

Abstract
The mammalian Hippo signaling pathway, through its effectors YAP and TAZ, coerces epithelial progenitor cell expansion for appropriate tissue development or regeneration upon damage. Its ability to drive rapid tissue growth explains why many oncogenic events frequently exploit this pathway to promote cancer phenotypes. Indeed, several tumor types including basal cell carcinoma (BCC) show genetic aberrations in the Hippo (or YAP/TAZ) regulators. Here, we uncover that while YAP is dispensable for homeostatic epidermal regeneration, it is required for BCC development. Our clonal analyses further demonstrate that the few emerging Yap‐null dysplasia have lower fitness and thus are diminished as they progress to invasive BCC. Mechanistically, YAP depletion in BCC tumors leads to effective impairment of the JNK‐JUN signaling, a well‐established tumor‐driving cascade. Importantly, in this context, YAP does not influence canonical Wnt or Hedgehog signaling. Overall, we reveal Hippo signaling as an independent promoter of BCC pathogenesis and thereby a viable target for drug‐resistant BCC.
Keywords: AP1, basal cell carcinoma, Hippo signaling, Jun, YAP
Subject Categories: Cancer, Signal Transduction, Transcription
Introduction
Initially discovered in Drosophila, the mammalian Hippo signaling pathway is a central modulator of progenitor cell state necessary for proper epithelial tissue regeneration. The culminating effect of the upstream Hippo kinases and/or cytoskeleton‐bound adaptor proteins is to tightly regulate the Hippo effectors’ (YAP and TAZ) interaction with the DNA‐binding TEA domain (TEAD) transcription factors (Patel et al, 2017). Coordinated YAP/TAZ‐TEAD interaction and resulting gene signature is vital for embryonic tissue development, as any perturbation in their function results in the depletion of progenitor cell pool and failed organogenesis (Varelas, 2014). The robust propensity of activated YAP and TAZ to enhance the progenitor cell state phenotype (i.e., self‐renewal, epithelial–mesenchymal transition, proliferation, and survival) in epithelial cells is believed to be the reason why cancer cells frequently hijack the Hippo pathway during tumorigenesis (Harvey et al, 2013). Indeed, conditional YAP overexpression results in hyperproliferation and tumor development in several epithelial tissues including skin epidermis. Short‐term epidermal YAP overexpression induces the expansion of undifferentiated/progenitor cells in the interfollicular epidermis (IFE), skin thickening, and development of basal cell carcinoma‐like (BCC) lesions (Schlegelmilch et al, 2011; Silvis et al, 2011; Zhang et al, 2011; Akladios et al, 2017).
Basal cell carcinoma is the most common sporadic tumor type in individuals of European descent, and its incidence has increased at a steady and alarming rate (Flohil et al, 2013; Wu et al, 2013). Histologically, BCCs closely resemble expanded IFE basal progenitor cells that have invaded into underlying dermis (Youssef et al, 2010; Sanchez‐Danes et al, 2016). Sporadic human BCC is initiated and driven by mutations in the Hedgehog pathway regulators, mainly the receptor Patched 1 (PTCH1) and G‐protein‐coupled receptor Smoothened (SMO). Mutation in PTCH1 or SMO relieves PTCH1 inhibitory effect on SMO and triggers constitutive activation of the GLI‐driven growth‐promoting programs (Johnson et al, 1996; Reifenberger et al, 2005; Epstein, 2008). Strong genetic evidence and near 100% mutation frequency of the Hedgehog pathway in basal cell nevus syndrome and BCC have led to successful development of the SMO inhibitor, vismodegib, for the treatment of these diseases. However, locally advanced and metastatic BCCs frequently (~50% recurrence) escape Hedgehog pathway inhibition, predominantly via the mutation of SMO within the drug‐binding pocket or the alternative potentiation of GLI signaling (Atwood et al, 2012, 2015; Sharpe et al, 2015; Zhao et al, 2015; Danhof et al, 2018). The development of new approaches to inhibit BCC tumor growth in parallel with vismodegib therapy would provide significant clinical benefits for patients with advanced BCC.
Phenotypic overlap in the skin with perturbed Hedgehog or Hippo signaling implies that these two pathways might cooperate in the transformation process (Nieuwenhuis et al, 2007; Villani et al, 2010; Schlegelmilch et al, 2011; Zhang et al, 2011). Indeed, YAP gene amplification and overexpression were found in Hedgehog‐driven medulloblastoma (Fernandez et al, 2009). The latest genomic analyses of human BCC samples revealed that up to 30% patients’ tumors harbor loss‐of‐function alterations in the known Hippo regulators (Bonilla et al, 2016). Hence, in this report we set out to investigate the functional relevance of Hippo pathway output in Hedgehog‐driven BCC development. Our rigorous in vitro and in vivo analyses demonstrate that YAP, but not TAZ, is required for BCC initiation and progression to advanced stage. Functionally, YAP potentiated the well‐established JNK‐JUN signaling network to drive BCC growth, without any impact on Wnt or Hedgehog signaling. Our data indicate that targeting the Hippo/YAP pathway could offer a novel therapeutic avenue for BCC patients with advanced disease.
Results
YAP is dispensable for normal epidermal homeostasis but upregulated in BCC
Previous studies from our laboratory and others have demonstrated the importance of the Hippo effector, YAP, in epidermal development during embryogenesis (Schlegelmilch et al, 2011; Silvis et al, 2011; Zhang et al, 2011). We sought to investigate YAP function in adult epidermis by conditional Yap deletion in 8‐week‐old mice. Compared to embryonic Yap knockout, the adult epidermis showed no evidence of macroscopic or histologic abnormalities in Yap‐null epidermis during unperturbed homeostatic tissue maintenance up to 12 weeks following Yap knockout (Fig EV1A). However, conditions such as wound healing, which require rapid basal progenitor cell expansion to regenerate damaged epidermis, critically depend on YAP function (Lee et al, 2014; Elbediwy et al, 2016). Thus, we explored the possibility that the Hippo effector, YAP, may be involved in BCC development, a process with uncontrolled expansion of basal progenitor cells (Youssef et al, 2010, 2012; Sanchez‐Danes et al, 2016). First, we assessed YAP expression pattern in 26 human BCC samples by immunohistochemistry (IHC). In 24 out of 26 BCC samples (~92%) analyzed, YAP was highly expressed and localized to the nucleus in the basally located epithelial tumor cells (i.e., tumor cells adjacent to surrounding dermis/mesenchyme) (Fig 1A). Next, we studied YAP expression in a mouse model of BCC (R26 LSL‐SmoM2YFP/+ :K14CreER), which develops invasive tumors in ear and tail epidermis 10 weeks postexpression of mutant Smoothened (Youssef et al, 2010). Similarly to human BCC, mouse tumors showed robust nuclear YAP localization in the basal tumor cells (Fig 1B). Using in situ hybridization, we further compared Yap and Taz mRNA expression patterns in mouse BCC. Whereas Yap mRNA was highly expressed in epithelial BCC cells with particular enrichment in the basal compartment of dermis invading tumor clones, Taz mRNA was predominately expressed in the surrounding mesenchyme (Figs 1C and EV1B). Thus, we speculated that YAP, but not TAZ, is more likely the Hippo effector contributing to the BCC epithelial cell phenotype. Although YAP nuclear versus cytoplasmic localization has been widely used as a measure of Hippo transcriptional output (i.e., Hippo kinases/adaptor proteins inactivity), we sought to better define in situ YAP activity within BCC tumors. We crossed transgenic mice with a Cyr61 BAC promoter driving eGFP expression (Cyr61 eGFP), a well‐established and canonical YAP target gene, with the R26 LSL‐SmoM2YFP/+ :K14CreER BCC model (Heintz, 2004; Galli et al, 2015; Stein et al, 2015; Zanconato et al, 2015). Although most normal basal keratinocytes show robust nuclear YAP localization, the eGFP (Cyr61 eGFP) marks rare basal keratinocytes and mostly cells within sebaceous gland (Figs 1D and EV1A). In BCC tumors, we observed a robust increase in the mosaic eGFP signal in Cyr61 eGFP :R26 SmoM2YFP compared to background uniform YFP expression in R26 SmoM2YFP (SmoM2‐YFP expression) alone (Figs 1D and EV1C). In other words, we could differentiate a subset of cells with transcriptionally active YAP (i.e., activated Cyr61 eGFP) within a tumor clone where each BCC cell uniformly expresses SmoM2‐YFP (Fig EV1C). In agreement with YAP localization observed by IHC, IF, and RNAscope (Figs 1B and D, and EV1B and C), a strong punctate eGFP signal in Cyr61 eGFP :R26 SmoM2YFP was preferentially found in basally located BCC cells. Overall, we demonstrate the dispensable nature of the Hippo effector, YAP, in adult epidermal homeostasis and specific upregulation of YAP nuclear localization and activity in both human and mouse BCC.
Figure EV1. Homeostatic YAP function in skin epidermis and Yap/Taz expression patterns in BCC .
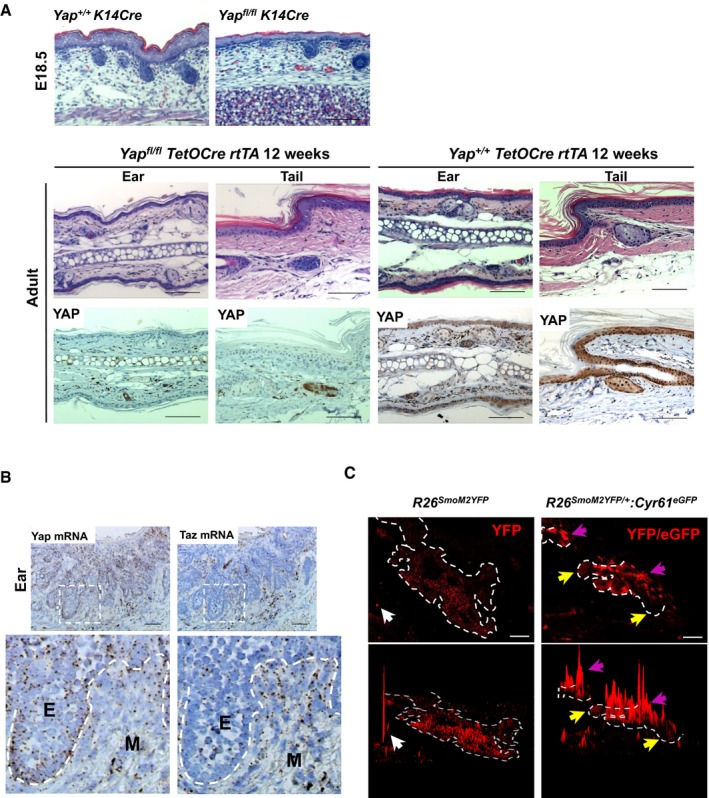
- Assessment of YAP requirement in embryonic back skin (upper panels) and adult ear and tail epidermis (lower panels). K14Cre is used to knockout Yap in embryonic epidermis; TetO‐Cre is activated in 8‐week‐old mouse using doxycycline for 1 week and tissue analyzed 12 weeks later. Scale bar is 100 μm.
- Yap and Taz mRNA expression (RNAscope) in ear BCC. Dashed lines outline epithelial cells (E). M denotes surrounding mesenchymal cells. Scale bar is 50 μm.
- Immunofluorescent YFP/eGFP pixel intensity histogram in the R26 SmoM2YFP/+ and R26 SmoM2YFP/+ :Cyr61 eGFP mice to visualize differential YFP/eGFP signal in BCC cells. White arrows indicate high YFP/eGFP peak in a non‐specific splotch (left panels). Yellow arrows indicate cells with low YFP/eGFP signal; purple arrows indicate cells with high YFP/eGFP signal (right panels). ZEN software was used to generate pixel intensity histograms. Dashed lines in the image outline BCC clones. Scale bar is 50 μm.
Figure 1. YAP is highly express and localized to nucleus in human and mouse BCC .
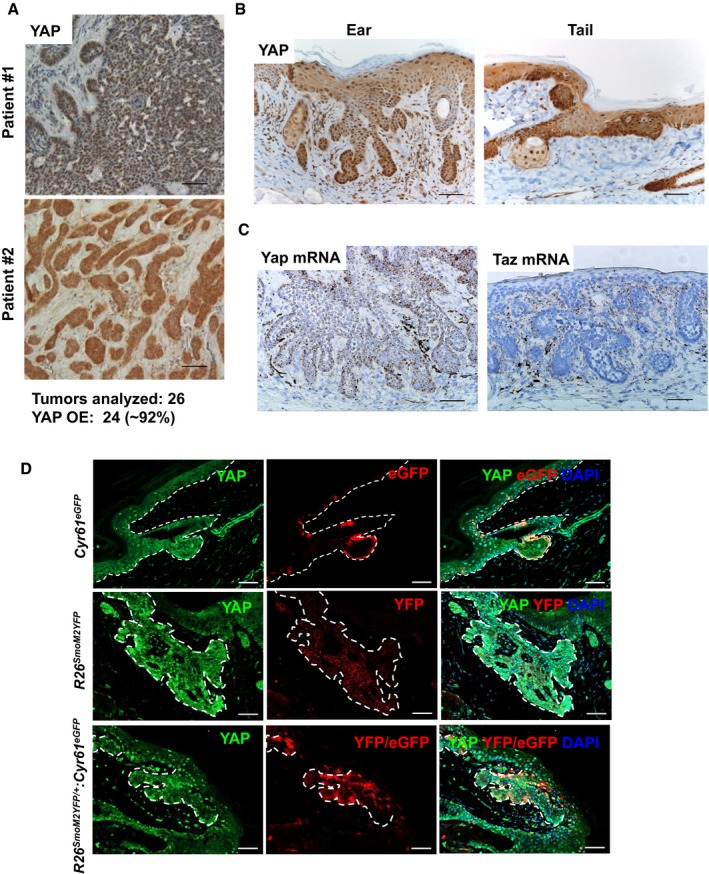
- Representative YAP IHC images from analyses of human BCC samples. 24 out of 26 (˜92%) BCC cases analyzed showed YAP overexpression (YAP OE) and nuclear localization. Scale bar is 50 μm.
- YAP IHC in ear and tail BCC tumors from the R26 LSL‐SmoM2YFP/+ :K14CreER (mutant Smo) mouse model. Scale bar is 50 μm.
- RNAscope detection of Yap and Taz mRNA in mouse ear BCC. Scale bar is 50 μm.
- YAP activity in BCC tumors assessed using Cyr61 eGFP transgenic reporter mouse. Cyr61 eGFP transgenic mouse (upper panels) was crossed with R26 LSL‐SmoM2YFP/+ :K14CreER (R26 SmoM2YFP/+ :Cyr61 eGFP; bottom panels) and compared with R26 LSL‐SmoM2YFP/+ :K14CreER (R26 SmoM2YFP/+; middle panels). YFP denotes SmoM2‐YFP fusion gene expression. eGFP denotes Cyr61 eGFP expression. Upper panels: Dashed lines mark separation between basal epidermal cells and dermis. Middle and lower panels: Dashed lines outline BCC clones. Scale bar is 50 μm.
BCC development and tumor progression critically depend on YAP‐TEAD interaction
In order to investigate YAP's contribution during BCC development, we crossed the R26 LSL‐SmoM2YFP/+ :K14CreER mice onto either Yap +/+ or Yap fl/fl backgrounds. The BCC tumors were induced using high‐dose tamoxifen starting at postnatal days P24–28 and monitored for visible tumor onset in ear and tail skin (Fig 2A). Whereas Yap +/+ mice developed macroscopic BCC in the ear epidermis starting at 8 weeks after tamoxifen administration, Yap loss (Yap fl/fl background) significantly rescued visible tumor burden (Fig 2B). In fact, Yap +/+ mice required euthanasia on average 12 weeks after tamoxifen administration due to necrotic ear epidermis, while Yap fl/fl succumbed to BCC after ~21.7 weeks (χ2 = 29.95; P < 0.0001, Fig 2B and C). The apparent reduction in tumor burden in Yap fl/fl versus Yap +/+ mice was also observed at a histologic level when compared at the stage where dysplastic clones appear (4 weeks after tamoxifen) as well as later (10 weeks after tamoxifen), at which point BCCs have invaded the underlying dermis (Figs 2D and EV2A). Importantly, all the Yap fl/fl mice eventually developed invasive BCC tumors that were Cre‐driven recombination escapers for Yap bi‐allelic knockout (i.e., YAP‐positive by IHC), further supporting YAP's critical function in BCC development and progression (Figs 2B and C, and EV2B). We further validated specificity of the Cyr61 eGFP reporter for YAP activity in BCC by assessing eGFP expression in Yap‐null tumor clones (Yap fl/fl :R26 SmoM2YFP/+ :K14CreER:Cyr61 eGFP). Indeed, YAP‐negative tumor clones or individual cells show baseline YFP/eGFP expression (i.e., equivalent to SmoM2‐YFP alone) compared to robust YFP/eGFP expression in a subset of YAP‐positive BCCs (Appendix Fig S1A).
Figure 2. Yap deletion significantly reduces tumor burden in BCC mouse model.
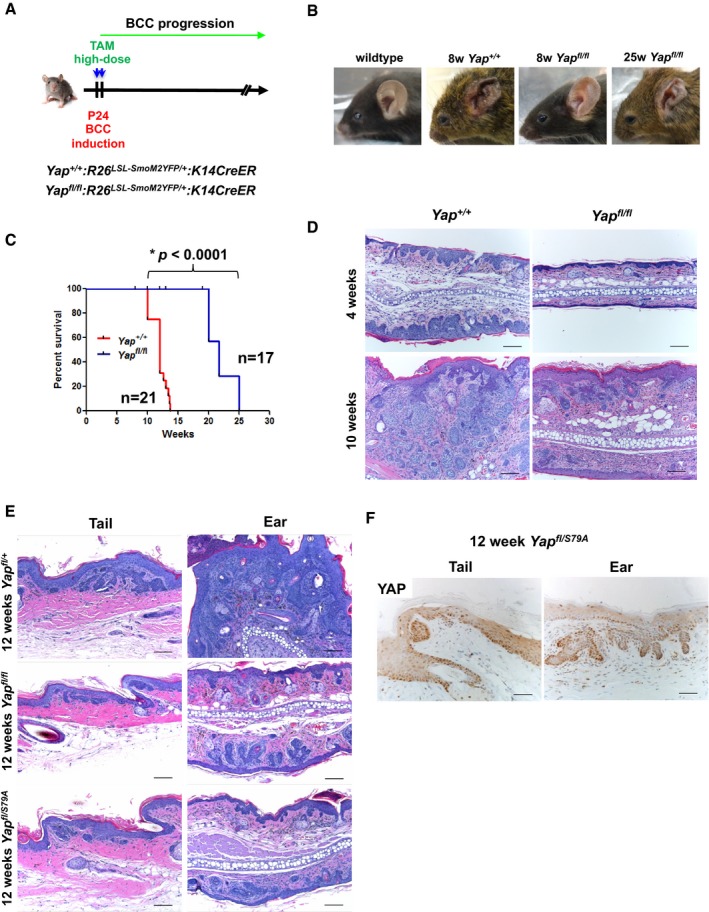
- Scheme representing the genetic strategy used to induce BCC in mouse epidermis.
- Representative images of macroscopic tumor burden in mouse ears from Yap +/+ and Yap fl/fl at 8 and 25 weeks after tamoxifen.
- Kaplan–Meier survival curve comparing Yap +/+ with Yap fl/fl mice. The groups were compared using log‐rank (Mantel–Cox) test. *P < 0.0001.
- Representative histological analysis of ear BCC from Yap +/+ and Yap fl/fl. Scale bar is 100 μm.
- BCC histology comparing Yap fl/+ , Yap fl/fl , and Yap fl/S79A mutant backgrounds (YAPS79A does not bind TEAD transcription factors). Scale bar is 100 μm.
- YAP IHC in Yap fl/S79A background demonstrating YAP expression and nuclear localization. Scale bar is 50 μm.
Figure EV2. YAP is required for in vivo and in vitro BCC growth.
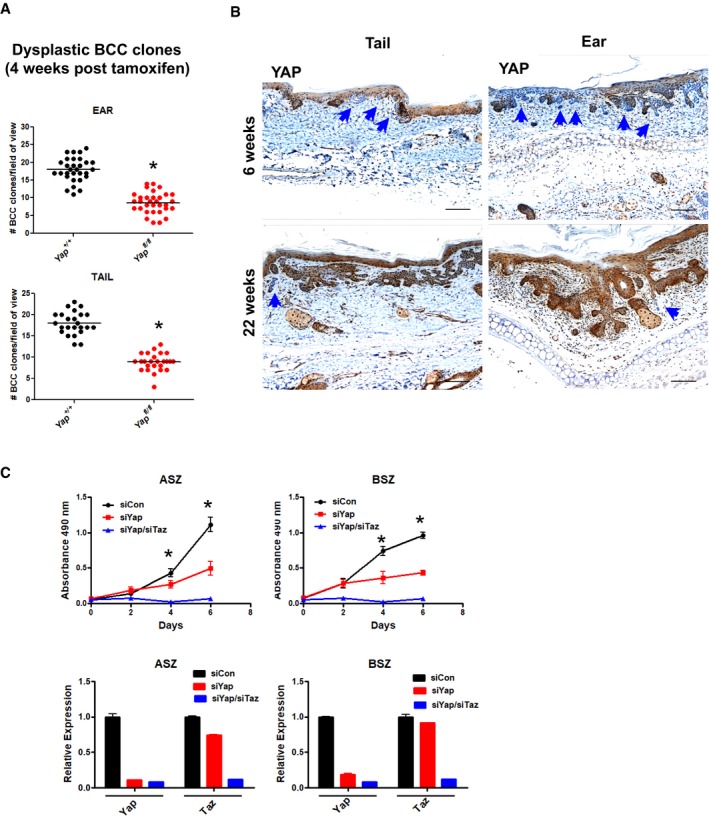
- Quantified number of dysplastic BCC clones per field of view (5× magnification) in ear and tail skin (Yap +/+and Yap fl/fl mice) 4 weeks after high‐dose tamoxifen administration. Total of three mice per genotype were used, and at least eight clones were counted per tissue section. The groups were compared using Student's t‐test. *P < 0.0001.
- YAP IHC in ear and tail BCC tumors from Yap fl/fl mice 6 and 25 weeks after high‐dose tamoxifen administration. Blue arrows indicate Yap‐null BCC clones. Scale bar is 100 μm.
- Yap and Yap/Taz RNAi impairs in vitro proliferation of ASZ and BSZ mouse BCC cell lines (upper panels). *P < 0.05. qPCR analysis of Yap and Taz knockdown efficiency in ASZ and BSZ mouse BCC cell lines (lower panels).
While Yap RNAi alone inhibited the proliferation of two mouse BCC cell lines (ASZ and BSZ), combined Yap/Taz RNAi provided additive growth suppression in vitro (Fig EV2C; So et al, 2006). Although Taz mRNA detection in the mesenchyme surrounding BCC suggested unlikely contribution to epithelial tumor growth, our in vitro data suggested that Taz might provide a compensatory mechanism in the Yap‐null BCC clones. To address TAZ contribution in BCC, we first validated YAP‐specific and YAP/TAZ antibodies using epidermis from TetO‐Cre rtTA Yap fl/fl mice to show a low but detectable signal with YAP/TAZ antibody (i.e., TAZ protein) in Yap‐null ear and tail basal keratinocytes (Appendix Fig S2A). Next, we performed IHC in the Yap fl/fl BCC serial sections with YAP and YAP/TAZ antibodies to demonstrate extremely low or undetectable TAZ signal with YAP/TAZ antibody in the Yap‐null BCC (Appendix Fig S2B). We confirmed a decrease in Taz mRNA expression in the Yap‐null BCC using FACS‐sorted tumors (Appendix Fig S2C). Finally, we crossed the R26 LSL‐SmoM2YFP/+ :K14CreER mice onto the Yap fl/fl and Taz fl/fl backgrounds to genetically assess TAZ contribution in BCC tumor initiation and progression. When compared at 8 weeks after tamoxifen administration, Yap/Taz or Yap knockout‐alone BCCs were indistinguishable upon histologic and tumor clone size analyses (Appendix Fig S2D–E). Hence, our data strongly support that YAP specifically drives BCC growth without significant compensation from TAZ in the Yap‐null tumors.
Since the majority of known YAP growth‐promoting properties have been attributed to its interaction with the DNA‐binding transcription factors, TEAD, we investigated the necessity of YAP‐TEAD interaction in BCC. We took advantage of the already established Yap S79A allele, a mutant YAP that does not bind TEAD, and crossed it onto the BCC model to establish Yap fl/S79A :R26 LSL‐SmoM2YFP/+ :K14CreER (Schlegelmilch et al, 2011). Thus, upon tamoxifen induction, the wild‐type Yap allele was floxed out, allowing only expression of the Yap S79A in BCC tumors, which closely phenocopied Yap fl/fl when compared to Yap +/+ BCC tumors (Fig 2E and F). Notably, no phenotypic differences were detected between Yap fl/+ and Yap +/+ backgrounds, indicating that YAP is not haplo‐insufficient for BCC development (data not shown). Therefore, our data strongly support the notion that YAP promotes BCC growth by direct interaction with TEAD transcription factors.
To more precisely evaluate the relative behavior of BCC clones, tumors in the Yap fl/fl :R26 LSL‐SmoM2YFP/+ :K14CreER background were induced with low‐dose tamoxifen (Fig 3A). The longitudinal tracking of the Yap‐null versus Yap‐positive clonal evolution began 4 weeks after tamoxifen induction, when dysplastic tumors appear, and was continued until 20 weeks, when most clones have already acquired the invasive phenotype (Fig 3B; Youssef et al, 2012; Sanchez‐Danes et al, 2016). Overall, we observed a significant and time‐dependent reduction in the percentage of Yap‐null BCC clones in ear and tail epidermis (Fig 3C). Our analysis of absolute Yap‐null clone numbers at each time point indicates that they are not only outcompeted by the Yap‐positive tumor cells, but also specifically depleted over time (Fig 3D). In other words, although we could detect a significant number of established dysplastic Yap‐null BCC clones at 4 weeks, their number was diminished as the tumors progressed to invasive BCC (10‐ to 20‐week time points). Lastly, we analyzed relative BCC clone size between Yap +/+, Yap fl/fl, and Yap fl/S79A backgrounds at the 12‐week time point. By further differentiating Yap‐null versus Yap‐positive BCC clones in the Yap fl/fl background using IHC, we demonstrate that YAP, and its interaction with TEAD, is critical for BCC clone expansion (Yap‐null vs. Yap‐positive; Yap‐null vs. Yap +/+; and Yap +/+ vs. Yap fl/S79A; P < 0.05; Fig 3E). Hence, our data reveal that YAP loss or disruption in YAP‐TEAD interaction significantly impairs BCC establishment and tumor progression from dysplasia to an invasive phenotype.
Figure 3. Clonal evolution of Yap‐null BCC tumors.
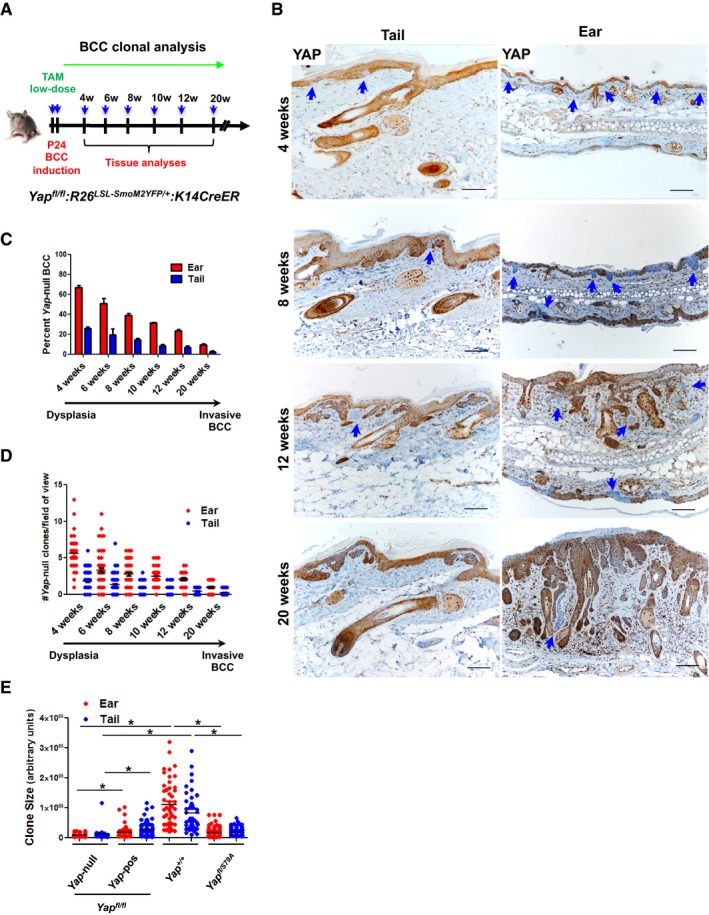
- Schematic depiction of clonal Yap‐null versus Yap‐positive BCC tracing over time.
- YAP IHC and histological analyses of ear and tail BCC tumors in Yap fl/fl background at indicated time points. Blue arrows indicate Yap‐null BCC clones. Scale bar is 100 μm.
- Quantification of YAP‐positive versus YAP‐negative clones at 4, 6, 8, 10, 12, and 20 weeks after low‐dose tamoxifen administration. At least three mice per group were analyzed. Error bars indicate SEM.
- Quantification of total number of Yap‐null clones per field of view in ear and tail skin. Total of three mice per time point were used, and at least eight random fields per mouse were counted for each tissue section. Error bars indicate SEM.
- Ear and tail BCC clone size quantification using ImageJ at 12 weeks after low‐dose tamoxifen administration. In Yap fl/fl background, YAP IHC analysis was used to determine Yap‐positive versus Yap‐null clones. Total of three mice per genotype were used and at least seven clones were counted per tissue section. The groups were compared using Student's t‐test. *P < 0.05.
YAP transcriptional gene signature drives BCC cell proliferation independent of Wnt or Hedgehog signaling
Following the observation that Yap loss leads to a strong growth‐deficit phenotype in BCC, we set out to understand the mechanistic nature by which YAP drives tumor growth. Using EdU pulse‐chase in vivo, we show that Yap‐null clones have impaired proliferation compared to their neighboring Yap‐positive tumor clones (Fig 4A and B). This observation is in line with the observed decrease in proliferation upon Yap knockdown in two BCC cell lines (Fig EV2C). The activity of the most commonly mutated pathway in BCC, Hedgehog, and its cooperation with Wnt signaling are considered to be absolutely necessary for BCC growth and survival (Yang et al, 2008; Atwood et al, 2012; Youssef et al, 2012). Hence, we foremost studied whether Yap loss has an impact on Wnt or Hedgehog signaling outputs and thereby impairs BCC growth. Using serial sections from the Yap fl/fl BCC tumor, we show no change in β‐catenin nuclear localization between YAP‐negative and YAP‐positive clones (Fig 4C). Wnt activity in BCC establishes an embryonic hair follicle (EHF) phenotype that is marked by an expression of PCDH, LEF1, CUX1, and LHX2, none of which were found affected in the Yap‐null BCC clones (Fig EV3A; Ito et al, 2007; Youssef et al, 2012). The canonical Wnt target genes, Axin2 and Lgr5, and a well‐established BCC stemness‐promoting transcription factor, SOX9, were also unaffected in YAP‐negative BCC (Fig EV3B; Youssef et al, 2012; Larsimont et al, 2015). Equally, Yap loss had no impact on the canonical Hedgehog target genes in BCC, Gli1 or Ptch1 (Fig 4D). Therefore, our data support the notion that YAP promotes BCC growth independent of Wnt and Hedgehog signaling.
Figure 4. YAP provides proliferative advantage to BCC tumors independent of Wnt or Hedgehog signaling.
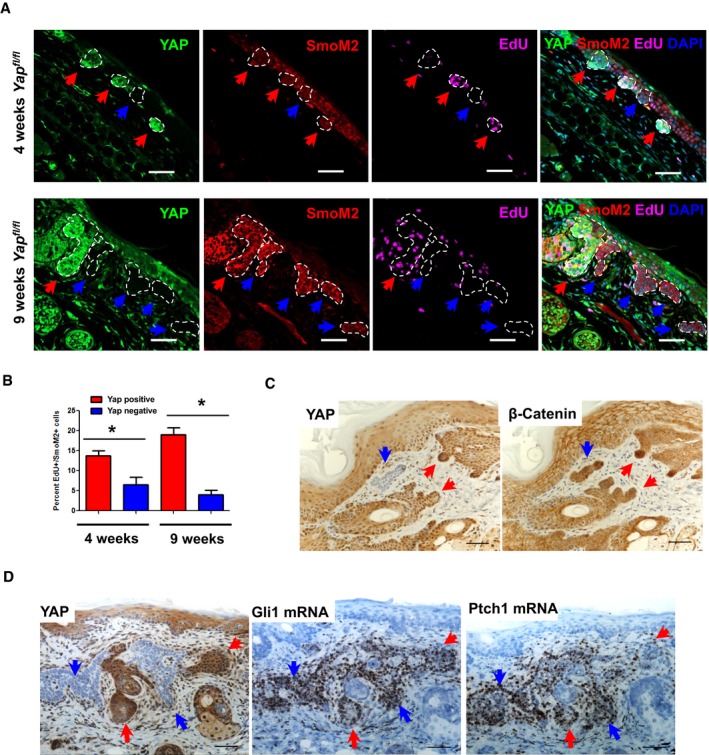
- Immunofluorescent imaging of EdU incorporation in the YAP‐negative versus YAP‐positive clones in Yap fl/fl ear BCC. Red arrows indicate YAP‐positive clones; blue arrows indicate YAP‐negative clones. Scale bar is 50 μm.
- Quantified EdU incorporation in YAP‐negative versus YAP‐positive dysplastic clones (4 and 9 weeks after tamoxifen). Total of three mice were used per time point, and at least 20 clones per tissue section were quantified. Error bars indicate SEM. The groups were compared using Student's t‐test *P < 0.05.
- YAP and β‐catenin IHC in Yap fl/fl BCC serial sections. Red arrows indicate YAP‐positive clones; blue arrows indicate YAP‐negative clones. Scale bar is 50 μm.
- YAP IHC and Gli1/Ptch1 RNAscope in Yap fl/fl BCC serial sections. Blue arrows indicate YAP‐negative BCC clones; red arrows indicate YAP‐positive BCC clones. Scale bar is 50 μm.
Figure EV3. YAP has no effect on Wnt signaling in BCC .
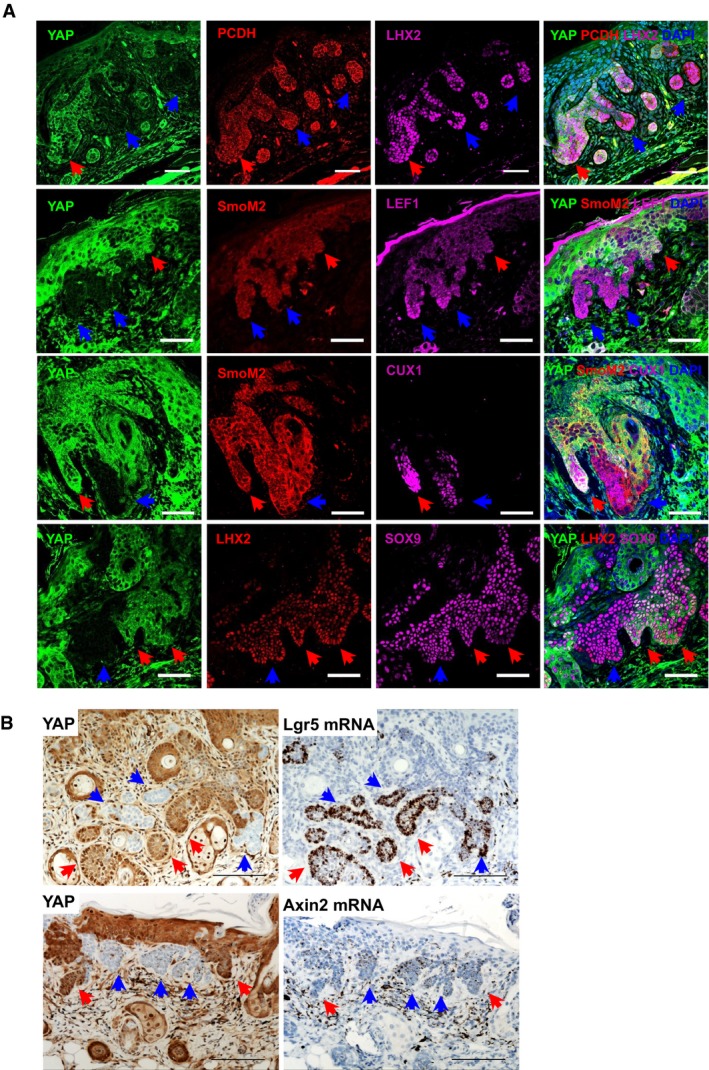
- Immunofluorescent staining of YAP with SOX9 or embryonic hair follicle phenotype genes (LEF1, CUX1, LHX2, PCDH) in Yap fl/fl BCC. Red arrows indicate YAP‐positive clones; blue arrows indicate YAP‐negative clones. Scale bar is 50 μm.
- YAP IHC and Lgr5 or Axin2 RNAscope in Yap fl/fl BCC serial sections. Red arrows indicate YAP‐positive clones; blue arrows indicate YAP‐negative clones. Scale bar is 100 μm.
In order to identify the YAP‐driven gene signature that contributes to the BCC phenotype, we performed RNAseq (Yap +/+ vs. Yap fl/fl) and ChIPseq (TEAD1, TEAD4, and H3K27Ac) analyses in the established BCC. First, we confirmed that combining tdTomato reporter with SmoM2‐YFP (R26LSL‐SmoM2YFP/LSL‐tdTom) allows us to purify BCC cells at 6 weeks after high‐dose tamoxifen, a time point when we observe the highest Yap knockout efficiency (~70%) in the ear BCCs (Appendix Fig S3A–C). Hence, the RNAseq was performed on the FACS‐sorted (DAPI/CD45/TER119 negative, tdTomato/α6‐integrin positive) Yap +/+ and Yap fl/fl ear BCC cells 6 weeks after high‐dose tamoxifen administration (Fig 5A). Differential gene expression analysis identified 97 genes that were significantly changed in the Yap‐null BCC, one of which is the YAP's canonical target gene, Cyr61 (Fig 5B, Table EV1). Of the 97 differentially expressed genes, 57 were bound by TEAD1 and TEAD4 within their TSS or enhancer region (Fig 5C, Table EV2). Thus, our RNAseq and ChIPseq data further support that a YAP‐TEAD‐driven gene signature directly contributes to BCC growth. Next, we performed Ingenuity Pathway Analysis—IPA (Qiagen), which ranked signaling pathways that are significantly affected by Yap loss. Interestingly, the JNK pathway, previously implicated in BCC, showed strongest changes upon Yap loss (P = 2.04 × 10−7, Fig 5D; Schnidar et al, 2009; Eberl et al, 2012). JNK (c‐Jun N‐terminal kinase) is the upstream kinase that phosphorylates and directly regulates transactivational activity of c‐JUN, a member of the AP1 transcriptional complex (Lopez‐Bergami et al, 2010). Our TEAD1, TEAD4, and H3K27Ac ChIPseq analyses reveal that TEAD strongly binds to known AP1‐regulated genes including c‐Jun (Fig 5E, Table EV3). In fact, Homer de novo motif analysis using TEAD1 and TEAD4 ChIPseq peaks confirms the previously described enrichment of the AP1 consensus motifs within TEAD1 and TEAD4 peaks (Fig EV4A; Zanconato et al, 2015; Liu et al, 2016). Therefore, our findings strongly suggest that YAP cooperates with c‐JUN and AP1 activity to induce the gene signature required for BCC initiation and progression.
Figure 5. Identification of YAP‐driven gene signature in BCC tumors.
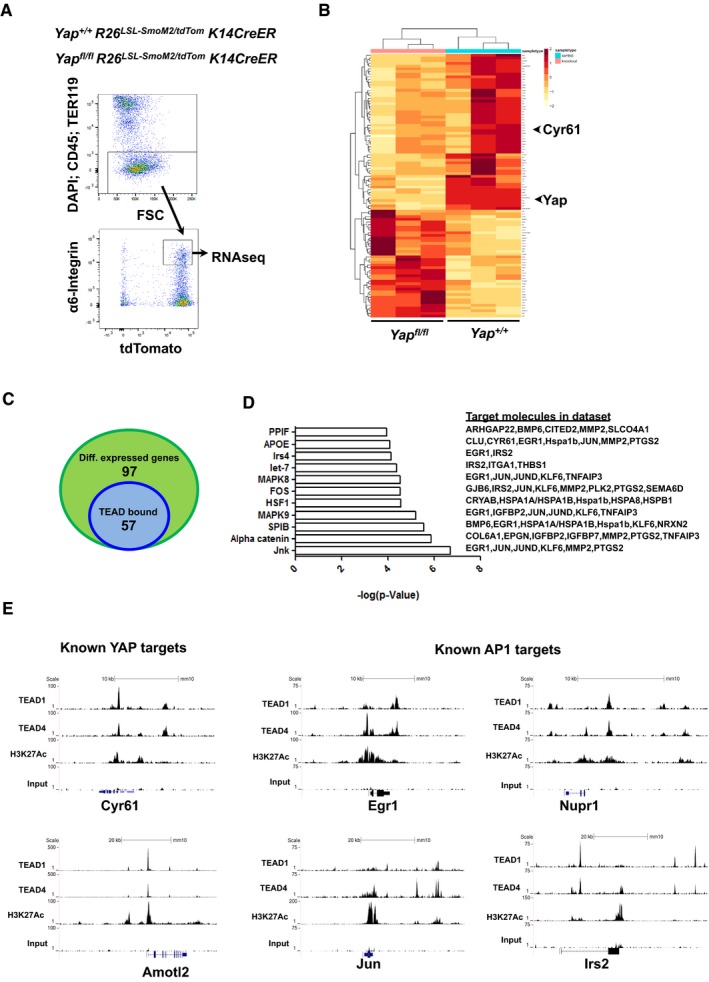
- FACS methodology to purify ear BCC cells from Yap +/+ and Yap fl/fl mice (6 weeks after high‐dose tamoxifen administration) for RNAseq analysis.
- RNAseq heatmap of differentially expressed genes between Yap +/+ and Yap fl/fl BCC.
- Venn diagram of TEAD1/TEAD4 peaks found within differentially expressed genes from Yap‐null BCC.
- Ingenuity Pathway Analysis of differentially expressed genes between Yap +/+ and Yap fl/fl BCC.
- TEAD1, TEAD4, H3K27Ac, and input (control) peaks associated with known YAP targets (Cyr61 and AmotL2) or known AP1 targets (Egr1, Jun, Nupr1, Irs2) from BCC ChIPseq.
Figure EV4. Bioinformatics analyses to define YAP‐dependent programs in BCC .
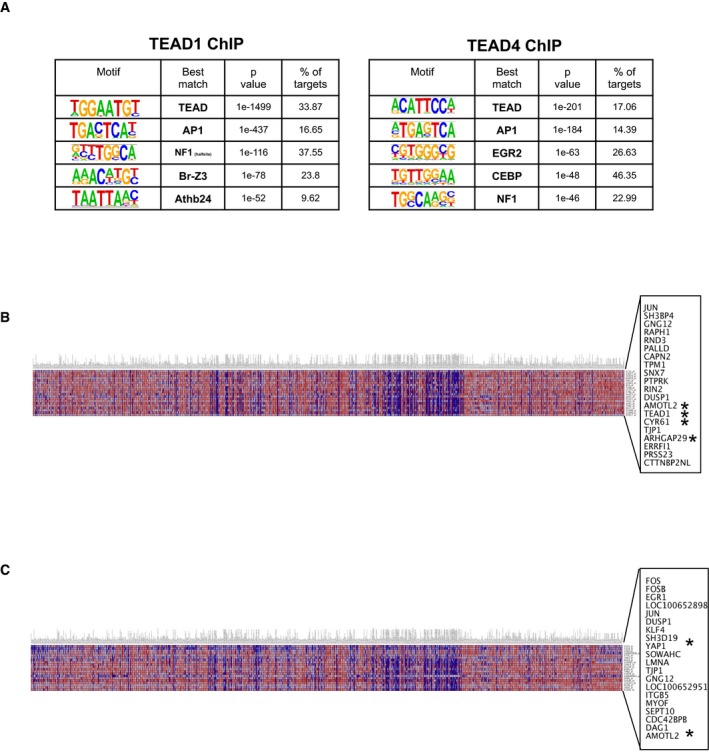
- Transcription factor motif enrichment from TEAD1 and TEAD4 ChIP in BCC (Homer de novo motifs).
- Top 20 genes co‐expressed with c‐JUN across various cancer cell lines in Gene Neighbors analysis from the Broad‐Novartis Cancer Cell Line Encyclopedia (CCLE). The x‐axis indicates cancer cell lines; y‐axis indicates target probes and their corresponding gene names. *denotes known YAP target genes.
- Top 20 genes co‐expressed with FOS across various cancer cell lines in Gene Neighbors analysis from the Broad‐Novartis Cancer Cell Line Encyclopedia (CCLE). The x‐axis indicates cancer cell lines; y‐axis indicates target probes and their corresponding gene names. *denotes known YAP target genes.
YAP potentiates JUN stability and activity in BCC to promote tumor growth
Since RNAseq analysis of the Yap‐null BCC suggested that YAP regulates known AP1 target genes and because we observed TEAD occupancy at those genes, including c‐Jun, we further assessed how YAP impacts c‐JUN activity. First, we validated RNAseq data by qPCR analyses of known YAP (Cyr61, AmotL2, Arhgap29) and AP1 (c‐Jun, Fos, Junb, Clu, Egr1, Irs2, Nupr1, Dusp5, Ptgs2, and Igfbp7) target genes, which were significantly downregulated in Yap‐null BCC (Fig. 6A; Jin & Howe, 1997; Goruppi et al, 2002; Troen et al, 2004; Cui et al, 2006; Hoffmann et al, 2008; Okamura et al, 2012; Mina et al, 2015). Likewise, in vitro Yap knockout in ASZ cells with doxycycline (Dox)‐inducible Yap sgRNA (TetON‐CRISPR‐YapKO) recapitulated reduction in AP1 (c‐JUN) regulated target genes (Fig EV5A). By taking advantage of transcriptomic cancer cell line data from the Broad‐Novartis Cancer Cell Line Encyclopedia (CCLE), we discovered strong c‐JUN and FOS mRNA correlation with an activated YAP signature (i.e., CYR61, AMOTL2, ARHGAP29, YAP) across various cancer types (Fig EV4B and C). To determine whether YAP directly affects c‐JUN activity, we studied c‐JUN expression and activating phosphorylation at two positions (Serine 63 and 73) in YAP‐negative and YAP‐positive BCCs (Lopez‐Bergami et al, 2010). Both of the c‐JUN phosphorylation sites and total c‐JUN were significantly reduced in the YAP‐negative BCC (Fig 6B). Next, we studied the status of c‐JUN phosphorylation in the BCC clones from Yap fl/fl mice. When compared with YAP‐positive clones, YAP‐negative BCC clones had significantly decreased c‐JUN phosphorylation on serines 63 and 73, the two well‐known JNK phosphorylation sites (Fig 6C). In agreement, phosphorylated JNK1/2 (pJNK1/2) was decreased in the same YAP‐negative clones relative to the neighboring YAP‐positive tumors. The YAP effect on the JNK‐JUN pathway activity was recapitulated in the TetON‐CRISPR‐YapKO ASZ and BSZ cell lines; YAP loss decreased phosphorylation levels of JNK and JUN (Fig 6D). Since the total c‐JUN protein, c‐JUN phosphorylation, and c‐Jun mRNA are simultaneously decreased in the YAP‐negative in vitro and in vivo BCC models, our data suggest that combination of impaired protein stability and gene expression contributes to reduced total c‐JUN protein (Figs 6A–D and EV5A). Notably, activated c‐JUN protein binds to its own promoter to increase c‐Jun mRNA expression, which further contributes to diminished total c‐JUN protein in the Yap‐null BCC (Angel et al, 1988).
Figure 6. YAP regulates JNK‐JUN activity in BCC .
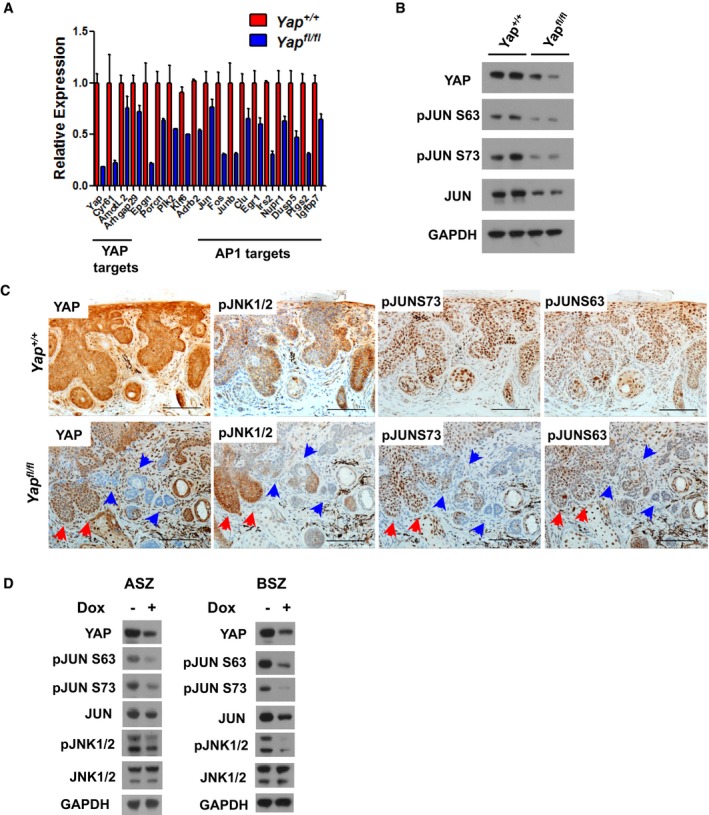
- qPCR validation of differentially expressed genes indicating that YAP affects AP1 (c‐JUN) target genes. Error bars represent SEM. Total of three biological replicates were used per genotype.
- Western blot analysis of JUN activity in Yap +/+ versus Yap fl/fl BCC at 9 weeks after high‐dose tamoxifen administration.
- IHC analysis of YAP and JNK‐JUN pathway activity in Yap fl/fl and Yap +/+ BCC serial sections. Red arrows indicate YAP‐positive clones; blue arrows indicate YAP‐negative clones. Scale bar is 100 μm.
- Western blot analysis of the JNK‐JUN activity in doxycycline (Dox)‐inducible Yap knockout (TetON‐CRISPR‐YapKO) in ASZ and BSZ cells.
Figure EV5. YAP regulates JNK‐JUN pathway in allograft and in vitro model of BCC .
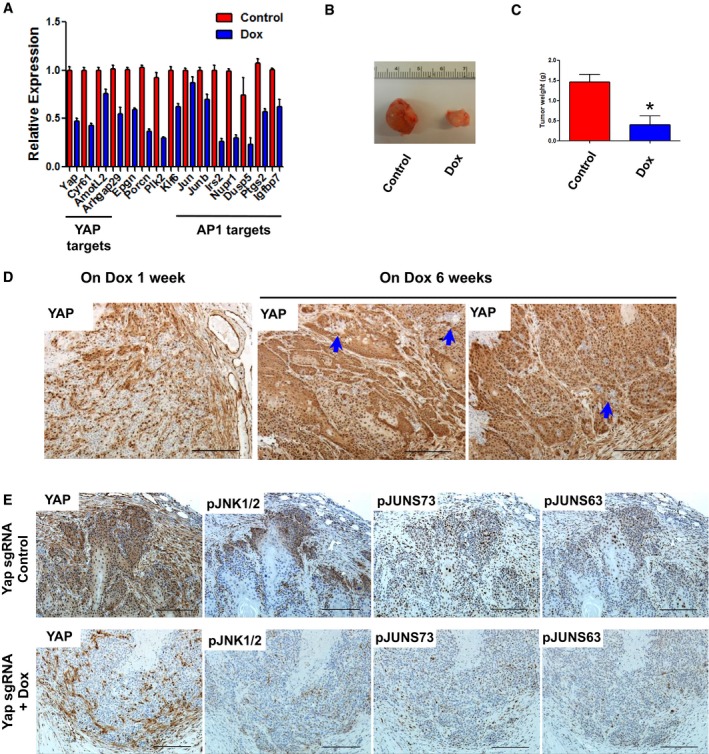
- qPCR analysis of YAP and AP1 target genes in Dox‐inducible Yap knockout (TetON‐CRISPR‐YapKO) ASZ cells. Error bars represent SEM. Total of three biological replicates were used per condition.
- Representative macroscopic images of TetON‐CRISPR‐YapKO ASZ allograft BCC tumors (Control versus Dox treatment for 6 weeks).
- Tumor weight quantification of Control versus Dox‐treated TetON‐CRISPR‐YapKO ASZ allograft BCC tumors. Error bars represent SEM. The groups were compared using Student's t‐test. *P < 0.05; n = 3 per group.
- YAP IHC in TetON‐CRISPR‐YapKO ASZ allograft tumors from mice receiving Dox for 1 week versus 6 weeks. Blue arrows indicate Yap‐null clones. Scale bar is 100 μm.
- IHC analysis of YAP and JNK‐JUN pathway activity in Control (Yap‐wt) and Dox (Yap‐null)‐treated TetON‐CRISPR‐YapKO ASZ allograft BCC serial sections. Scale bar is 100 μm.
To further validate YAP as a potential target in BCC and its effect on the JNK‐JUN signaling, we established subcutaneous allograft BCC tumors with TetON‐CRISPR‐YapKO ASZ cells in immunocompromised (nu/nu) mice. The injected TetON‐CRISPR‐YapKO ASZ cells were allowed to establish tumors before mice were randomized to either a control or Dox receiving group. Six weeks following randomization, control mice required euthanasia due to large tumor size; however, mice receiving Dox (induced Yap knockout) had significantly reduced tumor burden (Fig EV5B and C). When we analyzed Yap knockout efficiency in the mice receiving Dox for 6 weeks, we could detect only a small fraction of Yap‐null BCC cells relative to the tumors from mice receiving Dox for 1 week (Fig EV5D). This observation suggests that Yap‐null cells were specifically depleted over time while the cells escaping Yap bi‐allelic deletion outgrew and repopulated the tumor. Subsequently, we assessed JNK‐JUN pathway activity in tumor serial sections from the 1‐week Dox‐treated or control mice. The Yap‐null tumors exhibited significant reductions in pJNK1/2, pJUN S63, and pJUN S73 levels relative to the control tumors, further corroborating the link between YAP and JNK‐JUN activity (Fig EV5E). To assess sensitivity of BCC cell lines (ASZ and BSZ) to the JNK‐JUN pathway inhibition, we analyzed in vitro cell proliferation upon treatment with SP600125 (JNK1/2/3 inhibitor). Both of the BCC cell lines were completely impaired in their ability to grow upon SP600125 treatment, which was concomitant with decreased c‐JUN phosphorylation (Fig EV6A and B).
Figure EV6. YAP overexpression induces c‐JUN activation and interfollicular epidermis thickening.
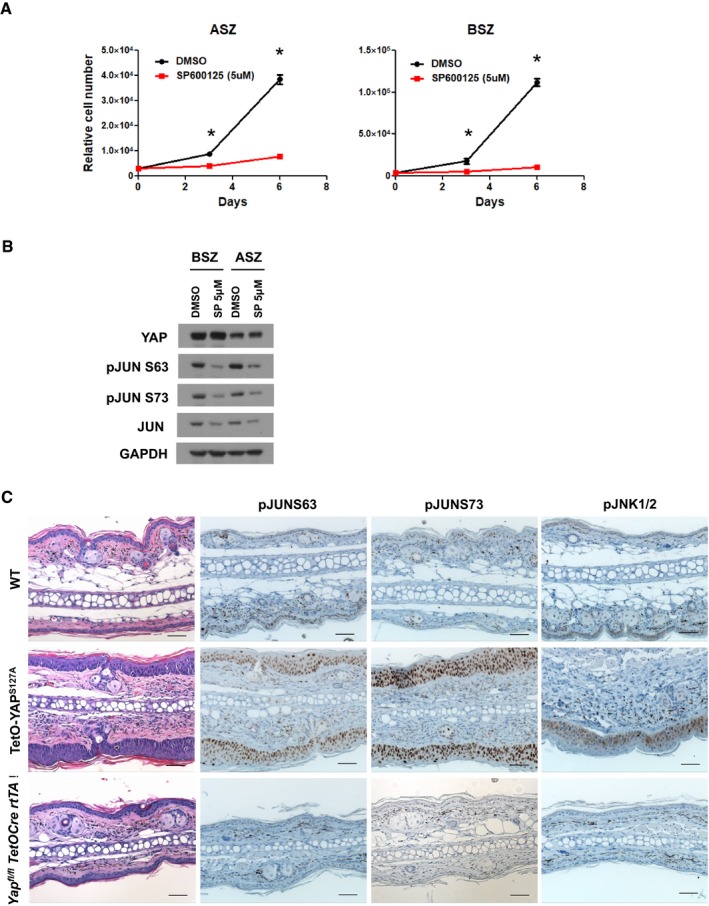
-
AJNK inhibitor (SP600125) blocks proliferation of BCC cell lines (ASZ and BSZ). The cells were treated in vitro with 5 μM SP600125 or DMSO over the course of 6 days, and growth was determined by cell counting. Total of three biological replicates were used per condition and time point. Error bars represent SEM. The groups were compared using ANOVA and Bonferroni post hoc test. *P < 0.01.
-
BWestern blot analysis of YAP and JUN activity (pJUN S63 and pJUN S73) in ASZ and BSZ cells 16 h after JNK inhibition (SP600125 5 μM).
-
CYAPS127A overexpression (Col1 TetO‐YAPS127A R26 LSL‐rtTA/+ K14Cre) in adult epidermis upon administration of doxycycline to 8‐week‐old mice for 7 days. YAPS127A induces interfollicular epidermis (IFE) thickening that closely resembles BCC. Representative H&E and IHC (pJUN S63, pJUN S73, pJNK1/2) images in WT, TetO‐YAP S127A, and Yap fl/fl TetO‐Cre rtTA IFE. Scale bar is 50 μm.
Since YAP overexpression is a potent inducer of IFE hyperproliferation, we set to examine the JNK‐JUN pathway activation in the YAP‐driven lesions. Indeed, doxycycline‐inducible YAPS127A (constitutively active) overexpression (Col1 TetO‐YAPS127A R26 LSL‐rtTA/+ K14Cre) in adult epidermis led to ear IFE thickening, which closely resembles BCC tumors. Our analyses demonstrate robust c‐JUN (S63 and S73), and weaker JNK1/2 phosphorylation increases in YAP‐driven lesions compared to the wild‐type ear epidermis (Fig EV6C). Conversely, both c‐JUN and JNK1/2 phosphorylation were almost undetectable in the Yap‐null ear epidermis (Yap fl/fl TetO‐Cre rtTA) when compared to the wild‐type control (Fig EV6C). Hence, our data provide genetic evidence that YAP regulates JNK‐JUN activity in vivo.
By using human tumor samples, genetically engineered mouse models, and in vitro cell lines, our study provides substantial evidence that: (i) YAP is dispensable for homeostatic epidermal regeneration; (ii) YAP activity is enriched in the basal tumor cells of BCC; (iii) YAP‐TEAD interaction is required for BCC initiation and tumor maintenance; and (iv) YAP potentiates c‐JUN activity to drive BCC growth.
Discussion
Basal cell carcinoma (BCC) is a skin tumor initiated and driven by activating mutations in the Hedgehog signaling pathway (Reifenberger et al, 2005; Epstein, 2008). Although the vast majority of BCC are curable with resection, vismodegib (SMO inhibitor) therapy, or a combination of both, the projected increase in BCC incidence over coming decades will result in a surge of advanced and vismodegib‐resistant cases (Danhof et al, 2018). Additionally, emerging findings suggest that advanced BCC could transdifferentiate into squamous cell carcinoma (SCC) with a much more aggressive phenotype (Orouji et al, 2014; Ransohoff et al, 2015; Zhao et al, 2015). Therefore, identification and validation of novel therapeutic strategies for patients with advanced BCC will be instrumental in preventing cancer‐associated death from this disease.
Recent detailed genomic analysis of human BCC tumors has identified alterations in additional pathways, which could contribute to initial tumor growth, invasion, and metastasis, or vismodegib resistance. One of the pathways identified with significant mutational propensity in its regulatory components was the Hippo, whose effectors, YAP and TAZ, have been shown to promote cancer phenotypes in a variety of tissues (Bonilla et al, 2016). Genetic alterations within the Hippo regulators have been generally low in other cancers, which elevates the significance of YAP activity in BCC (Harvey et al, 2013). Aside from cancer in adult tissues, YAP activity is critically required during emergency progenitor cell expansion for tissue repair following damage. This phenomenon has been observed in several epithelial tissues, such as skin, liver, intestine, heart, kidney, and pancreas (Patel et al, 2017). We speculated that skin epithelium might also exemplify a tissue where Hippo effectors, YAP and TAZ, are coerced by precise upstream cues to promote epithelial growth. Using the Hedgehog‐driven BCC as a model, we show that tumors, specifically those cells located basally within a growing clone, activate YAP's transcriptional role. Though TAZ has been speculated to carry overlapping cellular functions with YAP and might compensate in a YAP‐negative condition, we show that TAZ is meekly expressed in epidermis and its genetic deletion does not offer further change in BCC phenotype. However, YAP interaction with the DNA‐binding transcription factors, TEAD, and co‐activator function was required for BCC development. By inhibiting YAP expression in a different model where the tumor is already established, we also observe impaired BCC growth and depletion of Yap‐null tumor cells. Detailed longitudinal analyses of the Yap‐null BCC clones provided robust evidence that YAP is required not only for dysplastic BCC clone initiation and proliferation but also tumor survival until the invasive phenotype. Our data, however, do not distinguish whether Yap loss inhibits conversion of dysplastic clones to invasive tumors or whether YAP is required for the overall long‐term tumor survival regardless of its progression stage. Remarkably, YAP was completely dispensable for the normal adult epidermal homeostasis. These observations support the concept that conditions such as BCC, which resemble some aspects of tissue regeneration, engage YAP activity for epithelial cell growth and thus might offer a favorable therapeutic index for targeting this pathway in cancer.
The Hedgehog–Hippo interaction and how they influence each other have been studied in several tissues and contexts (Fernandez et al, 2009). Based on the published reports, we speculated that YAP might be directly affecting Hedgehog signaling output and the subsequent BCC phenotype. However, we saw no change in the expression of known Hedgehog target genes (Ptch1, Gli1, Gli2, or Hhip) in Yap‐null BCC as assessed by in situ hybridization or RNAseq analyses. Aside from the driver pathway, Hedgehog, Wnt signaling is the other well‐established cooperating pathway significantly contributing to BCC phenotype (Yang et al, 2008; Youssef et al, 2012). Likewise, Wnt and Hippo signaling components have been shown to engage each other in numerous circumstances. Once more, we demonstrated that Wnt signaling and its EHF phenotype are absolutely unaffected in the Yap‐null BCC. Our data show that activated YAP, independent of Hedgehog and Wnt signaling, plays a unique and vital role in driving BCC progression. Furthermore, our in vivo functional data explain elevated selective pressure to inactivate the Hippo regulators, LATS1/2 and PTPN14, in human BCC (Bonilla et al, 2016).
Using unbiased RNAseq and ChIPseq analyses to decipher the mechanisms by which YAP drives BCC development and progression, we found the strongest effect on the activity of AP1 transcription complex in Yap‐null BCC. The AP1 complex is composed of c‐JUN, FOS, ATF, and MAF family homo‐ and heterodimers giving them highly diverse function in tissue homeostasis (Lopez‐Bergami et al, 2010). In particular, c‐JUN and FOS activity is required for the transformation and growth of tumors including those originating in skin epidermis (Lopez‐Bergami et al, 2010). JUN depletion in BCC cells blocks their growth in vitro and in vivo (Eberl et al, 2012). Interestingly, YAP‐TEAD‐AP1 cooperation by directly interacting on the target gene promoters to regulate their expression has already been described (Zanconato et al, 2015; Liu et al, 2016). While our ChIPseq data also confirm that YAP‐TEAD and AP1 could bind in close proximity at the same target genes, we find that YAP regulates overall c‐JUN protein, mRNA, and transcriptional activity. The Yap‐null BCC had significant reduction in the c‐JUN phosphorylation on serines 63 and 73, two JNK phosphorylation sites that stabilize c‐JUN and increase its activity. JNK inhibition in BCC cell lines reduces c‐JUN phosphorylation and cell proliferation. Correspondingly, JNK activity was also diminished or increased in the tumors lacking YAP expression or YAP‐overexpressing IFE lesions, respectively. Recently published findings also propose that YAP could increase JNK activity in endothelial cells (Wang et al, 2016). Hence, our in vivo and in vitro analyses of the JNK‐JUN pathway strongly support that YAP regulates their activity in BCC. While the Yap‐null BCC tumors or BCC cell lines show consistent reduction in phospho‐JUN concomitant with decreased total c‐JUN protein, we also detect decrease in c‐Jun mRNA expression. Our ChIPseq data show that c‐Jun promoter/enhancer contains TEAD binding sites but also multiple AP1 consensus sites, which can be bound by c‐JUN protein to positively regulate its own expression (Angel et al, 1988). In fact, c‐JUN expression strongly correlated with the canonical YAP targets in various human cancer cell lines (CCLE). Thus, it is highly probable that Yap loss in BCC decreases YAP‐TEAD transcriptional activation of the c‐Jun promoter, JNK‐mediated c‐JUN phosphorylation, c‐JUN protein stability, and subsequent AP1 activity. Although here we demonstrate that YAP activity potentiates the AP1 signaling in BCC, upstream molecular mechanisms linking YAP and JNK‐JUN remain to be elucidated. Our RNAseq data suggest that YAP could impact MAPK signaling via transcriptional regulation of known upstream components (i.e., IRS2, IGFBP7, IGFPB2, EPGN, and DUSP5) and thus regulating JNK‐JUN activity. Others have recently shown that YAP could regulate actin cytoskeleton via RHO‐ROCK cascade, which also has been shown to directly control JNK‐JUN axis (Marinissen et al, 2004; Qiao et al, 2017). Hereafter, future work should explore how and under which circumstances YAP impacts MAPK signaling, cytoskeleton, and JNK‐JUN pathway in cancer.
In the clinic, the biggest challenge for BCC remains treatment of patients with advanced or metastatic disease, of which only about 50% respond to vismodegib therapy (Puig & Berrocal, 2015; Danhof et al, 2018). The mechanisms of resistance vary from emergence of the SMO‐mutant BCC clones and activation of kinase pathways to amplification of the GLI signaling via other mechanisms (Atwood et al, 2013, 2015; Sharpe et al, 2015). As our work demonstrates that YAP drives BCC growth in a Hedgehog‐independent fashion, we propose that a combination therapy of vismodegib and YAP inhibition could offer a new strategy to circumvent resistance while effectively killing BCC tumors. Furthermore, published work from our laboratory has demonstrated that sustained YAP overexpression in skin epidermis initially induces BCC‐like clonal growth which eventually evolves into SCC phenotype (Schlegelmilch et al, 2011; Silvis et al, 2011). Hence, it is possible that YAP/Hippo pathway actively participates in therapy‐induced BCC to SCC transdifferentiation observed in some patients. Future research should focus on developing transdifferentiation models for better understanding YAP's contribution in this phenotype.
Materials and Methods
Animals and tumor induction
The Yap fl/fl,Yap S79A, and Col1 TetO‐YAPS127A mice were previously described (Schlegelmilch et al, 2011). The Taz fl/fl (Wwtr1 fl/fl) mice were previously described in Xin, M., et al (Xin et al, 2013). The K14CreER, K14Cre, R26 LSL‐SmoM2YFP, R26 LSL‐tdTomato, TetO‐Cre, CAG‐rtTA3, and athymic nude (nu/nu) mice were obtained from The Jackson Laboratories. Embryonic Yap‐knockout skin tissue (Yap fl/fl:K14Cre) was collected at E18.5 for analyses. The adult Yap‐knockout (Yap fl/fl TetO‐Cre CAG‐rtTA3) or YAPS27A (Col1 TetO‐YAPS127A R26 LSL‐rtTA/+ K14Cre) overexpression was initiated at P56 (8 weeks of age) by administering doxycycline (1 g/l) with 5 g/l sucrose in drinking water for one 1 week. The Yap‐knockout mice were followed up to 12 weeks. BCC was induced in all mice at postnatal days P24–P28 with intraperitoneal (i.p.) and/or topical tamoxifen administration. The clonal BCC induction (“low‐dose” tamoxifen) was done with i.p. injections of 2.5 mg tamoxifen in 100 μl of corn oil for two consecutive days. The “high‐dose” tamoxifen protocol included i.p. injections as described for the “low‐dose” protocol plus simultaneous topical tamoxifen (2% tamoxifen in acetone) application on the ear and tail skin for 2 days. The ear and tail skin samples were analyzed at indicated time points following the last tamoxifen dose. To measure cell proliferation in vivo, mice were injected i.p. with 50 μg EdU (5‐ethynyl‐2′‐deoxyuridine) in saline 24 h prior to euthanasia. For the allograft experiments, 5 × 106 ASZ (TetON‐CRISPR‐YapKO) cells in 100 μl of Geltrex® (Thermo Fisher Scientific) were injected subcutaneously into flanks of 8‐week‐old athymic nu/nu mice. The tumors were allowed to engraft for 2 weeks, after which a cohort of mice received doxycycline (1 g/l) with 5 g/l sucrose in drinking water for the duration of the experiment. All of the experiments with mice were approved by IACUC at Boston Children's Hospital.
Antibodies
Following commercially available antibodies were used: YAP (D8H1X—Cell Signaling), YAP (1A12—Cell Signaling), YAP/TAZ (D24E4—Cell Signaling), GAPDH (D16H11—Cell Signaling), pJUNS73 (D47G9—Cell Signaling), pJUNS63 (54B3—Cell Signaling), pJNK Thr183/Tyr185 (81E11—Cell Signaling), JNK (sc7345—Santa Cruz), JUN (BD 610327), GFP (ab6673—Abcam) H3K27Ac (ab4729—Abcam), TEAD1 (BD 610922), TEAD4 (ab58310—Abcam), SOX9 (AB5535—Millipore), LHX2 (sc19344—Santa Cruz), PCDH (MAB761‐500 R&D Systems), LEF1 (C12A5—Cell Signaling), and CUX1 (sc13024—Santa Cruz).
Cell culture
The mouse BCC cell lines, ASZ and BSZ, were a kind gift from Ervin H. Epstein, Jr. (Children's Hospital Oakland Research Institute). The cells were grown in M154CF (Thermo Fisher Scientific) media supplemented with 0.05 mM CaCl2, penicillin–streptomycin, and 10 ml chelexed FBS. The Silencer Select (Thermo Fisher Scientific) siControl (AM4611), siYap (s76160), and siTaz (s97145) RNAi were transfected into cells using Lipofectamine® RNAiMAX (Invitrogen) according to the manufacturer's protocol. The cell proliferation was measured using the CellTiter 96® AQueous One Solution Cell Proliferation Assay from Promega. JNK1/2/3 were inhibited in vitro with 5 μM SP600125 or DMSO, and proliferation was determined by cell counting. Doxycycline‐inducible Yap knockout (TetON‐CRISPR‐YapKO) in ASZ or BSZ cells was achieved by lentiviral infection of plasmids carrying Yap sgRNA (FgH1UTG) and SpCas9 (lentiCRISPRv1; Sanjana et al, 2014; Aubrey et al, 2015). The infected cells were selected with 2 μg/ml puromycin (lentiCRISPRv1 selection) for 4 days and FACS‐sorted for GFP (FgH1UTG selection). Yap knockout in vitro was induced with 1 μg/ml doxycycline treatment in culture media for 6 days. The targeting sgRNA sequence (5′‐ACAACGATCAG ACAACAACA) used in our studies was directed against mouse Yap Exon 2.
Protein and RNA analyses
Total protein from cell lines was isolated with the ice‐cold RIPA buffer containing cOmplete™ protease and PhosSTOP™ phosphatase inhibitor cocktails (Roche Applied Science). The protein concentrations were quantified with Pierce™ BCA Protein Assay Kit, and equal protein amounts were resolved on NuPAGE® Novex® 4–12% Bis‐Tris protein gels (Invitrogen). Total RNA from the cell lines was isolated using TRIzol® (Invitrogen) according to the manufacturer's protocol. The RNA was converted to cDNA using iScript® (Bio‐Rad) cDNA synthesis kit for qPCR analyses with either Fast SYBR Green or Fast Advanced TaqMan Master Mixes (Applied Biosystems). The qPCR primer sequences used in this study are listed in Table EV4.
Histology, Immunohistochemistry, Immunofluorescence, and RNAscope
The isolated tissues were fixed in 10% neutral buffered formalin for at least 24 h, paraffin‐embedded, and sectioned onto slides to 5 μm thickness. Antigens were retrieved using Antigen Unmasking Solution (citric acid based) (Vector Labs) at 95°C for 1 h. All of the primary antibody incubations were done overnight at 4°C. For Immunohistochemistry, target protein was detected with secondary biotinylated Anti‐Rabbit or Anti‐Goat IgG antibodies (Vector Labs) followed by signal amplification with RTU Vectastain Elite ABC (Vector Labs). The slides were developed with DAB Peroxidase (HRP) Substrate Kit (Vector Labs), dehydrated, and mounted with Vectamount (Vector Labs). For Immunofluorescence (IF), secondary Donkey anti‐Rabbit, anti‐Goat, anti‐Rat, or anti‐Mouse Alexa Fluor®‐conjugated antibodies (Thermo Fisher Scientific) were used to detect primary antibodies. Tissue‐incorporated EdU was detected using the Click‐iT™ Plus EdU Alexa Fluor™ 647 Imaging Kit (Thermo Fisher Scientific). All of the IF slides were mounted with Prolong Gold anti‐fade with DAPI (Invitrogen) and imaged on Zeiss Axio Observer Z1 microscope equipped with Zeiss Apotome.2. The RNAscope® for Yap, Taz, Gli1, Ptch1, Axin2, and Lgr5 was done using the ACD inventoried probes and RNAscope® 2.5 HD Reagent Kit‐BROWN (ACD) detection system according to the manufacturer's instructions. De‐identified human BCC tissue microarray was purchased from US Biomax, Inc.
BCC cell isolation and FACS
The RNAseq and qPCR analyses were performed on FACS‐sorted ear BCC cells 6 weeks after high‐dose tamoxifen administration. For the purpose of FACS sorting, we crossed R26 LSL‐SmoM2YFP :K14CreER mice with R26 LSL‐tdTomato to use tdTomato expression as a marker for Cre recombined epidermal cells. Briefly, ear tissue from mice was minced and incubated at 37°C in 0.25% trypsin‐EDTA solution for 1 h. The tissue was then passed 5–6 times through 16‐ and 18‐gauge needle, trypsin was quenched with excess PBS containing 2% chelexed FBS, and entire solution was passed through a 70‐μm filter. The cells were pelleted by centrifugation at 200 g for 15 min (4°C), washed once with PBS containing 2% chelexed FBS, and re‐pelleted as before. Next, single cells were stained with indicated FACS antibodies (TER119‐eFluor450, CD45‐eFluor450, α6‐integrin‐APC) (eBiosciences) on ice for 30 min. BCC cells were FACS‐sorted based on their negativity for blood markers (CD45, TER119) and positivity for tdTomato and α6‐integrin. ChIPseq analysis was performed on the tail BCC 10 weeks after tamoxifen induction. First, dermal fat was scraped from the tail skin and the skin tissue was floated dermis side down onto 0.25% trypsin‐EDTA solution for 1 h at 37°C. The epidermal cells were then separated from the dermis by scraping and subjected to series of filtration and centrifugation steps as described for the ear BCC.
RNAseq and ChIPseq
The RNA from the FACS sorted ear BCC cells was isolated using the RNeasy® Plus Mini Kit (Qiagen). The libraries for the RNAseq analysis were prepared using the TruSeq RNA Library Preparation Kit v2 (Illumina) according to the manufacturer's protocol. The sample preparation and chromatin immunoprecipitations with Tead1, Tead4, and H3K27Ac antibodies for the ChIPseq analyses were done as described previously (Galli et al, 2015). The input DNA was used as a background for defining significant signal enrichment. The ChIPseq libraries were prepared with the NEBNext® Ultra DNA Library Prep Kit and NEBNext® Multiplex Oligos for Illumina (NEB) according to the manufacturer's recommendations. All of the libraries were sequenced on the NextSeq High 75 cycles (Illumina) sequencer at Harvard University Bauer Core Facility. Differential gene expression analysis was generated using the DESeq2 package (Bioconductor) with set thresholds to adjusted P‐value < 0.05 and fold change <1.5. Ingenuity Pathway Analysis (Qiagen) was done on the identified differentially expressed genes. The ChIPseq bioinformatics analysis was done as described previously (Galli et al, 2015). JUN or FOS co‐expression correlation with other genes in human cancer cell lines was done using the Gene Neighbors tool from the Broad‐Novartis Cancer Cell Line Encyclopedia (https://portals.broadinstitute.org/ccle_legacy/home).
Data accessibility
All of the RNAseq and ChIPseq data were deposited in the Gene Expression Omnibus under the accession number GSE115223.
Statistical analyses
All of the statistical tests and calculations were performed using GraphPad Prism software. Specific and appropriate statistical tests performed are indicated in the figure legends.
Author contributions
FDC, DM, and KS conceived hypotheses and generated research project plans. FDC helped write and review the manuscript. DM carried out in vitro and in vivo experiments, analyzed data, generated figures, and wrote the manuscript. KS generated preliminary data and reviewed the manuscript. AFMD helped with in vitro data generation and analyses. MD provided in vivo data and reviewed the manuscript. RP and RAC helped analyze and interpret RNAseq and ChIPseq data.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Acknowledgments
We are extremely grateful to Dr. Ervin H. Epstain Jr. (CHORI) for providing us with the mouse BCC cell lines. We thank Dr. Eric Olson (UT Southwestern) for kindly providing us with Taz fl/fl mice. We also thank all the valuable scientific and technical feedback from the current and previous Camargo laboratory members. Dejan Maglic has been supported by the NRSA Postdoctoral Fellowship from NCI (5F32CA192815‐03). This project was supported by the NIH/NIAMS awarded to Fernando Camargo (4R01AR064036‐05). [HHS|NIH|National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS); HHS|NIH|National Cancer Institute (NCI)]
The EMBO Journal (2018) 37: e98642
See also: https://doi.org/10.15252/embj.201899921 (September 2018)
References
- Akladios B, Mendoza Reinoso V, Cain JE, Wang T, Lambie DL, Watkins DN, Beverdam A (2017) Positive regulatory interactions between YAP and Hedgehog signalling in skin homeostasis and BCC development in mouse skin in vivo . PLoS One 12: e0183178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angel P, Hattori K, Smeal T, Karin M (1988) The jun proto‐oncogene is positively autoregulated by its product, Jun/AP‐1. Cell 55: 875–885 [DOI] [PubMed] [Google Scholar]
- Atwood SX, Chang AL, Oro AE (2012) Hedgehog pathway inhibition and the race against tumor evolution. J Cell Biol 199: 193–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood SX, Li M, Lee A, Tang JY, Oro AE (2013) GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature 494: 484–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, Ally MS, Kim J, Yao C, Chang ALS, Oro AE, Tang JY (2015) Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 27: 342–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O'Connor L, Milla L, Wilcox S, Tai L, Strasser A, Herold MJ (2015) An inducible lentiviral guide RNA platform enables the identification of tumor‐essential genes and tumor‐promoting mutations in vivo . Cell Rep 10: 1422–1432 [DOI] [PubMed] [Google Scholar]
- Bonilla X, Parmentier L, King B, Bezrukov F, Kaya G, Zoete V, Seplyarskiy VB, Sharpe HJ, McKee T, Letourneau A, Ribaux PG, Popadin K, Basset‐Seguin N, Ben Chaabene R, Santoni FA, Andrianova MA, Guipponi M, Garieri M, Verdan C, Grosdemange K et al (2016) Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Genet 48: 398–406 [DOI] [PubMed] [Google Scholar]
- Cui X, Kim HJ, Kuiatse I, Kim H, Brown PH, Lee AV (2006) Epidermal growth factor induces insulin receptor substrate‐2 in breast cancer cells via c‐Jun NH(2)‐terminal kinase/activator protein‐1 signaling to regulate cell migration. Cancer Res 66: 5304–5313 [DOI] [PubMed] [Google Scholar]
- Danhof R, Lewis K, Brown M (2018) Small molecule inhibitors of the hedgehog pathway in the treatment of basal cell carcinoma of the skin. Am J Clin Dermatol 19: 195–207 [DOI] [PubMed] [Google Scholar]
- Eberl M, Klingler S, Mangelberger D, Loipetzberger A, Damhofer H, Zoidl K, Schnidar H, Hache H, Bauer HC, Solca F, Hauser‐Kronberger C, Ermilov AN, Verhaegen ME, Bichakjian CK, Dlugosz AA, Nietfeld W, Sibilia M, Lehrach H, Wierling C, Aberger F (2012) Hedgehog‐EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour‐initiating pancreatic cancer cells. EMBO Mol Med 4: 218–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbediwy A, Vincent‐Mistiaen ZI, Spencer‐Dene B, Stone RK, Boeing S, Wculek SK, Cordero J, Tan EH, Ridgway R, Brunton VG, Sahai E, Gerhardt H, Behrens A, Malanchi I, Sansom OJ, Thompson BJ (2016) Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development 143: 1674–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein EH (2008) Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer 8: 743–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez LA, Northcott PA, Dalton J, Fraga C, Ellison D, Angers S, Taylor MD, Kenney AM (2009) YAP1 is amplified and up‐regulated in hedgehog‐associated medulloblastomas and mediates Sonic hedgehog‐driven neural precursor proliferation. Genes Dev 23: 2729–2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flohil SC, Seubring I, van Rossum MM, Coebergh JW, de Vries E, Nijsten T (2013) Trends in Basal cell carcinoma incidence rates: a 37‐year Dutch observational study. J Invest Dermatol 133: 913–918 [DOI] [PubMed] [Google Scholar]
- Galli GG, Carrara M, Yuan WC, Valdes‐Quezada C, Gurung B, Pepe‐Mooney B, Zhang T, Geeven G, Gray NS, de Laat W, Calogero RA, Camargo FD (2015) YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol Cell 60: 328–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goruppi S, Bonventre JV, Kyriakis JM (2002) Signaling pathways and late‐onset gene induction associated with renal mesangial cell hypertrophy. EMBO J 21: 5427–5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey KF, Zhang X, Thomas DM (2013) The Hippo pathway and human cancer. Nat Rev Cancer 13: 246–257 [DOI] [PubMed] [Google Scholar]
- Heintz N (2004) Gene expression nervous system atlas (GENSAT). Nat Neurosci 7: 483 [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Ashouri J, Wolter S, Doerrie A, Dittrich‐Breiholz O, Schneider H, Wagner EF, Troppmair J, Mackman N, Kracht M (2008) Transcriptional regulation of EGR‐1 by the interleukin‐1‐JNK‐MKK7‐c‐Jun pathway. J Biol Chem 283: 12120–12128 [DOI] [PubMed] [Google Scholar]
- Ito M, Yang Z, Andl T, Cui C, Kim N, Millar SE, Cotsarelis G (2007) Wnt‐dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature 447: 316–320 [DOI] [PubMed] [Google Scholar]
- Jin G, Howe PH (1997) Regulation of clusterin gene expression by transforming growth factor beta. J Biol Chem 272: 26620–26626 [DOI] [PubMed] [Google Scholar]
- Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH Jr, Scott MP (1996) Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272: 1668–1671 [DOI] [PubMed] [Google Scholar]
- Larsimont JC, Youssef KK, Sanchez‐Danes A, Sukumaran V, Defrance M, Delatte B, Liagre M, Baatsen P, Marine JC, Lippens S, Guerin C, Del Marmol V, Vanderwinden JM, Fuks F, Blanpain C (2015) Sox9 controls self‐renewal of oncogene targeted cells and links tumor initiation and invasion. Cell Stem Cell 17: 60–73 [DOI] [PubMed] [Google Scholar]
- Lee MJ, Byun MR, Furutani‐Seiki M, Hong JH, Jung HS (2014) YAP and TAZ regulate skin wound healing. J Invest Dermatol 134: 518–525 [DOI] [PubMed] [Google Scholar]
- Liu X, Li H, Rajurkar M, Li Q, Cotton JL, Ou J, Zhu LJ, Goel HL, Mercurio AM, Park JS, Davis RJ, Mao J (2016) Tead and AP1 coordinate transcription and motility. Cell Rep 14: 1169–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Bergami P, Lau E, Ronai Z (2010) Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat Rev Cancer 10: 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinissen MJ, Chiariello M, Tanos T, Bernard O, Narumiya S, Gutkind JS (2004) The small GTP‐binding protein RhoA regulates c‐Jun by a ROCK‐JNK signaling axis. Mol Cell 14: 29–41 [DOI] [PubMed] [Google Scholar]
- Mina M, Magi S, Jurman G, Itoh M, Kawaji H, Lassmann T, Arner E, Forrest ARR, Carninci P, Hayashizaki Y, Daub CO, the FC, Okada‐Hatakeyama M, Furlanello C (2015) Promoter‐level expression clustering identifies time development of transcriptional regulatory cascades initiated by ErbB receptors in breast cancer cells. Sci Rep 5: 11999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwenhuis E, Barnfield PC, Makino S, Hui C‐C (2007) Epidermal hyperplasia and expansion of the interfollicular stem cell compartment in mutant mice with a C‐terminal truncation of Patched1. Dev Biol 308: 547–560 [DOI] [PubMed] [Google Scholar]
- Okamura J, Huang Y, Moon D, Brait M, Chang X, Kim MS (2012) Downregulation of insulin‐like growth factor‐binding protein 7 in cisplatin‐resistant non‐small cell lung cancer. Cancer Biol Ther 13: 148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orouji A, Goerdt S, Utikal J, Leverkus M (2014) Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol 171: 431–433 [DOI] [PubMed] [Google Scholar]
- Patel SH, Camargo FD, Yimlamai D (2017) Hippo signaling in the liver regulates organ size, cell fate, and carcinogenesis. Gastroenterology 152: 533–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig S, Berrocal A (2015) Management of high‐risk and advanced basal cell carcinoma. Clin Transl Oncol 17: 497–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y, Chen J, Lim YB, Finch‐Edmondson ML, Seshachalam VP, Qin L, Jiang T, Low BC, Singh H, Lim CT, Sudol M (2017) YAP regulates actin dynamics through ARHGAP29 and promotes metastasis. Cell Rep 19: 1495–1502 [DOI] [PubMed] [Google Scholar]
- Ransohoff KJ, Tang JY, Sarin KY (2015) Squamous change in basal‐cell carcinoma with drug resistance. N Engl J Med 373: 1079–1082 [DOI] [PubMed] [Google Scholar]
- Reifenberger J, Wolter M, Knobbe CB, Kohler B, Schonicke A, Scharwachter C, Kumar K, Blaschke B, Ruzicka T, Reifenberger G (2005) Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br J Dermatol 152: 43–51 [DOI] [PubMed] [Google Scholar]
- Sanchez‐Danes A, Hannezo E, Larsimont JC, Liagre M, Youssef KK, Simons BD, Blanpain C (2016) Defining the clonal dynamics leading to mouse skin tumour initiation. Nature 536: 298–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F (2014) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods 11: 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, Camargo FD (2011) Yap1 acts downstream of alpha‐catenin to control epidermal proliferation. Cell 144: 782–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser‐Kronberger C, Regl G, Kroismayr R, Moriggl R, Sibilia M, Aberger F (2009) Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res 69: 1284–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe HJ, Pau G, Dijkgraaf GJ, Basset‐Seguin N, Modrusan Z, Januario T, Tsui V, Durham AB, Dlugosz AA, Haverty PM, Bourgon R, Tang JY, Sarin KY, Dirix L, Fisher DC, Rudin CM, Sofen H, Migden MR, Yauch RL, de Sauvage FJ (2015) Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 27: 327–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvis MR, Kreger BT, Lien WH, Klezovitch O, Rudakova GM, Camargo FD, Lantz DM, Seykora JT, Vasioukhin V (2011) alpha‐catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Sci Signal 4: ra33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- So PL, Langston AW, Daniallinia N, Hebert JL, Fujimoto MA, Khaimskiy Y, Aszterbaum M, Epstein EH Jr (2006) Long‐term establishment, characterization and manipulation of cell lines from mouse basal cell carcinoma tumors. Exp Dermatol 15: 742–750 [DOI] [PubMed] [Google Scholar]
- Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, Agarinis C, Schmelzle T, Bouwmeester T, Schübeler D, Bauer A (2015) YAP1 exerts its transcriptional control via TEAD‐mediated activation of enhancers. PLoS Genet 11: e1005465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troen G, Nygaard V, Jenssen TK, Ikonomou IM, Tierens A, Matutes E, Gruszka‐Westwood A, Catovsky D, Myklebost O, Lauritzsen G, Hovig E, Delabie J (2004) Constitutive expression of the AP‐1 transcription factors c‐jun, junD, junB, and c‐fos and the marginal zone B‐cell transcription factor Notch2 in splenic marginal zone lymphoma. J Mol Diagn 6: 297–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varelas X (2014) The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 141: 1614–1626 [DOI] [PubMed] [Google Scholar]
- Villani RM, Adolphe C, Palmer J, Waters MJ, Wainwright BJ (2010) Patched1 inhibits epidermal progenitor cell expansion and basal cell carcinoma formation by limiting Igfbp2 activity. Cancer Prev Res 3: 1222 [DOI] [PubMed] [Google Scholar]
- Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, Liu J, Deng D, Lau CW, Wan S, Ai D, Mak KK, Tong KK, Kwan KM, Wang N, Chiu JJ, Zhu Y, Huang Y (2016) Integrin‐YAP/TAZ‐JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 540: 579–582 [DOI] [PubMed] [Google Scholar]
- Wu S, Han J, Li WQ, Li T, Qureshi AA (2013) Basal‐cell carcinoma incidence and associated risk factors in US women and men. Am J Epidemiol 178: 890–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, Richardson JA, Sadek HA, Bassel‐Duby R, Olson EN (2013) Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci USA 110: 13839–13844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Andl T, Grachtchouk V, Wang A, Liu J, Syu LJ, Ferris J, Wang TS, Glick AB, Millar SE, Dlugosz AA (2008) Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/beta3‐catenin signaling. Nat Genet 40: 1130–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef KK, Van Keymeulen A, Lapouge G, Beck B, Michaux C, Achouri Y, Sotiropoulou PA, Blanpain C (2010) Identification of the cell lineage at the origin of basal cell carcinoma. Nat Cell Biol 12: 299–305 [DOI] [PubMed] [Google Scholar]
- Youssef KK, Lapouge G, Bouvree K, Rorive S, Brohee S, Appelstein O, Larsimont JC, Sukumaran V, Van de Sande B, Pucci D, Dekoninck S, Berthe JV, Aerts S, Salmon I, del Marmol V, Blanpain C (2012) Adult interfollicular tumour‐initiating cells are reprogrammed into an embryonic hair follicle progenitor‐like fate during basal cell carcinoma initiation. Nat Cell Biol 14: 1282–1294 [DOI] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S (2015) Genome‐wide association between YAP/TAZ/TEAD and AP‐1 at enhancers drives oncogenic growth. Nat Cell Biol 17: 1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Pasolli HA, Fuchs E (2011) Yes‐associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci USA 108: 2270–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Ponomaryov T, Ornell KJ, Zhou P, Dabral SK, Pak E, Li W, Atwood SX, Whitson RJ, Chang ALS, Li J, Oro AE, Chan JA, Kelleher JF, Segal RA (2015) RAS/MAPK activation drives resistance to Smo inhibition, metastasis and tumor evolution in Shh pathway‐dependent tumors. Can Res 75: 3623–3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Review Process File
Data Availability Statement
All of the RNAseq and ChIPseq data were deposited in the Gene Expression Omnibus under the accession number GSE115223.