

In addition to estrogens, ER function is modulated by other steroid receptors and multiple signaling pathways, and many of these pathways affect drug resistance and patient outcome. Here, Siersbæk et al. review the mechanisms through which these pathways impact ER function and drug resistance.
Keywords: breast cancer, cross-talk, cytokines, estrogen receptor, growth factors
Abstract
Estrogen receptor α (ER) is the major driver of ∼75% of breast cancers, and multiple ER targeting drugs are routinely used clinically to treat patients with ER+ breast cancer. However, many patients relapse on these targeted therapies and ultimately develop metastatic and incurable disease, and understanding the mechanisms leading to drug resistance is consequently of utmost importance. It is now clear that, in addition to estrogens, ER function is modulated by other steroid receptors and multiple signaling pathways (e.g., growth factor and cytokine signaling), and many of these pathways affect drug resistance and patient outcome. Here, we review the mechanisms through which these pathways impact ER function and drug resistance as well as discuss the clinical implications.
Breast cancer is now the most common cancer diagnosed in the United States, with an estimated 266,120 new cases of invasive breast cancer to be diagnosed in women in the United States in 2018 and an estimated 40,920 breast cancer deaths (https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2018/cancer-facts-and-figures-2018.pdf). The lifetime risk of developing breast cancer is now one in eight for women (Kohler et al. 2015). Approximately 75% of breast tumors are driven by the estrogen receptor α (ERα; referred to here as ER)-mediated transcriptional activity. A number of established endocrine therapies exist, including selective ER modulators (SERMs) such as tamoxifen, selective ER down-regulators (SERDs) such as fulvestrant, and aromatase inhibitors (AIs) such as letrozole, anastrazole (nonsteroidal AI), and exemestane (steroidal AI). More recently, the targeting of cell cycle progression with cyclin-dependent kinase 4/6 (CDK4/6) inhibitors in combination with anti-estrogen therapy has become the first line standard of care in de novo or recurrent metastatic disease. However, despite standard endocrine therapy, >20% of patients with early stage disease develop resistance to anti-estrogens and relapse with incurable metastatic disease (Mauri et al. 2006; Early Breast Cancer Trialists’ Collaborative Group [EBCTCG] 2011). While many different mechanisms of resistance have been described, recent data show that 11%–55% of metastatic cancers have point mutations in the ligand-binding domain of ER, especially in amino acids Y537 and D538, generating a constitutively active ER that is less dependent on estrogen for activity (Merenbakh-Lamin et al. 2013; Robinson et al. 2013; Toy et al. 2017). Compared with wild-type ER, mutant ER is resistant to estrogen deprivation and is less responsive to tamoxifen or fulvestrant. This highlights the pivotal role of the ER pathway in driving breast tumor progression as well as its clinical importance in late stage disease.
The activity of wild-type ER is largely controlled by the availability of estrogens, which bind to the ER ligand-binding domain and mediate homodimerization and binding of the receptor complex to chromatin, usually at distal regulatory enhancer sites. However, it is becoming increasingly clear that growth factors and signaling molecules from the tumor microenvironment play important roles in the progression of ER+ breast cancer, and many of these signaling pathways directly impact ER transcriptional activity and function. This has clinical implications and suggests that targeting these pathways may provide opportunities for the treatment of ER+ breast cancer patients. Here, we review the established molecular connections between the ER and signaling pathways initiated by growth factors, hormones, and cytokines from the tumor microenvironment and discuss the clinical opportunities raised by this insight.
Modulation of ER function by phosphorylation
Growth factors
Canonical activation of ER involves binding of estrogens such as estradiol to the ligand-binding domain, resulting in structural changes in ER and cofactor recruitment. However, it is well established that post-translational modifications such as phosphorylation, acetylation, SUMOylation, methylation, and ubiquitination can also modulate ER activity through multiple different mechanisms (Le Romancer et al. 2011). Phosphorylation of ER in particular is well described and plays an important role in ER activation—in some cases, in a ligand-independent manner (Anbalagan and Rowan 2015). The N terminus of the receptor is particularly highly phosphorylated by multiple different kinases, and these phosphorylation events can modulate ER activity (Le Goff et al. 1994; Kato et al. 1995; Joel et al. 1998b). Phosphorylation of Ser118 (S118) is one of the most well-characterized ER phosphorylation events, and phosphorylation of this residue is mediated through Cdk7 upon activation of ER by estradiol (Fig. 1; Chen et al. 2000, 2002; Harrod et al. 2017). Growth factors such as epidermal growth factor (EGF) can also induce phosphorylation of S118 through mitogen-activated protein kinase (MAPK), thereby activating ER independently of estrogens (Fig. 1; Kato et al. 1995; Bunone et al. 1996; Chen et al. 2002). More recently, EGF-induced S118 phosphorylation has been suggested to increase breast cancer cell proliferation by activating a specific ER chromatin-binding profile through cooperation with different transcription factor complexes, including AP-1 transcription factors and the pre-B-cell leukemia transcription factor 1 (PBX1) (Lupien et al. 2010; Magnani et al. 2015). This implies that growth factor activation of ER can alter the binding potential and target genes of this transcription factor complex, and, in some cases, this can occur in the absence of estrogen stimulation.
Figure 1.
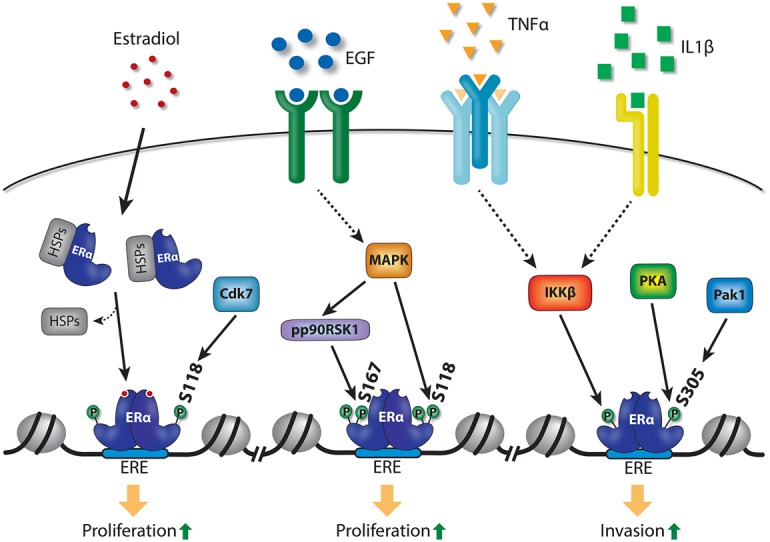
Activation of ER by phosphorylation induced by growth factor and cytokine signaling pathways. Estradiol can induce dimerization of ER and binding of the dimer to ER response elements (EREs) in chromatin, and, from these sites, ER drives a proproliferative gene program. In addition, multiple growth factor and cytokine signaling pathways can induce phosphorylation of ER at S167, S118, or S305, which can also activate the receptor and drive it onto chromatin in the absence of estradiol, thereby promoting cell proliferation.
The recently described Y537 and D538 ER mutants are constitutively phosphorylated on S118 by Cdk7 in an estrogen-independent manner, and this phosphorylation event is likely to play an important role in regulating the activity of the mutant receptors (Harrod et al. 2017; Jeselsohn et al. 2018). This highlights the importance of this phosphorylation event for ER activity and indicates that phosphorylation of S118 may play an important role in drug-resistant metastatic disease by potentiating transcriptional activity of mutant ER-driven cancer. Importantly, the Cdk7 inhibitor THZ1, which inhibits general RNA polymerase II-mediated transcription (as well as S118 phosphorylation of ER), suppresses growth in MCF7 breast cancer cells expressing either wild-type or mutant ER (Harrod et al. 2017; Jeselsohn et al. 2018). Although THZ1 is not specific for mutant ER and instead blocks a component of the general transcription machinery, it reveals a potential avenue for pharmacological intervention in patients with ER mutations.
In addition to S118, S167 is another major phosphorylation site in the N terminus of ER, which has been shown to be important for ER-mediated transcriptional activation (Joel et al. 1998a; Becker et al. 2011; Anbalagan and Rowan 2015). Phosphorylation of ER on this residue is induced by phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) and MAPK signaling in response to hormones such as insulin, insulin-like growth factor (IGF), and EGF (Joel et al. 1998a; Campbell et al. 2001; Yamnik et al. 2009; Yamnik and Holz 2010; Becker et al. 2011; Held et al. 2012). Interestingly, ectopic expression of AKT has been shown to redistribute the ligand-dependent binding of ER to chromatin, presumably through S167 phosphorylation of the receptor (Bhat-Nakshatri et al. 2008), thereby further supporting the notion that phosphorylation of ER can modulate receptor binding and target gene activation.
In addition to the N terminus, the hinge region of ER has also been shown to be phosphorylated at S305 by protein kinase-A (PKA) (Michalides et al. 2004) and Pak1 (Fig. 1; Wang et al. 2002). Phosphorylation of ER on S305 was shown to activate the receptor in the absence of estradiol (Wang et al. 2002) and drives receptor activity that is refractory to tamoxifen inhibition (Michalides et al. 2004). Using a phospho-specific antibody for transcription factor mapping (chromatin immunoprecipitation [ChIP] followed by deep sequencing [ChIP-seq]) analysis, PKA-mediated ER phosphorylation on S305 has been suggested to induce receptor binding to a distinct set of binding sites that are not typically seen following estradiol-induced ER binding, implying that these are nonclassic regulatory sites. It was suggested that this mechanism can mediate tamoxifen resistance, partly through expression of the oncogene c-MYC (de Leeuw et al. 2013). Together, this demonstrates that phosphorylation of ER can modulate receptor binding and activity in a ligand-independent and nonclassical manner.
Cytokines
The tumor microenvironment is composed of a complex ensemble of cell types, including fibroblasts, fat cells, and immune cells such as macrophages, neutrophils, and T cells (Artacho-Cordon et al. 2012). These cells play an important role in breast cancer progression. As an example, Finak et al. (2008) identified a gene signature from the stromal compartment of breast cancer patients that predicts outcome independent of other tumor markers such as ER and HER2. Paracrine signaling is likely to be the major mechanism through which stromal cells affect tumor cell function, as stromal cells secrete a variety of signaling molecules, including hormones and inflammatory cytokines, many of which have been shown to be associated with tumor progression in breast cancer patients, where they impinge on the function and phenotype of cancer epithelial cells. For example, coculture of fibroblasts with breast cancer cells has been shown to decrease expression of ER in the cancer cells (Brechbuhl et al. 2017; Huang et al. 2017; Morgan et al. 2018) and activate potent growth factor pathways (e.g., AKT and MAPK), thereby modulating the response of the epithelial cancer cells to anti-estrogen treatment (Brechbuhl et al. 2017; Huang et al. 2017). Furthermore, the adipokines leptin and interleukin-6 (IL-6) have been shown to be associated with increased tumor size and metastasis in ER+ breast cancer patients (Madeddu et al. 2014). However, the molecular mechanisms underlying potentially causal effects of paracrine signaling from the stromal cells in the tumor microenvironment on breast cancer progression are only starting to emerge and appear to involve modulation of ER action through different mechanisms (see also “Redirection of ER Function by Transcription Factors Downstream from Hormone and Cytokine Signaling”).
It has been shown recently that the cytokines tumor necrosis factor α (TNFα) and IL-1β, which can be produced by multiple different cell types, including immune cells such as macrophages, can induce phosphorylation of S305 on ER (Stender et al. 2017). Cytokine-induced phosphorylation of this residue is mediated by inhibitor of nuclear factor κ B kinase subunit β (IKKβ), rather than PKA or Pak1 (which have been shown previously to phosphorylate S305). Cytokine-induced S305 phosphorylation of ER results in ER binding to a subset of binding sites typically seen following estradiol induction, and the target genes regulated by cytokine-induced phosphorylation of S305 represent a subset of estradiol-induced genes (Stender et al. 2017). Importantly, the cytokine-induced gene program in MCF7 breast cancer cells can induce extravasation, an important part of the metastatic process, and this occurs through ER but independently of estradiol (Stender et al. 2017). Consistently, overexpressing a constitutive active form of IKKβ, which is a key kinase downstream from TNFα, increases invasion in vitro and in vivo in the presence of estradiol (El-Shennawy et al. 2018). Interestingly, MCF7 breast cancer cells also become resistant to tamoxifen in the presence of these cytokines, showing that extracellular stimuli, in the form of specific cytokines, can modulate drug responsiveness.
Transforming growth factor β (TGFβ) is another cytokine secreted by cancer cells as well as stromal cells in the tumor microenvironment such as fibroblasts and immune cells (e.g., macrophages and leukocytes) and is known to play an important role in tumor progression in many different cancer types (Papageorgis and Stylianopoulos 2015). TGFβ appears to have a dual function, where it represses early tumor growth but promotes metastasis in late stage disease. This takes place both through direct effects on the tumor cells (e.g., TGFβ can induce epithelial-to-mesenchymal transition [EMT]) (Deckers et al. 2006) and by modulating the tumor microenvironment (e.g., by inducing an immunosuppressive environment) (Papageorgis and Stylianopoulos 2015). Canonical TGFβ signaling induces phosphorylation of the transcription factors SMAD2 and SMAD3, which then associate with SMAD4 to activate TGFβ target genes. TGFβ signaling inhibits ER function in a SMAD4-dependent manner (Wu et al. 2003; Ren et al. 2009). Consistent with a repressive function of SMAD4 on ER function, the association between these proteins is induced by anti-estrogens such as tamoxifen (Wu et al. 2003). In the absence of SMAD4, activation of SMAD3 by TGFβ enhances ER activity (Wu et al. 2003; Ren et al. 2009). Thus, although the precise mechanisms through which these SMADs regulate ER activity are not clear, these findings indicate that TGFβ signaling regulates breast cancer progression at least partly by directly regulating ER function. Furthermore, ER also inhibits TGFβ signaling, which is likely to be another important mechanism through which cross-talk between these pathways regulates breast cancer progression (Band and Laiho 2011).
Taken together, this illustrates the potential power of cytokine secretion from cells in the tumor microenvironment to alter ER activity and endocrine responsiveness in breast cancer. It also suggests that targeting these pathways may provide novel approaches for treating resistant ER+ breast cancer. Validating these mechanisms in vivo and in patient samples will be an important next step to investigate the translatability of these findings.
Effect of drugging growth factor and cytokine pathways in ER+ breast cancer
The PI3K/AKT/mTOR pathway is commonly mutated in both primary and recurrent ER+ breast cancer (The Cancer Genome Atlas Network 2012; Ciruelos Gil 2014; Yates et al. 2017) and regulates activation of ER through phosphorylation as described above, and activation of this pathway can lead to acquired endocrine resistance (Campbell et al. 2001; Vivanco and Sawyers 2002; Miller et al. 2010; Sanchez et al. 2011; Cavazzoni et al. 2012). Consequently, drugs directed against these pathways have been assessed in clinical trials in combination with ER targeting compounds. Preclinical data in a breast cancer cell line and murine xenograft models demonstrating anti-proliferative effects of these drugs suggest that this approach is viable (Boulay et al. 2005; Crowder et al. 2009; Ghayad et al. 2010; Guichard et al. 2015).
EGFR inhibitors
The utility of the EGFR tyrosine kinase inhibitors (TKIs) gefitinib and lapatinib, in combination with endocrine therapy for the management of de novo or recurrent metastatic breast cancer, has been investigated in both the first and second line setting in four separate studies. With the exception of one study (Cristofanilli et al. 2010), no significant progression-free survival (PFS) improvements have been reported with EGFR TKIs (Osborne et al. 2011; Carlson et al. 2012; Burstein et al. 2014). Paradoxically, in the small phase II study where a PFS benefit was observed (Cristofanilli et al. 2010), the objective response rate was numerically lower with the EGFR TKI plus AI combination (2%) compared with the AI plus placebo (12%), further indicating that endocrine resistance is not delayed by EGFR inhibition.
mTOR inhibitors
Inhibition of the mTOR pathway has received substantial attention in recent years, due to the development of inhibitors and the preclinical data functionally linking mTOR activity in ER+ disease. Clinical trial evidence culminated in the approval of everolimus (an inhibitor of the mTOR complex 1) in combination with exemestane for the treatment of nonsteroidal AI-resistant metastatic ER+ breast cancer. In the BOLERO-2 study, a double-blind phase III trial (Yardley et al. 2013), investigator-assessed PFS (the primary end point) was more than doubled with the addition of everolimus alone (7.8 mo for everolimus compared with 3.2 mo for placebo; hazard ratio [HR] 0.45; 95% confidence interval [CI]; 0.38–0.54). However, median overall survival (OS) was not significantly improved when everolimus was combined with exemestane: 31 mo compared with 26.6 mo with placebo plus exemestane (HR 0.89; 95% CI; 0.73–1.10; P = 0.14) (Piccart et al. 2014). The phase 2 BOLERO-4 study then explored the utility of combining everolimus with an AI as first line treatment for ER+ HER2− advanced cancer, yielding a median PFS of 22.0 mo (95% CI; 18.1–25.1 mo) (Royce et al. 2018). While this is potentially impactful, cross-trial comparisons with studies of newer therapeutics, such as CDK4/6 inhibitors, in combination with AIs can be made only cautiously due to the single-arm open-label design of the BOLERO-4 study. Interestingly, retrospective evidence has now established the use of everolimus and exemestane as second line therapy following progression on CDK4/6 inhibitors plus AIs—the now established first line therapy for metastatic ER+ HER2− metastatic breast cancer (Dhakal et al. 2018).
AKT inhibitors
Given that AKT has been shown to activate ER through phosphorylation of S167 (Campbell et al. 2001) and that increased AKT activity is found in 20%–55% of breast cancers (Altomare and Testa 2005), which is associated with reduced OS in patients with ER+ cancer treated with tamoxifen (Kirkegaard et al. 2005), the impact of targeting AKT is also being explored. Preclinical evidence for combining AIs and AKT inhibitors in anastrozole-resistant cancer cells (Vilquin et al. 2013) has lead to the initiation of a clinical trial using this combination in the AI-resistant setting (NCT01344031) (Ma et al. 2016). Other novel AKT inhibitors such as AZD5363 (Banerji et al. 2018) are in early phase clinical trials.
PI3K inhibitors
Clinical trials assessing the pan-class 1 PI3K inhibitors buparlisib (BKM120) and pictilisib (GDC-0941) were limited by prohibitive dose-limiting toxicity and lack of efficacy, resulting in marginal PFS benefits in a cohort of AI-pretreated post-menopausal women with metastatic breast cancer (Krop et al. 2016; Baselga et al. 2017). More promisingly, the α-specific PI3K inhibitors alpelisib (BYL719) and taselisib (GDC-0032) have demonstrated efficacy in early phase trials and shown modest toxicity. While the results of the placebo-controlled phase III SOLAR-1 trials evaluating the potential improvement in PFS with the addition of alpelisib to fulvestrant (NCT02437318) among patients with ER+ metastatic breast cancer are awaited, results from the SANDPIPER trial using taselisib have now been reported (Baselga et al. 2018). A PFS benefit of only 2 mo was observed with the combination of taselisib plus fulvestrant (7.4 mo), compared with placebo plus fulvestrant (5.4 mo) in a population of patients harboring PIK3CA mutations (HR 0.7; P = 0.0037). In addition, this small benefit came at the expense of significant toxicity, with 32% of patients experiencing serious adverse events. The concept of targeting PIK3CA remains critically important, but recent data highlight the need to develop more α-specific targeted agents to maximally exploit this combination. Additionally, resistance to these α-specific inhibitors through acquisition of mutations in PTEN have already been described (Juric et al. 2015), potentially further limiting the clinical utility of these drugs.
Mechanism of action of PI3K/AKT/mTOR targeting drugs
The finding that some of the PI3K/AKT/mTOR inhibitors described above show efficacy in ER+ breast cancer has increased focus on the underlying mechanism of action. The fact that the PI3K/AKT/mTOR pathway has been shown to regulate ER activity directly through phosphorylation as described above raises the question of whether PI3K/AKT/mTOR targeting drugs inhibit tumor growth by directly modulating ER activity. Unexpectedly, in preclinical models, the α-specific PI3K inhibitor alpelisib was found to redistribute chromatin binding of the ER complex, thereby increasing ER activity (Toska et al. 2017), in stark contrast to what would be predicted from inhibition of an activating PI3K pathway. In contrast to this finding, we showed recently that the two mTOR inhibitors everolimus (inhibitor of mTOR complex 1) and vistusertib (AZD2014; inhibitor of mTOR complexes 1 and 2) do not affect binding of ER to chromatin despite robust inhibition of the mTOR pathway. This indicates that these drugs likely function through an ER-independent mechanism to control tumor growth. Indeed, this is consistent with findings from the BOLERO-3 study (Andre et al. 2014) and subsequent BOLERO-1 study (Hurvitz et al. 2015), where benefit from everolimus, when combined with trastuzumab (HER2 monoclonal antibody) and chemotherapy in HER2-positive breast cancer patients, was, in fact, observed to be greater in ER-negative patients compared with ER+ patients. This questions the direct impact of mTOR inhibition on ER transcriptional activity in ER+ disease.
Taken together, this raises two important points. First, given that many of the drugs targeting the PI3K/AKT/mTOR pathway show some efficacy in ER+ breast cancer, it is critical to delineate the precise mechanisms through which these drugs inhibit tumor growth. An essential part of this is to clarify whether these inhibitors genuinely modulate ER activity or are general cell cycle regulatory compounds. This information is required for rational design of combinatorial strategies to treat ER+ breast cancer patients. Second, due to the generally modest clinical efficacy of assessed PI3K/AKT/mTOR targeting drugs, it is important to evaluate the role and clinical utility of PI3K/AKT/mTOR inhibition compared with newer treatment modalities, such as CDK4/6 inhibition, that are rapidly altering the treatment landscape for ER+ breast cancer. Currently, beside PIK3CA mutational status (Mayer et al. 2017; Baselga et al. 2018), no biomarkers are used in the clinic to stratify patients to receive inhibitors of the PI3K/AKT/mTOR pathway, and attempts to identify such biomarkers have had limited success (Chandarlapaty et al. 2016; Hortobagyi et al. 2016). A significant requirement for maximal exploitation of PI3K/AKT/mTOR inhibitors is therefore the identification of specific biomarkers that are predictive of response to these compounds.
Cytokine pathway inhibitors
The potential for targeting cytokine pathways therapeutically is highlighted by a wealth of preclinical evidence supporting the capacity for cytokine secretion to not only promote growth, proliferation, and metastatic potential but also alter ER activity and endocrine responsiveness.
For example, given that activation of the nuclear factor κB (NF-κB) pathway has been implicated in endocrine resistance and poor outcomes in ER+ breast cancer (Oida et al. 2014), the role of combining anti-NF-κB and endocrine therapy has been explored. Bortezomib, a proteasome inhibitor that blocks the NF-κB pathway, was added to either an AI or tamoxifen in a small single-arm phase II study to investigate whether endocrine responsiveness could be re-established in a cohort of relapsed patients with progressive and measurable disease (Trinh et al. 2012). While no objective responses were observed, a clinical benefit rate of 22% was reported; however, this came at the expense of significant gastrointestinal toxicities.
Accumulating evidence also suggests that inhibition of the receptor activator of NF-κB ligand (RANKL), which activates the NF-κB pathway, not only increases bone mass and strength but also has anti-tumor effects. Preclinically, RANKL inhibition decreases mammary carcinogenesis and reduces metastatic tumor burden in bone and other tissues (de Groot et al. 2018). Denosumab, an antibody targeting RANKL, is licensed to treat osteoporosis and prevent skeletal-related events in patients with breast cancer bone metastases (Gnant et al. 2015) and has been shown recently to improve disease-free survival in patients (Gnant et al. 2018). Conversely, however, in the recently reported D-Care study, adjuvant denosumab failed to reduce rates of breast cancer recurrence or death in higher-risk patients receiving optimal loco-regional and “standard of care” systemic adjuvant therapy (Coleman et al. 2018).
Overall, these early signs of activity indicate that exploration of inhibition of cytokine pathways in ER+ breast cancer patients warrants further clinical investigation.
Redirection of ER function by transcription factors downstream from hormone and cytokine signaling
It has been demonstrated in breast cancer patient samples that differential binding profiles for ER are associated with clinical outcome (Ross-Innes et al. 2012). It has been hypothesized that the differential ER genomic binding patterns dictate activation of distinct target genes that are associated with treatment response. This highlights the functional importance of ER–chromatin interactions for tumor progression and the need for delineating the factors and pathways that influence ER binding to chromatin.
ER functions as part of a large transcriptional complex involving multiple transcription factors, including its pioneer factor, FOXA1 (Hurtado et al. 2011), as well as other cooperating factors; e.g., PBX1 (Magnani et al. 2011), the transcription factor AP-2γ (Tan et al. 2011), and GATA-binding protein 3 (GATA3) (Theodorou et al. 2013). Many of these modulate the activity of the ER pathway by directly affecting binding of ER to chromatin; e.g., by modulating the local accessibility of the chromatin, as has been suggested for FOXA1 (Hurtado et al. 2011) and PBX1 (Magnani et al. 2011, 2015).
The finding that multiple cytokines and hormones are associated with the outcome in ER+ breast cancer suggests that the downstream effectors of these stimuli may also modulate ER function. Indeed, it has been shown that a cocktail of cytokines and growth factors, including IL6, TNFα, IGF-1, and EGF, can redistribute binding of ER and its pioneer factor, FOXA1, to different sites even in the presence of estrogen (Ross-Innes et al. 2012). This suggests that the cytokine/hormone-induced redistribution of ER binding may contribute to altered transcriptional activity and therefore may play a functional role in tumor progression.
TNFα-mediated redistribution of ER binding through NF-κB
The role of TNFα in modulating ER binding to chromatin has been characterized recently in more detail at selected loci (Pradhan et al. 2012) and genome-wide (Franco et al. 2015). TNFα signaling drives phosphorylation and subsequent degradation of the repressor IκB, which releases the transcription factor complex NF-κB, which subsequently translocates to the nucleus and binds to target regions in chromatin (Hayden and Ghosh 2008). TNFα signaling has been shown to inhibit breast cancer cell proliferation induced by estradiol both in vitro and in vivo. Franco et al. (2015) showed that activation of NF-κB by TNFα redirects ER to a subset of NF-κB-binding sites, and this leads to increased production of noncoding enhancer RNA (eRNA) transcripts from these sites, which has been linked previously to enhancer activation by ER (Fig. 2A; Hah et al. 2013; Li et al. 2013; Lam et al. 2014). Importantly, this increase in enhancer activity is associated with activation of nearby protein-coding genes, which are associated with clinical outcome in breast cancer patients (Franco et al. 2015). This indicates that targeting of this pathway may have therapeutic potential. This mechanism of cooperativity between NF-κB and ER is consistent with detailed molecular analysis of the baculoviral inhibitor of apoptosis (IAP) repeat-containing 3 (BIRC3) target gene, showing that TNFα-induced binding of NF-κB to the BIRC3 promoter primes it for ER binding, which in turn potentiates TNFα-induced activation of BIRC3 (Pradhan et al. 2012). Taken together, this illustrates how signaling molecules from the tumor microenvironment can ultimately activate new enhancers and target genes in the tumor cells by co-opting ER function.
Figure 2.
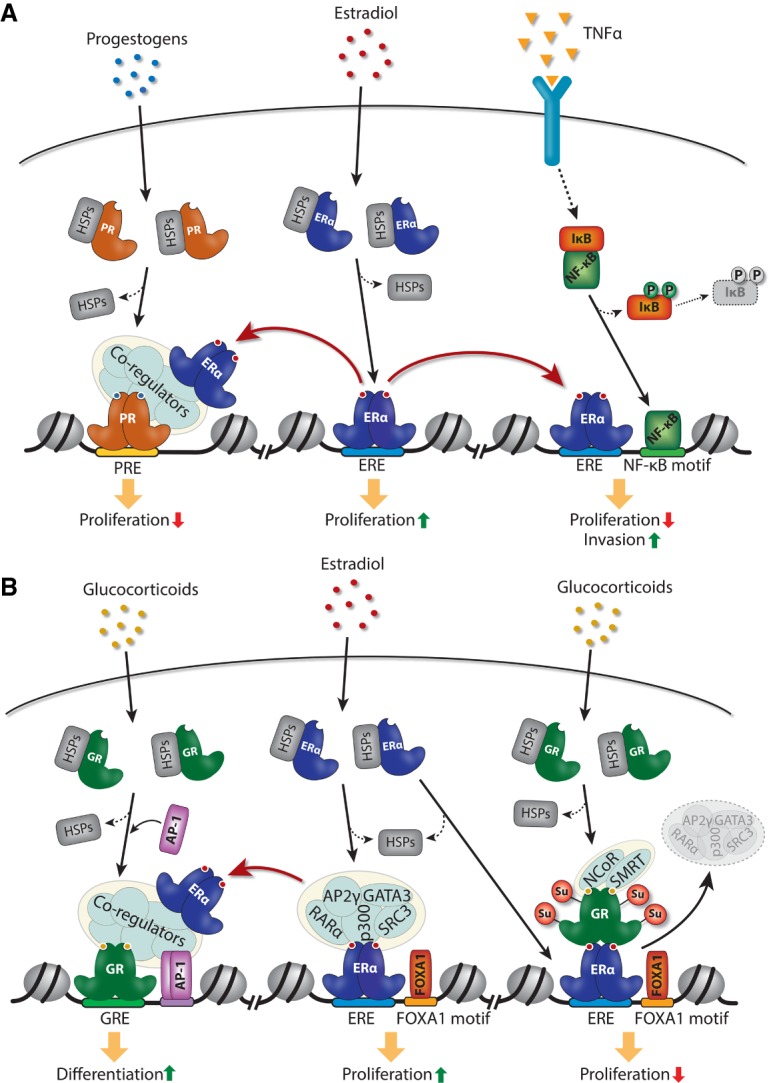
Redistribution of ER chromatin binding by other transcription factors. (A) Redistribution of ER binding to chromatin by progestogens and the cytokine TNFα through activation of progesterone receptor (PR) and NF-κB, respectively. (B) Modulation of ER binding and activity by glucocorticoids through activation of glucocorticoid receptor (GR).
Although TNFα can activate ER by phosphorylation in the absence of ER ligand, redistribution of ER binding to new enhancers by TNFα has been demonstrated only in the presence of estrogen-liganded ER (Franco et al. 2015; Stender et al. 2017). It is currently unclear whether NF-κB can redistribute tamoxifen-bound ER and whether ER will occupy these new NF-κB-primed ER-binding sites in response to TNFα in the absence of estrogens (e.g., in breast cancer patients successfully treated with AIs). However, some patients relapsing on AIs will have a functional ER pathway, such as those with the Y537 and D538 mutations in the ligand-binding domain of ER, which renders the receptor constitutively active (Robinson et al. 2013). It will be important to determine whether binding of mutant ER is also redistributed by TNFα, which would indicate that TNFα might also regulate ER function in the metastatic setting.
Cross-talk between ER and other steroid hormone receptors
Progesterone receptor (PR)
Although ER is the driving transcription factor in ER+ breast cancer, other steroid receptors have been shown to affect tumor progression by modulating ER function—most notably the PR, which is expressed in ∼75% of ER+ breast cancer. PGR is a classical ER target gene (Horwitz and McGuire 1978), and PR expression therefore has been used historically as a biomarker of an active ER pathway, which is predictive of patient outcome (Blows et al. 2010; Purdie et al. 2014). However, it is becoming increasingly clear that PR plays a more direct functional role in controlling progression of established tumors. Progestogens (i.e., natural and synthetic compounds that activate PR) have been shown in multiple studies to inhibit breast tumor growth in both preclinical models and patients (for review, see Carroll et al. 2017). More recently, our laboratory described the mechanism underlying the inhibitory effect of progestogens on tumor growth. This involves PR-directed redistribution of ER to PR-binding sites through an ER response element (ERE)-independent mechanism involving tethering of ER to chromatin through PR (Fig. 2A; Mohammed et al. 2015). This effectively sequesters ER away from its proproliferative gene targets, thereby inhibiting growth. This indicates that redirecting ER binding around the genome by progestogens may provide a mechanism to inhibit ER function in breast cancer (Carroll et al. 2017), which may be effective both in combination with SERMs, as suggested by preclinical xenograft experiments (Mohammed et al. 2015), and in tamoxifen-resistant cancer cells (Vignon et al. 1983).
Clinical benefit has been demonstrated from a single injection of progesterone administered before surgery (Badwe et al. 2011), and the use of a single agent, progestogen, has been consistently shown to clinically benefit patients as either first line therapy in de novo metastatic ER-positive breast cancer or in advanced disease when ER targeted endocrine agents have failed (Pannuti et al. 1979; Alexieva-Figusch et al. 1980; Izuo et al. 1981; Morgan 1985; Muss et al. 1988; Jonat et al. 1996; Buzdar et al. 1997, 2001; Abrams et al. 1999;). Notably, the progestin megestrol acetate was found to be efficacious in patients with ER-positive metastatic breast cancer after AI treatment failure (Bines et al. 2014). In this single-arm phase II trial, 48 post-menopausal women were administered 160 mg of megestrol acetate daily (a well-tolerated treatment yielding a clinical benefit rate of 40%), with a median treatment duration of 10 mo. Based on preclinical evidence, we speculate that this response to megesterol acetate results from PR activation and sequestration of ER binding and activity.
Most recently, the preclinical findings of functional cross-talk between ER and PR from our laboratory have now been translated into the clinic, driven by the hypothesis that the addition of a progesterone agonist will enhance the anti-proliferative effect of standard anti-estrogen therapy. This is based on the paradigm that PR activation will influence ER binding and change the target genes of this transcription factor (Fig. 2A). A phase II preoperative window study (NCT03306472) investigating the effect of combining megestrol acetate and an AI in post-menopausal women with early breast cancer is currently ongoing at our institute, while another ongoing study is evaluating this combination in a cohort of patients with advanced ER+ breast cancer (NCT03024580). In addition, two other studies in the UK and Australia using progestins in combination with ER targeting agents are scheduled to open soon.
It is important to acknowledge that, in contrast to the beneficial role of progestogens in breast cancer patients, certain specific progestogens have been shown to increase breast cancer incidence in healthy women (Hankinson et al. 2004; Chlebowski et al. 2013; Asi et al. 2016), although, importantly, this is not associated with long-term increased risk of all-cause mortality (Manson et al. 2017). Accordingly, the mechanism underlying the role of the PR axis in driving breast cancer risk in the mammary glands has been explored extensively (Graham et al. 2009; Lydon and Edwards 2009; Hilton et al. 2015). Taken together, while PR targeting agents in breast cancer patients remain a viable therapeutic approach (Carroll et al. 2017), the precise mechanisms through which PR regulates tumor compared with mammary gland proliferation warrants further exploration.
Androgen receptor (AR)
In addition to PR, functional cross-talk between ER and the AR has also been described in breast cancer cells. ER and AR are coexpressed in 80%–90% of ER+ breast cancer cells, and high AR expression is associated with good outcome in ER+, but not ER−, breast cancers (Peters et al. 2009). However, the role of AR in ER+ breast cancer is controversial, since both AR agonists and antagonists have been suggested to inhibit growth of breast cancer cells in preclinical models, at least partly by inhibiting ER function (Panet-Raymond et al. 2000; Greeve et al. 2004; Peters et al. 2009; Cochrane et al. 2014; D'Amato et al. 2016). This inhibitory effect of AR is mediated to a certain extent through binding of ER and AR to the same genomic regions (Need et al. 2012; D'Amato et al. 2016). Although this clearly implicates AR as an important steroid receptor in ER+ breast cancer, further work is needed to fully understand the mechanism through which this receptor works in breast cancer cells.
Clinically, the high level of coexpression of AR and PR in ER+ breast cancer makes these sex steroid receptors attractive targets for therapeutic intervention. Despite the historic demonstration of therapeutic benefits seen with androgen treatment in breast cancer (Tormey et al. 1983; Ingle et al. 1991), the use of androgens diminished due to the virilizing adverse effects of this class of agents, concerns regarding aromatization of androgens to estrogen, and the emergence of tamoxifen and AIs as effective therapies in ER+ breast cancer. However, given the resurgence of preclinical data supporting a role of AR in ER+ breast cancer described above, a focus on targeting of the AR signaling axis in these tumors has re-emerged, with both opposing agonistic and antagonistic strategies. Preclinical data suggesting a proproliferative role of AR in ER+ breast cancer (Cochrane et al. 2014; D'Amato et al. 2016), together with the growing evidence supporting efficacy of AR inhibition in AR+ triple-negative breast cancer (Bonnefoi et al. 2016; Traina et al. 2018), have lead to ongoing combination trials using the selective AR inhibitor bicalutamide (NCT02910050) and the anti-androgen enzalutamide (NCT02953860). However, AR inhibition strategies are likely to be limited to contexts in which AR is the driving nuclear receptor, as seen in the AR+ triple-negative breast cancer subtype, and the bulk of the evidence in ER+ breast cancer supports an anti-proliferative role of androgens. In this regard, great promise surrounds a new breed of selective AR modulators, such as enobosarm (GTx-024). Enobosarm is a potent AR agonist with reduced capacity for androgenization and without estrogenic properties (Coss et al. 2014). A small proof-of-concept study confirmed activity and tolerability in a cohort of heavily pretreated ER/AR+ patients with metastatic breast cancer (Overmoyer et al. 2014), and results from a follow-up phase II trial are awaited (NCT02463032). These recent findings support a role for AR agonists as anti-proliferative agents in ER+ breast cancer contexts.
Glucocorticoid receptor (GR)
GR is expressed in nearly 70% of ER+ breast cancers, and high GR expression is associated with low tumor grade (Abduljabbar et al. 2015) and, in some cases, has been associated with good clinical outcome (Pan et al. 2011; West et al. 2016). Mechanistic studies in mammary epithelial and breast cancer cell lines have convincingly demonstrated that both receptors can modulate chromatin binding of each other upon cotreatment with estradiol and the synthetic glucocorticoid dexamethasone (Fig. 2B; Miranda et al. 2013; West et al. 2016). This redistribution of receptor binding to chromatin occurs through an assisted loading mechanism involving chromatin remodeling by one receptor followed by recruitment of the other receptor (Miranda et al. 2013). Interestingly, GR-mediated redistribution of ER binding is not dependent on ER DNA-binding capacity. Instead, tethering of ER to chromatin at these sites is dependent on the transcription factor AP-1, which is recruited upon chromatin remodeling by GR (Fig. 2B; Miranda et al. 2013). Similarly, GR gets recruited indirectly to ER-binding sites upon costimulation with the ligands for these receptors (Fig. 2B). This occurs through a tethering mechanism likely involving a direct interaction between ER bound to its ERE and the DNA-binding domain of GR, although the DNA-binding capacity of GR is not required for GR recruitment to these sites (Karmakar et al. 2013; Miranda et al. 2013; West et al. 2016; Yang et al. 2017). This indirect recruitment of GR through ER is dependent on SUMOylation of GR, and, although it does not affect binding of ER and its pioneer factor, FOXA1, to these sites, it disrupts binding of other well-described ER cooperating factors such as AP2γ, GATA3, retinoic acid receptor α (RARα), p300, and steroid receptor coactivator-3 (SRC3) (Karmakar et al. 2013; Yang et al. 2017). Furthermore, GR recruits nuclear receptor corepressor (NCoR) and silencing mediator of retinoid and thyroid hormone (SMRT) receptor corepressor complexes, and this further alters the balance of coactivators and corepressors at these DNA regulatory sites (Fig. 2B). This leads to a repressive chromatin environment, resulting in less localized enhancer transcription from these sites (Yang et al. 2017).
Importantly, the transcriptional response to either dexamethasone or estradiol alone in MCF7 breast cancer cells is changed when both steroid hormones are added together, resulting in transcriptional changes in genes linked to cell proliferation and differentiation (West et al. 2016). Interestingly, some of the genes induced by costimulation of ER and GR were shown to inhibit cell proliferation of MCF7 cells. Furthermore, GR binding to ER enhancers represses a subset of ER target genes associated with poor outcome in breast cancer patients (Yang et al. 2017). This indicates that redistribution of ER and GR binding upon costimulation with glucocorticoids and estradiol can change the transcriptome to favor a less proliferative phenotype. Taken together, this demonstrates extensive genomic cross-talk between GR and ER, which is likely to drive the anti-proliferative effect of GR and glucocorticoids on ER+ tumor growth.
In addition to the direct cross-talk between ER and GR described above, GR activation can also affect tumor growth by modulating the metabolism of estradiol (Gong et al. 2008). GR activated by dexamethasone induces expression of the sulfotransferase SULT1E1 in both mouse livers and MCF7 breast cancer cells. SULT1E1 sulfonates and thereby inactivates estradiol, which results in decreased circulating estradiol levels and, consequently, inhibition of tumor growth in a mouse xenograft model (Gong et al. 2008).
Clinically, glucocorticoids are used ubiquitously in breast cancer patients in conjunction with chemotherapy to mitigate allergic reactions and for their anti-emetic and anti-inflammatory properties. While high tumor GR expression has been associated with a relatively poor outcome in ER-negative breast cancer, meta-analysis of genomic data sets has revealed that tumor GR mRNA expression is associated with improved recurrence-free survival in ER+ breast cancer, independent of PR expression (Pan et al. 2011). Despite the preclinical finding of GR activation reducing estrogen-induced proliferation in ER+ tumors, clinical studies have demonstrated varied effects of glucocorticoid use on breast cancer patient survival, with modest effects when used as a single agent and with no effect in combination with other drugs, including anti-estrogens in ER+ breast cancer patients (Keith 2008; Lietzen et al. 2014). However, given the in vitro evidence for an inhibitory effect of glucocorticoids on ER function and breast cancer cell proliferation (Fig. 2B) together with the proven role of glucocorticoids in AR-driven prostate cancer (Kach et al. 2015) and the widespread use of glucocorticoids in the supportive care of breast cancer patients, we feel that investigation of the full therapeutic potential of glucorticoids still needs to be explored.
Discussion
Seventy-five percent of breast cancers are driven by the steroid receptor ER, which is regulated by estrogenic hormones. Here, we discussed the link between the function of ER in tumor cells and signaling pathways triggered by other steroid hormones, cytokines, and growth factors, many of which originate from the many different cell types surrounding the cancer cells in the tumor microenvironment. It is clear that in addition to activation by classic estrogenic ligands, there are at least two levels of regulation of ER. First, chromatin binding and activity of ER can be regulated by phosphorylation in the absence of estrogens. Particularly, phosphorylation of S118 and S305 appears to be important for this estrogen-independent activation of ER, and several growth factor (e.g., EGF) and cytokine (e.g., TNFα) pathways can induce phosphorylation of these sites on ER. In addition, once ER is on chromatin, other transcription factors can redirect the genomic binding of ER, essentially reprogramming the transcriptional activity of ER to other target genes. The transcription factors that function downstream from cytokine signaling (e.g., NF-κB) and other steroid hormone receptors (e.g., PR) play important roles in regulating where in the genome ER binds.
The fact that multiple pathways triggered by signals from the tumor microenvironment impact ER function and have been linked to endocrine resistance clearly highlights the importance of paracrine signaling for tumor progression. This emphasizes the need to further explore the clinical potential of targeting tumor-extrinsic factors in the microenvironment in ER+ breast cancer. However, most of the mechanisms described above are based on in vitro experiments, and validation of these mechanisms in disease-relevant preclinical models (e.g., mouse xenograft models and patient-derived xenografts) and patient samples is critical in order to determine whether these pathways may provide clinical benefit to patients. In this regard, it is interesting to note the recent use of intraductal injections of human cancer cells into the mouse mammary gland to establish xenograft models from cell lines and patient samples (Sflomos et al. 2016). This method more accurately recapitulates the tumor progression observed in patients, with the development of metastases in organs such as the liver, lungs, brain, and bone. This is likely to increase the translatability of preclinical findings in the future. In addition to xenograft models, tumor explant methods that allow investigations of drug responses in patient samples (Centenera et al. 2012, 2013) are a powerful way to investigate the potential therapeutic impact of preclinical findings, as demonstrated for PR agonists (Mohammed et al. 2015). Validating mechanistic findings in preclinical models such as these is crucial to ensure translatability of the wealth of mechanistic in vitro findings.
It is particularly interesting to note the high degree of cross-talk between the different steroid receptors on chromatin. This group of transcription factors is highly targetable by small molecules, and numerous agonists and antagonists for these receptors are already Food and Drug Administration-approved, thereby significantly shortening the time from preclinical discoveries to clinical testing. In support of this is the rapid translation of findings linking PR agonists to ER function, with two clinical trials initiated (NCT03306472 and NCT03024580) and two more in development within 2 yr of publication of the biological discovery.
In conclusion, ER function and therefore breast cancer progression are directly modulated by both tumor-intrinsic and tumor-extrinsic factors, and many of the latter are promising candidates for targeted therapy aimed at improving survival for ER+ breast cancer patients.
Acknowledgments
We thank Sankari Nagarajan and Soleilmane Omarjee for critical reading of the manuscript. R.S. is funded by the Novo Nordisk Foundation (NNF15OC0014136). S.K. is funded by Cancer Research UK. J.S.C. is funded by Cancer Research UK, an ERC Consolidator Award, and a Komen Scholarship.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.316646.118.
References
- Abduljabbar R, Negm OH, Lai CF, Jerjees DA, Al-Kaabi M, Hamed MR, Tighe PJ, Buluwela L, Mukherjee A, Green AR, et al. 2015. Clinical and biological significance of glucocorticoid receptor (GR) expression in breast cancer. Breast Cancer Res Treat 150: 335–346. [DOI] [PubMed] [Google Scholar]
- Abrams J, Aisner J, Cirrincione C, Berry DA, Muss HB, Cooper MR, Henderson IC, Panasci L, Kirshner J, Ellerton J, et al. 1999. Dose-response trial of megestrol acetate in advanced breast cancer: cancer and leukemia group B phase III study 8741. J Clin Oncol 17: 64–73. [DOI] [PubMed] [Google Scholar]
- Alexieva-Figusch J, van Gilse HA, Hop WC, Phoa CH, Blonk-van der Wijst J, Treurniet RE. 1980. Progestin therapy in advanced breast cancer: megestrol acetate—an evaluation of 160 treated cases. Cancer 46: 2369–2372. [DOI] [PubMed] [Google Scholar]
- Altomare DA, Testa JR. 2005. Perturbations of the AKT signaling pathway in human cancer. Oncogene 24: 7455–7464. [DOI] [PubMed] [Google Scholar]
- Anbalagan M, Rowan BG. 2015. Estrogen receptor α phosphorylation and its functional impact in human breast cancer. Mol Cell Endocrinol 418: 264–272. [DOI] [PubMed] [Google Scholar]
- Andre F, O'Regan R, Ozguroglu M, Toi M, Xu B, Jerusalem G, Masuda N, Wilks S, Arena F, Isaacs C, et al. 2014. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol 15: 580–591. [DOI] [PubMed] [Google Scholar]
- Artacho-Cordon A, Artacho-Cordon F, Rios-Arrabal S, Calvente I, Nunez MI. 2012. Tumor microenvironment and breast cancer progression: a complex scenario. Cancer Biol Ther 13: 14–24. [DOI] [PubMed] [Google Scholar]
- Asi N, Mohammed K, Haydour Q, Gionfriddo MR, Vargas OL, Prokop LJ, Faubion SS, Murad MH. 2016. Progesterone vs. synthetic progestins and the risk of breast cancer: a systematic review and meta-analysis. Syst Rev 5: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badwe R, Hawaldar R, Parmar V, Nadkarni M, Shet T, Desai S, Gupta S, Jalali R, Vanmali V, Dikshit R, et al. 2011. Single-injection depot progesterone before surgery and survival in women with operable breast cancer: a randomized controlled trial. J Clin Oncol 29: 2845–2851. [DOI] [PubMed] [Google Scholar]
- Band AM, Laiho M. 2011. Crosstalk of TGF-β and estrogen receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia 16: 109–115. [DOI] [PubMed] [Google Scholar]
- Banerji U, Dean EJ, Perez-Fidalgo JA, Batist G, Bedard PL, You B, Westin SN, Kabos P, Garrett MD, Tall M, et al. 2018. A phase I open-label study to identify a dosing regimen of the pan-AKT inhibitor AZD5363 for evaluation in solid tumors and in PIK3CA-mutated breast and gynecologic cancers. Clin Cancer Res 24: 2050–2059. [DOI] [PubMed] [Google Scholar]
- Baselga J, Im SA, Iwata H, Cortes J, De Laurentiis M, Jiang Z, Arteaga CL, Jonat W, Clemons M, Ito Y, et al. 2017. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 18: 904–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Dent SF, Cortés J, Im Y-H, Diéras V, Harbeck N, Krop IE, Verma S, Wilson TR, Jin H, et al. 2018. Phase III study of taselisib (GDC-0032) + fulvestrant (FULV) v FULV in patients (pts) with estrogen receptor (ER)-positive, PIK3CA-mutant (MUT), locally advanced or metastatic breast cancer (MBC): primary analysis from SANDPIPER. J Clin Oncol 36: LBA1006. [Google Scholar]
- Becker MA, Ibrahim YH, Cui X, Lee AV, Yee D. 2011. The IGF pathway regulates ERα through a S6K1-dependent mechanism in breast cancer cells. Mol Endocrinol 25: 516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat-Nakshatri P, Wang G, Appaiah H, Luktuke N, Carroll JS, Geistlinger TR, Brown M, Badve S, Liu Y, Nakshatri H. 2008. AKT alters genome-wide estrogen receptor α binding and impacts estrogen signaling in breast cancer. Mol Cell Biol 28: 7487–7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bines J, Dienstmann R, Obadia RM, Branco LG, Quintella DC, Castro TM, Camacho PG, Soares FA, Costa ME. 2014. Activity of megestrol acetate in postmenopausal women with advanced breast cancer after nonsteroidal aromatase inhibitor failure: a phase II trial. Ann Oncol 25: 831–836. [DOI] [PubMed] [Google Scholar]
- Blows FM, Driver KE, Schmidt MK, Broeks A, van Leeuwen FE, Wesseling J, Cheang MC, Gelmon K, Nielsen TO, Blomqvist C, et al. 2010. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: a collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med 7: e1000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefoi H, Grellety T, Tredan O, Saghatchian M, Dalenc F, Mailliez A, L'Haridon T, Cottu P, Abadie-Lacourtoisie S, You B, et al. 2016. A phase II trial of abiraterone acetate plus prednisone in patients with triple-negative androgen receptor positive locally advanced or metastatic breast cancer (UCBG 12-1). Ann Oncol 27: 812–818. [DOI] [PubMed] [Google Scholar]
- Boulay A, Rudloff J, Ye J, Zumstein-Mecker S, O'Reilly T, Evans DB, Chen S, Lane HA. 2005. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin Cancer Res 11: 5319–5328. [DOI] [PubMed] [Google Scholar]
- Brechbuhl HM, Finlay-Schultz J, Yamamoto TM, Gillen AE, Cittelly DM, Tan AC, Sams SB, Pillai MM, Elias AD, Robinson WA, et al. 2017. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin Cancer Res 23: 1710–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunone G, Briand PA, Miksicek RJ, Picard D. 1996. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J 15: 2174–2183. [PMC free article] [PubMed] [Google Scholar]
- Burstein HJ, Cirrincione CT, Barry WT, Chew HK, Tolaney SM, Lake DE, Ma C, Blackwell KL, Winer EP, Hudis CA. 2014. Endocrine therapy with or without inhibition of epidermal growth factor receptor and human epidermal growth factor receptor 2: a randomized, double-blind, placebo-controlled phase III trial of fulvestrant with or without lapatinib for postmenopausal women with hormone receptor-positive advanced breast cancer—CALGB 40302 (Alliance). J Clin Oncol 32: 3959–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzdar AU, Jones SE, Vogel CL, Wolter J, Plourde P, Webster A. 1997. A phase III trial comparing anastrozole (1 and 10 milligrams), a potent and selective aromatase inhibitor, with megestrol acetate in postmenopausal women with advanced breast carcinoma. Arimidex Study Group. Cancer 79: 730–739. [PubMed] [Google Scholar]
- Buzdar A, Douma J, Davidson N, Elledge R, Morgan M, Smith R, Porter L, Nabholtz J, Xiang X, Brady C. 2001. Phase III, multicenter, double-blind, randomized study of letrozole, an aromatase inhibitor, for advanced breast cancer versus megestrol acetate. J Clin Oncol 19: 3357–3366. [DOI] [PubMed] [Google Scholar]
- Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. 2001. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor α: a new model for anti-estrogen resistance. J Biol Chem 276: 9817–9824. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network. 2012. Comprehensive molecular portraits of human breast tumours. Nature 490: 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson RW, O'Neill A, Vidaurre T, Gomez HL, Badve SS, Sledge GW. 2012. A randomized trial of combination anastrozole plus gefitinib and of combination fulvestrant plus gefitinib in the treatment of postmenopausal women with hormone receptor positive metastatic breast cancer. Breast Cancer Res Treat 133: 1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JS, Hickey TE, Tarulli GA, Williams M, Tilley WD. 2017. Deciphering the divergent roles of progestogens in breast cancer. Nat Rev Cancer 17: 54–64. [DOI] [PubMed] [Google Scholar]
- Cavazzoni A, Bonelli MA, Fumarola C, La Monica S, Airoud K, Bertoni R, Alfieri RR, Galetti M, Tramonti S, Galvani E, et al. 2012. Overcoming acquired resistance to letrozole by targeting the PI3K/AKT/mTOR pathway in breast cancer cell clones. Cancer Lett 323: 77–87. [DOI] [PubMed] [Google Scholar]
- Centenera MM, Gillis JL, Hanson AR, Jindal S, Taylor RA, Risbridger GP, Sutherland PD, Scher HI, Raj GV, Knudsen KE, et al. 2012. Evidence for efficacy of new Hsp90 inhibitors revealed by ex vivo culture of human prostate tumors. Clin Cancer Res 18: 3562–3570. [DOI] [PubMed] [Google Scholar]
- Centenera MM, Raj GV, Knudsen KE, Tilley WD, Butler LM. 2013. Ex vivo culture of human prostate tissue and drug development. Nat Rev Urol 10: 483–487. [DOI] [PubMed] [Google Scholar]
- Chandarlapaty S, Chen D, He W, Sung P, Samoila A, You D, Bhatt T, Patel P, Voi M, Gnant M, et al. 2016. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol 2: 1310–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Riedl T, Washbrook E, Pace PE, Coombes RC, Egly JM, Ali S. 2000. Activation of estrogen receptor α by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol Cell 6: 127–137. [PubMed] [Google Scholar]
- Chen D, Washbrook E, Sarwar N, Bates GJ, Pace PE, Thirunuvakkarasu V, Taylor J, Epstein RJ, Fuller-Pace FV, Egly JM, et al. 2002. Phosphorylation of human estrogen receptor α at serine 118 by two distinct signal transduction pathways revealed by phosphorylation-specific antisera. Oncogene 21: 4921–4931. [DOI] [PubMed] [Google Scholar]
- Chlebowski RT, Manson JE, Anderson GL, Cauley JA, Aragaki AK, Stefanick ML, Lane DS, Johnson KC, Wactawski-Wende J, Chen C, et al. 2013. Estrogen plus progestin and breast cancer incidence and mortality in the Women's Health Initiative Observational Study. J Natl Cancer Inst 105: 526–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruelos Gil EM. 2014. Targeting the PI3K/AKT/mTOR pathway in estrogen receptor-positive breast cancer. Cancer Treat Rev 40: 862–871. [DOI] [PubMed] [Google Scholar]
- Cochrane DR, Bernales S, Jacobsen BM, Cittelly DM, Howe EN, D'Amato NC, Spoelstra NS, Edgerton SM, Jean A, Guerrero J, et al. 2014. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res 16: R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RE, Finkelstein D, Barrios CH, Martin M, Iwata H, Glaspy JA, Zhou Y, Jandial D, Chan A. 2018. Adjuvant denosumab in early breast cancer: first results from the international multicenter randomized phase III placebo controlled D-CARE study. J Clin Oncol 36: 501–501. [Google Scholar]
- Coss CC, Jones A, Dalton JT. 2014. Selective androgen receptor modulators as improved androgen therapy for advanced breast cancer. Steroids 90: 94–100. [DOI] [PubMed] [Google Scholar]
- Cristofanilli M, Valero V, Mangalik A, Royce M, Rabinowitz I, Arena FP, Kroener JF, Curcio E, Watkins C, Bacus S, et al. 2010. Phase II, randomized trial to compare anastrozole combined with gefitinib or placebo in postmenopausal women with hormone receptor-positive metastatic breast cancer. Clin Cancer Res 16: 1904–1914. [DOI] [PubMed] [Google Scholar]
- Crowder RJ, Phommaly C, Tao Y, Hoog J, Luo J, Perou CM, Parker JS, Miller MA, Huntsman DG, Lin L, et al. 2009. PIK3CA and PIK3CB inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor-positive breast cancer. Cancer Res 69: 3955–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amato NC, Gordon MA, Babbs B, Spoelstra NS, Carson Butterfield KT, Torkko KC, Phan VT, Barton VN, Rogers TJ, Sartorius CA, et al. 2016. Cooperative dynamics of AR and ER activity in breast cancer. Mol Cancer Res 14: 1054–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckers M, van Dinther M, Buijs J, Que I, Lowik C, van der Pluijm G, ten Dijke P. 2006. The tumor suppressor Smad4 is required for transforming growth factor β-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res 66: 2202–2209. [DOI] [PubMed] [Google Scholar]
- de Groot AF, Appelman-Dijkstra NM, van der Burg SH, Kroep JR. 2018. The anti-tumor effect of RANKL inhibition in malignant solid tumors—a systematic review. Cancer Treat Rev 62: 18–28. [DOI] [PubMed] [Google Scholar]
- de Leeuw R, Flach K, Bentin Toaldo C, Alexi X, Canisius S, Neefjes J, Michalides R, Zwart W. 2013. PKA phosphorylation redirects ERα to promoters of a unique gene set to induce tamoxifen resistance. Oncogene 32: 3543–3551. [DOI] [PubMed] [Google Scholar]
- Dhakal A, Thomas RA, Levine EG, Brufsky A, Hanna MG, Miller A, Khoury T, Takabe K, Early AP, O'Connor T, et al. 2018. Outcome of everolimus based therapy in hormone receptor positive metastatic breast cancer patients after progression on palbociclib combination. J Clin Oncol 36: 1064–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). 2011. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 378: 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Shennawy L, Dubrovskyi O, Kastrati I, Danes JM, Zhang Y, Whiteley HE, Creighton CJ, Frasor J. 2018. Coactivation of estrogen receptor and IKKβ induces a dormant metastatic phenotype in ER-positive breast cancer. Cancer Res 78: 974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, et al. 2008. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 14: 518–527. [DOI] [PubMed] [Google Scholar]
- Franco HL, Nagari A, Kraus WL. 2015. TNFα signaling exposes latent estrogen receptor binding sites to alter the breast cancer cell transcriptome. Mol Cell 58: 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghayad SE, Vendrell JA, Ben Larbi S, Dumontet C, Bieche I, Cohen PA. 2010. Endocrine resistance associated with activated ErbB system in breast cancer cells is reversed by inhibiting MAPK or PI3K/Akt signaling pathways. Int J Cancer 126: 545–562. [DOI] [PubMed] [Google Scholar]
- Gnant M, Pfeiler G, Dubsky PC, Hubalek M, Greil R, Jakesz R, Wette V, Balic M, Haslbauer F, Melbinger E, et al. 2015. Adjuvant denosumab in breast cancer (ABCSG-18): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 386: 433–443. [DOI] [PubMed] [Google Scholar]
- Gnant M, Pfeiler G, Steger GG, Egle D, Greil R, Fitzal F, Wette V, Balic M, Haslbauer F, Melbinger-Zeinitzer E, et al. 2018. Adjuvant denosumab in early breast cancer: disease-free survival analysis of 3,425 postmenopausal patients in the ABCSG-18 trial. J Clin Oncol 36: 500–500. [Google Scholar]
- Gong H, Jarzynka MJ, Cole TJ, Lee JH, Wada T, Zhang B, Gao J, Song WC, DeFranco DB, Cheng SY, et al. 2008. Glucocorticoids antagonize estrogens by glucocorticoid receptor-mediated activation of estrogen sulfotransferase. Cancer Res 68: 7386–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JD, Mote PA, Salagame U, van Dijk JH, Balleine RL, Huschtscha LI, Reddel RR, Clarke CL. 2009. DNA replication licensing and progenitor numbers are increased by progesterone in normal human breast. Endocrinology 150: 3318–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greeve MA, Allan RK, Harvey JM, Bentel JM. 2004. Inhibition of MCF-7 breast cancer cell proliferation by 5α-dihydrotestosterone; a role for p21(Cip1/Waf1). J Mol Endocrinol 32: 793–810. [DOI] [PubMed] [Google Scholar]
- Guichard SM, Curwen J, Bihani T, Cruz CM, Yates JWT, Grondine M, Howard Z, Davies BR, Bigley G, Klinowska T, et al. 2015. AZD2014, an inhibitor of mTORC1 and mTORC2, is highly effective in ER+ breast cancer when administered using intermittent or continuous schedules. Mol Cancer Ther 14: 2508. [DOI] [PubMed] [Google Scholar]
- Hah N, Murakami S, Nagari A, Danko CG, Kraus WL. 2013. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res 23: 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankinson SE, Colditz GA, Willett WC. 2004. Towards an integrated model for breast cancer etiology: the lifelong interplay of genes, lifestyle, and hormones. Breast Cancer Res 6: 213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrod A, Fulton J, Nguyen VTM, Periyasamy M, Ramos-Garcia L, Lai CF, Metodieva G, de Giorgio A, Williams RL, Santos DB, et al. 2017. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 36: 2286–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. 2008. Shared principles in NF-kappaB signaling. Cell 132: 344–362. [DOI] [PubMed] [Google Scholar]
- Held JM, Britton DJ, Scott GK, Lee EL, Schilling B, Baldwin MA, Gibson BW, Benz CC. 2012. Ligand binding promotes CDK-dependent phosphorylation of ER-α on hinge serine 294 but inhibits ligand-independent phosphorylation of serine 305. Mol Cancer Res 10: 1120–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton HN, Graham JD, Clarke CL. 2015. Minireview: progesterone regulation of proliferation in the normal human breast and in breast cancer: a tale of two scenarios? Mol Endocrinol 29: 1230–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortobagyi GN, Chen D, Piccart M, Rugo HS, Burris HA III, Pritchard KI, Campone M, Noguchi S, Perez AT, Deleu I, et al. 2016. Correlative analysis of genetic alterations and everolimus benefit in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: results from BOLERO-2. J Clin Oncol 34: 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz KB, McGuire WL. 1978. Estrogen control of progesterone receptor in human breast cancer. Correlation with nuclear processing of estrogen receptor. J Biol Chem 253: 2223–2228. [PubMed] [Google Scholar]
- Huang J, Woods P, Normolle D, Goff JP, Benos PV, Stehle CJ, Steinman RA. 2017. Downregulation of estrogen receptor and modulation of growth of breast cancer cell lines mediated by paracrine stromal cell signals. Breast Cancer Res Treat 161: 229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. 2011. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet 43: 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurvitz SA, Andre F, Jiang Z, Shao Z, Mano MS, Neciosup SP, Tseng LM, Zhang Q, Shen K, Liu D, et al. 2015. Combination of everolimus with trastuzumab plus paclitaxel as first-line treatment for patients with HER2-positive advanced breast cancer (BOLERO-1): a phase 3, randomised, double-blind, multicentre trial. Lancet Oncol 16: 816–829. [DOI] [PubMed] [Google Scholar]
- Ingle JN, Twito DI, Schaid DJ, Cullinan SA, Krook JE, Mailliard JA, Tschetter LK, Long HJ, Gerstner JG, Windschitl HE, et al. 1991. Combination hormonal therapy with tamoxifen plus fluoxymesterone versus tamoxifen alone in postmenopausal women with metastatic breast cancer. An updated analysis. Cancer 67: 886–891. [DOI] [PubMed] [Google Scholar]
- Izuo M, Iino Y, Endo K. 1981. Oral high-dose medroxyprogesterone acetate (MAP) in treatment of advanced breast cancer. A preliminary report of clinical and experimental studies. Breast Cancer Res Treat 1: 125–130. [DOI] [PubMed] [Google Scholar]
- Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, Xiao T, Li W, Qiu X, Buchwalter G, et al. 2018. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell 33: 173–186 e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joel PB, Smith J, Sturgill TW, Fisher TL, Blenis J, Lannigan DA. 1998a. pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol Cell Biol 18: 1978–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joel PB, Traish AM, Lannigan DA. 1998b. Estradiol-induced phosphorylation of serine 118 in the estrogen receptor is independent of p42/p44 mitogen-activated protein kinase. J Biol Chem 273: 13317–13323. [DOI] [PubMed] [Google Scholar]
- Jonat W, Howell A, Blomqvist C, Eiermann W, Winblad G, Tyrrell C, Mauriac L, Roche H, Lundgren S, Hellmund R, et al. 1996. A randomised trial comparing two doses of the new selective aromatase inhibitor anastrozole (Arimidex) with megestrol acetate in postmenopausal patients with advanced breast cancer. Eur J Cancer 32A: 404–412. [DOI] [PubMed] [Google Scholar]
- Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, Ebbesen SH, Ainscough BJ, Ramu A, Iyer G, et al. 2015. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature 518: 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kach J, Conzen SD, Szmulewitz RZ. 2015. Targeting the glucocorticoid receptor in breast and prostate cancers. Sci Transl Med 7: 305ps319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar S, Jin Y, Nagaich AK. 2013. Interaction of glucocorticoid receptor (GR) with estrogen receptor (ER) α and activator protein 1 (AP1) in dexamethasone-mediated interference of ERα activity. J Biol Chem 288: 24020–24034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, et al. 1995. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 270: 1491–1494. [DOI] [PubMed] [Google Scholar]
- Keith BD. 2008. Systematic review of the clinical effect of glucocorticoids on nonhematologic malignancy. BMC Cancer 8: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkegaard T, Witton CJ, McGlynn LM, Tovey SM, Dunne B, Lyon A, Bartlett JM. 2005. AKT activation predicts outcome in breast cancer patients treated with tamoxifen. J Pathol 207: 139–146. [DOI] [PubMed] [Google Scholar]
- Kohler BA, Sherman RL, Howlader N, Jemal A, Ryerson AB, Henry KA, Boscoe FP, Cronin KA, Lake A, Noone AM, et al. 2015. Annual report to the nation on the status of cancer, 1975–2011, featuring incidence of breast cancer subtypes by race/ethnicity, poverty, and state. J Natl Cancer Inst 107: djv048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krop IE, Mayer IA, Ganju V, Dickler M, Johnston S, Morales S, Yardley DA, Melichar B, Forero-Torres A, Lee SC, et al. 2016. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 17: 811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam MT, Li W, Rosenfeld MG, Glass CK. 2014. Enhancer RNAs and regulated transcriptional programs. Trends Biochem Sci 39: 170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goff P, Montano MM, Schodin DJ, Katzenellenbogen BS. 1994. Phosphorylation of the human estrogen receptor. Identification of hormone-regulated sites and examination of their influence on transcriptional activity. J Biol Chem 269: 4458–4466. [PubMed] [Google Scholar]
- Le Romancer M, Poulard C, Cohen P, Sentis S, Renoir JM, Corbo L. 2011. Cracking the estrogen receptor's posttranslational code in breast tumors. Endocr Rev 32: 597–622. [DOI] [PubMed] [Google Scholar]
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. 2013. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498: 516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lietzen LW, Ahern T, Christiansen P, Jensen AB, Sorensen HT, Lash TL, Cronin-Fenton DP. 2014. Glucocorticoid prescriptions and breast cancer recurrence: a Danish nationwide prospective cohort study. Ann Oncol 25: 2419–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien M, Meyer CA, Bailey ST, Eeckhoute J, Cook J, Westerling T, Zhang X, Carroll JS, Rhodes DR, Liu XS, et al. 2010. Growth factor stimulation induces a distinct ERα cistrome underlying breast cancer endocrine resistance. Genes Dev 24: 2219–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydon JP, Edwards DP. 2009. Finally! A model for progesterone receptor action in normal human breast. Endocrinology 150: 2988–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CX, Sanchez C, Gao F, Crowder R, Naughton M, Pluard T, Creekmore A, Guo Z, Hoog J, Lockhart AC, et al. 2016. A phase I study of the AKT inhibitor MK-2206 in combination with hormonal therapy in postmenopausal women with estrogen receptor-positive metastatic breast cancer. Clin Cancer Res 22: 2650–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeddu C, Gramignano G, Floris C, Murenu G, Sollai G, Maccio A. 2014. Role of inflammation and oxidative stress in post-menopausal oestrogen-dependent breast cancer. J Cell Mol Med 18: 2519–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani L, Ballantyne EB, Zhang X, Lupien M. 2011. PBX1 genomic pioneer function drives ERα signaling underlying progression in breast cancer. PLoS Genet 7: e1002368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani L, Patten DK, Nguyen VT, Hong SP, Steel JH, Patel N, Lombardo Y, Faronato M, Gomes AR, Woodley L, et al. 2015. The pioneer factor PBX1 is a novel driver of metastatic progression in ERα-positive breast cancer. Oncotarget 6: 21878–21891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson JE, Aragaki AK, Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Chlebowski RT, Howard BV, Thomson CA, Margolis KL, et al. 2017. Menopausal hormone therapy and long-term all-cause and cause-specific mortality: the women's health initiative randomized trials. JAMA 318: 927–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauri D, Pavlidis N, Polyzos NP, Ioannidis JP. 2006. Survival with aromatase inhibitors and inactivators versus standard hormonal therapy in advanced breast cancer: meta-analysis. J Natl Cancer Inst 98: 1285–1291. [DOI] [PubMed] [Google Scholar]
- Mayer IA, Abramson VG, Formisano L, Balko JM, Estrada MV, Sanders ME, Juric D, Solit D, Berger MF, Won HH, et al. 2017. A phase Ib study of alpelisib (BYL719), a PI3Kα-specific inhibitor, with letrozole in ER+/HER2− metastatic breast cancer. Clin Cancer Res 23: 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, Yelensky R, Brown M, Miller VA, Sarid D, et al. 2013. D538G mutation in estrogen receptor-α: a novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res 73: 6856–6864. [DOI] [PubMed] [Google Scholar]
- Michalides R, Griekspoor A, Balkenende A, Verwoerd D, Janssen L, Jalink K, Floore A, Velds A, van't Veer L, Neefjes J. 2004. Tamoxifen resistance by a conformational arrest of the estrogen receptor α after PKA activation in breast cancer. Cancer Cell 5: 597–605. [DOI] [PubMed] [Google Scholar]
- Miller TW, Hennessy BT, Gonzalez-Angulo AM, Fox EM, Mills GB, Chen H, Higham C, Garcia-Echeverria C, Shyr Y, Arteaga CL. 2010. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest 120: 2406–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda TB, Voss TC, Sung MH, Baek S, John S, Hawkins M, Grontved L, Schiltz RL, Hager GL. 2013. Reprogramming the chromatin landscape: interplay of the estrogen and glucocorticoid receptors at the genomic level. Cancer Res 73: 5130–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed H, Russell IA, Stark R, Rueda OM, Hickey TE, Tarulli GA, Serandour AA, Birrell SN, Bruna A, Saadi A, et al. 2015. Progesterone receptor modulates ERα action in breast cancer. Nature 523: 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan LR. 1985. Megestrol acetate v tamoxifen in advanced breast cancer in postmenopausal patients. Semin Oncol 12: 43–47. [PubMed] [Google Scholar]
- Morgan MM, Livingston MK, Warrick JW, Stanek EM, Alarid ET, Beebe DJ, Johnson BP. 2018. Mammary fibroblasts reduce apoptosis and speed estrogen-induced hyperplasia in an organotypic MCF7-derived duct model. Sci Rep 8: 7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muss HB, Wells HB, Paschold EH, Black WR, Cooper MR, Capizzi RL, Christian R, Cruz JM, Jackson DV, Powell BL, et al. 1988. Megestrol acetate versus tamoxifen in advanced breast cancer: 5-year analysis—a phase III trial of the Piedmont Oncology Association. J Clin Oncol 6: 1098–1106. [DOI] [PubMed] [Google Scholar]
- Need EF, Selth LA, Harris TJ, Birrell SN, Tilley WD, Buchanan G. 2012. Research resource: interplay between the genomic and transcriptional networks of androgen receptor and estrogen receptor α in luminal breast cancer cells. Mol Endocrinol 26: 1941–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oida K, Matsuda A, Jung K, Xia Y, Jang H, Amagai Y, Ahn G, Nishikawa S, Ishizaka S, Jensen-Jarolim E, et al. 2014. Nuclear factor-kB plays a critical role in both intrinsic and acquired resistance against endocrine therapy in human breast cancer cells. Sci Rep 4: 4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne CK, Neven P, Dirix LY, Mackey JR, Robert J, Underhill C, Schiff R, Gutierrez C, Migliaccio I, Anagnostou VK, et al. 2011. Gefitinib or placebo in combination with tamoxifen in patients with hormone receptor-positive metastatic breast cancer: a randomized phase II study. Clin Cancer Res 17: 1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overmoyer B, Sanz-Altamira P, Taylor RP, Hancock ML, Dalton JT, Johnston M, Steiner MS. 2014. Enobosarm: a targeted therapy for metastatic, androgen receptor positive, breast cancer. J Clin Oncol 32: 568–568. [Google Scholar]
- Pan D, Kocherginsky M, Conzen SD. 2011. Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res 71: 6360–6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panet-Raymond V, Gottlieb B, Beitel LK, Pinsky L, Trifiro MA. 2000. Interactions between androgen and estrogen receptors and the effects on their transactivational properties. Mol Cell Endocrinol 167: 139–150. [DOI] [PubMed] [Google Scholar]
- Pannuti F, Martoni A, Di Marco AR, Piana E, Saccani F, Becchi G, Mattioli G, Barbanti F, Marra GA, Persiani W, et al. 1979. Prospective, randomized clinical trial of two different high dosages of medroxyprogesterone acetate (MAP) in the treatment of metastatic breast cancer. Eur J Cancer 15: 593–601. [DOI] [PubMed] [Google Scholar]
- Papageorgis P, Stylianopoulos T. 2015. Role of TGFβin regulation of the tumor microenvironment and drug delivery (review). Int J Oncol 46: 933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AA, Buchanan G, Ricciardelli C, Bianco-Miotto T, Centenera MM, Harris JM, Jindal S, Segara D, Jia L, Moore NL, et al. 2009. Androgen receptor inhibits estrogen receptor-α activity and is prognostic in breast cancer. Cancer Res 69: 6131–6140. [DOI] [PubMed] [Google Scholar]
- Piccart M, Hortobagyi GN, Campone M, Pritchard KI, Lebrun F, Ito Y, Noguchi S, Perez A, Rugo HS, Deleu I, et al. 2014. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2dagger. Ann Oncol 25: 2357–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan M, Baumgarten SC, Bembinster LA, Frasor J. 2012. CBP mediates NF-κB-dependent histone acetylation and estrogen receptor recruitment to an estrogen response element in the BIRC3 promoter. Mol Cell Biol 32: 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdie CA, Quinlan P, Jordan LB, Ashfield A, Ogston S, Dewar JA, Thompson AM. 2014. Progesterone receptor expression is an independent prognostic variable in early breast cancer: a population-based study. Br J Cancer 110: 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Wu L, Frost AR, Grizzle W, Cao X, Wan M. 2009. Dual effects of TGF-β on ERα-mediated estrogenic transcriptional activity in breast cancer. Mol Cancer 8: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, Kalyana-Sundaram S, Wang R, Ning Y, Hodges L, et al. 2013. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 45: 1446–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. 2012. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481: 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royce M, Bachelot T, Villanueva C, Ozguroglu M, Azevedo SJ, Cruz FM, Debled M, Hegg R, Toyama T, Falkson C, et al. 2018. Everolimus plus endocrine therapy for postmenopausal women with estrogen receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: a clinical trial. JAMA Oncol 4: 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez CG, Ma CX, Crowder RJ, Guintoli T, Phommaly C, Gao F, Lin L, Ellis MJ. 2011. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res 13: R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sflomos G, Dormoy V, Metsalu T, Jeitziner R, Battista L, Scabia V, Raffoul W, Delaloye JF, Treboux A, Fiche M, et al. 2016. A preclinical model for ERα-positive breast cancer points to the epithelial microenvironment as determinant of luminal phenotype and hormone response. Cancer Cell 29: 407–422. [DOI] [PubMed] [Google Scholar]
- Stender JD, Nwachukwu JC, Kastrati I, Kim Y, Strid T, Yakir M, Srinivasan S, Nowak J, Izard T, Rangarajan ES, et al. 2017. Structural and molecular mechanisms of cytokine-mediated endocrine resistance in human breast cancer cells. Mol Cell 65: 1122–1135.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SK, Lin ZH, Chang CW, Varang V, Chng KR, Pan YF, Yong EL, Sung WK, Cheung E. 2011. AP-2γ regulates oestrogen receptor-mediated long-range chromatin interaction and gene transcription. EMBO J 30: 2569–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodorou V, Stark R, Menon S, Carroll JS. 2013. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res 23: 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tormey DC, Lippman ME, Edwards BK, Cassidy JG. 1983. Evaluation of tamoxifen doses with and without fluoxymesterone in advanced breast cancer. Ann Intern Med 98: 139–144. [DOI] [PubMed] [Google Scholar]
- Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, Dickler MN, Scaltriti M, Leslie CS, Armstrong SA, et al. 2017. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 355: 1324–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, Mazzola AM, Smith A, Wilson J, Morrow C, Wong WL, et al. 2017. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov 7: 277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traina TA, Miller K, Yardley DA, Eakle J, Schwartzberg LS, O'Shaughnessy J, Gradishar W, Schmid P, Winer E, Kelly C, et al. 2018. Enzalutamide for the treatment of androgen receptor-expressing triple-negative breast cancer. J Clin Oncol 36: 884–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh XB, Sas L, Van Laere SJ, Prove A, Deleu I, Rasschaert M, Van de Velde H, Vinken P, Vermeulen PB, Van Dam PA, et al. 2012. A phase II study of the combination of endocrine treatment and bortezomib in patients with endocrine-resistant metastatic breast cancer. Oncol Rep 27: 657–663. [DOI] [PubMed] [Google Scholar]
- Vignon F, Bardon S, Chalbos D, Rochefort H. 1983. Antiestrogenic effect of R5020, a synthetic progestin in human breast cancer cells in culture. J Clin Endocrinol Metab 56: 1124–1130. [DOI] [PubMed] [Google Scholar]
- Vilquin P, Villedieu M, Grisard E, Ben Larbi S, Ghayad SE, Heudel PE, Bachelot T, Corbo L, Treilleux I, Vendrell JA, et al. 2013. Molecular characterization of anastrozole resistance in breast cancer: pivotal role of the Akt/mTOR pathway in the emergence of de novo or acquired resistance and importance of combining the allosteric Akt inhibitor MK-2206 with an aromatase inhibitor. Int J Cancer 133: 1589–1602. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. 2002. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2: 489–501. [DOI] [PubMed] [Google Scholar]
- Wang RA, Mazumdar A, Vadlamudi RK, Kumar R. 2002. P21-activated kinase-1 phosphorylates and transactivates estrogen receptor-α and promotes hyperplasia in mammary epithelium. EMBO J 21: 5437–5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West DC, Pan D, Tonsing-Carter EY, Hernandez KM, Pierce CF, Styke SC, Bowie KR, Garcia TI, Kocherginsky M, Conzen SD. 2016. GR and ER coactivation alters the expression of differentiation genes and associates with improved ER+ breast cancer outcome. Mol Cancer Res 14: 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Wu Y, Gathings B, Wan M, Li X, Grizzle W, Liu Z, Lu C, Mao Z, Cao X. 2003. Smad4 as a transcription corepressor for estrogen receptor α. J Biol Chem 278: 15192–15200. [DOI] [PubMed] [Google Scholar]
- Yamnik RL, Holz MK. 2010. mTOR/S6K1 and MAPK/RSK signaling pathways coordinately regulate estrogen receptor α serine 167 phosphorylation. FEBS Lett 584: 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamnik RL, Digilova A, Davis DC, Brodt ZN, Murphy CJ, Holz MK. 2009. S6 kinase 1 regulates estrogen receptor α in control of breast cancer cell proliferation. J Biol Chem 284: 6361–6369. [DOI] [PubMed] [Google Scholar]
- Yang F, Ma Q, Liu Z, Li W, Tan Y, Jin C, Ma W, Hu Y, Shen J, Ohgi KA, et al. 2017. Glucocorticoid Receptor:MegaTrans switching mediates the repression of an ERα-regulated transcriptional program. Mol Cell 66: 321–331.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardley DA, Noguchi S, Pritchard KI, Burris HA, Baselga J, Gnant M, Hortobagyi GN, Campone M, Pistilli B, Piccart M, et al. 2013. Everolimus plus exemestane in postmenopausal patients with HR+ breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther 30: 870–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, Alexandrov LB, Van Loo P, Haugland HK, Lilleng PK, et al. 2017. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell 32: 169–184.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]