

Abstract
Membrane blebbing‐dependent (blebby) amoeboid migration can be employed by lymphoid and cancer cells to invade 3D‐environments. Here, we reveal a mechanism by which the small GTPase RhoB controls membrane blebbing and blebby amoeboid migration. Interestingly, while all three Rho isoforms (RhoA, RhoB and RhoC) regulated amoeboid migration, each controlled motility in a distinct manner. In particular, RhoB depletion blocked membrane blebbing in ALL (acute lymphoblastic leukaemia), melanoma and lung cancer cells as well as ALL cell amoeboid migration in 3D‐collagen, while RhoB overexpression enhanced blebbing and 3D‐collagen migration in a manner dependent on its plasma membrane localization and down‐stream effectors ROCK and Myosin II. RhoB localization was controlled by endosomal trafficking, being internalized via Rab5 vesicles and then trafficked either to late endosomes/lysosomes or to Rab11‐positive recycling endosomes, as regulated by KIF13A. Importantly, KIF13A depletion not only inhibited RhoB plasma membrane localization, but also cell membrane blebbing and 3D‐migration of ALL cells. In conclusion, KIF13A‐mediated endosomal trafficking modulates RhoB plasma membrane localization to control membrane blebbing and blebby amoeboid migration.
Keywords: amoeboid cell migration, intracellular trafficking, KIF13A, membrane blebbing, RhoB
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Membrane & Intracellular Transport
Introduction
Amoeboid migration, characterized by weak or no adhesion to the extracellular matrix (ECM) and little or no ECM proteolysis, is a rapid migration mode driven by hydrostatically generated membrane blebs (blebby amoeboid migration), actin‐rich pseudopodia and/or highly contractile uropods (Sahai, 2005; Paluch & Raz, 2013; Liu et al, 2015; Ruprecht et al, 2015). Utilized by Dictyostelium amoeba, leucocytes and some cancer cells, amoeboid migration exhibits several advantages when compared with mesenchymal motility. For example, no matrix metalloproteinase (MMP) activity is needed for cancer cell invasion via this modality, and higher migration efficiency can be obtained when adhesion to the substrate is limited, as may occur in a 3D‐matrix (Sahai, 2005; Charras & Paluch, 2008; Fackler & Grosse, 2008; Ridley, 2011; Paluch & Raz, 2013; Paluch et al, 2016; van Helvert et al, 2018). Amoeboid migration is also involved in embryo development (Fackler & Grosse, 2008; Jung et al, 2013) and is physiologically employed by immune cells to execute immune defence functions (Khajah & Luqmani, 2016), underlining the fundamental importance of this migration strategy.
Blebby amoeboid migration is used to drag and push the cell forward within 3D‐ECM environments and can be particularly useful for a cell to migrate through space‐restricted local ECM niches (Sahai, 2005; Charras & Paluch, 2008; Fackler & Grosse, 2008). As a main morphological feature of blebby amoeboid migration, membrane blebbing has attracted increasing recent attention (Fackler & Grosse, 2008; Ridley, 2011; Laser‐Azogui et al, 2014; Gebala et al, 2016). Membrane blebs are initiated by local weakening of the interaction between the plasma membrane (PM) and the underlying actin filament cortex (Ridley, 2011). The small GTPase Rho family and its down‐stream effector Rho‐kinase (ROCK) play essential roles in controlling actomyosin contractility by stimulating myosin light chain (MLC) phosphorylation of Myosin II, leading to its activation (Charras et al, 2006; Ridley, 2011). Activated Myosin II can in turn increase the local hydrostatic pressure, leading to focal rupture of the cortical actin cytoskeleton from the membrane (Charras et al, 2005). This leads to a cytoplasmic fluid flow rapidly pushing the membrane outwards, thereby driving the formation of blebs (Charras et al, 2005; Tinevez et al, 2009). Once membrane blebs form, actin polymerizes on the bleb membrane to form a new actin cortex, leading to re‐engagement of the PM with the newly formed cortical actin filaments, thus promoting the retraction of membrane blebs together with Myosin II‐mediated actomyosin contraction (Charras et al, 2006; Charras & Paluch, 2008; Fackler & Grosse, 2008).
Although it has been shown that Rho regulates membrane blebbing by increasing actomyosin contractility through ROCK and Myosin II, it remains elusive which of the three Rho subfamily members (RhoA, RhoB and/or RhoC) may govern membrane blebbing (Ridley, 2011). While it has been suggested that membrane blebbing is controlled by RhoA, this is largely based on the use of unselective reagents that are unable to pinpoint which specific Rho family member is involved (Gadea et al, 2007; Godin & Ferguson, 2010; Cartier‐Michaud et al, 2012; Takahara et al, 2014; Bang et al, 2015; Aoki et al, 2016), and the potential involvement of RhoB and RhoC has not been specifically addressed.
Here, we show that RhoB controls membrane blebbing and blebby amoeboid migration, since depletion of RhoB blocked membrane blebbing and 3D‐amoeboid cell migration, while RhoB overexpression enhanced blebbing and 3D‐migration. Notably, all three Rho isoforms were involved in the regulation of amoeboid migration by controlling different aspects of motility. We also detail a mechanism regulating this RhoB function, involving KIF13A‐mediated re‐distribution of RhoB to the PM, where RhoB engages ROCK and Myosin II to drive bleb formation.
Results
RhoB depletion inhibits cell membrane blebbing
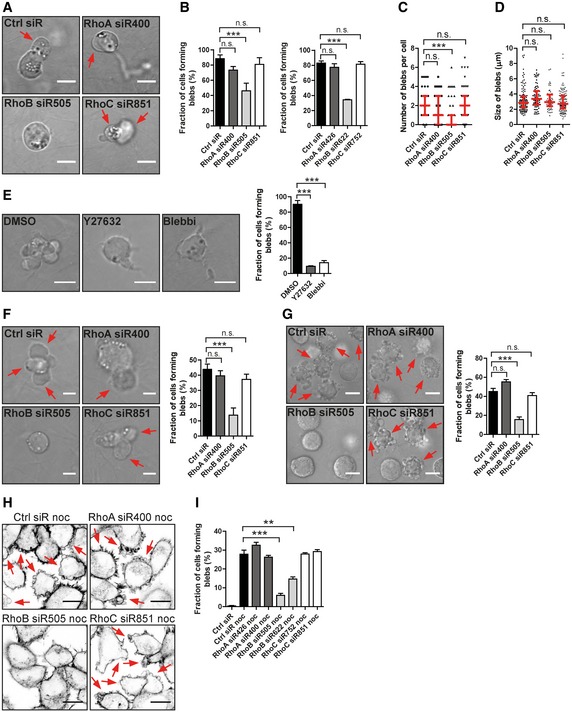
To explore the regulation of membrane blebbing in the context of amoeboid migration (Trendowski, 2015), we selected two acute lymphoblastic leukaemia cell lines, T‐ALL and Reh. These two cell lines displayed high or moderate blebbing activity, respectively, comprised of large membrane blebs (Fig 1), consistent with previous observations (Haston & Shields, 1984; Charras & Paluch, 2008). Interestingly, knock‐down of RhoB caused a significant decrease in T‐ALL cell membrane blebbing within 3D‐collagen, while knock‐down of RhoA or RhoC did not and the size of blebs was not affected (Figs 1A–D, and EV1A and B; Movie EV1). Both the ROCK inhibitor, Y27632, and the Myosin II inhibitor, Blebbistatin, blocked blebbing in T‐ALL cells (Fig 1E), suggesting that RhoB‐mediated blebbing may depend on ROCK and Myosin II, consistent with previous reports on ROCK and Myosin II dependence (Charras et al, 2006; Ridley, 2011). A similar selective dependence on RhoB for membrane blebbing was detected in Reh ALL cells (Figs 1F and EV1C). Knock‐down of RhoB also inhibited membrane blebbing in A375M2 human melanoma cells, which was previously used as a model of innate membrane blebbing in 3D‐environments (Tozluoglu et al, 2013; Figs 1G and EV1D).
Figure 1. RhoB depletion impairs cell membrane blebbing.
-
A–DT‐ALL leukaemia cells were electroporated with different siRNAs and replated in a 2.5 mg/ml 3D‐Collagen type I gel. (A) Arrows indicate blebs on the cell surface. (B) The fraction of cells forming blebs was quantified. Two different siRNAs were used for each Rho protein. (C, D) Quantification of number of blebs per cell and the bleb size. Red bars show the median and quartiles.
-
ET‐ALL cells were replated in 3D‐collagen as above and treated with DMSO, 1 μM Y27632 or 10 μM Blebbistatin (Blebbi). The fraction of cells forming blebs was quantified.
-
F, GReh leukaemia cells (F) and A375M2 DS melanoma cells (G) were transfected with different siRNAs and replated in 3D‐collagen as above. Arrows indicate blebs of the cells (F) or blebbing cells (G). The fraction of cells forming blebs was quantified.
-
H, IH1299 cells were transfected with different siRNAs and treated with or without 1 μM nocodazole (noc) for 30 min and labelled for F‐actin. (H) Arrows indicate noc‐induced cell membrane blebbing. (I) The fraction of cells forming blebs was quantified.
Figure EV1. Effects of RNAi‐mediated knock‐down of Rho family members on cell morphological features and migration.
-
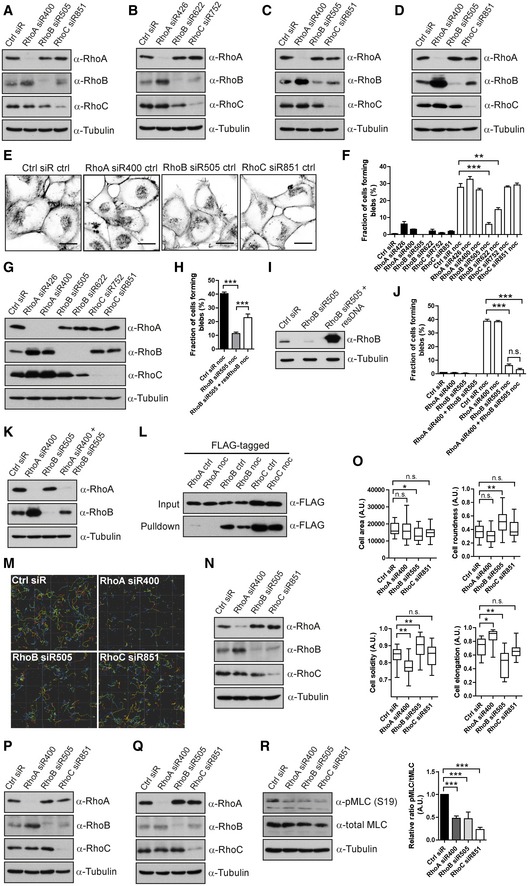
A–DKnock‐down efficiency controls for Fig 1. (A, B) T‐ALL cells; (C) Reh cells; or (D) A375M2 DS cells transfected with Ctrl siRNA or siRNAs against RhoA, RhoB or RhoC; the knock‐down efficiency and potential effect on other Rho proteins were detected by immunoblotting.
-
E, FH1299 cells transfected with Ctrl, RhoA‐, RhoB‐ or RhoC‐siRNAs, treated with or without 1 μM nocodazole (noc) for 30 min, were labelled for F‐actin. (E) Images show H1299 cells treated without nocodazole (Images of H1299 cells treated with nocodazole are shown in Fig 1H). Scale bars, 20 μm. (F) The fraction of cells forming blebs was quantified. Bars show mean ± SEM (n = 3 experiments). Dunnett's multiple comparison test derived P‐values: **P < 0.01; ***P < 0.001. The right half of this graph is also displayed in Fig 1I.
-
GH1299 cells transfected with different Ctrl, RhoA‐, RhoB‐ or RhoC‐siRNAs; the effect of knock‐down of individual siRNAs was detected by immunoblotting.
-
H, IH1299 cells transfected with either RhoB‐siRNA alone or RhoB‐siRNA together with an siRNA‐resistant FLAG‐tagged RhoB construct, treated with nocodazole and labelled for F‐actin. (H) The fraction of cells forming blebs was quantified. Bars show mean ± SEM (n = 4 experiments). Dunnett's multiple comparison test derived P‐values: ***P < 0.001. (I) The level of RhoB was detected by immunoblotting.
-
J, KH1299 cells transfected with Ctrl, RhoA‐, RhoB‐ or RhoA+RhoB‐siRNAs, treated with or without 1 μM nocodazole (noc) for 30 min and labelled for F‐actin. (J) The fraction of cells forming blebs was quantified. Bars show mean ± SEM (n = 3 experiments). Dunnett's multiple comparison test derived P‐values: n.s. = non‐significant; ***P < 0.001. (K) The levels of RhoA and RhoB were detected by immunoblotting.
-
LH1299 cells transfected with FLAG‐tagged RhoA, RhoB or RhoC, treated with or without 1 μM nocodazole (noc) for 30 min. Rho GTPases were pulled down with GST‐RBD protein, and the GTP‐binding activities of RhoA, RhoB and RhoC were detected by immunoblotting with anti‐FLAG antibody.
-
M, NT‐ALL cells electroporated with Ctrl, RhoA‐, RhoB‐ or RhoC‐siRNA, replated in 2.5 mg/ml 3D‐Collagen type I matrix. The cells were imaged, and their migration speed was measured (same data set as in Fig 2A). (M) A representative example of Imaris analysis of T‐ALL cell migration with different Rho protein knock‐down is displayed. (N) Immunoblotting knock‐down efficiency control for Fig 2A.
-
O, PT‐ALL cells electroporated with Ctrl, RhoA‐, RhoB‐ or RhoC‐siRNA, replated in 2.5 mg/ml 3D‐Collagen type I matrix (same data set as in Fig 2D). (O) Cellular features, that is cell area, roundness, solidity and elongation were analysed. The boxes show the median and quartiles, and the whiskers display 5 and 95 percentiles. Kruskal–Wallis test derived P‐values: *P < 0.05; **P < 0.01; n.s.—non‐significant. Data shown are one representative example among three independent experiments. (P) Immunoblotting knock‐down efficiency control for Fig 2D.
-
Q, RT‐ALL cells electroporated with Ctrl, RhoA‐, RhoB‐ or RhoC‐siRNA, replated on FN. The knock‐down efficiency of individual Rho proteins, as well as the levels of phosphorylated and total MLC, was detected by immunoblotting. Bar graph shows quantification of the signal ratio between phospho‐MLC and total MLC as mean ± SD (n = 3 experiments). t‐test (paired, two‐tailed) derived P‐values: ***P < 0.001.
Source data are available online for this figure.
We further investigated membrane blebbing in human H1299 non‐small cell lung carcinoma cells (NSCLC) by using the widely employed strategy of nocodazole treatment to induce blebbing (Hagmann et al, 1999; Takesono et al, 2010). Again, RhoB‐directed siRNAs, but not RhoA‐ or RhoC‐siRNAs, inhibited nocodazole‐induced cell membrane blebbing (Figs 1H and I, and EV1E–G). Overexpression of siRNA‐resistant RhoB partially rescued membrane blebbing caused by RhoB knock‐down, verifying a specific role of RhoB (Fig EV1H and I). Consistent with previous observations, RhoA knock‐down increased RhoB levels (Vega et al, 2011; Fig EV1G). However, combined RhoA and RhoB knock‐down produced no further blebbing inhibition compared to RhoB knock‐down alone (Fig EV1J and K). The addition of RhoA‐siRNA increased RhoB levels as compared to RhoB‐siRNA alone, but without increasing blebbing (Fig EV1J and K). This leaves the possibility that, while RhoB appears essential for blebbing, RhoA may also have a function in blebbing that could be compensated by increased RhoB upon RhoA knock‐down. Further, nocodazole was reported to activate RhoA in CCRF‐CEM cells (Takesono et al, 2010). However, we could not detect any activation of RhoA by nocodazole in our system (Fig EV1L), possibly due to use of different cell lines and/or our significantly lower dosing of nocodazole treatment. Interestingly, we found that RhoB (and RhoC) showed intrinsic GTP‐binding activity in resting cells (Fig EV1L), suggesting that an activation step may not be necessary for RhoB to execute its functions. In summary, RhoB knock‐down caused defective membrane blebbing in four cancer cell lines of distinct origin, indicating that RhoB may be widely required for cell membrane blebbing.
RhoA and RhoB control 3D cell migration through distinct mechanisms
As membrane blebs are important cellular protrusions in amoeboid migration, we examined the role of RhoB in 3D amoeboid cell migration of T‐ALL cells. Interestingly, siRNAs to RhoA, RhoB and RhoC all inhibited cell migration of these leukaemia cells in 3D‐collagen (Figs 2A, and EV1M and N). Importantly, the T‐ALL cells appeared to utilize a combination of membrane blebs and membrane ruffles for efficient migration (Fig 2B; Movie EV2), and approximately 80–90% of the T‐ALL cells formed membrane blebs in 3D‐collagen during 10 min of imaging (Fig 2C). Based on live cell imaging of totally 748 T‐ALL cells, we observed that upon perturbation, a fraction of the cells displayed defects in protrusion and/or retractions and/or blebbing with associated difficulties in movement (Fig 2B; [Link], [Link]). Notably, quantitative analysis showed that RhoB‐siRNA caused a strong decrease in membrane blebbing, RhoA‐siRNA caused elongated tails with impaired retraction, while RhoC‐siRNA caused no obvious effect on the observed features (Fig 2C). These data suggest that RhoA and RhoB play distinct roles in amoeboid migration, mediated by impacts upon different cellular processes, where RhoA appeared to modulate retraction activities while RhoB controlled membrane blebbing. Potential mechanism(s) of RhoC action in this context remain unclear.
Figure 2. Distinct functions of RhoA and RhoB in 3D cell migration of T‐ALL cells.

-
AT‐ALL cells electroporated with different siRNAs and replated in 3D‐collagen. Left graph: boxes show the median and quartile migration speed, whiskers display 5 and 95 percentiles based on pooled data from three independent experiments. Right graph: Ctrl siRNA cell median speed was defined as 1, and the relative migration speed was calculated and binned into three groups to display three speed distributions: 0–1, slow moving cells; 1–3, moderate moving cells; > 3, fast moving cells.
-
BExamples of different migratory features captured by high‐resolution live cell imaging of 748 cells. Red arrows, membrane blebs; blue arrows, membrane ruffles; green arrow, tail with retraction problem.
-
CThe fractions of T‐ALL cells with different morphological features were quantified and plotted. Two different siRNAs were used for each Rho protein.
-
D, ET‐ALL cells electroporated with different siRNAs and replated in 3D‐collagen. (D) Cells were stained for F‐actin, and a Z‐stack image was produced (top panel); the images of the middle focus plane were overlaid with a phase contrast projection in the bottom panel. White arrows, membrane blebs; green arrows, filopodia; yellow arrow, long tail. (E) The fractions of cells with different F‐actin features were quantified.
-
F, GT‐ALL cells electroporated with different siRNAs and replated onto FN. (F) Cells were immunolabelled for pMLC and stained for F‐actin. (G) Quantification of pMLC signals. Boxes show the median and quartile pMLC intensity, and whiskers display the 5 and 95 percentiles among 102–139 cells per condition.
We also visualized F‐actin in T‐ALL cells within 3D‐collagen, performing high‐resolution 3D imaging of more than 100 cells for each condition. Similar to our morphological observations, RhoB‐siRNA led to a strong decrease in membrane bleb‐labelling F‐actin structures, along with decreased cell area and elongation, as well as increased cell roundness and solidity (a parameter defining whether cells have concave or convex profiles; Figs 2D and E, and EV1O and P). In contrast, RhoA knock‐down cells had unaffected blebbing structures, but instead displayed elongated tails, often associated with long actin filaments at the end of the tail (Fig 2D and E). This correlates with the long rear tails observed by live cell imaging upon RhoA knock‐down (Fig 2B and C and [Link], [Link]) and resulted in decreased cell solidity and increased cell elongation (Fig EV1O). RhoC knock‐down caused no detectable F‐actin alterations (Figs 2D and E, and EV1O and P). Together, this suggests that knock‐down of RhoB inhibited formation of F‐actin‐based blebbing structures, while inhibition of RhoA caused retraction problems.
As RhoB appeared to mediate membrane blebbing via the down‐stream ROCK to Myosin II pathway (Fig 1E), we analysed myosin light chain (MLC) activity indicated by phosphorylation of myosin light chain 2 at Ser19 (pMLC) in T‐ALL cells. While knock‐down of each of the three Rho isoforms inhibited MLC activity, knock‐down of RhoB displayed the strongest inhibition (Figs 2F and G, and EV1Q and R). This supports our indications that RhoB controls membrane blebbing in T‐ALL cells through the ROCK‐Myosin II pathway, while RhoA and RhoC may control 3D amoeboid cell migration through distinct actinomyosin‐dependent processes.
Plasma membrane‐localized RhoB induces membrane blebs and promotes cell migration and invasion
As membrane blebbing is a highly localized event caused by local weaknesses in cortical F‐actin‐to‐plasma membrane interaction, it is plausible that RhoB localization is important for its role in membrane blebbing. Consistently, in T‐ALL and Reh leukaemia cells that form spontaneous blebs, endogenous RhoB mainly localized at the PM and to a low extent to cytoplasmic vesicles and the nucleus (Figs 3A and EV2A–C). However, in H1299 cells, endogenous RhoB as well as exogenous FLAG‐tagged RhoB mainly localized in vesicle‐like structures with a small amount of RhoB at the PM (Fig 3B). This is consistent with previous reports from adherent cells displaying mainly vesicular RhoB localization (Adamson et al, 1992b). Interestingly, the predominant vesicular localization of RhoB in H1299 cells correlated with lack of innate blebbing and FLAG‐RhoB overexpression in these cells also did not induce blebbing (Fig 3B). However, mutation of the RhoB DYDR motif within the Switch2 domain to NFNQ re‐directed RhoB to the PM and induced blebbing, suggesting that redirection of RhoB to the PM caused blebbing (Fig 3C). Consistently, removal of the RhoB CAAX‐box (ΔCKVL) in the NFNQ mutant to deplete its membrane association led to a predominantly cytoplasmic RhoB localization, and blebbing could no longer be observed (Figs 3C and EV2D). Overall, these results are in agreement with previous findings that RhoB localization varies between different models and conditions (Michaelson et al, 2001; Wherlock et al, 2004; Garcia‐Mariscal et al, 2017). Our findings now extend on this by indicating correspondences between innate differences in RhoB localization and blebbing activity, with high RhoB localization at the PM coupled to spontaneous blebbing behaviour.
Figure 3. Plasma membrane‐localized RhoB induces membrane blebs and promotes migration and invasion.
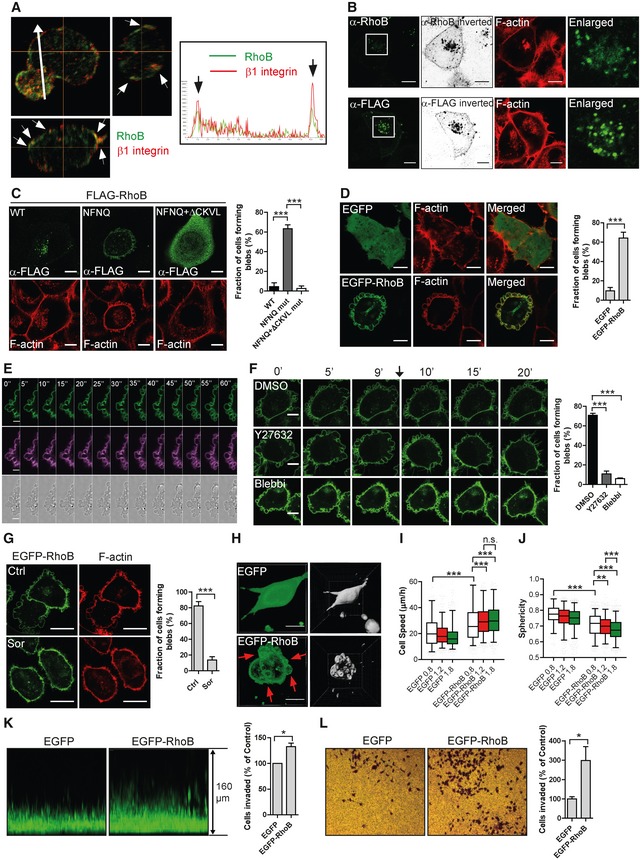
-
AT‐ALL cells replated on FN‐coated surface and immunolabelled for RhoB (green) and β1 integrin (red); Z‐stack with middle focus plane (top left), Y–Z axis projection (top right) and X–Z axis projection (bottom left). Arrowheads indicate co‐localization of RhoB and β1 integrin at the cell periphery. Arrow indicates the direction for the fluorescence intensity quantification along this line shown in the right box. Arrows in the box indicate the RhoB and β1 integrin signals at cell boundaries.
-
BH1299 cells labelled for F‐actin and immunolabelled either for endogenous RhoB (top) or transfected with FLAG‐RhoB and labelled for FLAG‐tag (bottom). The RhoB/FLAG labelling was imaged in a saturated manner and displayed in an inverted b/w projection. The boxed regions are enlarged and shown to the right.
-
C, DF‐actin labelled H1299 cells (C) transfected with FLAG‐RhoB WT or different mutants and labelled for FLAG‐tag or (D) stably expressing EGFP or EGFP‐RhoB. Bleb‐positive cells were quantified using the F‐actin channel.
-
ELive cell imaging time series of EGFP‐RhoB H1299 cell of EGFP‐RhoB (green), CellMask DeepRed plasma membrane dye (violet) and bright field (bottom).
-
FEGFP‐RhoB H1299 cells were imaged for 10 min, then DMSO, 1 μM Y27632 or 10 μM Blebbistatin (Blebbi) were added and cells continued to be imaged. The arrow indicates the time point of adding inhibitors. The fraction of cells forming blebs was quantified.
-
GEGFP‐RhoB H1299 cells were treated with or without 0.5 M sorbitol (Sor) for 30 min, fixed and labelled for F‐actin. The fraction of cells forming blebs was quantified.
-
HEGFP or EGFP‐RhoB H1299 cells replated in 1.8 mg/ml 3D‐Collagen type I gel and imaged. Arrows indicate membrane blebs. The segmentation by Imaris is shown to the right.
-
I, JEGFP or EGFP‐RhoB H1299 cells in 3D‐Collagen type I gels of different densities (0.8, 1.2 and 1.8 mg/ml) with their migratory behaviours (I, cell speed; J, sphericity) analysed. Boxes show the median and quartiles, and whiskers display the 5 and 95 percentiles.
-
KEGFP or EGFP‐RhoB H1299 cells invaded into 1.8 mg/ml 3D‐Collagen type I were imaged with a Z‐stack.
-
LEGFP or EGFP‐RhoB H1299 cells were allowed to invade into Matrigel using transwell chambers. The numbers of invaded cells were normalized.
Figure EV2. Effects of RhoB localization on membrane blebbing.

-
A, BT‐ALL cells electroporated with Ctrl or RhoB‐siRNA were replated on FN and immunolabelled for RhoB. (A) RhoB intensity quantification. Red bars show the median and quartiles based on pooled data from three independent experiments. Mann–Whitney test derived P‐value: ***P < 0.001. (B) Immunoblotting detecting RhoB knock‐down efficiency.
-
CReh cells labelled with CellTracker Red Dye (cytoplasmic label) and Hoechst 33342 (nucleus) and immunostained for RhoB. Scale bars, 5 μm. The fluorescent spectra for RhoB and CellTracker labelling are shown below the images. The white arrow indicates the direction for the fluorescence intensity quantification along this line; green and red arrows indicate the RhoB and CellTracker signal, respectively, close to the cell boundaries.
-
DH1299 cells were transfected with FLAG‐RhoB WT or different mutants and labelled for FLAG‐tag and F‐actin (same data set as in Fig 3C). Cells with different intracellular localization and with blebs were quantified. Bars show mean ± SD (n = 3 experiments). Dunnett's multiple comparison test derived P‐values: **P < 0.01; ***P < 0.001; n.s. = non‐significant.
-
ECell viability (relative cell number) of EGFP or EGFP‐RhoB stable H1299 cells was analysed by MTT assay. Bars show mean with 95% CI (n = 3 experiments).
-
FImmunoblotting analysed cleaved caspase‐3 in EGFP and EGFP‐RhoB stable H1299 cells, with puromycin treated cells as positive control.
-
GEGFP‐RhoB H1299 cells transfected with Ctrl or EGFP‐siRNA, labelled for F‐actin. The fraction of cells forming blebs was quantified. Bars show mean ± SEM (n = 3 experiments). t‐test (unpaired, two‐tailed) derived P‐value: ***P < 0.001.
-
HEGFP‐RhoB cells treated with DMSO, 1 μM Y27632 or 10 μM Blebbistatin for 30 min, fixed and labelled for F‐actin. Scale bars, 10 μm.
-
I, JCOS‐7, MCF‐7, PC‐3, HeLa, NIH3T3 and MDCK cells were transfected with EGFP or EGFP‐RhoB and labelled for F‐actin (I). Scale bars, 10 μm. (J) Quantification of fractions of cells forming blebs; cell number used for each cell line is shown in the graph.
-
KEGFP or EGFP‐RhoB stable cells were replated in 3D‐Collagen type I gels of different densities (0.8, 1.2 and 1.8 mg/ml) and migration straightness analysed (same data set as in Fig 3I and J). Boxes show median and quartiles, and whiskers display the 5 and 95 percentiles based on pooled data from three independent experiments. Kruskal–Wallis test derived P‐values: n.s. = non‐significant.
-
LUntransfected H1299 cells (left), FLAG‐tagged RhoA or RhoC transfected (middle) and EGFP‐RhoA or RhoC transfected (right). Cells were immunolabelled for RhoA or RhoC (left), or for FLAG‐tag (middle), and stained for F‐actin. Scale bars, left and middle: 20 μm, right: 10 μm.
-
MH1299 cells with or without stable expression of EGFP, EGFP‐RhoA, EGFP‐RhoB or EGFP‐RhoC were labelled for F‐actin. Bleb‐positive cells were quantified using the F‐actin channel. Bars show mean ± SD (n = 6 experiments). Dunnett's multiple comparison test derived P‐values: **P < 0.01; ***P < 0.001; n.s. = non‐significant.
-
NH1299 cells stably expressing EGFP‐tagged RhoA, RhoB or RhoC, or EGFP were pulled down with GST‐RBD protein, and the GTP‐binding activities were detected by immunoblotting with anti‐GFP antibody.
Source data are available online for this figure.
Previous observations show that EGFP‐fused RhoB is PM‐localized in Madin Darby canine kidney (MDCK) epithelial cells, implying that EGFP tagging of RhoB may shift the relative distribution of RhoB localization from vesicles towards the PM (Michaelson et al, 2001). Consistent with this, EGFP‐RhoB mainly localized at the PM in H1299 cells, with a small portion in vesicles. Importantly, unlike overexpression of FLAG‐tagged RhoB (predominant vesicular localization), EGFP‐RhoB expression did induce membrane blebbing (Fig 3D). These membrane blebs were not apoptotic (Fig EV2E and F) and were blocked by an siRNA against EGFP (Fig EV2G), indicating that the blebbing activity was specifically caused by EGFP‐RhoB. EGFP‐RhoB induced very dynamic membrane blebs in 2D culture (Fig 3E; Movie EV3), which were blocked by Y27632 or Blebbistatin treatment (Figs 3F and EV2H; Movie EV4), further supporting a role for the ROCK to Myosin II pathway in RhoB‐induced membrane blebbing. The dynamic bleb protrusion–retraction process (Fig 3E; Movie EV3), together with results from sorbitol treatment (Laser‐Azogui et al, 2014; providing a hyperosmotic condition that depletes intracellular fluid pressure; Fig 3G; Movie EV4), verified that the EGFP‐RhoB‐induced blebs were bona fide membrane blebs. Further, overexpression of EGFP‐RhoB in six additional epithelial and mesenchymal adherent cell lines caused a predominant plasma membrane EGFP‐RhoB localization and also induced membrane blebbing (Fig EV2I and J), indicating that blebbing induction is a common effect of membrane‐localized RhoB.
Given that EGFP‐RhoB also induced very dynamic membrane blebs in 3D‐collagen (Fig 3H; Movie EV5), we tested whether EGFP‐RhoB also affected 3D cell migration. Indeed, EGFP‐RhoB caused a significant increase in migration speed of H1299 cells within 3D‐Collagen type I (Fig 3I). Interestingly, the effect of EGFP‐RhoB on migration speed was enhanced with increased 3D‐matrix density, without altering migration straightness (Figs 3I and EV2K). Also cell sphericity, a parameter determined by the ratio of cell volume to surface area, decreased in EGFP‐RhoB cells as compared to EGFP‐expressing cells (Fig 3J; Movie EV5). This is consistent with cells forming blebs that increase cell surface area relative to volume. Similarly, cell sphericity also decreased in EGFP‐RhoB cells within increased 3D‐matrix density (Fig 3J). These EGFP‐RhoB‐induced changes in migration speed and cell sphericity within high‐density 3D‐matrix reflect an increased capacity for cell deformation enabling efficient migration in 3D‐environments. This is a key feature of blebby amoeboid motility. Supporting these indications, expression of EGFP‐RhoB also promoted 3D‐migration of H1299 cells in two additional assays: a 3D‐Collagen type I invasion assay (Fig 3K) and a 3D‐Matrigel invasion assay (Fig 3L). As distinct from EGFP‐RhoB, EGFP‐tagged RhoA or RhoC showed similar cytoplasmic localizations to their endogenous and FLAG‐tagged counterparts (Fig EV2L) and had no effect on membrane blebbing (Fig EV2M) despite similar GTP‐binding activity to EGFP‐tagged RhoB (Fig EV2N). In summary, while FLAG‐RhoB overexpression mainly localized in vesicular structures and did not affect blebbing in H1299 cells, EGFP‐RhoB predominantly localized to the PM where it induced membrane blebbing and promoted 3D cell migration and invasion.
RhoB GTP‐binding activity and C‐terminal geranylgeranylation are required for RhoB plasma membrane localization and blebbing
GTP‐binding activity is critical for small GTPases to execute their functions (Wennerberg & Der, 2004; Hodge & Ridley, 2016). We found that RhoB displayed intrinsic GTP‐binding activity similar to that of the constitutively active RhoB mutant (63L), while the dominant‐negative (19N) RhoB mutant showed no detectable activity (Fig EV3A and B). This indicates that RhoB has strong inherent GTP‐binding activity, consistent with previous observations (Dubois et al, 2016; Arsic et al, 2017). While WT and GTP‐binding mutant FLAG‐RhoB displayed similar vesicular localization and did not affect F‐actin structures (Fig 4A), EGFP‐tagged RhoB (WT) and (63L) both localized mainly at the PM and both induced blebs, whereas EGFP‐RhoB (19N) predominantly localized in vesicles and caused no membrane blebbing (Fig 4B). These results suggest that the RhoB GTP‐binding activity is required for PM localization and thus for membrane blebbing.
Figure EV3. GTP‐binding activities of RhoB and its mutants.

- H1299 cells transfected with FLAG‐tagged RhoB wild‐type (WT), a dominant‐negative mutant (19N) or a constitutively active mutant (63L). Rho GTPases were pulled down with GST‐RBD protein, and the GTP‐binding activities were detected by immunoblotting with an anti‐FLAG antibody.
- H1299 cells transfected with EGFP‐tagged RhoB (WT), (19N) or (63L). Rho GTPases were pulled down with GST‐RBD protein, and the GTP‐binding activities were detected by immunoblotting with an anti‐GFP antibody.
- H1299 cells stably expressing EGFP‐tagged RhoB (WT), (192S), (193S), (CLLL) or (ΔCKVL), or EGFP were pulled down with GST‐RBD protein, and the GTP‐binding activities were detected by immunoblotting with an anti‐GFP antibody.
Source data are available online for this figure.
Figure 4. RhoB GTP‐binding activity and geranylgeranylation of the CAAX‐box are required for RhoB cell membrane localization and blebbing.

- H1299 cells transiently transfected with FLAG‐tagged RhoB wild‐type (WT), a dominant‐negative mutant (19N) or a constitutively active mutant (63L), labelled for FLAG‐tag and F‐actin.
- H1299 cells stably expressing EGFP‐RhoB (WT), (19N) or (63L), labelled for F‐actin.
- H1299 cells stably expressing different CKVL motif (CAAX‐box) mutants of EGFP‐tagged RhoB labelled for F‐actin.
- H1299 cells transiently transfected with EGFP‐RhoB (WT) or (63L), or their CKVL motif‐deletion (ΔCKVL) variants, labelled for F‐actin.
Rho family proteins contain a C‐terminal CAAX‐box whose prenylation determines membrane insertion and anchoring. The RhoB CAAX‐box can be palmitoylated at C189/C192 and either geranylgeranylated or farnesylated at C193 (Adamson et al, 1992a; Baron et al, 2000). Point mutations at these sites did not inhibit EGFP‐RhoB GTP‐binding activity (Fig EV3C). The EGFP‐RhoB‐CLLL mutant [preventing farnesylation but retaining geranylgeranylation (Baron et al, 2000)] localized at the PM and induced blebs, while expression of the C193S mutant that abolishes both farnesylation and geranylgeranylation of RhoB displayed a diffuse localization and lack of bleb formation (Fig 4C). Together, this indicates a role for RhoB geranylgeranylation, but not farnesylation, in PM localization and bleb formation. Consistently, the C192S mutant, which blocks geranylgeranylation, but not farnesylation of RhoB on C193 (Adamson et al, 1992a), localized in the cytoplasm and did not induce blebs (Fig 4C). However, as C192 is also one of the RhoB palmitoylation sites, the C192S mutation may also affect RhoB palmitoylation, and therefore, palmitoylation of RhoB may also regulate its localization, which is consistent with a previous report (Michaelson et al, 2001). Both the RhoB CAAX‐box deletion mutant (ΔCKVL) and the C193S mutant localized diffusely throughout the cell with no membrane blebs observed (Fig 4C). Importantly, addition of 63L RhoB mutation was not sufficient to re‐localize the ΔCKVL mutant to the PM (Fig 4D), indicating a dominant role for the CAAX‐box in controlling the PM localization of RhoB. In conclusion, geranylgeranylation of the CAAX‐box is needed for RhoB PM localization and RhoB‐induced blebbing (Fig 4E).
Regulation of the intracellular localization of RhoB
While the cellular localization of RhoB appears critical for its functions and while RhoB may regulate endocytic processes (Fernandez‐Borja et al, 2005; Marcos‐Ramiro et al, 2016), the intracellular trafficking of RhoB itself has remained poorly characterized. We therefore examined RhoB vesicular localization in H1299 cells. Endogenous RhoB and FLAG‐tagged RhoB predominantly co‐localized with the late endosome (LE)/lysosome (LY) markers Rab7 and lysosomal‐associated membrane protein 1 (LAMP1; Wherlock et al, 2004), but not with the fast recycling endosome marker Rab4 or early endosome (EE) marker Rab5 (Figs 5A and B, and EV4A). The endosome acidification inhibitor chloroquine was employed to block early endosome maturation, causing a shift in the localization of RhoB from Rab7‐positive to Rab5‐positive vesicles (Figs 5C and EV4B). This suggests that LE/LY‐localized RhoB derived from Rab5‐positive early endosomes. Similar effect on RhoB localization was observed using a constitutively active mutant of Rab5, which blocks normal endosome maturation (Fig EV4C). To avoid any potential effect of EGFP‐Rab7 expression on RhoB localization, we also utilized Rab7‐siRNAs. Similar to chloroquine treatment, a shift in the localization of endogenous RhoB from LAMP1‐positive to early endosome antigen 1 (EEA1)‐positive vesicles was observed (Figs 5D and EV4D), again indicating that LE/LY‐localized RhoB derives from early endosomes. Failure of RhoB transport to LE/LY led to increased total RhoB protein levels (Fig EV4D). The transfer process of RhoB from Rab5‐positive to Rab7‐positive vesicles was directly visualized by live cell imaging of EGFP‐RhoB, CFP‐Rab7 and TagRFP‐Rab5 (Fig 5E; Movie EV6).
Figure 5. RhoB is internalized by endocytosis and localizes in Rab5 and Rab7 vesicles.

-
A, BH1299 cells transfected with (A) EGFP‐Rab4, ‐Rab5 or ‐Rab7, or (B) FLAG‐RhoB, immunolabelled for RhoB (A) or RhoB and LAMP1 (B).
-
CH1299 cells transfected with EGFP‐Rab4, ‐Rab5 or ‐Rab7, and treated with 50 μM chloroquine (CQ) for 6 h, immunolabelled for RhoB.
-
DH1299 cells transfected with Ctrl or Rab7‐siRNAs, and immunolabelled for RhoB and EEA1 (top) or LAMP1 (bottom).
-
EImaging time series of EGFP‐RhoB H1299 cells transfected with TagRFP‐Rab5 and CFP‐Rab7. The boxed region is enhanced to the right and displayed over 130 s. The arrows show the transfer of RhoB from a Rab5‐ to a Rab7‐positive vesicle.
-
FImaging time series of EGFP‐RhoB H1299 cells. The top image shows time zero and the boxed region within is enhanced at the bottom and displayed over 420 s. The arrows show the dynamic process of internalization of RhoB by PM invagination.
-
GH1299 cells transfected with PAGFP‐RhoB and TagRFP‐Rab5. The boxed region of the cell was photoactivated and imaged during a time series. The arrows show the co‐localization of photoactivated RhoB with Rab5‐positive vesicles.
-
H, IEGFP‐RhoB (19N) vesicles were bleached, and the fluorescence recovery process was imaged (H). Top, whole vesicle was bleached. Bottom, half of the vesicle was bleached. Arrows indicate vesicles before and after bleaching. (I) Quantification of the recovery. Bars show mean ± SD. A total number of 15 vesicles from 15 different cells were imaged and analysed.
Figure EV4. Internalization and intracellular trafficking of RhoB.

-
A, BH1299 cells co‐transfected with EGFP‐tagged Rab4, Rab5 or Rab7, together with FLAG‐tagged RhoB. Cells were treated without (A) or with (B) 50 μM CQ for 6 h, fixed and immunolabelled for FLAG‐tag. Scale bars, 10 μm.
-
CH1299 cells co‐transfected with EGFP‐Rab5 wild‐type (WT), a dominant‐negative (DN) Rab5 mutant, or a constitutively active (CA) Rab5 mutant, together with TagRFP‐RhoB (WT). Scale bars, 10 μm.
-
DH1299 cells transfected with Ctrl or Rab7‐siRNAs. The levels of Rab7 and RhoB were detected by immunoblotting as a control for Fig 5D.
Source data are available online for this figure.
We next explored how RhoB may reach LE/LY from the PM and observed EGFP‐RhoB in dynamic PM invaginations, indicating internalization from the PM (Fig 5F; Movie EV7). We therefore tracked RhoB originating at the PM by utilizing photoactivatable GFP (PAGFP)‐tagged RhoB, which was activated by laser specifically at the PM (Fig 5G; Movie EV8). Interestingly, activated PAGFP‐RhoB rapidly spread along the PM, indicating that RhoB diffuses in the PM (Fig 5G; Movie EV8). Importantly, PAGFP‐RhoB activated at the PM also rapidly appeared in Rab5‐positive vesicles (Fig 5G; Movie EV8), indicating that RhoB is endocytosed from the PM to early endosomes. Further, FRAP experiments indicated that the internalized RhoB trafficked in a vesicle‐bound manner with little or no exchange occurring between vesicles and the cytoplasm, as revealed by low recovery of fluorescence on vesicles after photobleaching (Fig 5H and I; Movie EV9). Together, these results indicate that RhoB can be internalized from the PM via endocytosis to be trafficked through Rab5‐positive EE to reach Rab7‐positive LE/LY.
KIF13A mediates re‐distribution of RhoB to the plasma membrane, membrane blebbing and amoeboid migration
Interestingly, while most RhoB localized in Rab7‐positive vesicles, a small portion of RhoB co‐localized with the recycling endosome (RE) marker Rab11 (Fig 6A). We therefore asked whether RhoB may be trafficked from early endosomes to recycling endosomes as an alternative to the early endosome to late endosome path. Indeed, live cell imaging revealed transfer of EGFP‐RhoB from Rab5‐positive to Rab11‐positive vesicles (Fig 6B; Movie EV10). The Rab11 vesicle pointed out in Fig 6B, into which RhoB was transferred from a Rab5 compartment, then rapidly moved adjacent to the PM, after which it was not detected (Fig 6B and Movie EV10).
Figure 6. KIF13A controls RhoB plasma membrane localization and membrane blebbing.
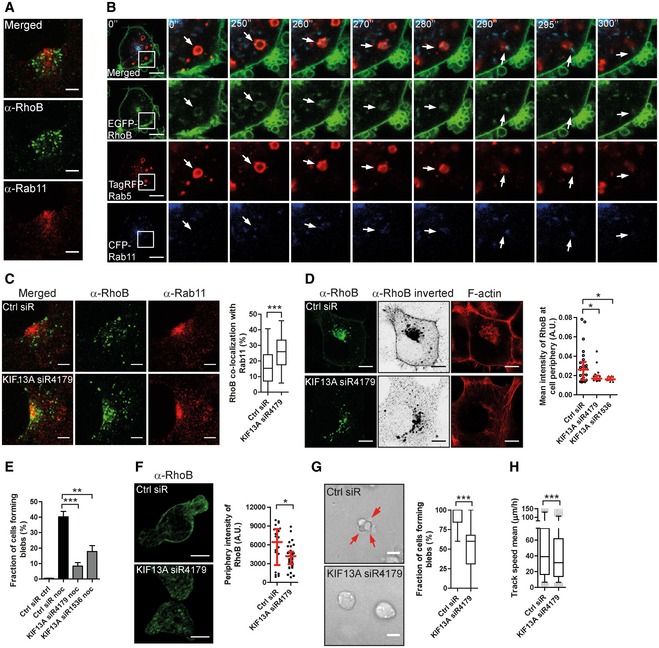
-
AH1299 cells fixed and immunolabelled for RhoB and Rab11.
-
BImaging time series of EGFP‐RhoB H1299 cells transfected with TagRFP‐Rab5 and CFP‐Rab11. The boxed region is enhanced to the right and displayed over 300 s. The arrows show the transfer of RhoB from a Rab5‐ to a Rab11‐positive vesicle.
-
CH1299 cells transfected with Ctrl or KIF13A‐siRNA immunolabelled for RhoB and Rab11. The co‐localization of RhoB with Rab11 was quantified.
-
DH1299 cells transfected with FLAG‐tagged RhoB, together with Ctrl or KIF13A‐siRNAs, immunostained for RhoB. Cells were imaged in a saturated manner for the RhoB channel and displayed in an inverted between projection. The mean intensity of RhoB at the cell periphery (within 1 μm from cell border) was quantified.
-
EH1299 cells were transfected with Ctrl or KIF13A‐siRNAs and treated with or without 1 μM nocodazole (noc) for 30 min and stained for F‐actin. Cells forming blebs were quantified.
-
FT‐ALL cells were electroporated with Ctrl or KIF13A‐siRNA, replated on FN‐coated surface and immunostained for RhoB. The mean intensity of RhoB at the cell periphery (within 0.5 μm from cell border) was quantified.
-
G, HT‐ALL cells electroporated with Ctrl or KIF13A‐siRNA replated in a 2.5 mg/ml 3D‐Collagen type I gel. The fraction of cells forming blebs (G) and 3D cell migration speed (H) was quantified. Red arrows indicate membrane blebs.
Considering also the well‐established role of Rab11 vesicles in recycling to the PM (Stenmark, 2009), we hypothesized that disruption of this trafficking route may accumulate RhoB in recycling vesicles and thereby inhibit RhoB PM localization. Interestingly, KIF13A belongs to the kinesin‐3 subfamily of kinesin motors and is known to mediate recycling from the endosomal recycling compartment (ERC) to the PM (Nakagawa et al, 2000; Hirokawa et al, 2009; Perez Bay et al, 2013). We tested the effect of KIF13A knock‐down on RhoB localization and found that depletion of KIF13A in H1299 cells increased the co‐localization of RhoB with Rab11 (Figs 6C and EV5A), indicating that KIF13A controls RhoB trafficking in Rab11‐positive recycling endosomes. Importantly, knock‐down of KIF13A also depleted the small fraction of endogenous and FLAG‐tagged RhoB localized at the PM (visualized by saturated imaging; Fig 6D). This is consistent with the accumulation of RhoB in Rab11 recycling endosomes and supports a role for KIF13A in RhoB recycling to the PM. Further, KIF13A‐siRNA also blocked nocodazole‐induced membrane blebbing in H1299 cells (Figs 6E and EV5B), suggesting that the effect of KIF13A depletion on membrane blebbing may be linked to its regulation of RhoB localization. Considering that nocodazole disrupts the microtubule system, including the microtubule association of kinesins like KIF13A, kinesins will lose their transport function upon nocodazole treatment. Therefore, knock‐down effects in this experiment reflect differences in kinesin function accumulated over the 48 h of KIF13A knock‐down prior to the 30‐min acute nocodazole treatment, for example in trafficking of RhoB to the PM. To test whether the KIF13A knock‐down may act specifically on RhoB to block membrane blebbing, we induced blebbing in H1299 cells (that otherwise do not bleb) by overexpressing the constitutively active ROCK‐Δ3 mutant (Figs 1L and EV5C). ROCK acts down‐stream of RhoB in blebbing (Figs 1E and 3F); and therefore, ROCK‐Δ3 should induce blebbing in a RhoB‐independent manner. Consistent with this hypothesis, knock‐down of KIF13A did not affect ROCK‐induced blebbing (Fig EV5C and D), showing that KIF13A knock‐down selectively inhibits RhoB‐dependent blebbing. Together, this suggests that KIF13A can support blebbing by mediating RhoB recycling to the PM.
Figure EV5. Effects of KIF13A knock‐down on RhoB localization and membrane blebbing.

-
AKnock‐down efficiency control for Fig 6C. H1299 cells transfected with Ctrl or KIF13A‐siRNA. The knock‐down efficiency was analysed by immunoblotting. Arrow indicates the KIF13A bands and asterisk indicates unspecific bands.
-
BKnock‐down efficiency control for Fig 6D. H1299 cells transfected with Ctrl or KIF13A‐siRNAs. The knock‐down efficiency was analysed by immunoblotting. Arrow indicates the KIF13A bands, and asterisk indicates unspecific bands.
-
C, DH1299 cells transfected with Ctrl or KIF13A‐siRNA, together with a myc‐tagged active ROCK mutant (ROCK‐Δ3). The fraction of cells forming blebs was quantified (C), and the levels of myc‐ROCK‐Δ3 and KIF13A were detected by immunoblotting (D). Bars show mean ± SEM (n = 3 experiments). t‐test (unpaired, two‐tailed) derived P‐value: n.s. = non‐significant.
-
EKnock‐down efficiency control for Fig 6F. T‐ALL cells electroporated with Ctrl or KIF13A‐siRNA. The knock‐down efficiency was analysed by immunoblotting. Arrow indicates the KIF13A bands, and asterisk indicates unspecific bands.
-
F, GT‐ALL cells electroporated with Ctrl or KIF13A‐siRNA and replated in 2.5 mg/ml 3D‐Collagen type I gel. A representative example of Imaris tracking analysis is displayed (F). The knock‐down efficiency was analysed by immunoblotting (G). Corresponds to Fig 6H.
Source data are available online for this figure.
Importantly, knock‐down of KIF13A in T‐ALL cells decreased the PM localization of endogenous RhoB (Figs 6F and EV5E) and significantly inhibited both cell membrane blebbing and 3D cell migration in these cells (Figs 6G and H, and EV5F and G; Movie EV11). Together, this implies that KIF13A is required for RhoB recycling and PM localization, thereby promoting cell membrane blebbing and amoeboid migration.
Based on our results, we present a hypothetical model for the role of RhoB in membrane blebbing and blebby amoeboid migration (Fig 7). Membrane blebbing and blebby amoeboid migration depend on geranylgeranylation‐dependent localization at the PM of GTPase‐active RhoB. PM‐localized RhoB can be internalized by endocytosis and can then traffic from early endosomes to either late endosomes/lysosomes or to a Rab11‐positive endocytic recycling compartment from where KIF13A mediates RhoB return to the PM. At the PM, RhoB induces membrane blebs through local ROCK and Myosin II‐mediated F‐actin contraction, thereby promoting blebby amoeboid cell migration (Fig 7).
Figure 7. Hypothetical model for RhoB trafficking and control of membrane blebbing.
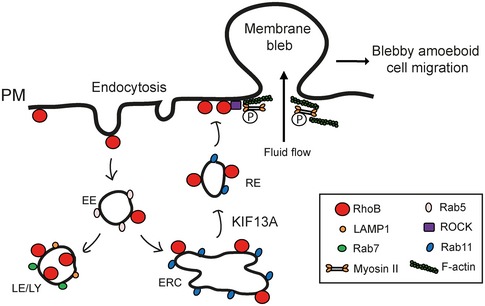
RhoB localized at the plasma membrane (PM) via its C‐terminal CAAX‐box can be internalized by endocytosis. Internalized RhoB can then either be trafficked from early endosomes (EE) to late endosomes/lysosomes (LE/LY) or sorted for recycling through Rab11‐positive vesicles back to the PM dependent on KIF13A. Localized at the PM, local enrichment of RhoB, with its intrinsic GTPase activity, induces membrane blebs through ROCK and Myosin II, thereby promoting blebby amoeboid cell migration.
Discussion
In this study, we define integrated localization and signalling‐based mechanisms by which the small GTPase RhoB drives membrane blebbing, thereby accelerating blebby amoeboid migration in cancer cells within 3D‐environments. While many Rho family GTPases are modulated by regulation both of their GTPase activity and subcellular localization (Wennerberg & Der, 2004; Hodge & Ridley, 2016), mechanisms controlling RhoB function have remained elusive. We now present evidence that regulating the PM localization of RhoB is critical to its novel function in cell membrane blebbing.
Our results indicate that alternative routes of RhoB intracellular vesicular trafficking determine RhoB's localization and function. From the PM, RhoB is internalized and trafficked via Rab5‐positive early endosomes to either Rab7‐positive late endosomes/lysosomes or to Rab11‐positive recycling endosomes. From recycling endosomes, RhoB can re‐distribute to the PM in a KIF13A‐dependent manner. KIF13A has a role in anterograde trafficking (trans‐golgi network to PM; Nakagawa et al, 2000) and also cooperates with Rab11 to transport‐associated tubulation of recycling endosome membranes (Delevoye et al, 2014). These findings are consistent with our evidence that KIF13A has a key role in regulating RhoB transport to the PM from recycling endosomes. The effects of KIF13A on RhoB PM content are important because PM localization of RhoB is critical to its bleb‐inducing activity, although we do not exclude the possibility that RhoB PM levels may be subject to additional mechanisms, such as altered PM retention. Collectively, our finding that KIF13A is necessary for RhoB‐induced membrane blebbing and 3D amoeboid migration of leukaemia cells now places importance on this KIF13A‐mediated RhoB recycling pathway in the context of cancer cell motility.
Endosomal trafficking has previously been shown to control cell migration (Rainero et al, 2012; Macpherson et al, 2014; Ratcliffe et al, 2016). For example, endosomal trafficking of integrins is critical for mesenchymal migration and relates to cancer cell invasion (Rainero et al, 2012; Ratcliffe et al, 2016). RhoB has also been reported to regulate mesenchymal cell migration, with reports suggesting a mixture of activating or inhibitory effects (Wheeler & Ridley, 2007; Bousquet et al, 2016; Dubois et al, 2016; Vega & Ridley, 2016). Here, we link intracellular trafficking to the control of amoeboid cell migration, where re‐distribution of RhoB to the PM enables cells to utilize blebby amoeboid migration, thereby promoting migration in 3D‐ECM microenvironments. This is consistent with evidence that inhibition of endocytosis promotes membrane blebbing (Holst et al, 2017), since this would be expected to increase PM‐localized RhoB. Blebby amoeboid migration contributes to leucocyte infiltration and dissemination of leukaemia and other cancer cells (Charras & Paluch, 2008; Fackler & Grosse, 2008; Paluch & Raz, 2013; Khajah & Luqmani, 2016). Our findings thus highlight mechanisms whose inhibition may perturb amoeboid migration, providing possibilities to limit inflammation and cancer dissemination. However, the net impact of RhoB on cancer is likely to be context‐dependent since RhoB also controls other processes including cell proliferation, apoptosis, migration and invasion; some of which promote tumour progression while others are inhibitory (Wennerberg & Der, 2004; Wheeler & Ridley, 2004; Kazerounian et al, 2013; Vega & Ridley, 2016). Any targeting of RhoB, including through its trafficking, must therefore be assessed in the full context of other RhoB functions and effects. Interestingly, RhoB itself regulates endosomal dynamics (Fernandez‐Borja et al, 2005; Marcos‐Ramiro et al, 2016) and controls the intracellular transport of membrane receptors and intracellular kinases (Gampel et al, 1999; Adini et al, 2003; Sandilands et al, 2004; Wherlock et al, 2004; Neel et al, 2007). Considering the potential for RhoB autoregulation of its own transport, we find that, although RhoB does internalize by endocytosis, the equivalent intracellular distributions of GTP‐binding negative RhoB(19N) and wild‐type RhoB suggest that RhoB internalization is independent of its own GTP‐binding activity.
Both endogenous and EGFP‐tagged RhoB reside in two pools: an LE/LY pool and a PM pool. Importantly, the RhoB distribution can be tuned by specific perturbations (Michaelson et al, 2001; Wherlock et al, 2004; Garcia‐Mariscal et al, 2017), suggesting that a dynamic balance between these two pools is determined by factors such as prenylation state, trafficking, sorting and recycling machineries. In line with this, we reveal that geranylgeranylation, but not farnesylation, is required for RhoB PM localization. Alternate RhoB variants do however display differences in distribution proportion between the PM and vesicular pools (Michaelson et al, 2001; Wherlock et al, 2004), something that we also observed here. Interestingly, we observed large differences in RhoB distribution between epithelial‐ and leucocyte‐derived cells, suggesting recurrent differences in these underlying localization‐determining factors.
In agreement with previous reports (Dubois et al, 2016; Arsic et al, 2017), we observed substantial endogenous RhoB GTP‐binding activity, supporting the notion that intrinsic RhoB GTP‐binding activity is high and may not require extrinsic activation. This is consistent with our findings that regulation of RhoB intracellular localization rather than of its activity controls membrane blebbing‐mediated amoeboid motility. We also demonstrate that RhoB causes membrane blebbing through the well‐established ROCK‐Myosin II pathway, which acts down‐stream of several Rho isoforms, including RhoB, and has previously been implicated in membrane blebbing (Charras et al, 2006; Ridley, 2011).
RhoB is the most divergent of the Rho subfamily of small GTPases (Wheeler & Ridley, 2004; Vega & Ridley, 2016) and differences in palmitoylation and prenylation patterns compared to RhoA and RhoC support the notion that RhoB may regulate distinct cellular and physiological functions. This is in line with our finding that RhoA and RhoB controlled different aspects of 3D amoeboid leukaemia cell migration, with RhoA being important for cell rear retraction, while RhoB controlled membrane blebbing. This contrasts with previous studies that have suggested a role for RhoA in cell membrane blebbing (Gadea et al, 2007; Godin & Ferguson, 2010; Cartier‐Michaud et al, 2012; Tozluoglu et al, 2013; Takahara et al, 2014; Bang et al, 2015; Aoki et al, 2016). However, in most of these studies, experimental methods (e.g. non‐specific drug‐based inhibition) did not provide clear indications as to which Rho family member is responsible for effects and in other cases, blebbing function was inferred from gross cell morphology rather than direct visualization of blebbing per se (Gadea et al, 2007; Pinner & Sahai, 2008; Godin & Ferguson, 2010; Cartier‐Michaud et al, 2012; Tozluoglu et al, 2013; Takahara et al, 2014; Bang et al, 2015; Aoki et al, 2016). In any case, our finding that depletion of RhoB impaired membrane blebbing in four distinct cell models of different origin shows that RhoB is critical for membrane blebbing under a variety of conditions. However, we cannot exclude a function for RhoA in membrane blebbing, since such a function may be rescued by upregulation of RhoB upon RhoA knock‐down. This possibility is supported by a recent study showing that upregulation of RhoB upon loss of RhoA can functionally replace RhoA with respect to invasion, clonogenic growth and survival (Garcia‐Mariscal et al, 2017).
In conclusion, we here propose that KIF13A‐regulated RhoB vesicular recycling to the PM, in conjunction with geranylgeranylation of RhoB, is needed for membrane blebbing and blebby amoeboid migration of cancer cells. Given that amoeboid motility is involved in many physiological processes, extending this understanding of how RhoB regulates membrane blebbing is now of fundamental importance.
Materials and Methods
Cell culture
H1299 (human non‐small cell lung carcinoma cell; ATCC# CRL‐5803), Reh (B‐cell acute lymphoblastic leukaemia cell; ATCC# CRL‐8286) and T‐ALL (T‐cell acute lymphoblastic leukaemia cell) cells (Malyukova et al, 2007) were cultured in RPMI 1640 medium (Gibco) supplemented with 1 mM l‐Glutamine and 10% foetal bovine serum (FBS, Gibco) at 37°C, 5% CO2. H1299 cells stably transfected with EGFP, EGFP‐RhoB or EGFP‐RhoB(19N) were maintained in RPMI 1640 culture medium supplemented with 200 μg/ml geneticin (G‐418 sulfate, Gibco). A375M2 DS human melanoma cells, stably expressing LifeAct and H2B, were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% FBS and 100 μg/ml geneticin.
Constructs, siRNAs and chemicals
EGFP‐Rab4, EGFP‐Rab5 and EGFP‐Rab7 were described previously (Plantard et al, 2010). Human RhoB cDNA was obtained by PCR amplification of a human lung cDNA library and inserted into pEGFP‐C3 vector (Clontech) and pTagRFP‐C vector (Evrogen). PCR‐amplified Rab5 was inserted into pTagRFP‐C vector, and PCR‐amplified Rab7 or Rab11 was inserted into PS‐CFP2‐C vector (Evrogen). PAGFP‐RhoB was obtained by replacing EGFP of EGFP‐RhoB with PAGFP (from pPAGFP‐N1, Addgene, #11909). All the mutants were generated by PCR‐based mutagenesis (QuikChange Site‐Directed Mutagenesis Kit, Stratagene) and confirmed by sequencing (Macrogen Europe, the Netherlands). All siRNAs (sequences listed in Appendix Table S1) were obtained from GenePharma (Shanghai, China). Chemicals were purchased from Sigma‐Aldrich: nocodazole (M1404), blebbistatin (B0560), Y27632 (Y0503), D‐sorbitol (S1876), MTT (M2128), chloroquine (C6628) and DMSO (D2650).
Transfection
For lipid‐mediated siRNA transfection, 2 × 104 H1299 cells per well were seeded into a 24‐well plate in 1 ml culture medium. 24 h later, 30 pmol siRNA was transfected into H1299 cells with 1.5 μl RNAiMAX (Thermo Fisher Scientific), according to the manufacturer's protocol. For electroporation‐mediated siRNA transfection, Reh or T‐ALL cells were harvested and adjusted to 6 × 106 per ml. Cells were electroporated by the Neon Transfection System (Thermo Fisher Scientific) with 100 μl tips. The settings for electroporation were as follows: 1,450 V, 10 ms and four pulses. Electroporated cells were plated into a 6‐well plate with pre‐warmed culture medium without antibiotics. After 48 h at 37°C, cells were harvested for live cell imaging or for immunoblotting.
For plasmid transfection, 8 × 104 H1299 cells per well were seeded into a 24‐well plate in 1 ml culture medium. 24 h later, 0.5 μg DNA was transfected into H1299 cells with 0.6 μl lipofectamine 3000 and 1.2 μl P3000 reagent (Thermo Fisher Scientific), according to the manufacturer's protocol. After 24 h‐incubation, cells were harvested for live cell imaging or for immunoblotting. COS‐7, MCF‐7, PC‐3, Hela, NIH3T3 and MDCK cells were transfected in the same way.
Immunocytochemistry
Cells transfected with FLAG‐tagged vectors were fixed in 4% paraformaldehyde for 10 min at RT, washed with PBS and permeabilized with 0.1% Triton X‐100 for 5 min. Following 3× washing, cells were blocked by incubation with 3% BSA/PBS. Then, cells were incubated with an anti‐FLAG antibody (1:100, Cell Signaling Technology) for 60 min, washed 3× with PBS and incubated with FITC‐conjugated goat anti‐mouse IgG (1:300, Thermo Fisher Scientific, USA) for 45 min at RT. F‐actin or nuclei were in some experiments labelled by simultaneous incubation with Alexa 568 phalloidin (Thermo Fisher Scientific) or Hoechst 33342. Cells were washed with PBS 3× before imaging by confocal microscopy as described below. The same procedure of immunocytochemistry was conducted for cells immunostained with anti‐pMLC (Ser19, 1:100, #3671, Cell Signaling Technology), anti‐LAMP1 (1:100, #9091, Cell Signaling Technology), anti‐EEA1 (1:100, #3288, Cell Signaling Technology) or anti‐Rab11 (1:100, #5589, Cell Signaling Technology) antibodies. For the immunolabelling of endogenous RhoB, the method by Adamson et al (1992b) was followed. Briefly, after fixation and permeabilization, cells were incubated with 10% FCS in PBS for 30 min, followed by incubation with anti‐RhoB antibody (sc‐8048, Santa Cruz) at 1:100 dilution in incubation buffer (50 mM Tris.Cl, pH 7.6, 1% NP‐40, 0.5% sodium deoxycholate and 5 mM EDTA), which was also used for washing and FITC‐conjugated goat anti‐mouse IgG dilution. Cells were finally washed 3× with PBS. For the co‐localization of RhoB and β1 integrin, an anti‐integrin α5β1 antibody (1:100, MAB1969, clone JBS5, Millipore) was used.
2D cell blebbing
EGFP or EGFP‐RhoB cells in 96‐well glass‐bottom plates (145 μm optical glass, 5241‐20, Zell Kontakt, Germany) were incubated with CellMask Deep Red Plasma membrane Stain (1:5,000, Thermo Fisher Scientific) for 10 min, and then, the dye was washed out. Cells were imaged at 5 s interval for 5 min with an oil‐immersion objective (PlanApo VC 60×/1.4 NA) of a Nikon A1 confocal microscope.
3D‐Collagen type I gel cell blebbing and migration
For 2.5 mg/ml Collagen type I gel, 20 μl 10 × RPMI, 23 μl 0.1 M NaOH and 6 μl 7.5% NaHCO3 were mixed thoroughly, and then, 75 μl Collagen Type I (rat tail, 4.48 mg/ml, 08‐115, Millipore) was added and mixed thoroughly again. 1 × 105 T‐All, Reh or different H1299 stable cells suspended in 10 μl serum‐free culture medium without antibiotics were added and mixed thoroughly by pipetting. For different concentrations of Collagen type I gels, the amounts of gradients were adjusted accordingly. Cells containing Collagen I gel was plated into 96‐well glass‐bottom plate, and the plate was gently tapped to let the gel form a flat surface. The plate was put in the incubator for 45 min, and then, 100 μl normal culture medium was added on the top. One hour later, cells were subjected to live cell imaging at 37°C and 5% CO2.
Live cell images for blebbing were acquired with a Nikon A1 confocal microscope using an oil‐immersion objective (PlanApo VC 60×/1.4 NA), at 10 s interval for 10 min with 512 × 512 pixel resolution. Quantification was performed by determining the number of cells forming blebs during the entire imaging time. For A375M2 DS cells, 1 h after incubation with normal culture medium, cells in the Collagen type I gel were fixed with 4% paraformaldehyde and imaged, and cells forming blebs were counted.
For cell migration imaging, T‐All cells were incubated with CellTracker Red CMTPX Dye (1:5,000, Thermo Fisher Scientific) for 10 min and then the dye was washed out. Cells were imaged with a 10× objective (Plan Apo 10× DIC L) of Nikon A1 confocal microscope, with pinhole as 3 (48.53 μm). Images were acquired at 5‐min interval for 6 h with 512 × 512 pixel resolution, and cells in a 300 μm vertical range were imaged by obtaining z‐stacks (5.925 μm/step, Ti ZDrive). Images were analysed and quantified using Imaris (Version 8, Bitplane, Switzerland).
Confocal cell fluorescence imaging
Cells in 96‐well glass‐bottomed plates were imaged with a Nikon A1R laser scanning confocal microscope (Nikon Laboratory Imaging) equipped with an Argon‐Laser Multiline (458/488/514 nm) and 405 nm, 561 nm and 647 nm lasers. Images were acquired with NIS‐Elements AR Imaging Software 4.12.01.
Image analysis by Imaris
Images of 3D‐collagen gel migration and invasion were analysed using Imaris software. Briefly, firstly a “surface” was added to the images as the basis for object segmentation, and then a small image field was chosen for tentative analysis to optimize parameter setting, and finally these settings were applied to analyse the whole data set. Background subtraction was set to 15 μm, and MaxDistance as 20 μm, Max Gap Size as 3. Autoregression motion was selected, and number of voxels for filtering was set to 10.
Endocytosis of photoactivatable RhoB
H1299 cells were co‐transfected with PAGFP‐RhoB and TagRFP‐Rab5. Cells were imaged with the 488 nm (0.7% power) and 561 nm (0.5% power) lasers once, and then, the PAGFP‐RhoB at a defined small region of the cell membrane was activated by the 405 nm laser (20% power, 1 s). The cells were then imaged during a time series of 3 min with an interval of 3 s, with the same laser powers as the imaging before activation.
Fluorescence recovery after photobleaching (FRAP)
EGFP‐RhoB(19N) stable cells were plated into 96‐well glass‐bottom plates and cultured for 24 h. Cells were treated with 50 μM CQ for 24 h and 1 ng/ml (0.0033 μM) nocodazole for 1 h. A defined RhoB‐positive vesicle was imaged three times with 512 × 512 pixel resolution using a Nikon A1R confocal microscope equipped with a Plan Apo VC 60× Oil N/A 1.4 objective, followed by being bleached at high laser power (40% power, 32 mW, 40 iterations) using the 488 nm line of an Argon laser. Recovery of fluorescence was monitored by scanning the vesicle at low laser power (0.8% power, 1.1 mW) for 120 s at an interval of 3 s. Images were analysed using Fiji (ImageJ; https://fiji.sc/). Quantification was performed by measuring the background fluorescence intensity of the cell and the fluorescence intensity of the bleached vesicle before, directly after and during recovery of bleaching. Fluorescence relative intensity (RI) was calculated according to the equation: RI = (Ibg[0]/Ible[0]) × (Ible[t]/Ibg[t]), where Ibg(0) is the mean intensity of background area before bleaching, Ible(0) the mean intensity of the bleached area before bleaching, Ible(t) the mean intensity of the bleached area at time t and Ibg(t) the mean intensity of the background area at time t.
Co‐localization of RhoB with Rab11
H1299 cells were transfected with different siRNAs, and 48 h later, cells were fixed and immunostained with anti‐RhoB and anti‐Rab11 antibodies. Cells were imaged using a Nikon A1R confocal microscope, and the co‐localization of RhoB with Rab11 was analysed and quantified with the Co‐localization Pipeline of CellProfiler software 2.2.0 (http://cellprofiler.org/examples/#Colocalization).
Statistical analysis
All statistical analyses were carried out with GraphPad Prism 5 (GraphPad Software, San Diego, USA). Each statistical method used is specified in the figure legends. P‐values < 0.05 were considered statistically discernable (*P < 0.05; **P < 0.01; ***P < 0.001; n.s.—non‐significant).
Data availability
The authors declare that the data supporting the findings of this study are available within the article and from the authors upon request.
Author contributions
XG conceived of, designed and performed the research, analysed and interpreted data, and wrote the paper. YD analysed images with CellProfiler. JGL contributed to data interpretation. SS performed research design, interpreted data, wrote the paper and provided financing. SS and XG supervised the study. All authors read and approved the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Movie EV9
Movie EV10
Movie EV11
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We wish to thank Dr. Dan Grander, Karolinska Institutet, Sweden, for providing T‐ALL and Reh cells and Dr. Erik Sahai, The Francis Crick Institute, United Kingdom, for the A375M2 DS cells. We thank Dr. Shuh Narumiya, Kyoto University, Japan, for the pCAG‐myc‐ROCK Δ3 construct. This work was supported by grants to SS from the Swedish Childhood Cancer Foundation (PR2015‐0038), the EU‐FP7‐Systems Microscopy Network of Excellence (HEALTH‐F4‐2010‐258068), EU‐H2020‐Multimot (EU Grant agreement #634107), The Strategic Research Foundation of Sweden (SB16‐0046), The Swedish Research Council (521‐2012‐3180) and the Swedish Cancer Society. Imaging was performed at the Live cell imaging facility and Nikon centre of excellence at the Department of Biosciences and Nutrition, Karolinska Institutet, supported by grants from the Knut and Alice Wallenberg Foundation, the Swedish Research Council, the Centre for Innovative Medicine and the Jonasson donation to the School of Technology and Health, Royal Institute of Technology, Sweden. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
The EMBO Journal (2018) 37: e98994
Contributor Information
Xiaowei Gong, Email: xiaowei.gong@ki.se.
Staffan Strömblad, Email: staffan.stromblad@ki.se.
References
- Adamson P, Marshall CJ, Hall A, Tilbrook PA (1992a) Post‐translational modifications of p21rho proteins. J Biol Chem 267: 20033–20038 [PubMed] [Google Scholar]
- Adamson P, Paterson HF, Hall A (1992b) Intracellular localization of the P21rho proteins. J Cell Biol 119: 617–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adini I, Rabinovitz I, Sun JF, Prendergast GC, Benjamin LE (2003) RhoB controls Akt trafficking and stage‐specific survival of endothelial cells during vascular development. Genes Dev 17: 2721–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki K, Maeda F, Nagasako T, Mochizuki Y, Uchida S, Ikenouchi J (2016) A RhoA and Rnd3 cycle regulates actin reassembly during membrane blebbing. Proc Natl Acad Sci USA 113: E1863–E1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsic N, Ho‐Pun‐Cheung A, Evelyne C, Assenat E, Jarlier M, Anguille C, Colard M, Pezet M, Roux P, Gadea G (2017) The p53 isoform delta133p53ss regulates cancer cell apoptosis in a RhoB‐dependent manner. PLoS One 12: e0172125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang J, Jang M, Huh JH, Na JW, Shim M, Carlson BA, Tobe R, Tsuji PA, Gladyshev VN, Hatfield DL, Lee BJ (2015) Deficiency of the 15‐kDa selenoprotein led to cytoskeleton remodeling and non‐apoptotic membrane blebbing through a RhoA/ROCK pathway. Biochem Biophys Res Commun 456: 884–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron R, Fourcade E, Lajoie‐Mazenc I, Allal C, Couderc B, Barbaras R, Favre G, Faye JC, Pradines A (2000) RhoB prenylation is driven by the three carboxyl‐terminal amino acids of the protein: evidenced in vivo by an anti‐farnesyl cysteine antibody. Proc Natl Acad Sci USA 97: 11626–11631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet E, Calvayrac O, Mazieres J, Lajoie‐Mazenc I, Boubekeur N, Favre G, Pradines A (2016) RhoB loss induces Rac1‐dependent mesenchymal cell invasion in lung cells through PP2A inhibition. Oncogene 35: 1760–1769 [DOI] [PubMed] [Google Scholar]
- Cartier‐Michaud A, Malo M, Charriere‐Bertrand C, Gadea G, Anguille C, Supiramaniam A, Lesne A, Delaplace F, Hutzler G, Roux P, Lawrence DA, Barlovatz‐Meimon G (2012) Matrix‐bound PAI‐1 supports cell blebbing via RhoA/ROCK1 signaling. PLoS One 7: e32204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras GT, Yarrow JC, Horton MA, Mahadevan L, Mitchison TJ (2005) Non‐equilibration of hydrostatic pressure in blebbing cells. Nature 435: 365–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras GT, Hu CK, Coughlin M, Mitchison TJ (2006) Reassembly of contractile actin cortex in cell blebs. J Cell Biol 175: 477–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras G, Paluch E (2008) Blebs lead the way: how to migrate without lamellipodia. Nat Rev Mol Cell Biol 9: 730–736 [DOI] [PubMed] [Google Scholar]
- Delevoye C, Miserey‐Lenkei S, Montagnac G, Gilles‐Marsens F, Paul‐Gilloteaux P, Giordano F, Waharte F, Marks MS, Goud B, Raposo G (2014) Recycling endosome tubule morphogenesis from sorting endosomes requires the kinesin motor KIF13A. Cell Rep 6: 445–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois F, Keller M, Calvayrac O, Soncin F, Hoa L, Hergovich A, Parrini MC, Mazieres J, Vaisse‐Lesteven M, Camonis J, Levallet G, Zalcman G (2016) RASSF1A suppresses the invasion and metastatic potential of human non‐small cell lung cancer cells by inhibiting YAP activation through the GEF‐H1/RhoB pathway. Cancer Res 76: 1627–1640 [DOI] [PubMed] [Google Scholar]
- Fackler OT, Grosse R (2008) Cell motility through plasma membrane blebbing. J Cell Biol 181: 879–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Borja M, Janssen L, Verwoerd D, Hordijk P, Neefjes J (2005) RhoB regulates endosome transport by promoting actin assembly on endosomal membranes through Dia1. J Cell Sci 118: 2661–2670 [DOI] [PubMed] [Google Scholar]
- Gadea G, de Toledo M, Anguille C, Roux P (2007) Loss of p53 promotes RhoA‐ROCK‐dependent cell migration and invasion in 3D matrices. J Cell Biol 178: 23–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gampel A, Parker PJ, Mellor H (1999) Regulation of epidermal growth factor receptor traffic by the small GTPase rhoB. Curr Biol 9: 955–958 [DOI] [PubMed] [Google Scholar]
- Garcia‐Mariscal A, Li H, Pedersen E, Peyrollier K, Ryan KM, Stanley A, Quondamatteo F, Brakebusch C (2017) Loss of RhoA promotes skin tumor formation and invasion by upregulation of RhoB. Oncogene 37: 847–860 [DOI] [PubMed] [Google Scholar]
- Gebala V, Collins R, Geudens I, Phng LK, Gerhardt H (2016) Blood flow drives lumen formation by inverse membrane blebbing during angiogenesis in vivo . Nat Cell Biol 18: 443–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin CM, Ferguson SS (2010) The angiotensin II type 1 receptor induces membrane blebbing by coupling to Rho A, Rho kinase, and myosin light chain kinase. Mol Pharmacol 77: 903–911 [DOI] [PubMed] [Google Scholar]
- Hagmann J, Burger MM, Dagan D (1999) Regulation of plasma membrane blebbing by the cytoskeleton. J Cell Biochem 73: 488–499 [PubMed] [Google Scholar]
- Haston WS, Shields JM (1984) Contraction waves in lymphocyte locomotion. J Cell Sci 68: 227–241 [DOI] [PubMed] [Google Scholar]
- van Helvert S, Storm C, Friedl P (2018) Mechanoreciprocity in cell migration. Nat Cell Biol 20: 8–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S (2009) Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol 10: 682–696 [DOI] [PubMed] [Google Scholar]
- Hodge RG, Ridley AJ (2016) Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol 17: 496–510 [DOI] [PubMed] [Google Scholar]
- Holst MR, Vidal‐Quadras M, Larsson E, Song J, Hubert M, Blomberg J, Lundborg M, Landstrom M, Lundmark R (2017) Clathrin‐independent endocytosis suppresses cancer cell blebbing and invasion. Cell Rep 20: 1893–1905 [DOI] [PubMed] [Google Scholar]
- Jung JJ, Inamdar SM, Tiwari A, Ye D, Lin F, Choudhury A (2013) Syntaxin 16 regulates lumen formation during epithelial morphogenesis. PLoS One 8: e61857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazerounian S, Gerald D, Huang M, Chin YR, Udayakumar D, Zheng N, O'Donnell RK, Perruzzi C, Mangiante L, Pourat J, Phung TL, Bravo‐Nuevo A, Shechter S, McNamara S, Duhadaway JB, Kocher ON, Brown LF, Toker A, Prendergast GC, Benjamin LE (2013) RhoB differentially controls Akt function in tumor cells and stromal endothelial cells during breast tumorigenesis. Cancer Res 73: 50–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajah MA, Luqmani YA (2016) Involvement of membrane blebbing in immunological disorders and cancer. Med Princ Pract 25(Suppl 2): 18–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laser‐Azogui A, Diamant‐Levi T, Israeli S, Roytman Y, Tsarfaty I (2014) Met‐induced membrane blebbing leads to amoeboid cell motility and invasion. Oncogene 33: 1788–1798 [DOI] [PubMed] [Google Scholar]
- Liu YJ, Le Berre M, Lautenschlaeger F, Maiuri P, Callan‐Jones A, Heuze M, Takaki T, Voituriez R, Piel M (2015) Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell 160: 659–672 [DOI] [PubMed] [Google Scholar]
- Macpherson IR, Rainero E, Mitchell LE, van den Berghe PV, Speirs C, Dozynkiewicz MA, Chaudhary S, Kalna G, Edwards J, Timpson P, Norman JC (2014) CLIC3 controls recycling of late endosomal MT1‐MMP and dictates invasion and metastasis in breast cancer. J Cell Sci 127: 3893–3901 [DOI] [PubMed] [Google Scholar]
- Malyukova A, Dohda T, von der Lehr N, Akhoondi S, Corcoran M, Heyman M, Spruck C, Grander D, Lendahl U, Sangfelt O (2007) The tumor suppressor gene hCDC4 is frequently mutated in human T‐cell acute lymphoblastic leukemia with functional consequences for Notch signaling. Cancer Res 67: 5611–5616 [DOI] [PubMed] [Google Scholar]
- Marcos‐Ramiro B, Garcia‐Weber D, Barroso S, Feito J, Ortega MC, Cernuda‐Morollon E, Reglero‐Real N, Fernandez‐Martin L, Duran MC, Alonso MA, Correas I, Cox S, Ridley AJ, Millan J (2016) RhoB controls endothelial barrier recovery by inhibiting Rac1 trafficking to the cell border. J Cell Biol 213: 385–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson D, Silletti J, Murphy G, D'Eustachio P, Rush M, Philips MR (2001) Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J Cell Biol 152: 111–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Setou M, Seog D, Ogasawara K, Dohmae N, Takio K, Hirokawa N (2000) A novel motor, KIF13A, transports mannose‐6‐phosphate receptor to plasma membrane through direct interaction with AP‐1 complex. Cell 103: 569–581 [DOI] [PubMed] [Google Scholar]
- Neel NF, Lapierre LA, Goldenring JR, Richmond A (2007) RhoB plays an essential role in CXCR2 sorting decisions. J Cell Sci 120: 1559–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluch EK, Raz E (2013) The role and regulation of blebs in cell migration. Curr Opin Cell Biol 25: 582–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluch EK, Aspalter IM, Sixt M (2016) Focal adhesion‐independent cell migration. Annu Rev Cell Dev Biol 32: 469–490 [DOI] [PubMed] [Google Scholar]
- Perez Bay AE, Schreiner R, Mazzoni F, Carvajal‐Gonzalez JM, Gravotta D, Perret E, Lehmann Mantaras G, Zhu YS, Rodriguez‐Boulan EJ (2013) The kinesin KIF16B mediates apical transcytosis of transferrin receptor in AP‐1B‐deficient epithelia. EMBO J 32: 2125–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinner S, Sahai E (2008) PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat Cell Biol 10: 127–137 [DOI] [PubMed] [Google Scholar]
- Plantard L, Arjonen A, Lock JG, Nurani G, Ivaska J, Stromblad S (2010) PtdIns(3,4,5)P(3) is a regulator of myosin‐X localization and filopodia formation. J Cell Sci 123: 3525–3534 [DOI] [PubMed] [Google Scholar]
- Rainero E, Caswell PT, Muller PA, Grindlay J, McCaffrey MW, Zhang Q, Wakelam MJ, Vousden KH, Graziani A, Norman JC (2012) Diacylglycerol kinase alpha controls RCP‐dependent integrin trafficking to promote invasive migration. J Cell Biol 196: 277–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe CD, Sahgal P, Parachoniak CA, Ivaska J, Park M (2016) Regulation of cell migration and beta1 integrin trafficking by the endosomal adaptor GGA3. Traffic 17: 670–688 [DOI] [PubMed] [Google Scholar]
- Ridley AJ (2011) Life at the leading edge. Cell 145: 1012–1022 [DOI] [PubMed] [Google Scholar]
- Ruprecht V, Wieser S, Callan‐Jones A, Smutny M, Morita H, Sako K, Barone V, Ritsch‐Marte M, Sixt M, Voituriez R, Heisenberg CP (2015) Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell 160: 673–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E (2005) Mechanisms of cancer cell invasion. Curr Opin Genet Dev 15: 87–96 [DOI] [PubMed] [Google Scholar]
- Sandilands E, Cans C, Fincham VJ, Brunton VG, Mellor H, Prendergast GC, Norman JC, Superti‐Furga G, Frame MC (2004) RhoB and actin polymerization coordinate Src activation with endosome‐mediated delivery to the membrane. Dev Cell 7: 855–869 [DOI] [PubMed] [Google Scholar]
- Stenmark H (2009) Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 10: 513–525 [DOI] [PubMed] [Google Scholar]
- Takahara Y, Maeda M, Hasegawa H, Ito S, Hyodo T, Asano E, Takahashi M, Hamaguchi M, Senga T (2014) Silencing of TBC1D15 promotes RhoA activation and membrane blebbing. Mol Cell Biochem 389: 9–16 [DOI] [PubMed] [Google Scholar]
- Takesono A, Heasman SJ, Wojciak‐Stothard B, Garg R, Ridley AJ (2010) Microtubules regulate migratory polarity through Rho/ROCK signaling in T cells. PLoS One 5: e8774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez JY, Schulze U, Salbreux G, Roensch J, Joanny JF, Paluch E (2009) Role of cortical tension in bleb growth. Proc Natl Acad Sci USA 106: 18581–18586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozluoglu M, Tournier AL, Jenkins RP, Hooper S, Bates PA, Sahai E (2013) Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nat Cell Biol 15: 751–762 [DOI] [PubMed] [Google Scholar]
- Trendowski M (2015) The inherent metastasis of leukaemia and its exploitation by sonodynamic therapy. Crit Rev Oncol Hematol 94: 149–163 [DOI] [PubMed] [Google Scholar]
- Vega FM, Fruhwirth G, Ng T, Ridley AJ (2011) RhoA and RhoC have distinct roles in migration and invasion by acting through different targets. J Cell Biol 193: 655–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega FM, Ridley AJ (2016) The RhoB small GTPase in physiology and disease. Small GTPases 10.1080/21541248.2016.1253528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennerberg K, Der CJ (2004) Rho‐family GTPases: it's not only Rac and Rho (and I like it). J Cell Sci 117: 1301–1312 [DOI] [PubMed] [Google Scholar]
- Wheeler AP, Ridley AJ (2004) Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res 301: 43–49 [DOI] [PubMed] [Google Scholar]
- Wheeler AP, Ridley AJ (2007) RhoB affects macrophage adhesion, integrin expression and migration. Exp Cell Res 313: 3505–3516 [DOI] [PubMed] [Google Scholar]
- Wherlock M, Gampel A, Futter C, Mellor H (2004) Farnesyltransferase inhibitors disrupt EGF receptor traffic through modulation of the RhoB GTPase. J Cell Sci 117: 3221–3231 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Movie EV9
Movie EV10
Movie EV11
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the article and from the authors upon request.