacity to produce the proteome in appropriate quality and quantity requires continuous removal from biosynthetic compartments of polypeptides that fail to attain the native structure. Paradoxically, the ER does not contain catabolic devices in its lumen. Misfolded ER clients are dislocated in the cytosol for clearance by the ubiquitin‐proteasome system (UPS; Guerriero & Brodsky, 2012; Pisoni & Molinari, 2016). Yet, misfolding may in certain cases cause polypeptide aggregation that impairs dislocation and UPS intervention. The disease‐causing polymerogenic E342K (ATZ) variant of alpha1 antitrypsin (AT) (Teckman & Perlmutter, 2000; Kamimoto et al, 2006; Kroeger et al, 2009), various serpin mutants (Kroeger et al, 2009), the E90K mutant of the gonadotropin‐releasing hormone receptor (GnRHR; Houck et al, 2014), the β subunits of thyrotrophic hormone (Noda & Farquhar, 1992), proalpha1(I) chains of type I collagen (Ishida et al, 2009), and the L1341P mutant of dysferlin (Fujita et al, 2007) are reported cases of proteasome‐resistant misfolded proteins generated in the ER and degraded with the contribution of the lysosomal system. Core components of the macroautophagy machinery required for membrane‐tethering of the cytosolic ubiquitin‐like LC3 and/or GABARAP proteins (e.g., ATG5, ATG7, or VPS34/BCN1) are involved in lysosomal clearance of these proteasome‐resistant polypeptides (Teckman & Perlmutter, 2000; Kamimoto et al, 2006; Fujita et al, 2007; Ishida et al, 2009; Kroeger et al, 2009; Gelling et al, 2012; Houck et al, 2014). Originally, LC3 lipidation was reported to occur at the phagophore membrane, as an early event in biogenesis of double‐membrane autophagosomes regulating macroautophagy (Suzuki et al, 2007; Mizushima et al, 2011). It has therefore been inferred that macroautophagy regulates clearance of proteasome‐resistant aggregates from the ER, as it does for cytosolic aggregates (Lamark & Johansen, 2012; Bento et al, 2016; Hurley & Young, 2017; Menzies et al, 2017). However, luminal aggregates are shielded by the ER membrane from the macroautophagy machinery that operates in the cell cytosol. Thus, an intervention of macroautophagy in clearance of luminal aggregates would imply their dislocation across the ER membrane, or the capture of ER portions containing them by autophagosomes (Marciniak et al, 2016). This latter option is particularly intriguing in view of the recent identification of ER‐resident, LC3‐binding proteins proposed to selectively label ER subdomains for lysosomal clearance via processes collectively defined as ER‐phagy (Khaminets et al, 2015; Fumagalli et al, 2016; Grumati et al, 2017; Fregno & Molinari, 2018; Loi et al, 2018; Smith et al, 2018). Conventional macroautophagy inducers such as rapamycin and starvation enhance clearance of cytosolic inclusions (Lamark & Johansen, 2012; Bento et al, 2016; Hurley & Young, 2017; Menzies et al, 2017) and of some proteasome‐resistant, large polypeptides misfolding in the ER lumen such as proalpha1(I) chains of type I collagen (Ishida et al, 2009) and mutant dysferlin (Fujita et al, 2007). For ATZ and other serpin polymers and for the E90K GnRHR, however, lysosomal clearance in cellular and animal disease models is not affected, or it is actually impaired, on conventional macroautophagy activation upon mTOR inhibition (Teckman et al, 2002; Kroeger et al, 2009; Hidvegi et al, 2010; Houck et al, 2014). Thus, it can be speculated that macroautophagic (rapamycin‐responsive) and non‐macroautophagic (rapamycin‐unresponsive) catabolic pathways operate in mammalian cells to back‐up the UPS for clearance of compartmentalized aggregates. In such a scenario, the LC3 lipidation machinery that intervenes in clearance of rapamycin‐insensitive substrates could do so in a non‐canonical fashion (Bestebroer et al, 2013; Ktistakis & Tooze, 2016). To explore this notion, we monitored the fate of proteasome‐resistant ATZ polymers, whose lysosomal clearance is not enhanced by rapamycin or nutrient deprivation.
Results
ATZ polymers are delivered to LAMP1/RAB7‐positive endolysosomes for clearance
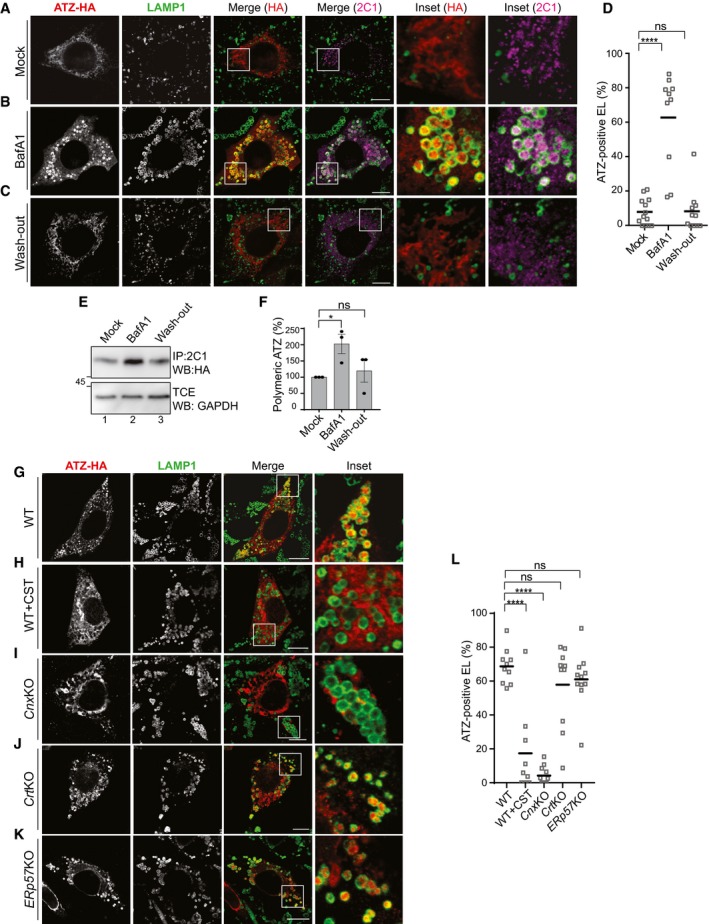
To understand lysosomal clearance of proteasome‐resistant ATZ polymers, we first identified the degradative organelle by transiently inhibiting lysosomal activity with bafilomycin A1 (BafA1; Klionsky et al, 2008). Confocal laser scanning microscopy (CLSM) of mouse embryonic fibroblasts (MEF) expressing HA‐tagged ATZ shows that in cells exposed to BafA1 ATZ accumulates in endolysosomes (EL, as defined in Huotari & Helenius, 2011) that display LAMP1 [Fig 1A (mock‐treated cells) vs. B (cells exposed to BafA1) and D] and RAB7 (Fig EV1) at the limiting membrane. The ATZ accumulating in inactive endolysosomes is decorated both with the anti‐HA (Fig 1B, Inset HA) and with the 2C1 antibody (Inset 2C1), which is specific for the polymeric form of ATZ (Fig EV2A; Miranda et al, 2010). The wash‐out of BafA1 readily re‐establishes the clearance of polymeric ATZ as shown by the disappearance of the ATZ‐specific signal in the LAMP1‐positive organelles (Fig 1C, Insets, D). Biochemical analyses where the intracellular level of ATZ polymers immunoisolated with the 2C1 antibody from cell lysates is checked in Western blots (WB) confirm the increase in the polymer level on cell exposure to BafA1 (Fig 1E, lane 1 vs. 2, F) and its return to steady‐state level 4 h after BafA1 wash‐out (Fig 1E, lane 3, F). All in all, LAMP1‐positive endolysosomes are the degradative organelles, where polymeric ATZ is delivered from the ER for clearance. These organelles are also visible in non‐transfected cells [e.g., cells in the upper left and right corners, Fig 1B, panels LAMP1, Merge (HA), and Merge (2C1)]. This shows that their formation is not induced by the luminal expression of proteasome‐resistant misfolded polypeptides such as ATZ.
Figure 1. Reversible accumulation of polymeric ATZ within LAMP1‐positive endolysosomes on acute lysosomal inhibition.
-
AIntracellular localization of total (HA) and polymeric ATZ (2C1) in WT MEF mock‐treated, confocal laser scanning microscopy (CLSM).
-
BSame as (A) for MEF exposed to 50 nM BafA1 for 12 h.
-
CSame as (B), 4 h after BafA1 wash‐out.
-
DQuantification of ATZ‐positive, LAMP1‐positive endolysosomes (EL) (n = 13, 10, 11 cells, respectively). One‐way ANOVA and Dunnett's multiple comparisons test, ns P > 0.05, ****P < 0.0001.
-
EATZ polymers immunoisolated from lysates of WT MEF mock‐treated (lane 1), incubated for 12 h with BafA1 (lane 2) and 4 h after BafA1 wash‐out (lane 3). Immunoprecipitation (IP) of ATZ polymers with polymer‐specific 2C1 antibody, transfer on PVDF membrane, revealed with anti‐HA antibody on Western blot (WB).
-
FQuantification of (E), n = 3, mean ± SEM. Unpaired two‐tailed t‐test, ns P > 0.05, *P < 0.05.
-
G–KSame as (B) in WT MEF, in cells exposed to 20 mM CST and in Cnx‐, Crt‐, and ERp57‐KO MEF.
-
LQuantification of ATZ‐positive EL (n = 10, 9, 11, 10, 11 cells, respectively). One‐way ANOVA and Dunnett's multiple comparisons test, ns P > 0.05, ****P < 0.0001.
Figure EV1. Endolysosomes display LAMP1 and GFP‐RAB7 at the limiting membrane.
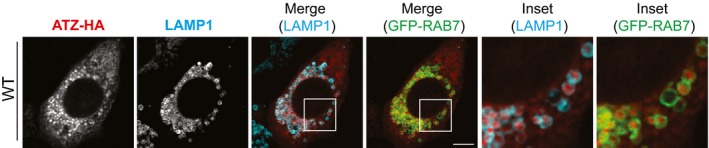
CLSM analysis showing LAMP1 (cyan) and GFP‐RAB7 (green) co‐localization at the limiting membrane of endolysosomes containing ATZ‐HA (red). Scale bar: 10 μm.
Figure EV2. ATZ, but not NHK, forms 2C1‐positive polymers.
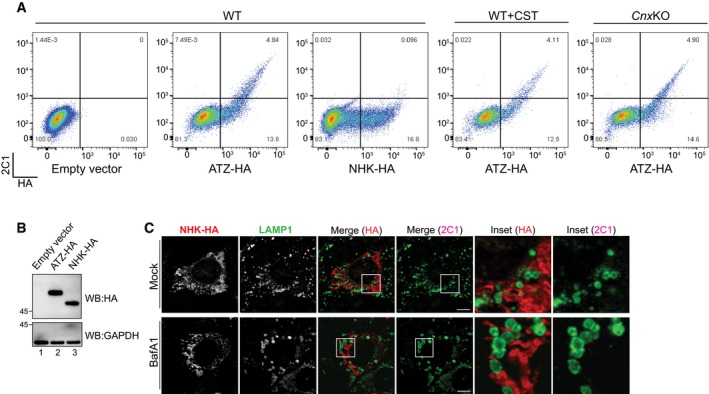
- Flow cytofluorimetric analyses to assess HA (x‐axis) and 2C1 (y‐axis) immunoreactivity in MEF mock‐transfected (first panel), expressing ATZ‐HA (second panel) or NHK‐HA (third panel). HA/2C1 double staining is revealed only in ATZ‐HA‐expressing cells. Same as panel 2 in CST‐treated and in CnxKO MEFs (fourth and fifth panels, respectively).
- Control of ATZ‐HA (lane 2) and NHK‐HA (lane 3) expression by WB with anti‐HA antibodies (upper panel). Loading control (lower panel).
- CLSM analyses showing distribution of the ERAD substrate NHK in WT MEF (upper panels). On BafA1 treatment (lower panels), NHK does not accumulate in LAMP1‐positive endolysosomes. Scale bars: 10 μm.
Source data are available online for this figure.
Another disease‐causing variant of the SERPINA1 gene, the folding‐defective AT variant NHK, is also retained in the ER lumen. At steady state, NHK and ATZ are present at similar intraluminal levels based on the intensities of the HA signal determined in cytofluorimetry and by WB of total cell lysates (Fig EV2A and B). However, in contrast to ATZ (Fig EV2A, second panel), NHK does not form polymers as revealed by the absence of 2C1 immunoreactivity (third panel). NHK also does not accumulate within LAMP1‐positive endolysosomes on lysosomal inactivation (Fig EV2C, BafA1). In fact, NHK is eventually dislocated across the ER membrane to be degraded by cytosolic proteasomes (Liu et al, 1999). Thus, delivery to endolysosomes for clearance is not a common fate of misfolded proteins generated in the ER, most of which are degraded by ER‐associated degradation (ERAD; Guerriero & Brodsky, 2012; Pisoni & Molinari, 2016). It rather occurs to remove proteasome‐resistant species (Teckman & Perlmutter, 2000; Kamimoto et al, 2006; Fujita et al, 2007; Ishida et al, 2009; Kroeger et al, 2009; Gelling et al, 2012; Houck et al, 2014), polymeric ATZ being a paradigmatic example.
Delivery of polymeric ATZ to endolysosomes requires the ER lectin CNX
The involvement of ER‐resident chaperones in selection of folding‐defective polypeptides for ERAD is well understood (Guerriero & Brodsky, 2012; Pisoni & Molinari, 2016). In contrast, their involvement in selection of proteasome‐resistant aberrant proteins for lysosomal clearance is not studied. The only exception, to the best of our knowledge, shows that pharmacologic inactivation of the calnexin (CNX)/calreticulin (CRT)/ERp57 chaperone system with the plant alkaloid castanospermine (CST; Elbein, 1991; Hebert et al, 1995) prevents lysosomal delivery and disposal of GnRHRE90K (Houck et al, 2014). The membrane‐bound lectin chaperone CNX, its soluble and functional homolog CRT, and the associated oxidoreductase ERp57 catalyze oxidative and reductive reactions during polypeptide folding and unfolding in the ER (Caramelo & Parodi, 2007). It has therefore been speculated that their intervention in lysosomal clearance is required to attain a GnRHRE90K structure competent for lysosomal delivery (Houck et al, 2014).
As reported for GnRHRE90K, CST also abolishes delivery of ATZ to LAMP1‐positive endolysosomes (Fig 1G vs. H and L). Our tests in cells lacking CNX reveal defective delivery of ATZ to the endolysosomes (Fig 1I and L). Ablation of CRT (Fig 1J and L) or ERp57 (Fig 1K and L) has in contrast no consequence. We have extensively studied ER‐chaperone‐assisted protein biogenesis and clearance. The folding machinery is highly redundant and pharmacologic inactivation of the CNX/CRT/ERp57 chaperone system on cell exposure to CST modestly impact on folding efficiency for most cellular proteins (Denzel et al, 2002; Molinari et al, 2004, 2005; Pieren et al, 2005; Soldà et al, 2006, 2007). On the same line, less invasive approaches, such as the ablation of either CNX or CRT or ERp57, are efficiently compensated in culture cells by the activation of surrogate pathways (Soldà et al, 2006). For that reason, we consider unlikely that the block of ATZ delivery to the endolysosomes on CST treatment, which is recapitulated in cells lacking CNX, results from a specific involvement of CNX in attainment of a delivery‐competent ATZ structure. Consistently, the generation of ATZ polymers is unperturbed in cells exposed to CST or lacking CNX (Fig EV2A, fourth and fifth panels, respectively). Rather, we hypothesize a role of CNX as a membrane‐bound receptor that segregates ATZ polymers in ER subdomains to be delivered to endolysosomes for clearance.
ATZ expression favors formation of a CNX:FAM134B:LC3II complex
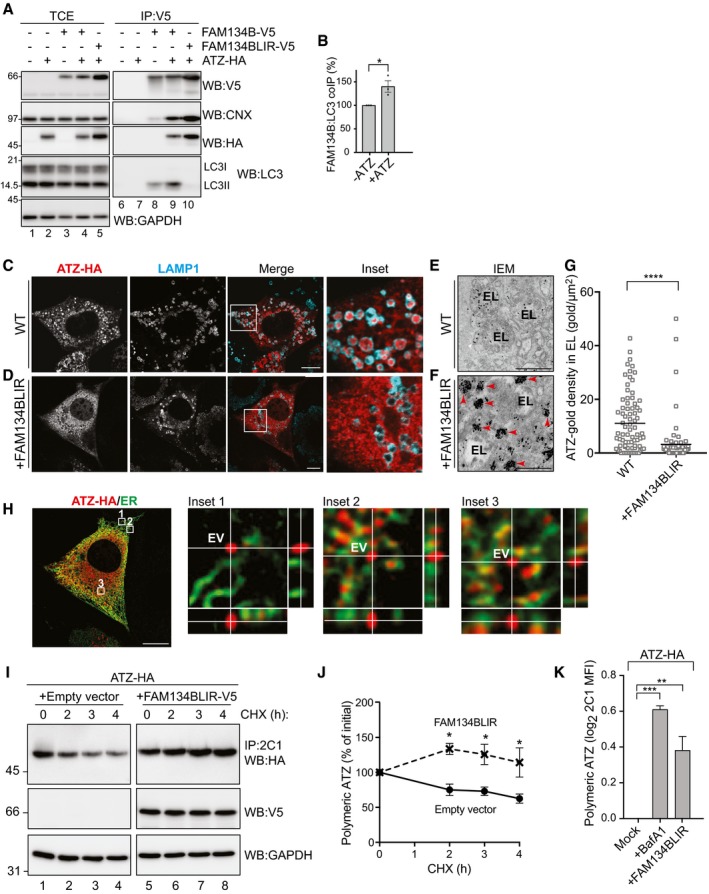
Our hypothesis that CNX participates in a membrane receptor to segregate ATZ polymers from the ER lumen is corroborated by recent literature indicating CNX as an interactor of FAM134B (Grumati et al, 2017). FAM134B is a recently characterized ER‐phagy receptor (i.e., an ER‐resident, LC3‐binding protein involved in ER fragmentation and delivery of ER subdomains to LAMP1‐positive endolysosomes for clearance; Fregno & Molinari, 2018; Loi et al, 2018). The biological significance of CNX association with FAM134B is not known, as this interaction is not required for FAM134B‐regulated, ER stress‐induced, or starvation‐induced ER‐phagy (unpublished and Fumagalli et al, 2016; Khaminets et al, 2015). Immunoisolation of epitope‐tagged FAM134B from lysates of HEK293 cells confirms the association of endogenous CNX and of endogenous LC3II (Fig 2A, lane 8). Significantly, the co‐expression of ATZ‐HA substantially enhances the co‐precipitation of endogenous CNX and LC3II with V5‐tagged FAM134B (Fig 2A, lane 9, B). FAM134BLIR, where the LC3‐binding function has been inactivated on replacement of six residues in the cytosolic LC3‐interacting region (LIR) of FAM134B (‐453DDFELL458‐ to ‐453AAAAAA458‐), also associates with ATZ and CNX but, as expected, it does not engage LC3 (Fig 2A, lane 10). Under these experimental conditions, i.e., ectopic expression of FAM134BLIR, endolysosomal delivery of ATZ is blocked (Fig 2C vs. D). Our analyses by immuno‐electron microscopy (IEM, immunogold‐labeled ATZ, Fig 2E–G) and by CLSM (Fig 2H) reveal that ATZ is released in ER‐derived vesicles (red arrowheads in Fig 2F, EV in the insets of Fig 2H and see below). However, the ATZ‐containing EV remain dispersed in the cytosol, thus failing to release their content within endolysosomes (Fig 2C vs. D, E vs. F and G). As a consequence, clearance of polymeric ATZ is substantially delayed as determined by a cycloheximide (CHX) chase (Fig 2I and J), thus resulting in intracellular accumulation of ATZ polymers at levels comparable to those obtained by cell exposure to BafA1 as quantified by cytofluorimetry (Fig 2K). All in all, the LC3‐binding function of FAM134B is dispensable for formation of a complex with CNX and ATZ and for the generation of ATZ‐containing EV, but it is required to target EV to endolysosomes where their content must be released for clearance. We postulate, and show below, that FAM134B association with LC3 promotes EV docking to the endolysosomal membrane that precedes membrane:membrane fusion events eventually leading to delivery of polymeric ATZ within endolysosomes for clearance.
Figure 2. Disposal of polymeric ATZ requires a functional LIR domain of FAM134B.
-
AHEK293 cells transfected with empty vector (lanes 1, 6), ATZ‐HA (2, 7), FAM134B‐V5 (3 and 8), FAM134B‐V5 and ATZ‐HA (4, 9), or FAM134BLIR‐V5 and ATZ‐HA (5, 10), incubated for 6 h with 100 nM BafA1 and then treated with the cross‐linker DSP before lysis as described in Materials and Methods. Lanes 1–5, WB of the total cell extract (TCE); lanes 6–10, WB of anti‐V5 immunoprecipitates to isolate complexes containing ectopically expressed FAM134B or FAM134BLIR. The membranes were probed with anti‐V5 (upper panels), anti‐CNX, anti‐HA, and anti‐LC3 antibodies.
-
BQuantification of LC3 co‐precipitating with FAM134B‐V5 (A, lanes 8 and 9). Mean ± SEM, n = 3, unpaired two‐tailed t‐test, *P < 0.05.
-
CSame as Fig 1B, WT MEF. Scale bar: 10 μm.
-
DEctopic expression of FAM134BLIR in WT MEF inhibits ATZ delivery to endolysosomes. Scale bar: 10 μm.
-
E, FDistribution of gold‐labeled ATZ‐HA by IEM in BafA1‐treated WT MEF and WT MEF overexpressing FAM134BLIR, respectively. EV, ER‐derived vesicles, red arrowheads; EL, endolysosome.
-
GQuantification of ATZ‐gold density of (E, F) (n = 75 and 79 EL, respectively). Unpaired two‐tailed t‐test, ****P < 0.0001.
-
HMax projection of the same cell as in (D) after deconvolution. Insets show orthogonal section of select regions. Scale bar: 10 μm.
-
IDecay of ATZ polymers (CHX chase, upper panel) immunoisolated with the polymer‐specific 2C1 antibody (visualized with anti‐HA in WB) in HEK293 cells mock‐transfected (lanes 1–4) or expressing FAM134BLIR‐V5 (lanes 5–8). Middle panel, expression of FAM134BLIR‐V5 assessed by WB; lower panel, loading control.
-
JQuantification of (I) (Mean ± SEM, n = 3, unpaired two‐tailed t‐test, *P < 0.05).
-
KFlow cytometry analysis of ATZ‐HA polymer levels in MEFs mock‐treated, exposed to BafA1, and co‐expressing FAM134LIR. MFI: mean fluorescence intensity (Mean ± SEM, n = 5, unpaired two‐tailed t‐test, ns P > 0.05, **P < 0.01, ***P < 0.001).
Source data are available online for this figure.
FAM134B is required for delivery of polymeric ATZ to endolysosomes
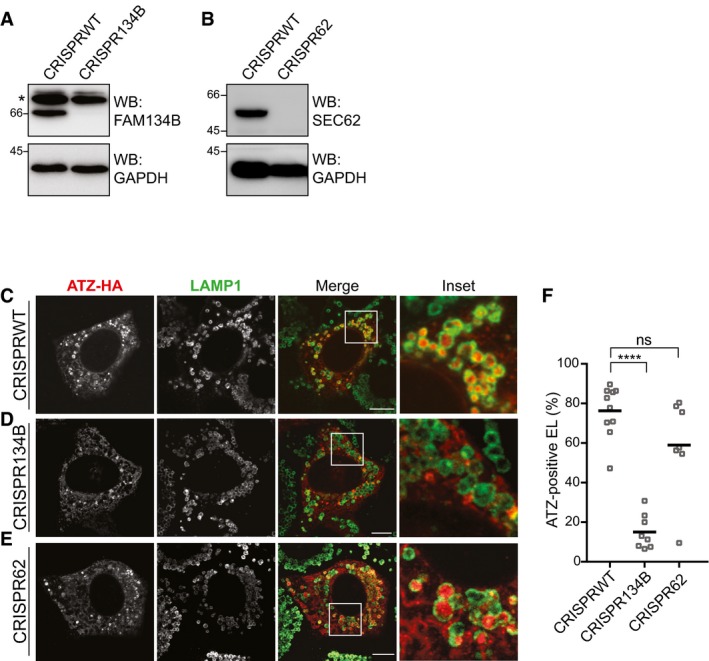
To confirm the role of FAM134B in clearance of proteasome‐resistant polymeric ATZ from the ER lumen, delivery of ATZ polymers within endolysosomes was analyzed in WT MEF and in MEF ablated of FAM134B by use of CRISPR/Cas9 genome editing technology (Fig 3A). CLSM analyses confirm delivery of ATZ in LAMP1‐positive endolysosomes in WT MEF (Fig 3C and F), which is compromised in FAM134B‐ablated cells (Fig 3D and F). Deletion of SEC62 (Fig 3B), another ER‐resident, LC3‐binding protein that regulates ER‐phagy after termination of an ER stress (Fumagalli et al, 2016; Fregno & Molinari, 2018; Loi et al, 2018) does not perturb endolysosomal delivery of ATZ polymers for clearance (Fig 3E and F).
Figure 3. Delivery of polymeric ATZ to LAMP1‐positive endolysosomes and clearance of ATZ polymers requires FAM134B.
-
AWB analysis showing KO efficiency for CRISPR134B MEF. Asterisks, cross‐reacting bands.
-
BSame as (A) for CRISPR62 MEF.
-
C–ESame as Fig 1B in CRISPR WT MEF, CRISPR134B MEF, CRISPR62 MEF, respectively. Scale bars: 10 μm.
-
FQuantification of (C–E) (n = 10, 8, 7 cells, respectively). One‐way ANOVA and Dunnett's multiple comparisons test, ns P > 0.05, ****P < 0.0001.
Source data are available online for this figure.
Delivery of polymeric ATZ to the endolysosomes requires a functional LC3 lipidation machinery
FAM134B associates with lipidated LC3II. The LC3‐binding function of FAM134B is required to deliver ER fragments within autophagolysosomes during ER‐phagy (Khaminets et al, 2015) and for delivery of ATZ polymers to endolysosomes for clearance (Fig 2). To assess whether the LC3 lipidation machinery participates in clearance of ATZ polymers, we monitored their delivery to LAMP1‐positive endolysosomes in MEF with defective LC3 lipidation due to ablation of ATG4B (Marino et al, 2010) or ATG7 (Komatsu et al, 2005; Fig EV3A and B). Indeed, lysosomal delivery of ATZ such as occurs in WT MEF (Fig 4A, Insets, I) is defective in cells lacking ATG4B (Fig 4B and I) or ATG7 (Fig 4C and I). On the same line, SAR405, a specific inhibitor of VPS34 that prevents LC3 lipidation and autophagosome biogenesis (Ronan et al, 2014; Bento et al, 2016), fully blocks delivery of proteasome‐resistant misfolded ATZ from the ER lumen to the endolysosomes (Fig EV4).
Figure EV3. Macroautophagy activity in Atg KO cell lines used in the study.
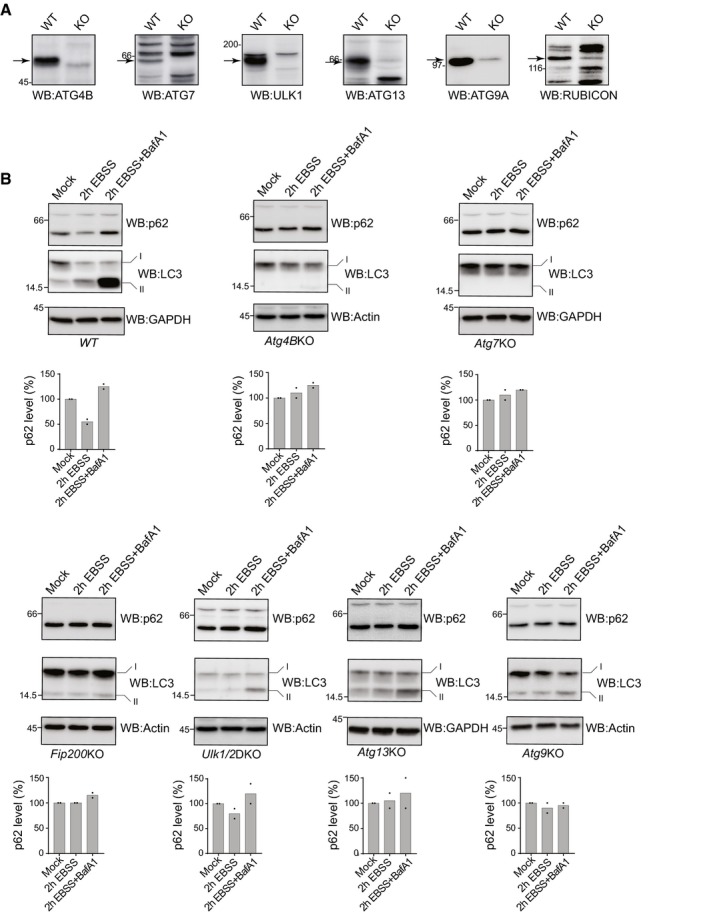
- KO efficiencies were controlled for ATG4B, ATG7, ULK1, ATG13, ATG9A, and RUBICON (arrows).
- Macroautophagy activity on nutrient deprivation is assessed by monitoring variation of the levels of the macroautophagy substrate p62 by WB in WT, Atg4BKO, Atg7KO, Fip200KO Ulk1/2DKO, Atg13KO, Atg9KO MEFs (the values for two independent experiments are given). For each cell line, also LC3 levels are shown. All the controls performed in this figure confirm the results published by the groups sharing these cell lines (cited in the text and in the Acknowledgements section).
Source data are available online for this figure.
Figure 4. Delivery of polymeric ATZ to endolysosomes in cells lacking LC3 lipidation or autophagosome biogenesis.
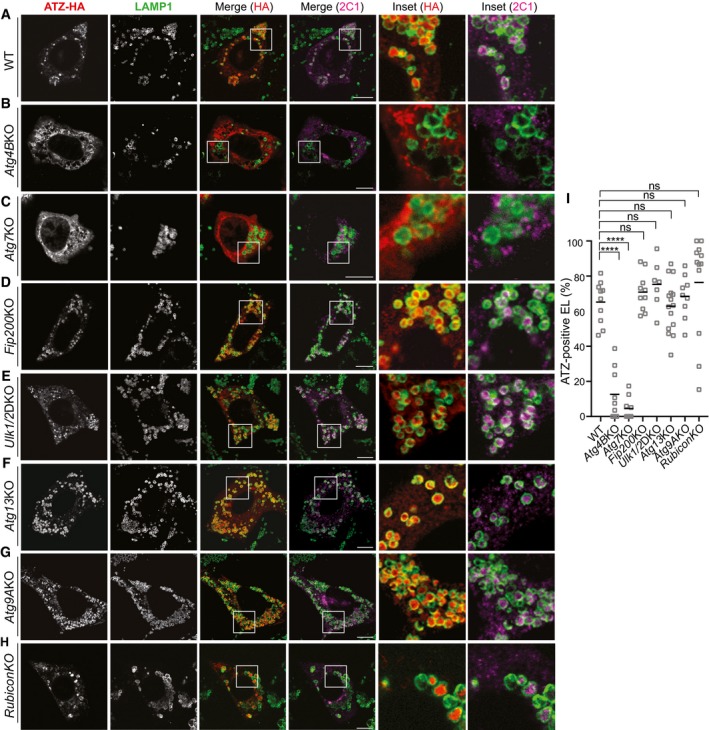
-
A–HIntracellular localization of LAMP1‐positive EL, total (HA), and polymeric (2C1) ATZ in WT MEF exposed to BafA1 for 12 h (A); in Atg4BKO MEF (B); in Atg7KO MEF (C); in Fip200KO MEF (D); in Ulk1/2 double‐KO MEF (E); in Atg13KO MEF (F); in Atg9KO MEF (G); in RubiconKO MEF (H). Scale bars: 10 μm.
-
IQuantifications of (A–H) (n = 10, 9, 9, 10, 7, 15, 9, 12 cells). One‐way ANOVA and Dunnett's multiple comparisons test, ns P > 0.05, ****P < 0.0001.
Figure EV4. VPS34 is required for polymeric ATZ delivery to EL .
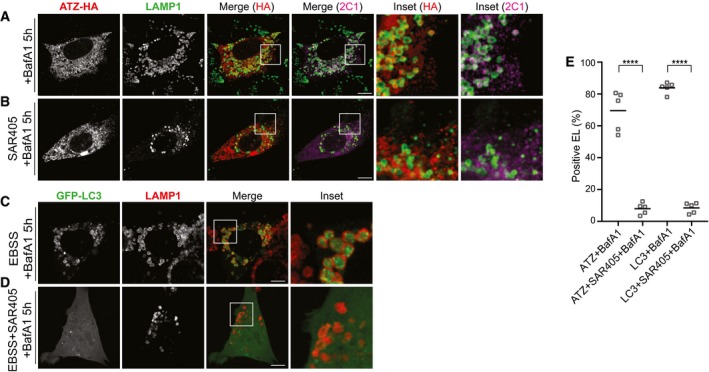
-
A, BIntracellular localization of total (HA) and polymeric ATZ (2C1) in WT MEF exposed to 100 nM BafA1 for 5 h and in cells exposed to 5 μM Sar405 and 100 nM BafA1 for 5 h, respectively.
-
C, DAs control for Sar405 treatment, WT MEF transfected with GFP‐LC3 were treated as in (A and B), respectively, on nutrient deprivation.
-
EQuantification of ATZ‐ or GFP‐LC3‐positive endolysosomes as shown in (A–D) (n = 5). Unpaired two‐tailed t‐test, ****P < 0.0001.
Delivery of polymeric ATZ to the endolysosomes does not require a functional autophagosome biogenesis machinery
So far, our data on endolysosomal delivery of ATZ polymers echo the findings on nutrient deprivation‐induced, FAM134B‐dependent, and LC3‐dependent ER‐phagy. Yet, nutrient deprivation‐induced ER‐phagy critically depends on FIP200 (Khaminets et al, 2015), a core component of the ULK kinase complex that is required for the generation of autophagosomes that eventually capture and deliver ER fragments to the lysosomal compartments (Hara et al, 2008; Hosokawa et al, 2009; Smith et al, 2018). Strikingly, our analyses reveal that the FAM134B‐dependent ATZ delivery to endolysosomes is not affected in cells lacking FIP200 (Fig 4D and I, and EV3B) or other components of the autophagosome biogenesis machinery such as ULK1 and ULK2 (Fig 4E and I, and EV3A and B), ATG13 (Fig 4F and I, and EV3A and B) or ATG9 (Fig 4G and I, and EV3A and B; Saitoh et al, 2009; Shang et al, 2011; McAlpine et al, 2013; Bento et al, 2016; Kaizuka & Mizushima, 2016; Hurley & Young, 2017; Kakuta et al, 2017). Thus, defective autophagosome biogenesis impairs several types of receptor‐mediated ER‐phagy (Khaminets et al, 2015; Grumati et al, 2017; Fregno & Molinari, 2018; Loi et al, 2018; Smith et al, 2018) and conventional macroautophagy (Fig EV3B; Hara et al, 2008; Hosokawa et al, 2009; Kakuta et al, 2017). However, it does not affect delivery of proteasome‐resistant ATZ polymers to endolysosomes for clearance. Dispensability of ULK1/ULK2 (with requirement of the LC3 conjugation machinery) has previously been reported, for example, for LC3‐associated phagocytosis (LAP; Martinez et al, 2015). Like delivery of polymeric ATZ to endolysosomes, LAP does not require the activity of the pre‐initiation complex and autophagosome biogenesis (ULK1/2, ATG13, FIP200 are dispensable for both pathways, the LC3 conjugation complex is required for both pathways). However, dispensability of Rubicon (Fig 4H and I, and EV3A) distinguishes ATZ clearance from LAP.
Vesicular delivery of proteasome‐resistant ATZ from the ER to endolysosomes
To better appreciate the intracellular fate of ATZ, we performed ultrastructural analyses by IEM in WT MEF, which revealed the presence of ATZ in the ER (Fig 5A, C, D and F, Movie EV1), its segregation in ER subdomains (ES, Fig 5D and F, Movie EV1) and in single‐membrane, ER‐derived vesicles (EV) that dock onto endolysosomes (EL) and deliver ATZ within their lumen (Fig 5A, B, E and F, Movie EV1, red asterisks in Fig 5B and E show the opening at the contact sites between EV and EL). Autophagosomes identified in correlative light‐electron microscopy for their double‐membrane heavily decorated with GFP‐LC3 (green fluorescence and immunogold labeling, Fig 5G and insets) do not contain ATZ, which is stained in red (Fig 5G). This is consistent with dispensability of the autophagosome biogenesis machinery for delivery of ATZ to the endolysosomes (Fig 4D–G and I).
Figure 5. Dissecting ATZ delivery to LAMP1‐positive endolysosomes.
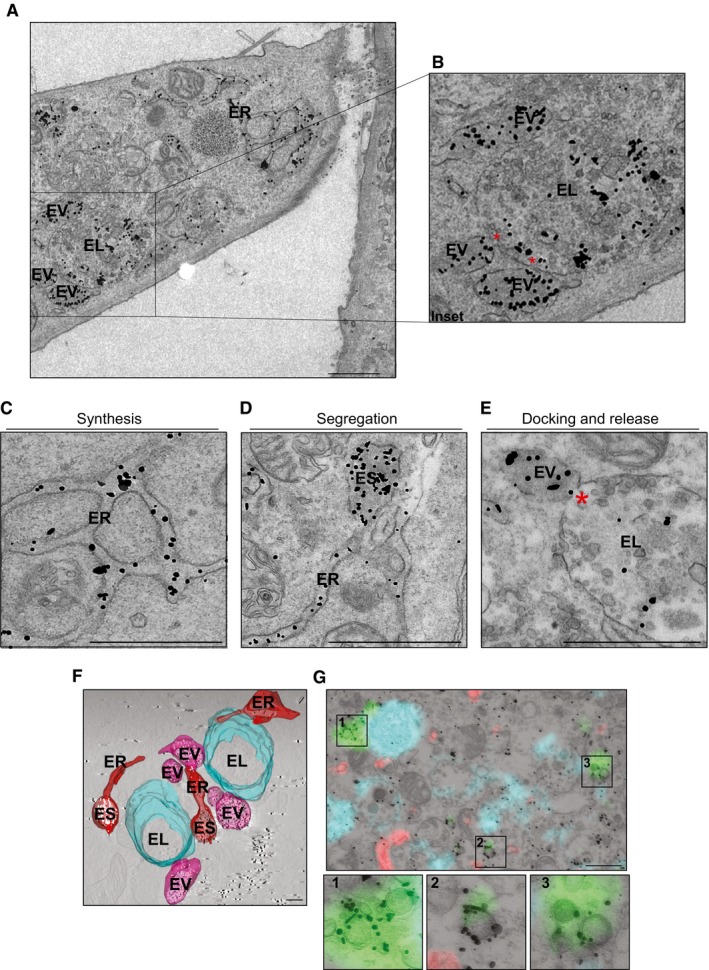
-
ADistribution of gold‐labeled ATZ‐HA by IEM in WT MEF exposed to BafA1 (the cell on the right is not transfected, as specificity control for gold labeling). ER, endoplasmic reticulum; EV, ER‐derived vesicle; EL, endolysosomes. Scale bar: 1 μm.
-
BInset of A. Red asterisks show site of EV:EL membranes fusion.
-
C–EDistribution of gold‐labeled ATZ‐HA by IEM in WT MEF. ES, ER subdomains. Red asterisk shows site of EV:EL membranes fusion. Scale bar: 1 μm.
-
F3D visualization by means of electron tomography of organelles containing gold‐labeled ATZ‐HA (visualized as white dots, see Movie EV1). ES, ER subdomains. Scale bar: 500 nm.
-
GCLEM showing ATZ (red), LAMP1‐positive EL (cyan), and GFP‐derived fluorescence (green) in correspondence to heavily labeled vesicles, which were identified as autophagosomes for their double membrane (insets 1–3). GFP‐LC3 was also stained by immunogold. Scale bar: 1 μm.
ATZ is delivered within endolysosomes in conjunction with ER luminal, but not ER membrane proteins
To elucidate the sequential steps of delivery of proteasome‐resistant polymeric ATZ from the ER lumen to LAMP1‐positive endolysosomes, we set up an original HaloTag pulse‐chase protocol. HaloTag is an inactive bacterial hydrolase, whose active site has been modified to facilitate covalent and irreversible binding of synthetic ligands, including cell‐permeable ligands that are coupled to fluorescent probes (England et al, 2015). We first validated that the HaloTag, which was placed at the N‐terminus of ATZ, did not affect polymerization and lysosomal clearance of ATZ (Dickens et al, 2016 and Fig EV5A). We then employed the following pulse‐chase setup: WT MEF expressing HaloTag‐ATZ were incubated for 30 min with a cell‐permeable “black ligand” (i.e., 6‐Chlorohexanol) to quench fluorescent detection of the pre‐existing pool of HaloTag‐ATZ. Next, we replaced the “black ligand” by a cell‐permeable fluorescent HaloTag ligand to label newly synthesized HaloTag‐ATZ for 15 min (fluorescent pulse). Finally, we washed out the fluorescent ligand and replaced it with the “black ligand” again to interrupt fluorescent labeling of HaloTag‐ATZ (chase). As such, we followed the fate of the cohort of fluorescently pulse‐labeled HaloTag‐ATZ for various chase times: 0, 30, 120, 180, and 300 min (Fig 6A–F). Early upon being synthesized, fluorescently labeled HaloTag‐ATZ co‐localized with the luminal ER marker (superfolder) sfGFP‐KDEL within the ER (Fig 6A and B, M1, M2, 0–30 min of chase and Fig 6G). Interestingly, the co‐localization of HaloTag‐ATZ with sfGFP‐KDEL persisted throughout the 300 min of chase (Fig 6G and H) and was observed in the ER (Fig 6A and B, M1–M3), in the EV (M4) and within the LAMP1‐positive endolysosomes (M5, M5A, M5B, and Fig EV5B). In mock‐transfected cells or in cells expressing the folding‐defective protein NHK, which relies on the UPS for clearance, delivery of sfGFP‐KDEL from the ER into LAMP1‐positive endolysosomes is below detection level (Fig EV5C and D). Thus, accumulation of proteasome‐resistant ATZ in the ER lumen activates (or strongly enhances) a vesicular transport pathway that delivers ATZ polymers and ER luminal content within endolysosomes.
Figure EV5. Various controls.
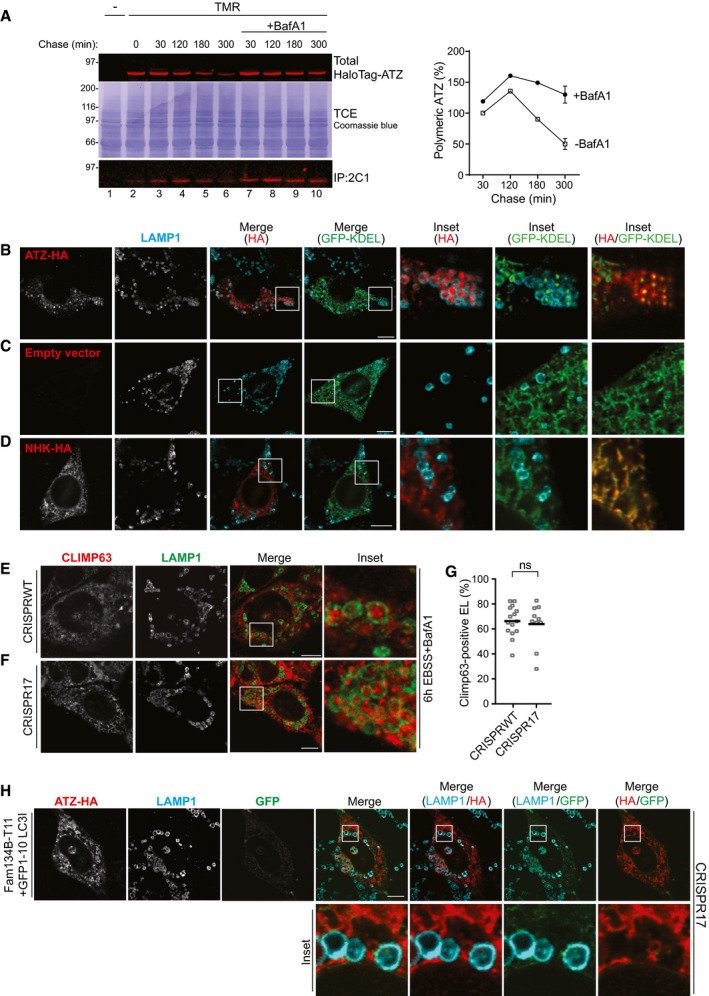
-
AKinetics of total (upper panel) and of polymeric (lower panel, IP:2C1) TMR ligand‐labeled HaloTag‐ATZ clearance in mock‐ (lanes 2–6) and in BafA1‐treated HEK293 cells (lanes 7–10). Quantification, n = 3 for 30’, 180’, 300’, 300’+BafA1. Mean ± SEM.
-
B–DCLSM analyses of BafA1‐treated WT cells showing co‐localization of ATZ‐HA (red) and sfGFP‐KDEL (green) in LAMP1‐positive (cyan) endolysosomes (Inset HA/GFP‐KDEL) (B); same in cells expressing only sfGFP‐KDEL (C) and in cells co‐expressing NHK‐HA and sfGFP‐KDEL (D). Scale bars: 10 μm. A time course is shown in Fig 6A and B.
-
E, FDelivery of the endogenous ER‐phagy marker CLIMP63 within LAMP1‐positive endolysosomes in WT and in CRISPR17 MEF, respectively, exposed to BafA1 on nutrient deprivation. Scale bars: 10 μm.
-
GQuantification of (E, F) (n = 15, 10 cells). Unpaired two‐tailed t‐test, ns P > 0.05, ****P < 0.0001).
-
HGFP reconstitution in ATZ‐HA‐transfected CRISPR17 MEF expressing FAM134B‐T11 and non‐lipidable GFP1–10‐LC3I. Scale bar: 10 μm.
Source data are available online for this figure.
Figure 6. Time course by HaloTag pulse‐chase of ATZ delivery to endolysosomes.
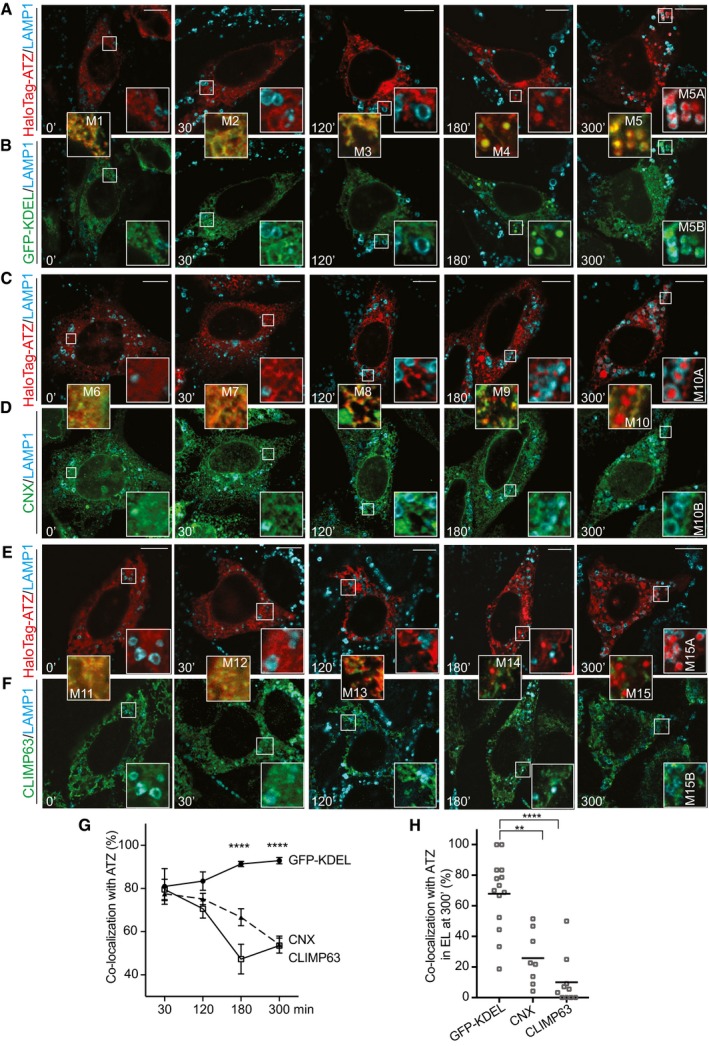
-
A, BTriple staining of cells with LAMP1 (cyan), HaloTag‐ATZ (red), and the luminal ER marker sfGFP‐KDEL (green).
-
C, DSame as (A, B), with CNX as ER membranes marker.
-
E, FSame as (A, B), with CLIMP63 as ER membranes marker.
-
GQuantification of ER markers and HaloTag‐ATZ co‐localization in time (sfGFP‐KDEL n = 10, 10, 12, 13, CNX n = 8, 10, 9, 8, CLIMP63 n = 8, 10, 6, 10 cells for corresponding time points). Mean ± SEM, unpaired two‐tailed t‐test, ****P < 0.0001.
-
HQuantification of double‐positive endolysosomes at 300’ (ATZ:sfGFP‐KDEL n = 14, ATZ:CNX n = 8, ATZ:CLIMP63 n = 10 cells. One‐way ANOVA and Dunnett's multiple comparisons test, **P < 0.01, ****P < 0.0001).
In contrast, co‐localization of HaloTag‐ATZ with markers of the ER membrane such as CNX and CLIMP63 is seen only early after pulse‐labeling, when HaloTag‐ATZ is still in the ER (Fig 6C and D, M6, M7 for CNX; 6E and F, M11, M12 for CLIMP63). At later chase times, co‐localization is progressively lost (Fig 6C and D, M8, M9, M10, M10A, M10B for CNX; Fig 6E and F, M13, M14, M15, M15A, M15B for CLIMP63 and Fig 6G). All in all, ATZ (Fig 6, M5A, M10A, M15A) and sfGFP‐KDEL (Fig 6, M5B) are co‐delivered within LAMP1‐positive endolysosomes. The ER membrane markers CNX (Fig 6, M10B) and CLIMP63 (Fig 6, M15B) are not. These data further highlight the mechanistic distinction with receptor‐regulated ER‐phagy pathways, which are activated by nutrient deprivation or ER stress to deliver ER fragments (i.e., luminal and membrane ER proteins) within lysosomal compartments (Khaminets et al, 2015; Fumagalli et al, 2016; Grumati et al, 2017; Fregno & Molinari, 2018; Loi et al, 2018; Smith et al, 2018).
The ER‐resident SNARE STX17 and the endolysosomal SNARE VAMP8 are required for ATZ delivery to endolysosomes for clearance
The observation of fusion events occurring between single‐membrane‐bound ER‐derived vesicles containing ATZ and endolysosomes (Fig 5) prompted us to assess the involvement in this catabolic pathway of the ER‐resident SNARE STX17, which is involved in smooth ER membrane dynamics and fusion events (Steegmaier et al, 2000) and of the endolysosomal SNARE VAMP8 (Ktistakis & Tooze, 2016). To this end, we generated cells lacking STX17 or VAMP8 (Fig 7A and B) using the CRISPR/Cas9 genome editing technology. In contrast to WT MEF, in which ATZ polymers accumulate in inactive endolysosomes (Fig 7C, Insets, F and G), STX17‐ablated MEF display ATZ‐containing vesicles (EV) that peripherally dock onto the endolysosomal (EL) membrane but fail to release their content in the EL lumen (Fig 7D, Insets, F and H). Likewise, deletion of VAMP8 abolishes ATZ delivery within endolysosomes (Fig 7E, F and I). Notably, starvation‐induced FAM134B‐regulated ER‐phagy proceeds normally in cells lacking STX17 as is evident from the delivery of CLIMP63 into the LAMP1‐positive endolysosomes (Fig EV5E–G).
Figure 7. STX17 and VAMP8 are required for ATZ delivery within endolysosomes.
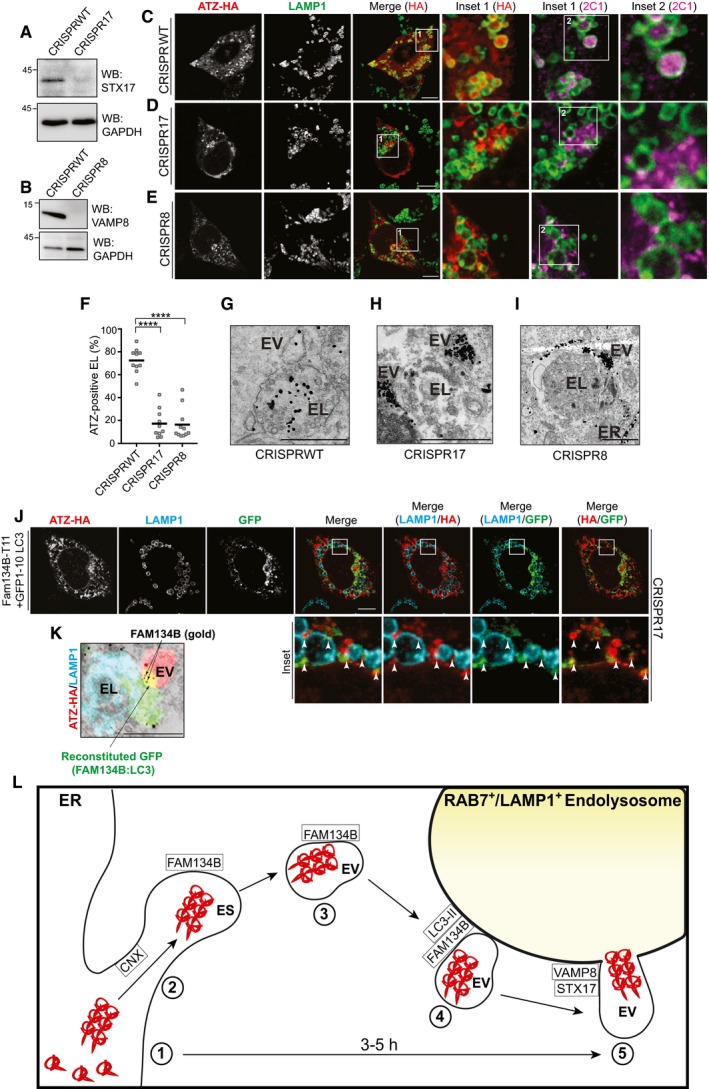
-
AKO efficiency in CRISPR17 MEF.
-
BSame as (A) for CRISPR8.
-
C–ECLSM analysis of ATZ delivery to LAMP1‐positive endolysosomes in BafA1‐treated CRISPRWT MEF (C); in CRISPR17 MEF(D); in CRISPR8 MEF (E). Scale bars: 10 μm.
-
FQuantification of (C–E) (n = 10, 10, 11 cells, respectively). One‐way ANOVA and Dunnett's multiple comparisons test, ****P < 0.0001.
-
G–IDistribution of gold‐labeled ATZ‐HA by IEM in CRISPRWT MEF exposed to BafA1 (G); in CRISPR17 (H); in CRISPR8. (I). ER, endoplasmic reticulum; EV, ER‐derived vesicles; EL, endolysosomes. Scale bars: 1 μm.
-
JGFP reconstitution in ATZ‐HA‐transfected CRISPR17 MEF expressing FAM134B‐T11 and GFP1–10‐LC3. Scale bar: 10 μm.
-
KSame as (J) by CLEM. ATZ‐HA staining in red, LAMP1‐positive endolysosomes in cyan. FAM134B:LC‐induced GFP reconstitution in green. FAM134B in gold is shown. Scale bar: 1 μm.
-
LERLAD schematics. 1. Synthesis and CNX‐independent polymers formation; 2. CNX‐regulated polymers segregation in ER subdomains (ES); 3. FAM134B‐dependent (LIR‐independent) formation of ER‐derived vesicles (EV); 4. FAM134B:LC3II‐mediated EV docking to RAB7/LAMP1‐positive endolysosomes (EL); 5. STX17/VAMP8‐controlled fusion of EV:EL membranes and polymers release in the EL lumen for clearance.
Source data are available online for this figure.
FAM134B:LC3II foci at the ER‐derived vesicles/endolysosomes interface
To monitor the formation of FAM134B:LC3II complexes, we used a bi‐partite split GFP complementation assay (Cabantous et al, 2005; Cieri et al, 2018). We tagged the C‐terminus of FAM134B with the eleventh β strand of GFP (T11) and the N‐terminus of LC3 with the remaining portion of GFP (GFP1–10), which lacks fluorescence ability (Cabantous et al, 2005; Cieri et al, 2018). Foci of GFP reconstitution thus report on active FAM134B:LC3II complexes, which co‐localize with ATZ‐HA and cluster at the limiting membrane of endolysosomes (Fig 7J, arrowheads in Insets). We confirmed by correlative light‐electron microscopy (CLEM) that foci of GFP reconstitution represent functional FAM134B:LC3II complexes (green and yellow signal in Fig 7K) that localize at the interface between ATZ‐containing ER‐derived vesicles (EV, red in Fig 7K) and LAMP1‐positive endolysosomes (cyan in Fig 7K, where FAM134B‐T11 is also labeled with gold). These foci are absent in cells co‐transfected with FAM134B‐T11 and non‐lipidable GFP1–10‐LC3I (Fig EV5H). Thus, we conclude that localization of the FAM134B:LC3II complex at sites where ER‐derived, ATZ transporting vesicles dock onto the endolysosomes is a prerequisite for the ensuing STX17/VAMP8‐mediated fusion.
Discussion
Our analyses reveal that accumulation of proteasome‐resistant ATZ polymers in the ER lumen (Fig 7L, step 1) activates a catabolic process, where ATZ polymers are first segregated into ER subdomains under control of the ER lectin chaperone CNX (step 2) and then in single‐membrane ER‐derived vesicles with intervention of the ER‐resident, LC3‐binding protein FAM134B (step 3). FAM134B association with membrane‐bound LC3II promotes EV docking to RAB7/LAMP1‐positive endolysosomes (step 4). A subsequent membrane:membrane fusion event controlled by the ER‐resident SNARE STX17 and the endolysosomal SNARE VAMP8 releases ATZ polymers within endolysosomes for clearance (step 5). We name this pathway ER‐to‐lysosome‐associated degradation (ERLAD) to distinguish it from catabolic pathways named ER‐phagy, where ER fragments whose production is enhanced upon nutrient deprivation or ER stresses are cleared from cells with the intervention of double‐membrane autophagosomes (Khaminets et al, 2015; Fumagalli et al, 2016; Grumati et al, 2017; Fregno & Molinari, 2018; Loi et al, 2018; Smith et al, 2018). ERLAD shares with one of the ER‐phagy pathways identified so far, namely with starvation‐induced ER sheets turnover (Khaminets et al, 2015), the requirement of the LC3‐binding, ER‐resident protein FAM134B, and of the LC3 lipidation machinery. However, in sharp contrast with ER‐phagy and other mTOR‐dependent catabolic pathways, the autophagosome biogenesis machinery is dispensable for ERLAD. In ERLAD, ER‐derived vesicles containing material to be removed from cells are not captured by autophagosomes but rather fuse with endolysosomes. Intriguingly, “non‐autophagic clearance from the ER of aggregated proteins by direct conversion of ER cisternae into lysosomes” has been reported by Marilyn Farquhar over 25 years ago for aggregated β subunits of thyrotrophic hormone in the mouse thyroid gland (Noda & Farquhar, 1992). We believe that our characterization of the ERLAD pathway supports and offers a mechanistic explanation to these early observations.
Misfolded ATZ associates with FAM134B. The interaction is indirect since FAM134B does not display significant luminal domains and occurs via the FAM134B‐interactor CNX. Apparently, the condensation of ATZ aggregates and the ensuing concentration of FAM134B in ER subdomains activate FAM134B to drive ER fragmentation. In ER‐phagy, ER fragmentation seemingly requires the association of the cytosolic domain of FAM134B with LC3II possibly displayed at the membrane of growing phagophores (Khaminets et al, 2015). Not so in ERLAD, where inactivation of the FAM134B's LIR does not prevent generation of ATZ‐containing ER‐derived vesicles. However, these remain dispersed in the cytosol and fail to dock to and to release their content within endolysosomes. This, and the observation in CLEM that GFP reconstitution on formation of FAM134B:LC3II complexes occurs at the interface between endolysosomes and ATZ‐containing ER‐derived vesicles, lead us to speculate that FAM134B decorates the ER vesicles containing ATZ and that LC3 is lipidated on the target, endolysosomal membrane. How this is regulated is matter of future investigation.
An original HaloTag pulse‐chase protocol used here for the first time allowed to monitor in CLSM the sequence of events that, with a half‐time of about 3 h, lead newly synthesized ATZ to form proteasome‐resistant polymers that are eventually delivered within endolysosomes for clearance (Fig 7L). These data reveal the progressive segregation of ATZ from the ER membrane marker proteins CNX and CLIMP63 that, as an additional difference with FAM134B‐regulated ER‐phagy, are not delivered within endolysosomes. The case of CNX (required for ATZ segregation and delivery within endolysosomes, but not itself transported to the degradative organelles) testifies a regulated dissociation of this lectin chaperone from ERLAD substrates (i.e., ATZ polymers). This reminds the regulated dissociation of CNX from folding‐defective polypeptides to be cleared by ERAD, where CNX selects the misfolded polypeptides and these are eventually handed off to the BiP chaperone system before their dislocation across the ER membrane and proteasomal clearance (Molinari et al, 2005). For ERAD, interruption of misfolded polypeptides association with CNX that precedes clearance from the ER lumen is regulated by the progressive de‐mannosylation of oligosaccharides displayed on misfolded polypeptides (Molinari et al, 2003; Oda et al, 2003; Olivari & Molinari, 2007). For ERLAD, this remains to be established.
There are clear genetic correlations between autophagy impairment and neurodegenerative diseases for which the root cause is the accumulation of proteasome‐resistant aggregates and polymers in the cytosol (Menzies et al, 2017). Excitingly, rapamycin, rapalogs, and other small and non‐small molecules that induce autophagic activity help to boost clearance of aggregates in various cellular and animal models of these types of disease. In fact, these strategies of enhancing autophagy are so promising that they are currently progressing into clinical validation (Menzies et al, 2017). There are also examples of proteasome‐resistant inclusions in the ER lumen such as those formed by procollagen and dysferlin, whose degradation is reportedly enhanced upon macroautophagy activation (Fujita et al, 2007; Ishida et al, 2009). Based on our findings, we would argue that removal of these inclusions is likely regulated by ER‐phagy and relies on autophagosome intervention. In contrast, rapamycin (Kroeger et al, 2009; Hidvegi et al, 2010), fasting (Teckman et al, 2002), or other classical macroautophagy activators are ineffective or even detrimental as therapeutic approaches both in cell lines and in mouse models for ATZ, other serpinopathies, and for the GnRHRE90K (Teckman et al, 2002; Kroeger et al, 2009; Hidvegi et al, 2010; Houck et al, 2014). Our data on vesicular delivery of ATZ polymers from the ER to endolysosomes and on dispensability of autophagosome intervention may explain why strategies that boost macroautophagy fail to induce clearance of ATZ and ATZ‐like aggregates. In this context, the reported benefits on ATZ disposal of pharmacologic [e.g., carbamazepine (Chu et al, 2014; Hidvegi et al, 2010; Kroeger et al, 2009)] or genetic approaches [transcription factor EB (TFEB) gene transfer (Pastore et al, 2013)] should/could be ascribed to ill‐defined consequences of the reduction in the inositol levels as a consequence of carbamazepine exposure, or to the enhanced number and function of lysosomes as a consequence of TFEB expression (Sardiello et al, 2009), respectively, rather than on induction of macroautophagy.
Finally, removal of misfolded proteins from the ER is crucial to maintain proteostasis. It is assumed that most aberrant polypeptides generated in the ER lumen are degraded by cytosolic 26S proteasomes by sequential processes collectively defined as ERAD. ERAD relies on dozens of ER‐resident and cytosolic chaperones and enzymes that sequentially regulate interruption of futile folding attempts in the ER lumen, deliver terminally misfolded polypeptides to translocons at the ER membrane, dislocate them into the cytosol, ensure poly‐ubiquitylation, and proteasomal degradation (Guerriero & Brodsky, 2012; Pisoni & Molinari, 2016). Degradation of aberrant polypeptides that fail to enter the ERAD pathways is much less studied. However, we anticipate similar mechanistic complexity, which is highlighted, as one example, by the substrate‐dependent sensitivity to activators of macroautophagy. Our study paves the way for a better understanding of the pathways ensuring removal of compartmentalized proteasome‐resistant aggregates defined here as ER‐to‐lysosome‐associated degradation (ERLAD). Since aggregate propagation is toxic to tissues and organisms and is linked to an expanding number of severe conformational diseases, these studies will be of medical relevance as they may lead to the identification of novel pharmacological targets.
Materials and Methods
Expression plasmids and antibodies
ATZ was subcloned in pcDNA3.1 expression plasmid with addition of a C‐terminus HA tag or N‐terminus HaloTag. FAM134B‐HA and FAM134BLIR‐HA (DDFELL to AAAAAA) expression plasmids were purchased from GenScript. HA tag was replaced by a V5 tag. Plasmids encoding GFP‐LC3 and sfGFP‐KDEL were a gift from N. Mizushima and E. Snapp, respectively. For the generation of Split GFP construct, FAM134B was tagged at the C‐terminus with a linker (italic) of 28 amino acids followed by the eleventh β strand of GFP (T11,GCGGCCGCCGGCGGACCCGGGAGCGGCGGTGAGGGCTCAGCCGGCGGAGGACCGGTCGGAGGCGGAAGCGGGATATCAGGTTCA GAGAAGAGGGACCACATGGTGCTGCTGGAGTACGTGACCGCCGCCGGCATCACCGACGCCTC) and the N‐terminus of LC3 with the remaining portion of GFP (GFP1–10), which lacks fluorescence ability. Commercial antibodies used in this study were against polymeric ATZ (2C1, HycultBiotech), GAPDH (Merck), HA (Sigma), HA probe F7 (Santa Cruz Biotech.), LAMP1 (Hybridoma Bank, 1D4B was deposited to the DSHB by August, J.T.), LC3 (Sigma), STX17 (Sigma), VAMP8 (Abcam), V5 (Invitrogen), CLIMP63 (Proteintech), p62 (MBL), Actin (Santa Cruz Biotech.), LC3B (Novus), GFP (Abcam), ATG4B (Sigma), ATG7 (Sigma), ATG1/ULK1 (Sigma), ATG13 (Sigma), ATG9A (Sigma), Rubicon (Abcam). Alexa‐conjugated secondary antibodies were purchased from Thermo Fisher Scientific, HRP‐conjugated from Jackson ImmunoResearch, and Protein A‐conjugated from Invitrogen. The anti‐SEC62 was a kind gift from R. Zimmermann, the anti‐CNX from A. Helenius, and the anti‐FAM134B from I. Kurth.
Cell culture, transient transfection, and inhibitors
Mouse embryonic fibroblasts and HEK293 cells were grown in DMEM supplemented with 10% FCS at 37°C and 5% CO2. Transient transfections were performed using JetPrime transfection reagent (PolyPlus) following the manufacturer's protocol. BafilomycinA1 (BafA1, Calbiochem) was used at 50 nM for 12 h if not otherwise specified. Castanospermine (Sigma) used at 20 mM for 4 h as pre‐treatment and then for additional 12 h together with BafA1 50 nM. SAR405 (Sigma) used at 5 μM for 5 h together with BafA1 100 nM.
CRISPR‐Cas9 engineered MEF cell lines
SEC62‐deficient MEF cells (CRISPR62) were engineered using CRISPR/Cas9 system as previously described (Fumagalli et al, 2016). FAM134B, STX17, and VAMP8 KO cells were generated as follows: for construction of the guideRNA‐Cas9 plasmid, lentiCRISPRv2‐puro system (Addgene52961) was obtained from Addgene (http://www.addgene.org). Guide sequences were obtained from the Cas9 target design tools (crispr.mit.edu:8079 and/or http://www.addgene.org/pooled-library). All protocols and information can be found at the website https://www.addgene.org/crispr. The target sequences for guide RNA were synthesized by Microsynth. Two annealed oligonucleotides (5′‐GGGGTCATACTCAGCTACCT‐3′, 5′‐AGGTAGCTGAGTATGACCCC‐3′ for murine FAM134B, 5′‐GCGCTCCAATATCCGAGAAA‐3′, 5′‐TTTCTCGGATATTGGAGCGC‐3′ for murine STX17, 5′‐CCACCTCCGAAACAAGACAG‐3′, 5′‐CTGTCTTGTTTCGGAGGTGG‐3′ for murine VAMP8) were inserted into the lentiCRISPRv2‐puro vector using the BsmBI restriction site. MEF cells were transfected with the plasmid JetPrime (Polyplus) according to the manufacturer's instructions to generate CRISPR134B and CRISPR17 lines. The cells were cultured in DMEM supplemented with 10% FBS. Two days after transfection, the medium was supplemented with 2 μg/ml puromycin. Puromycin‐resistant clones were picked after 10 days, and gene KO was verified by WB.
Protein cross‐linking with DSP
After respective treatment, HEK293 cells were washed with PBS followed by addition of 1 mM DSP (from a 100× solution in dimethylsulfoxide) in PBS. The dishes were incubated with DSP for 30 min at room temperature. The reaction was stopped by adding 1 M Tris, pH 7.8 to a final concentration of 20 mM and incubated for 15 min at room temperature. Cells were washed with PBS containing 20 mM NEM and lysed with RIPA buffer (1% Triton X‐100, 0.1% SDS, 0.5% sodium deoxycholate in HBS, pH 7.4) for 20 min on ice. After a 10 min centrifugation at 10,600 g, PNS were collected and eventually used for IP. SDS‐polyacrylamide gels were run under reducing conditions.
Cell lysis, immunoprecipitation, and Western blot
After the respective treatments, cells were washed with ice‐cold PBS containing 20 mM NEM then lysed with 1% NP‐40 (in HBS pH 7.4) or RIPA buffer (1% Triton X‐100, 0.1% SDS, 0.5% sodium deoxycholate in HBS, pH 7.4) supplemented with protease inhibitors. PNS were collected after centrifugation at 10,600 g for 10 min.
Immuno‐precipitations were performed diluting PNS with respective lysis buffer and incubating it with Protein G beads (VWR, 1:10 w/v, swollen in PBS) and the antibody against the protein of interest or V5‐conjugated beads (Sigma), at 4°C. After three washes of the immunoprecipitates with 0.5% Triton X‐100, beads were denatured for 5 min at 95°C and subjected to SDS–PAGE. Proteins were transferred to PVDF membranes using the Trans‐Blot Turbo Transfer System (Bio‐Rad). Membranes were blocked with 10% (w/v) non‐fat dry milk (Bio‐Rad) in TBS‐T and stained with primary antibodies diluted in TBS‐T followed by HRP‐conjugated secondary antibodies or Protein A diluted in TBS‐T. Membranes were developed using Luminata Forte ECL detection system (Millipore) and signals captured on an Amersham Imager 680 system or LAS4000 (GE Healthcare Life Sciences). Images were quantified with the Multi Gauge Analysis software (Fujifilm). Membrane stripping for probing additional antigens was done using Re‐Blot Plus Strong Solution (Millipore) following the manufacturer's instructions.
Confocal laser scanning microscopy
Mouse embryonic fibroblasts plated on alcian blue‐treated glass coverslips were DMSO or BafA1 treated for 12 h. Cells were then washed twice in PBS and fixed at room temperature for 20 min in 3.7% formaldehyde diluted in PBS. Antigen accessibility was enhanced by 15‐min incubation with permeabilization solution (PS, 0.05% saponin, 10% goat serum, 10 mM HEPES, 15 mM glycine). Cells were incubated with the primary antibodies diluted 1:100 in PS for 90 min, washed for 15 min in PS, and then incubated with Alexa Fluor‐conjugated secondary antibodies diluted 1:300 in PS for 45 min. Cells were rinsed with PS and water and mounted with Vectashield (Vector Laboratories) supplemented with 40,6‐diamidino‐2‐phenylindole (DAPI). Confocal pictures were acquired on a Leica TCS SP5 microscope with a Leica HCX PL APO lambda blue 63.0 × 1.40 OIL UV objective. Figure 2H was acquired with Leica HCS PL APO CS 100 × 1.44 OIL UV objective with a XY pixel size of 37 nm and XZ pixel size of 83 nm and pinhole 0.8 AU. Image was then deconvolved with Autoquant 3.1.1 (Media Cybernetics) with spherical aberration correction. Image analysis and quantification were performed with FIJI (Schindelin et al, 2012) after blinded randomization. Image processing was also done with Photoshop (Adobe).
Flow cytometry
Mouse embryonic fibroblasts were plated in a 12‐well plate and transfected with JetPrime reagent following the manufacturer's protocol. Seventeen hours after transfection, cells were treated with the respective drugs or mock‐treated for 12 h. Cells were detached and collected, washed with PBS, fixed with 3.7% PFA in PBS for 20 min at RT, washed three times in PBS, and permeabilized with saponin solution (5% goat serum, 15 mM glycine, 0.05% saponin, 10 mM HEPES in PBS). Primary antibodies were diluted in saponin solution and added for 1 h at RT, followed by three washes in PBS after which fluorophore‐conjugated secondary antibodies diluted in saponin solution were added for 45 min at RT. Finally, cells were washed three times in PBS, resuspended in MACS buffer (PBS with 2% FCS and 2 mM EDTA), and run on a FACSCanto II flow cytometer (BD Biosciences). Data were analyzed and annotated with using FlowJo software (FlowJo LLC).
Halo pulse‐chase analysis
For CLSM analysis, mouse embryonic fibroblasts cells were plated on alcian blue coverslip and transfected with HaloTag‐ATZ or with HaloTag‐ATZ and sfGFP‐KDEL as reported above. Seventeen hours after transfection, cells were incubated for 30 min with 15 μM 6‐Chlorohexanol (Sigma) in DMEM 10% FCS, a cell‐permeable black ligand that irreversibly occupied the HaloTag‐binding pocket. After three washes in DMEM 10% FCS, cells were incubated for 15 min with 1 μM PBI 5030 far red ligand (Promega) which exclusively enters the HaloTag ligand‐binding pocket of newly synthesized HaloTag‐ATZ. After three washes in DMEM 10% FCS, the fluorescent ligand is again replaced with 15 μM 6‐Chlorohexanol to block incorporation of the fluorescent ligand in the newly synthesized HaloTag‐ATZ. For biochemical analysis shown in Fig EV5A, HEK293 cells were transfected with HaloTag‐ATZ and pulsed with TMR (Promega). Cells were fixed or lysed after 30, 120, 180, and 300 min of chase and processed for CLSM or for SDS–PAGE as reported above. Time‐course quantification of fluorescence co‐localization between HaloTag‐ATZ, sfGFP‐KDEL, CNX, or CLIMP63 shown in Fig 6G was made using a custom‐developed CellProfiler (Carpenter et al, 2006) pipeline (segmentation of the two markers, followed by the measurement of the area of colocalizing pixels). For biochemical analysis shown in Fig EV5A, after SDS–PAGE gels were scanned with the Typhoon FLA 9500 (Software Version 1.0). Bands were quantified using the ImageQuant software (Molecular Dynamics, GE Healthcare).
Immunogold electron microscopy
Cells were plated on alcian blue coverslips, transfected and fixed with 3.7% formaldehyde as reported above. After washes in PBS and 50 mM glycine, cells were permeabilized with 0.25% saponin, 0.1% BSA, and blocked in blocking buffer (0.2% BSA, 5% goat serum, 50 mM NH4Cl, 0.1% saponin, 20 mM PO4 buffer, 150 mM NaCl). Staining with primary antibodies and nanogold‐labeled secondary antibodies (Nanoprobes) was performed in blocking buffer at room temperature. Cells were re‐fixed in 1% glutaraldehyde, and nanogold was enlarged with gold enhancement solution (Nanoprobes) according to the manufacturer's instructions. Cells were post‐fixed with osmium tetroxide, embedded in epon, and processed into ultrathin slices. After contrasting with uranyl acetate and lead citrate, sections were analyzed with a Zeiss LEO 512 electron microscope. Images were acquired by a 2k bottom‐mounted slow‐scan Proscan camera controlled by EsivisionPro 3.2 software. Image analysis and quantification were performed with FIJI.
Electron tomography
For electron tomography, 200‐ to 250‐nm‐thick sections were collected on formvar‐coated copper slot grids, and gold fiducials (10 nm) were applied on both surfaces of the grids. The samples were imaged in a 200 kV Tecnai G2 20 electron microscope (FEI, Eindhoven, The Netherlands) at magnification of 9.6k resulting in a pixel size of 2.29 nm. Tilted images (+60/−60 according to a Saxton scheme) were acquired using Xplorer 3D (FEI) with an Eagle 2k × 2k CCD camera (FEI). Tilted series alignment and tomography reconstruction were done with the IMOD software package (Mastronarde, 1997). Segmentation and 3D visualization were done with Microscopy Image Browser (Belevich et al, 2016) and IMOD software packages.
Correlative light‐electron microscopy (CLEM)
Cells were grown on finder grids and prepared for IEM as described above. Z‐stacks of cells of interest were taken with the PerkinElmer UltraView ERS confocal microscope. The coordinates of the cells on the finder grid were determined by bright‐field microscopy. Cells were fixed in 1% glutaraldehyde in 0.1 M cacodylate buffer (Sigma) and post‐fixed with 1.5% potassium ferricyanide, 1% OsO4 in 0.1 M cacodylate buffer. Cells were stained with 0.5% uranyl acetate overnight, dehydrated in ethanol, and embedded in epon. After baking for 48 h at 60°C, the resin was released from the glass coverslip by temperature shock in liquid nitrogen. Serial sections (70–90 nm) were collected on carbon‐coated formvar slot grids and imaged with a Zeiss LEO 512 electron microscope. Images were acquired by a 2k × 2k bottom‐mounted slow‐scan Proscan camera controlled by EsivisionPro 3.2 software. Immunofluorescence and IEM images were aligned using Icy bioimage analysis.
Amino acid starvation
To induce autophagy, cells were washed three times with Earle's balanced salt solution (EBSS, Thermo Fisher) and then incubated in EBSS as indicated.
Statistical analysis
Plots and statistical analyses were performed using GraphPad Prism 7 (GraphPad Software Inc.). In this study, one‐way ANOVA with Dunnett's multiple comparisons test and unpaired two‐tailed t‐test were used to assess statistical significance. An adjusted P‐value < 0.05 (for one‐way ANOVA with Dunnett's multiple comparisons test) or P‐value < 0.05 (for t‐test) was considered as statistically significant.
Author contributions
Conceptualization: MM, IF, EF, and EvA; Methodology: IF, EF, AR, RD'A, DM, AD, PP, EvA, and MM; Investigation: IF, EF, TJB, ML, TS, CG, and MM; Writing—original draft: MM; Writing—review & editing: IF, EF, ML, PP, EvA, and MM; Supervision: MM.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1E
Source Data for Figure 2A and I
Source Data for Figure 3A and B
Source Data for Figure 7A and B
Acknowledgements
We thank G. Hämmerling, J.‐L. Guan, D. Green, J. Gruenberg, A. Helenius, M. Komatsu, I. Kurth, G. Marino, M. Michalak, N. Mizushima, F. Reggiori, T. Saitoh, C. Settembre, E. Snapp, S. Tooze, R. Zimmermann, G. Van der Goot, X. Wang, and the members of Molinari's laboratory for gift of reagents, discussions, and critical reading of the manuscript. We are grateful to the ALEMBIC facility at San Raffaele Scientific Institute, Milan, Italy, for the help in electron microscopy analysis. M.M. is supported by Signora Alessandra, AlphaONE Foundation, Foundation for Research on Neurodegenerative Diseases, the Novartis Foundation, Swiss National Science Foundation (SNF), and Comel and Gelu Foundations. P.P. is supported by SNF.
The EMBO Journal (2018) 37: e99259
References
- Belevich I, Joensuu M, Kumar D, Vihinen H, Jokitalo E (2016) Microscopy image browser: a platform for segmentation and analysis of multidimensional datasets. PLoS Biol 14: e1002340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M, Menzies FM, Rubinsztein DC (2016) Mammalian autophagy: how does it work? Annu Rev Biochem 85: 685–713 [DOI] [PubMed] [Google Scholar]
- Bestebroer J, V'Kovski P, Mauthe M, Reggiori F (2013) Hidden behind autophagy: the unconventional roles of ATG proteins. Traffic 14: 1029–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantous S, Terwilliger TC, Waldo GS (2005) Protein tagging and detection with engineered self‐assembling fragments of green fluorescent protein. Nat Biotechnol 23: 102–107 [DOI] [PubMed] [Google Scholar]
- Caramelo JJ, Parodi AJ (2007) How sugars convey information on protein conformation in the endoplasmic reticulum. Semin Cell Dev Biol 18: 732–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM (2006) Cell Profiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7: R100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu AS, Perlmutter DH, Wang Y (2014) Capitalizing on the autophagic response for treatment of liver disease caused by alpha‐1‐antitrypsin deficiency and other genetic diseases. Biomed Res Int 2014: 459823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieri D, Vicario M, Giacomello M, Vallese F, Filadi R, Wagner T, Pozzan T, Pizzo P, Scorrano L, Brini M, Cali T (2018) SPLICS: a split green fluorescent protein‐based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ 25: 1131–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzel A, Molinari M, Trigueros C, Martin JE, Velmurgan S, Brown S, Stamp G, Owen MJ (2002) Early postnatal death and motor disorders in mice congenitally deficient in calnexin expression. Mol Cell Biol 22: 7398–7404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickens JA, Ordonez A, Chambers JE, Beckett AJ, Patel V, Malzer E, Dominicus CS, Bradley J, Peden AA, Prior IA, Lomas DA, Marciniak SJ (2016) The endoplasmic reticulum remains functionally connected by vesicular transport after its fragmentation in cells expressing Z‐alpha(1)‐antitrypsin. FASEB J 30: 4083–4097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbein AD (1991) Glycosidase inhibitors: inhibitors of N‐linked oligosaccharide processing. FASEB J 5: 3055–3063 [DOI] [PubMed] [Google Scholar]
- England CG, Luo HM, Cai WB (2015) HaloTag technology: a versatile platform for biomedical applications. Bioconjug Chem 26: 975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregno I, Molinari M (2018) Endoplasmic reticulum turnover: ER‐phagy and other flavors in selective and non‐selective ER clearance. F1000Res 7: 454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, Hayashi YK, Momoi T (2007) Two endoplasmic reticulum‐associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum Mol Genet 16: 618–629 [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Noack J, Bergmann TJ, Presmanes EC, Pisoni GB, Fasana E, Fregno I, Galli C, Loi M, Soldà T, D'Antuono R, Raimondi A, Jung M, Melnyk A, Schorr S, Schreiber A, Simonelli L, Varani L, Wilson‐Zbinden C, Zerbe O et al (2016) Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat Cell Biol 18: 1173–1184 [DOI] [PubMed] [Google Scholar]
- Gelling CL, Dawes IW, Perlmutter DH, Fisher EA, Brodsky JL (2012) The endosomal protein‐sorting receptor sortilin has a role in trafficking alpha‐1 antitrypsin. Genetics 192: 889–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumati P, Morozzi G, Holper S, Mari M, Harwardt MI, Yan R, Muller S, Reggiori F, Heilemann M, Dikic I (2017) Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. eLife 6: e25555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero CJ, Brodsky JL (2012) The delicate balance between secreted protein folding and endoplasmic reticulum‐associated degradation in human physiology. Physiol Rev 92: 537–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Takamura A, Kishi C, Iemura SI, Natsume T, Guan JL, Mizushima N (2008) FIP200, a ULK‐interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 181: 497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert DN, Foellmer B, Helenius A (1995) Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell 81: 425–433 [DOI] [PubMed] [Google Scholar]
- Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S, Michalopoulos G, Perlmutter DH (2010) An autophagy‐enhancing drug promotes degradation of mutant alpha 1‐antitrypsin Z and reduces hepatic fibrosis. Science 329: 229–232 [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, Guan JL, Oshiro N, Mizushima N (2009) Nutrient‐dependent mTORC1 association with the ULK1‐Atg13‐FIP200 complex required for autophagy. Mol Biol Cell 20: 1981–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houck SA, Ren HY, Madden VJ, Bonner JN, Conlin MP, Janovick JA, Conn PM, Cyr DM (2014) Quality control autophagy degrades soluble ERAD‐resistant conformers of the misfolded membrane protein GnRHR. Mol Cell 54: 166–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huotari J, Helenius A (2011) Endosome maturation. EMBO J 30: 3481–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, Young LN (2017) Mechanisms of autophagy initiation. Annu Rev Biochem 86: 225–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Yamamoto A, Kitamura A, Lamande SR, Yoshimori T, Bateman JF, Kubota H, Nagata K (2009) Autophagic elimination of misfolded procollagen aggregates in the endoplasmic reticulum as a means of cell protection. Mol Biol Cell 20: 2744–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaizuka T, Mizushima N (2016) Atg13 is essential for autophagy and cardiac development in mice. Mol Cell Biol 36: 585–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakuta S, Yamaguchi J, Suzuki C, Sasaki M, Kazuno S, Uchiyama Y (2017) Small GTPase Rab1B is associated with ATG9A vesicles and regulates autophagosome formation. FASEB J 31: 3757–3773 [DOI] [PubMed] [Google Scholar]
- Kamimoto T, Shoji S, Hidvegi T, Mizushima N, Umebayashi K, Perlmutter DH, Yoshimori T (2006) Intracellular inclusions containing mutant alpha1‐antitrypsin Z are propagated in the absence of autophagic activity. J Biol Chem 281: 4467–4476 [DOI] [PubMed] [Google Scholar]
- Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, Mauthe M, Katona I, Qualmann B, Weis J, Reggiori F, Kurth I, Hubner CA, Dikic I (2015) Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522: 354–358 [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC (2008) Does bafilomycin A(1) block the fusion of autophagosomes with lysosomes? Autophagy 4: 849–850 [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T (2005) Impairment of starvation‐induced and constitutive autophagy in Atg7‐deficient mice. J Cell Biol 169: 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeger H, Miranda E, MacLeod I, Perez J, Crowther DC, Marciniak SJ, Lomas DA (2009) Endoplasmic reticulum‐associated degradation (ERAD) and autophagy cooperate to degrade polymerogenic mutant serpins. J Biol Chem 284: 22793–22802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ktistakis NT, Tooze SA (2016) Digesting the expanding mechanisms of autophagy. Trends Cell Biol 26: 624–635 [DOI] [PubMed] [Google Scholar]
- Lamark T, Johansen T (2012) Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol 2012: 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Choudhury P, Cabral CM, Sifers RN (1999) Oligosaccharide modification in the early secretory pathway directs the selection of a misfolded glycoprotein for degradation by the proteasome. J Biol Chem 274: 5861–5867 [DOI] [PubMed] [Google Scholar]
- Loi M, Fregno I, Guerra C, Molinari M (2018) Eat it right: ER‐Phagy and RecovER‐Phagy. Biochem Soc Trans 46: 699–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Ordonez A, Dickens JA, Chambers JE, Patel V, Dominicus CS, Malzer E (2016) New concepts in alpha‐1 antitrypsin deficiency disease mechanisms. Ann Am Thorac Soc 13(Suppl 4): 289–296 [DOI] [PubMed] [Google Scholar]
- Marino G, Fernandez AF, Cabrera S, Lundberg YW, Cabanillas R, Rodriguez F, Salvador‐Montoliu N, Vega JA, Germana A, Fueyo A, Freije JMP, Lopez‐Otin C (2010) Autophagy is essential for mouse sense of balance. J Clin Invest 120: 2331–2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Malireddi RKS, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan JL, Tan HY, Peng JM, Kanneganti TD, Virgin HW, Green DR (2015) Molecular characterization of LC3‐associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol 17: 893–906 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mastronarde DN (1997) Dual‐axis tomography: an approach with alignment methods that preserve resolution. J Struct Biol 120: 343–352 [DOI] [PubMed] [Google Scholar]
- McAlpine F, Williamson LE, Tooze SA, Chan EYW (2013) Regulation of nutrient‐sensitive autophagy by uncoordinated 51‐like kinases 1 and 2. Autophagy 9: 361–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Fullgrabe J, Jackson A, Sanchez MJ, Karabiyik C, Licitra F, Ramirez AL, Pavel M, Puri C, Renna M, Ricketts T, Schlotawa L, Vicinanza M, Won H, Zhu Y et al (2017) Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93: 1015–1034 [DOI] [PubMed] [Google Scholar]
- Miranda E, Perez J, Ekeowa UI, Hadzic N, Kalsheker N, Gooptu B, Portmann B, Belorgey D, Hill M, Chambers S, Teckman J, Alexander GJ, Marciniak SJ, Lomas DA (2010) A novel monoclonal antibody to characterize pathogenic polymers in liver disease associated with alpha(1)‐antitrypsin deficiency. Hepatology 52: 1078–1088 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27: 107–132 [DOI] [PubMed] [Google Scholar]
- Molinari M, Calanca V, Galli C, Lucca P, Paganetti P (2003) Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 299: 1397–1400 [DOI] [PubMed] [Google Scholar]
- Molinari M, Eriksson KK, Calanca V, Galli C, Cresswell P, Michalak M, Helenius A (2004) Contrasting functions of calreticulin and calnexin in glycoprotein folding and ER quality control. Mol Cell 13: 125–135 [DOI] [PubMed] [Google Scholar]
- Molinari M, Galli C, Vanoni O, Arnold SM, Kaufman RJ (2005) Persistent glycoprotein misfolding activates the glucosidase II/UGT1‐driven calnexin cycle to delay aggregation and loss of folding competence. Mol Cell 20: 503–512 [DOI] [PubMed] [Google Scholar]
- Noda T, Farquhar MG (1992) A non‐autophagic pathway for diversion of ER secretory proteins to lysosomes. J Cell Biol 119: 85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda Y, Hosokawa N, Wada I, Nagata K (2003) EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299: 1394–1397 [DOI] [PubMed] [Google Scholar]
- Olivari S, Molinari M (2007) Glycoprotein folding and the role of EDEM1, EDEM2 and EDEM3 in degradation of folding‐defective glycoproteins. FEBS Lett 581: 3658–3664 [DOI] [PubMed] [Google Scholar]
- Pastore N, Blomenkamp K, Annunziata F, Piccolo P, Mithbaokar P, Maria Sepe R, Vetrini F, Palmer D, Ng P, Polishchuk E, Iacobacci S, Polishchuk R, Teckman J, Ballabio A, Brunetti‐Pierri N (2013) Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha‐1‐anti‐trypsin deficiency. EMBO Mol Med 5: 397–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieren M, Galli C, Denzel A, Molinari M (2005) The use of calnexin and calreticulin by cellular and viral glycoproteins. J Biol Chem 280: 28265–28271 [DOI] [PubMed] [Google Scholar]
- Pisoni GB, Molinari M (2016) Five questions (with their Answers) on ER‐associated degradation. Traffic 17: 341–350 [DOI] [PubMed] [Google Scholar]
- Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, Bachelot MF, Lamberton A, Mathieu M, Bertrand T, Marquette JP, El‐Ahmad Y, Filoche‐Romme B, Schio L, Garcia‐Echeverria C, Goulaouic H, Pasquier B (2014) A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol 10: 1013–1019 [DOI] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, Yamamoto N, Kawai T, Ishii K, Takeuchi O, Yoshimori T, Akira S (2009) Atg9a controls dsDNA‐driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA 106: 20842–20846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A (2009) A gene network regulating lysosomal biogenesis and function. Science 325: 473–477 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR (2000) Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404: 770–774 [DOI] [PubMed] [Google Scholar]
- Shang L, Chen S, Du F, Li S, Zhao L, Wang X (2011) Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci USA 108: 4788–4793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, von Kriegsheim A, Behrends C, Wilkinson S (2018) CCPG1 is a non‐canonical autophagy cargo receptor essential for ER‐phagy and pancreatic ER proteostasis. Dev Cell 44: 217–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldà T, Garbi N, Hammerling GJ, Molinari M (2006) Consequences of ERp57 deletion on oxidative folding of obligate and facultative clients of the calnexin cycle. J Biol Chem 281: 6219–6226 [DOI] [PubMed] [Google Scholar]
- Soldà T, Galli C, Kaufman RJ, Molinari M (2007) Substrate‐specific requirements for UGT1‐dependent release from calnexin. Mol Cell 27: 238–249 [DOI] [PubMed] [Google Scholar]
- Steegmaier M, Oorschot V, Klumperman J, Scheller RH (2000) Syntaxin 17 is abundant in steroidogenic cells and implicated in smooth endoplasmic reticulum membrane dynamics. Mol Biol Cell 11: 2719–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Kubota Y, Sekito T, Ohsumi Y (2007) Hierarchy of Atg proteins in pre‐autophagosomal structure organization. Genes Cells 12: 209–218 [DOI] [PubMed] [Google Scholar]
- Teckman JH, Perlmutter DH (2000) Retention of mutant alpha(1)‐antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am J Physiol Gastrointest Liver Physiol 279: G961–G974 [DOI] [PubMed] [Google Scholar]
- Teckman JH, An JK, Loethen S, Perlmutter DH (2002) Fasting in alpha1‐antitrypsin deficient liver: constitutive activation of autophagy. Am J Physiol Gastrointest Liver Physiol 283: G1156–G1165 [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T et al (2015) Tissue‐based map of the human proteome. Science 347: 1260419 [DOI] [PubMed] [Google Scholar]
- Vabulas RM, Hartl FU (2005) Protein synthesis upon acute nutrient restriction relies on proteasome function. Science 310: 1960–1963 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Movie EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1E
Source Data for Figure 2A and I
Source Data for Figure 3A and B
Source Data for Figure 7A and B