

D'Amico et al. show that loss of STAT3 preferentially associates with the acquisition of mesenchymal-like phenotypes and more aggressive tumor behavior.
Keywords: epithelial carcinogenesis, inflammation, context specificity, metastasis
Abstract
A dichotomy exists regarding the role of signal transducer and activator of transcription 3 (STAT3) in cancer. Functional and genetic studies demonstrate either an intrinsic requirement for STAT3 or a suppressive effect on common types of cancer. These contrasting actions of STAT3 imply context dependency. To examine mechanisms that underlie STAT3 function in cancer, we evaluated the impact of STAT3 activity in KRAS-driven lung and pancreatic cancer. Our study defines a fundamental and previously unrecognized function of STAT3 in the maintenance of epithelial cell identity and differentiation. Loss of STAT3 preferentially associates with the acquisition of mesenchymal-like phenotypes and more aggressive tumor behavior. In contrast, persistent STAT3 activation through Tyr705 phosphorylation confers a differentiated epithelial morphology that impacts tumorigenic potential. Our results imply a mechanism in which quantitative differences of STAT3 Tyr705 phosphorylation, as compared with other activation modes, direct discrete outcomes in tumor progression.
Signal transducer and activator of transcription 3 (STAT3) is a member of the STAT family of latent transcription factors, which play pivotal roles in relaying extracellular signals from cell surface receptors to the nucleus. In response to cytokines and growth factors, STATs are phosphorylated by Janus kinases (JAKs) or cognate receptors and form dimers able to bind DNA and activate expression of genes involved in differentiation, development, proliferation, and apoptosis (Schindler et al. 2007; Stark and Darnell 2012). STAT3 function has been characterized following phosphorylation of two different residues: Tyr705 (Y705) and Ser727 (S727). Of these, Y705 phosphorylation is generally regarded as the dominant actuator of cytokine signaling through the JAK/STAT3 pathway, while the role of S727 phosphorylation is still being ascertained (Shen et al. 2004; Androutsellis-Theotokis et al. 2006; Qin et al. 2008; Gough et al. 2009). The activation of STAT3 by tyrosine phosphorylation has been observed in various human malignancies (Yu et al. 2014). It has been hypothesized that STAT3 represents a driving force behind tumor progression; however, a broad consensus regarding the exact role of STAT3 in epithelial carcinogenesis has not been achieved.
Murine cancer models with tissue-specific deletions of STAT3 have provided contrasting results regarding the requirement of STAT3 in tumor development. Studies have demonstrated a requirement for STAT3 in the early development of skin, colon, bladder, liver, stomach, and pancreatic cancer (Chan et al. 2004; Ernst et al. 2008; Bollrath et al. 2009; Grivennikov et al. 2009; He et al. 2010; Corcoran et al. 2011; Fukuda et al. 2011; Lesina et al. 2011; Ho et al. 2012; Putoczki et al. 2013), whereas other studies have demonstrated that STAT3 behaves as a tumor suppressor in brain, breast, colorectal, lung, prostate, and thyroid cancer (de la Iglesia et al. 2008; Musteanu et al. 2010; Couto et al. 2012; Lee et al. 2012; Grabner et al. 2015; Pencik et al. 2015; Jhan and Andrechek 2016). Some of the studies have implied an anti-metastatic role for STAT3 (Lee et al. 2012; Pencik et al. 2015). In contrast, other findings have demonstrated that persistent activation of STAT3 in tumor cells and tumor-associated immune cells promotes cancer metastasis (Yu et al. 2014). Experimental conditions have varied with tissue, oncogene, and cancer stage, and so, together, the observations suggest the existence of context dependency mechanisms that underlie the differential roles of STAT3 in cancer. Characterization of these context-specific dependencies, once understood and considered, could be exploited therapeutically.
In this study, we sought to examine the underlying differential roles found for STAT3 in cancer using a model of endogenous expression of oncogenic KRASG12D in mouse lung and pancreatic cells. KRAS is the most frequently mutated oncogene in human cancer, and KRAS mutant mice have been used extensively to model human disease. While loss of cellular identity and the acquisition of phenotypic features of undifferentiated cells are characteristic of KRAS-induced tumors in mice (Kim et al. 2005; Winslow et al. 2011; Roy and Hebrok 2015), our study defines a fundamental and previously unrecognized function of STAT3 in the maintenance of epithelial cell identity and differentiation. Loss of STAT3 preferentially associates with the acquisition of mesenchymal-like phenotypes and more aggressive tumor behavior, while persistent STAT3 activation (i.e., Y705 phosphorylation) confers a differentiated epithelial morphology on cells that impacts their tumorigenic potential. Our results implicate STAT3 as a general tumor modifier. Mechanistically, we found that STAT3 and the transcription factor TP63 jointly regulate expression of genes that are specifically involved in epithelial cell commitment and differentiation. These data provide a strong foundation for dissecting mechanisms of context-dependent functions of STAT3 signaling in various cancer types.
Results
STAT3 expression varies at different stages of pancreatic carcinogenesis
Activation of the JAK/STAT3 pathway has been reported to be a general cancer dependency, evidenced by chronic Y705 phosphorylation of STAT3 (Yu et al. 2014; Buchert et al. 2016). For this reason, we used RNAseqV2 and reverse-phase protein array (RPPA) data from The Cancer Genome Atlas (TCGA) to assess the relationship between STAT3 gene copy aberrations, mRNA, and protein expression among >800 cases from three common cancer types; namely, pancreatic ductal adenocarcinoma (PDAC), lung adenocarcinoma (ADC), and lung squamous cell carcinoma (SCC). STAT3 gene alterations are found in only a low percentage of cancer cases (Supplemental Fig. S1A), and basal STAT3 phospho-Tyr705 (pY705) activity is only moderately altered in tumor samples across three cancer types (Supplemental Fig. S1B). Based on these observations, we evaluated STAT3 status by immunohistochemistry (IHC) of pancreatic tissue microarrays (TMAs) containing normal, precancerous pancreatic intraepithelial neoplasia (PanIN), PDAC, and metastatic tissues (Fig. 1). Staining for STAT3 and STAT3 pY705 was scored by intensity and calculated as percentage of total. The vast majority (94%) of normal tissue samples (50 out of 53) stained positive for STAT3, most staining moderately or strongly positive but with little STAT3 pY705 staining except in some inflamed areas. Likewise, 26 out of 31 precancerous PanIN lesions stained positive for STAT3, with about half of those also positive for STAT3 pY705, consistent with its possible role in early neoplasia. Comparatively, only 36 out of 84 of the PDAC samples stained positive for total STAT3, with the majority of those being only weakly positive (Fig. 1B). In those weakly positive tissues, staining was often heterogeneous, with a subset of cancer cells losing STAT3 positivity entirely. This apparent loss of STAT3 function associated with progression to cancer was made even more evident by only 11 out of 84 PDAC samples staining positive for STAT3 pY705 (Fig. 1C). There was a trend toward decreased STAT3 activity in poorly differentiated or undifferentiated carcinomas through loss of either Y705 phosphorylation or STAT3 expression entirely (Supplemental Fig. S1C). This corroborates data from the Human Protein Atlas (http://www.proteinatlas.org) reporting weak or no expression of STAT3 in more than half (65%) of all human malignancies (Supplemental Fig. S1D). Conversely, although the sample size was low, PDAC metastases generally stained positive for STAT3 (nine out of 11) (Fig. 1A). STAT3 expression in metastases is in accordance with the differentiation that commonly accompanies metastatic formation (McDonald et al. 2012).
Figure 1.

STAT3 expression varies at different stages of pancreatic carcinogenesis. (A) Representative IHC images of STAT3 and STAT3 pY705 staining of human pancreatic TMAs containing normal controls, precancerous lesions (PanIN), primary PDAC tumor samples, and metastatic tumors. Bar, 200 µm. (B) Quantitative analysis of total STAT3 staining for each tissue. (C) Statistical analysis of staining for STAT3 pY705 in the percentage of each sample positive for STAT3.
STAT3 is required for the maintenance of epithelial differentiation in KRAS-induced PDAC
To determine the influence of STAT3 in pancreatic cancer, we evaluated a genetically tractable murine model system. PDAC is the most common type of pancreatic cancer and is almost invariably associated with mutations in the KRAS gene (Perez-Mancera et al. 2012). PDAC is thought to arise either through KRAS-induced acinar-to-ductal metaplasia or from the expansion of duct-derived progenitor cells (Guerra et al. 2007; Zhu et al. 2007; Gidekel Friedlander et al. 2009; Roy et al. 2015). Using the KRASG12D p53 knockout mouse model of pancreatic tumorigenesis, we previously identified an epithelial population of cells in the adult pancreas expressing pancreas/duodenoum homeobox protein 1 (PDX1) that can be clonally expanded in culture and can form tumors in mice (Ischenko et al. 2014b). Cell lines generated from the cultured cells represent a premalignant stage with a potential for neoplastic progression. They have stable phenotypes and genetic characteristics and were chosen to investigate the role of STAT3 in KRAS-driven tumorigenesis. For STAT3 loss of function, we used the CRISPR/Cas9 system to generate stable knockouts for STAT3 (Fig. 2A). For gain of function, cells were transduced with lentiviruses expressing wild-type or hyperactive STAT3 alleles (Supplemental Fig. S2). Of these, the hyperactive mutants STAT3C (A662C/N664C), STAT3 Y640F, and STAT3 K658Y are reported to dimerize spontaneously and activate transcription in a Y705 phosphorylation-dependent manner (Y640F and K658Y are naturally occurring mutations) (Bromberg et al. 1999; Pilati et al. 2011; Crescenzo et al. 2015). The activity of STAT3 S727E is independent of Y705 phosphorylation (Qin et al. 2008). Mutants were expressed in pancreatic KRASG12D cell lines no more than fivefold higher than endogenous STAT3 (Fig. 2A). In line with previous reports, hyperactive STAT3 Y640F and STAT3C displayed increased levels of STAT3 pY705 regardless of cell culture conditions (Fig. 2A). Neither wild-type STAT3 nor its mutants exhibited strong S727 phosphorylation (data not shown).
Figure 2.

STAT3 is required for the maintenance of differentiated cell identity in KRAS-induced PDAC. (A) Western blot analysis of nuclear and cytoplasmic extracts from control and STAT3 knockout KRASG12D p53 knockout pancreatic cells (left panel) and cells transduced with wild-type or mutant STAT3 alleles (middle and right panels). (B) FACS analysis of CD133 and SCA1 expression in KRASG12D p53 knockout pancreatic cells and their STAT3 mutant derivatives. (C) Growth and viability of KRASG12D p53 knockout pancreatic cells in two-dimensional (2D) monolayer (left panel) and three-dimensional (3D) nonadherent (right panel) cultures. The cells were cultured for 1 wk. Relative cumulative cell numbers are shown. Values correspond to average and SD. Asterisks indicate a statistically significant difference compared with wild-type controls. (D) Subcutaneous tumor formation in nude mice by KRASG12D p53 knockout pancreatic cells and their mutant STAT3 derivatives (104 cells per injection site). n ≥ 4 for each cell type. (E) H&E and IHC (KRT19 and SOX9) staining of tumors arising from KRASG12D p53 knockout cells shown in D. Representative images are shown. Bar, 200 µm.
When cultured in vitro, STAT3-deficient (STAT3 knockout) KRASG12D p53 knockout cells retained many of their parental characteristics, as judged by morphology and by continuous expression of the ubiquitous and pancreas-specific transcription factors MYC, PDX1, SOX9, and HES1 (Fig. 2A). However, flow cytometry indicated that the STAT3 knockout cells acquired a less differentiated CD133−SCA1− phenotype associated with poorly differentiated or undifferentiated (anaplastic) tumor appearance (Supplemental 2B). SCA1 and CD133 expression has been shown previously to mark differentiated pancreatic ductal cells (Ischenko et al. 2014b; Roy et al. 2015). Ectopic expression of wild-type STAT3 or hyperactive STAT3 Y640F induced a shift toward a more differentiated CD133+SCA1+ phenotype (Fig. 2B). The persistent activation of STAT3 Y640F was found to retard cell growth in vitro in either two-dimensional (2D) or nonadherent cell culture conditions (Fig. 2C).
Following subcutaneous transplantation into mice, the KRASG12D pancreatic cells expressing the hyperactive STAT3 Y640F mutant were markedly delayed in their ability to form tumors in comparison with control (vector) cells (Fig. 2D). Therefore, persistent phosphorylation of STAT3 rather than ablation impeded tumorigenesis. Of particular importance, the amount and activation status of STAT3 present in the cultured cells correlated with the histological and immunohistological features of tumors. The histology of both heterotopic tumors and orthotopic pancreatic tumors arising from parental control cells was consistent with that of moderately differentiated PDAC (Fig. 2E; Supplemental Fig. S3A; data not shown). In contrast, the histology of tumors derived from STAT3 knockout cells showed characteristics of anaplastic (sarcomatoid) carcinoma, also known as epithelial–mesenchymal transition (EMT) tumors (Nieto et al. 2016; Lambert et al. 2017; Brabletz et al. 2018). The predominant sarcomatous elements in the STAT3 knockout tumors lacked a differentiated morphology and were negative for markers of ductal differentiation, such as keratin 19 (KRT19) and E-cadherin (CDH1), while inducing the mesenchymal marker smooth muscle actin (SMA; ACTA2) (Fig. 2E; Supplemental Fig. S3B). Markers of pancreatic ductal progenitor cells (e.g., SOX9) remained unchanged. In sharp contrast, tumors arising from cells overexpressing wild-type STAT3 or hyperactive STAT3 (STAT3C and STAT3 Y640F) appeared well differentiated. These tumors were strongly positive for STAT3 pY705, SOX9, CDH1, and KRT19, while ACTA2 expression was restricted to infiltrating stromal cells (Fig. 2E; Supplemental Fig. S3B). Because PDAC frequently displays TP53 mutations, giving rise to a stable mutant p53 protein, we also investigated pancreatic cells derived from PDX1-Cre KRASG12D p53R172H mice (Hingorani et al. 2005). Control KRASG12D p53R172H cells developed moderately to well-differentiated tumors organized into typical glandular structures, whereas the loss of STAT3 resulted in a less differentiated or poorly differentiated disease, further inferring a causal relationship between STAT3 status and the pathogenesis of PDAC (Supplemental Fig. S3C). Thus, using STAT3 pY705 as a measure of STAT3 activity with epithelial markers such as CDH1, KRT19, and PDX1, our data indicate that strong and persistent activation of STAT3 in KRASG12D pancreatic cells stabilizes their epithelial identity. These cells in turn give rise to well-differentiated tumors resembling human PDAC.
Activation of STAT3 at S727 directs loss of epithelial identity and partial EMT
In addition to tyrosine phosphorylation, phosphorylation of S727 has been reported to enhance STAT3 transcriptional activity, influence cell fate transitions, and promote oncogenic transformation (Wen et al. 1995; Shen et al. 2004; Androutsellis-Theotokis et al. 2006; Gough et al. 2009; Zhang et al. 2013). We measured S727 phosphorylation levels in parental KRASG12D p53 knockout cell lines grown in 2D and three-dimensional (3D) conditions in the absence of exogenously added cytokine (Fig. 3A). STAT3 pY705 was detectable only in 3D culture conditions, whereas low levels of STAT3 S727 phosphorylation (pS727) were discernible in all culture conditions and distributed in both nuclear and cytoplasmic fractions. The level of STAT3 pS727 was not affected by overexpression of activated RAS or RAS effectors in these cells with endogenous KRASG12D (Supplemental Fig. S4A). Since antibodies to STAT3 pS727 do not have sufficient sensitivity for IHC, we used the phosphorylation-mimetic STAT3 S727E as a genetic tool to study the role of STAT3 pS727 in cancer (Androutsellis-Theotokis et al. 2006; Qin et al. 2008). Remarkably, forced expression of STAT3 S727E conferred on KRASG12D cells clear features of partial EMT. Analysis of the transition over time showed that STAT3 S727E cells down-regulated markers of pancreatic epithelium (EpCAM and CD133) (Fig. 3B; Supplemental Fig. S4B) and transitioned to a fibroblast-like morphology (Supplemental Fig. S4C). Mesenchymal markers, such as ACTA2 and vimentin, were not induced, suggesting partial rather than complete EMT (Yang and Weinberg 2008). Notably, activation of an EMT program by STAT3 S727E occurred without additional activation of the classical EMT-inducing pathway, TGFβ/SMAD (Supplemental Fig. S4D,E). When cells expressing STAT3 S727E were transplanted into mice, they gave rise to morphologically mixed tumors composed of epithelial and mesenchymal components (Fig. 3C). Since no evidence of EMT was found in hyperactive STAT3-expressing cells (Fig. 2E), cells were evaluated that express a STAT3 double mutant that lacks the activating Y705 but has the S727 phosphomimetic (Y705F/S727E). The KRASG12D pancreatic cells expressing STAT3 Y705F/S727E gave rise to uniformly mesenchymal-like tumors (Fig. 3C). Therefore, we conclude that the STAT3 S727E mutation partially mimics the effects of STAT3 loss in promoting a mesenchymal-like phenotype, and this is fully revealed in the absence of STAT3 pY705 activation. The results further support our finding that phosphorylation of STAT3 Y705 confers on tumor cells a differentiated epithelial morphology.
Figure 3.
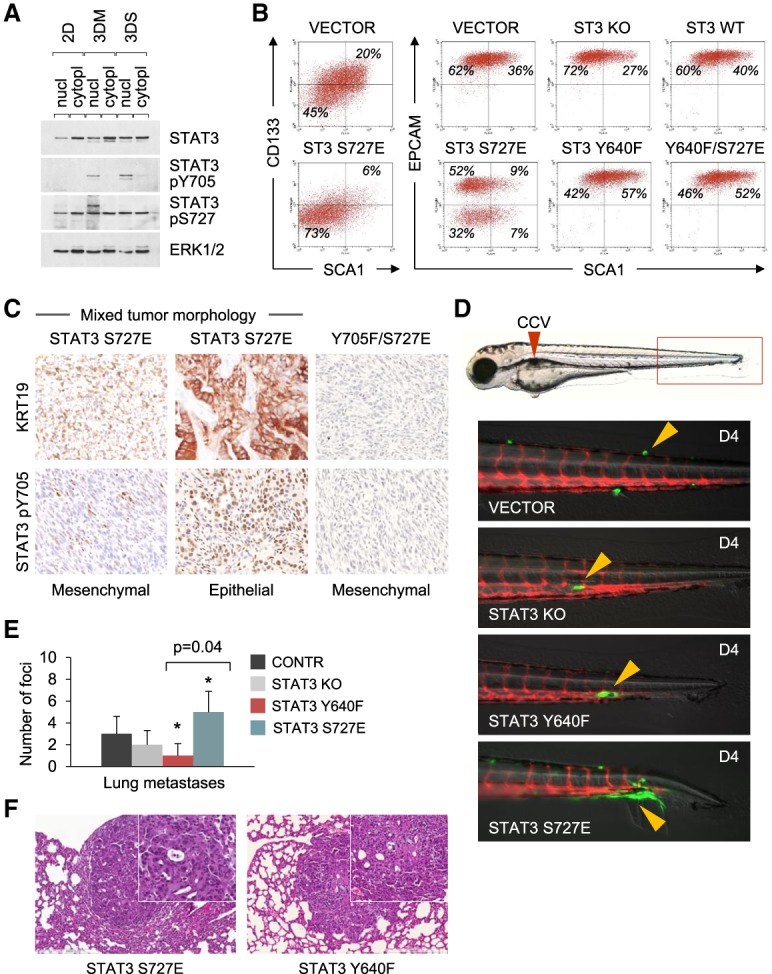
Activation of STAT3 by S727E directs loss of epithelial identity and partial EMT. (A) Western blot analysis of STAT3 Y705 and S727 phosphorylation in nuclear and cytoplasmic lysates from KRASG12D p53 knockout pancreatic cells maintained in serum-free 2D, 3D Matrigel (3DM), and 3D suspension (3DS) cultures. (B) FACS analysis of CD133, EpCAM, and SCA1 expression in KRASG12D p53 knockout pancreatic cells and their STAT3 derivatives. (C) IHC (KRT19 and pY705 STAT3) staining of mixed tumors arising from KRASG12D pancreatic cells expressing STAT3 S727E and the tyrosine phosphorylation-deficient STAT3 Y705F/S727E double mutant. (D) Representative images of zebrafish xenografts containing KRASG12D cells or their STAT3 derivatives at 4 d after injection. Red fluorescence indicates zebrafish blood vessels, and green fluorescence is transplanted mouse cells. Arrowheads indicate cells with various invasion capabilities. (E) Metastatic tumor formation in the lungs of nude mice injected by tail vein with KRASG12D p53 knockout pancreatic cells and their STAT3 derivatives. n = 4 for each cell type. Values correspond to average and SD. Asterisks indicate a statistically significant difference compared with the STAT Y640F group. (F) H&E staining of metastatic tumors in the lungs of nude mice injected with KRASG12D p53 knockout cells and their STAT3 derivatives (shown in E).
To extend this analysis to the process of metastasis, we used two different model systems: murine tail vein injection and zebrafish cardinal vein injection. The zebrafish model of metastasis allows direct visualization of fluorescently labeled cancer cells at single-cell resolution (Marques et al. 2009). The assay is highly reproducible and requires only a small number of injected cells. KRASG12D p53 knockout pancreatic parental cells and their STAT3 knockout, STAT3 Y640F, and STAT3 S727E derivatives (50–100 cells of each type) were injected into the common cardinal vein of kdrl:RFP zebrafish embryos 2 d after fertilization. The kdrl:RFP transgene labels the blood vessels with red fluorescence, allowing the simultaneous visualization of injected GFP-labeled cells. Of the four cell lines tested, STAT3 S727E cells displayed a greater ability for rapid extravasation and migration away from blood vessels along tissue boundaries, such as the intersomitic furrow, and within tissues, such as the ventral fin fold (Fig. 3D). Consistent with in vitro cell morphology, the STAT3 S727E cells exhibited an elongated mesenchymal phenotype after extravasation (Fig. 3D). While the other cells lines also exhibited the capacity to extravasate, when they did so, the cells maintained a rounded epithelioid shape and associated closely with the blood vessel from which they emerged. These cells apparently remained dormant (i.e., not expanding in mass), suggesting that dissemination alone is not sufficient to cause metastatic growth. To examine the behavior of the cells in a murine model of metastasis, the respective cell lines expressing GFP were injected into the tail veins of nude mice. After 6 wk, lung metastatic foci were observed in each injection group. Quantification of GFP-positive lung foci showed that STAT3 S727E cells surpassed STAT3 Y640F cells in terms of the number of foci (Fig. 3E). Of particular note, the metastatic tumors retained epithelial characteristics, as is commonly observed in metastatic PDAC (Fig. 3F; McDonald et al. 2012). These data indicate that STAT3 S727E (and, by analogy, STAT3 pS727) promotes extravasation and early initiation of experimental metastases.
Loss of STAT3 accelerates KRAS-induced lung cell tumorigenesis
To investigate the impact of STAT3 in KRAS-induced tumorigenesis in a distinct tissue type, we used the KRASG12D model of non-small cell lung (NSCLC) cancer. NSCLC is the most common type of lung cancer and is conventionally subdivided into ADC, SCC, and undifferentiated forms. Among these, mutations in KRAS prevail in lung ADCs (>30%), while PI3K pathway mutations prevail in lung SCCs (>40%) (The Cancer Genome Atlas Research Network 2012, 2014). We previously established primary cultures of KRASG12D p53 knockout cells from the lungs of adult mice (Ischenko et al. 2014a). Rather than being restricted to one tumor type, these cells can give rise to several types of cancer, including ADC and SCC. The cells can also be converted from one cancer type to another, and their plasticity depends on the expression of lineage-determining transcription factors (Ischenko et al. 2014a). We generated STAT3-specific knockouts in these lung epithelial cells using CRISPR/Cas9 technology and then assessed expression of STAT3-responsive genes with tumor-promoting or tumor-suppressing activities by Western blot, including MYC, TP63, and SOX2 (Fig. 4A; Foshay and Gallicano 2008; Ma et al. 2010; Tadokoro et al. 2014). Among these proteins, MYC is a common driver of solid tumors (Dang 2012), while TP63 and SOX2 play major roles in lung SCC differentiation (The Cancer Genome Atlas Research Network 2012; Liu et al. 2013). Knockout of STAT3 in the KRASG12D lung cells had marginal effects on the expression of these proteins (Fig. 4A). In contrast, overexpression of wild-type or activated STAT3 induced the up-regulation of TP63 and MYC but not SOX2 (Fig. 4A). The relative difference in the expression of these genes suggests that STAT3 controls their inducible, but not basal, expression. STAT3 S727E was a notable exception, as it failed to stimulate expression of genes traditionally associated with STAT3 activation (e.g., MYC, TP63, and SOCS3) (Fig. 4A).
Figure 4.

Loss of STAT3 accelerates KRAS-induced lung cell tumorigenesis. (A) Western blot analysis of nuclear and cytoplasmic extracts from control and STAT3 knockout KRASG12D p53 knockout lung epithelial cells (left panel) and cells transduced with wild-type or mutant STAT3 alleles (middle and right panels). (B) Growth and viability of KRASG12D p53 knockout lung epithelial cells in 2D monolayer (left panel) and 3D nonadherent (right panel) cultures. Relative cumulative cell numbers are shown in cultures for 1 wk. Values correspond to average and SD. Asterisks indicate a statistically significant difference compared with wild-type controls. (C) Subcutaneous tumor formation in nude mice by KRASG12D p53 knockout lung epithelial cells and mutant STAT3 derivatives (104 cells per injection site). n ≥ 4 for each cell type. (D,E) H&E and IHC (KRT17/19, ΔNP63, and CD45) staining of tumors arising from KRASG12D p53 knockout lung epithelial cells shown in C. Representative images are shown. Bar, 200 µm.
We next examined the effects of STAT3 on KRAS-driven lung cell transformation and tumorigenesis. Cells were maintained in tissue culture in defined serum-free medium for epithelial cells. Knockout of STAT3 had no adverse effects on growth and viability of parental KRASG12D p53 knockout lung cells maintained in 2D or 3D conditions, whereas cells expressing the hyperactive STAT3 Y640F allele exhibited a reduced ability to proliferate (Fig. 4B). Moreover, when these cells were subcutaneously transplanted into mice, the STAT3 knockout cells gave rise to tumors with a shorter latency compared with control-injected mice (Fig. 4C). In stark contrast, ectopic expression of wild-type STAT3 or persistently active STAT3 Y640F resulted in a marked reduction in tumorigenicity compared with cells transduced with vector alone. Notably, persistent activation of STAT3 extended tumor latency even beyond that of wild type (Fig. 4C). The effects of STAT3 in lung tumor latency was more considerable than in pancreatic tumors, and this could be due to the extensive stroma associated with the pancreatic tumors.
The tumors formed by control KRASG12D p53 knockout lung cells were composed of different histological components, as reported previously; i.e., ADC and sarcomatoid carcinoma (Fig. 4D, vector; Ischenko et al. 2014a). These tumors displayed variable and diffuse immunoreactivity to epithelial KRT14–19 and weak reactivity to TP63. Remarkably, all tumors arising from STAT3 knockout cells were carcinosarcomas, consisting of undifferentiated spindle cells. No immunoreactivity to markers of epithelial differentiation was detected. Likewise, STAT3 S727E partly mimicked the effects of STAT3 loss by producing the formation of EMT-like tumors (Fig. 4D). In contrast to the effects of STAT3 loss, cells expressing wild-type or hyperactive STAT3 Y640F formed tumors with prominent squamous morphology and robust expression of KRT17, KRT19, and TP63, consistent with the histological features of SCC but not ADC (Fig. 4D; Supplemental Fig. S5). The tumors with active STAT3 exhibited areas of infiltration by CD45-positive leukocytes that may contribute to their slower growth rate and increase in necrosis (Fig. 4E; Supplemental Fig. S5). Notably, STAT3 knockout had little or no effect on immune cell recruitment. Prolonged activation of STAT3 therefore appears disadvantageous for lung cancer cell growth and survival. In sum, both in vitro and in vivo assays revealed that a high level of wild-type STAT3 or hyperactive STAT3 delays tumorigenesis initiated by mutant KRAS and shifts tumor morphology toward an exclusive squamous epithelial phenotype.
STAT3 and TP63 cooperatively regulate expression of epithelial-specific genes
KRASG12D lung epithelial cells with persistent activation of STAT3 produced tumors with histological features of SCC and characteristic elevated levels of TP63 (Fig. 4; Dotto and Rustgi 2016). TP63 is critical for epithelial development in mice and humans and encodes two main isoforms: TAP63 and ΔNP63 (Crum and McKeon 2010; Melino et al. 2015). The TAP63 isoform is generally thought to be tumor-suppressive, while the ΔNP63 isoform can play a tumor-promoting function in the early stages of cancer. Pertinent to our study, reports have indicated that ΔNP63 is a direct downstream target of STAT3 and is expressed in SCCs (Chu et al. 2008; Ma et al. 2010; Ripamonti et al. 2013; Tadokoro et al. 2014). To verify ΔNP63 as a putative transcriptional target of STAT3 in KRASG12D p53 knockout lung epithelial cells, quantitative RT–PCR was performed. Analysis revealed that KRASG12D cells express TP63 mainly as the ΔNP63 isoform (data not shown). The parental and STAT3 knockout cells were found to express similar levels of ΔNP63 mRNA, whereas ΔNP63 mRNA was elevated in cells expressing hyperactive STAT3 (Fig. 5A). SOCS3, a known STAT3 target, was used as a positive control.
Figure 5.

STAT3 and TP63 cooperatively regulate expression of epithelial-specific genes. (A) Quantitative RT–PCR of SOCS3 and ΔNP63 mRNA levels in control, STAT3 knockout, and STAT3 Y640F transduced KRASG12D p53 knockout lung epithelial cells. Relative expression levels are shown normalized to GAPDH. (B) Gene ontology analysis of control, STAT3 knockout-expressing, and STAT3 Y640F-expressing tumors derived from transcriptome analysis of KRASG12D p53 knockout lung epithelial cells. (C) Heat map of RNA sequencing (RNA-seq) data representing a subset of differentially expressed genes specifically involved in EMT and epithelial identity in control, STAT3 knockout-expressing, and STAT3 Y640F-expressing tumors derived from KRASG12D p53 knockout lung epithelial cells. (D) Heat maps of RPPA data representing differentially expressed proteins in KRASG12D p53 knockout lung epithelial cells and their STAT3 derivatives. (E) Signaling perturbations in KRAS-related pathways derived from RPPA analysis by loss or gain of STAT3 function in KRASG12D p53 knockout lung epithelial cells. (F) H&E and IHC (ΔNP63) of tumors derived from fully formed murine lung ADC tumor cells following their culture and modification to express STAT3 knockout or ectopic wild-type STAT3. (G) Histological features of tumors arising from KRASG12D lung epithelial cells with STAT3, TP63, and TGFBR2 genotypes. (*) Mixed epithelial–mesenchymal tumor phenotype consisting of ≤10% mesenchymal component. (**) Mixed epithelial–mesenchymal tumor phenotype consisting of ≤10% epithelial component. (H) Model representing the mechanism by which STAT3 and TP63 regulate epithelial cell commitment and differentiation in KRAS-driven carcinogenesis.
To gain insight into the mechanistic role of STAT3 activation in KRAS-induced tumorigenesis, we performed whole-transcriptome sequencing of tumor tissues derived from control parental KRASG12D p53 knockout lung cells and their STAT3 knockout or hyperactive STAT3 Y640F derivatives (Fig. 5B,C). The analysis revealed >1000 genes whose expression was altered greater than twofold in the presence or absence of STAT3 (Supplemental Fig. S6A,B). The characteristic gene expression patterns corresponded to the morphological features identified in Figure 4. The gene ontology enrichment analysis showed modest but significant changes in expression of genes involved in mesenchymal cell differentiation (EMT), acute-phase inflammatory response, and blood microparticles in the STAT3 knockout lung tumors (Fig. 5B; Supplemental Fig. S6C–E). Collectively, these changes associate with enhancement of metastasis and poor cancer outcomes (Varon and Shai 2015; De Cock et al. 2016; Nieto et al. 2016; Brabletz et al. 2018). Specifically, EMT-promoting genes, such as ZEB1, ZEB2, SMADs, and Vimentin, were up-regulated in STAT3 knockout tumors (Fig. 5C; Supplemental Fig. S6C–E). In contrast, expression of epithelial differentiation genes (such as TP63, KLF4), epithelial tight junctions, keratins, and keratin-associated genes (>100 genes) was significantly up-regulated in STAT3 Y640F-expressing tumors (Fig. 5B,C; Supplemental Fig. S6C–E).
Additionally, we used RPPA to track the interactions and activities of proteins of KRASG12D p53 knockout lung and pancreatic cultured cells and their mutant STAT3 derivatives. The antibody panel used for the RPPA included >300 antibodies for total and modified proteins involved in cancer. Two main observations emerged from this analysis. First, we identified >40 potential STAT3 targets based on their differential expression in STAT3 knockout cells as compared with their respective STAT3-positive counterparts (Fig. 5D; Supplemental Fig. S6F,G). With persistently active STAT3 Y640F, ∼30% of the STAT3-regulated targets were shared between lung and pancreatic cells, suggesting the involvement of cell type-specific cofactors. Second, the expression profile of STAT3-induced proteins in premalignant lung cells more closely matched the histological subtype of corresponding tumors, as some of the STAT3 targets (e.g., AKT1, PTEN, ARID1A, and SOX2) overlap with components of PI3K/AKT and TP63 pathways. Activation of the PI3K and TP63 pathways is a hallmark of lung SCC (The Cancer Genome Atlas Research Network 2012) and was found in KRASG12D lung cells with elevated STAT3 Y705 phosphorylation (Fig. 5D,E, Supplemental Fig. S6G,H). Expression of a subset of proteins was validated by Western blot (Supplemental Fig. S6I,J). In contrast to lung cells, induction of TP63 by STAT3 was not seen in KRASG12D pancreatic cells, supporting the notion of the cell type specificity of STAT3 effects.
STAT3 and TP63 function nonredundantly to promote KRAS mutant lung cancer cell differentiation
To genetically validate our findings that STAT3 and TP63 converge on pathways relevant to epithelial cell commitment and differentiation, we used two complementary approaches. First, cell lines were established from fully formed lung ADC produced from parental KRASG12D p53 knockout lung cells, and the effects of STAT3 modification were evaluated. The cultured cells established from these tumors retained their original phenotypic characteristics and gene expression profiles (Ischenko et al. 2014a). The CRISPR/Cas9 system was used to generate STAT3 knockout cells, and lentiviral infection was used to overexpress wild-type STAT3. All cells produced tumors in mice, but the histological types were distinct (Fig. 5F). Parental ADC cells gave rise to only ADC pathology (vector). In contrast, stable knockout of STAT3 expression resulted in the formation of anaplastic (sarcomatoid) carcinomas, while ectopic expression of wild-type STAT3 induced ADC-to-SCC transdifferentiation coupled with an elevation in TP63 expression.
The cooperation of STAT3 and TP63 in the maintenance of epithelial identity was evaluated by ablation of the respective genes—either individually or in combination—in KRASG12D p53 knockout lung cells (Fig. 5G). Tumors derived from parental cells were mostly differentiated (Fig. 4), whereas tumors derived from STAT3 knockout or from TP63 knockout lung cells were anaplastic carcinomas (undifferentiated) and devoid of squamous differentiation (Fig. 5G). To evaluate a functional link between STAT3 and TP63, we ectopically expressed TP63 in the STAT3 knockout cells. Since TP63 codes for two isoforms with either tumor-suppressive (TAP63) or differentiation-promoting (ΔNP63) properties, we tested the effect of their individual expression on the phenotype of tumors produced by STAT3 knockout cells (Dotto and Rustgi 2016). Overexpression of ΔNP63 in STAT3 knockout cells recapitulated the differentiated epithelial morphology seen in parental tumors, while TAP63 was consistently less efficient (Fig. 5G). Conversely, ectopic expression of wild-type STAT3 in the TP63 knockout cells was completely incapable of driving epithelial differentiation. Likewise, STAT3 Y640F in the TP63 knockout cells was significantly impaired in its ability to promote squamous differentiation. These results indicate that STAT3 requires TP63 for its ability to direct epithelial differentiation of these KRASG12D tumor cells. Ablation of the TGFβ pathway by CRISPR-mediated receptor knockout was used as a control for SCC (Adorno et al. 2009; Wakefield and Hill 2013; Wang et al. 2016). Together, these data are consistent with the tenet that TP63 induction is downstream from STAT3 activity in lung differentiation and oncogenic transformation (Chu et al. 2008; Ma et al. 2010; Ripamonti et al. 2013; Tadokoro et al. 2014).
Finally, we assessed whether STAT3 and TP63 act in an additive or epistatic manner in pancreatic tumorigenesis. In contrast to the lung, TP63 expression did not increase following ectopic expression of STAT3 Y640F in tumors derived from KRASG12D p53 knockout pancreatic cells (Supplemental Fig. S7A). Since TP63 expression did not appear to be a direct response to STAT3 in the mutant KRAS pancreatic cells, the cell context regulation by STAT3 is evident. To ascertain a cooperative role of STAT3 and TP63 in pancreatic tumors, we generated KRASG12D p53 knockout pancreatic cells containing single and compound TP63 mutations (Supplemental Fig. S7B,C). Tumors derived from cells lacking TP63 were anaplastic carcinomas of spindle cell type, exhibiting aggressive features of TAP63/TP53-null tumors (Supplemental Fig. S7C; Tan et al. 2014; Bailey et al. 2016). As shown previously, pancreatic tumors lacking STAT3 were also anaplastic carcinomas (Fig. 2). Ectopic STAT3 had no discernible effect on the growth and morphology of TP63 knockout tumors, and ectopic expression of TAP63 or ΔNP63 had only a marginal effect on the malignant phenotype of STAT3 knockout tumors (Supplemental Fig. S7C). These results suggest that TP63 has incomplete penetrance in the absence of STAT3. STAT3 and TP63 both play essential and overlapping roles in pancreatic tumor progression, with their loss contributing independently to EMT.
Discussion
We have made the unprecedented discovery that STAT3 regulates epithelial differentiation of pancreatic and lung cell tumors driven by oncogenic KRAS. In this manner, STAT3 acts as a tumor modifier that regulates cellular plasticity and inhibits the EMT that is often linked to metastasis. Our results redefine the role of STAT3 in cancer and identify the activation of STAT3 as a keystone in the regulation of lineage plasticity.
The study initiated with an assessment of information available in major cancer data sets, including TCGA, the Cancer Genome Characterization Initiative (CGCI), and the International Cancer Genome Consortium (ICGC), that provide the frequency of genetic aberrations and levels of protein expression and activation for STAT3 in several human tumor types. Typically, STAT3 mutations are rare in most solid tumors, and protein levels are not higher than normal tissue. Additionally and somewhat counterintuitively, activation of STAT3 by specific Y705 phosphorylation in PDAC, lung ADC, and lung SCC does not significantly deviate from the mean (Supplemental Fig. S1). Based on these data, we used IHC of human pancreatic TMAs to interrogate STAT3 protein levels and tyrosine phosphorylation at different stages of pancreatic carcinogenesis. Unexpectedly, STAT3 activity was found to inversely correlate with PDAC (Fig. 1). The mechanisms that drive the loss of STAT3 expression in PDAC remain to be determined. However, the results are in accord with parallel mapping of gene expression patterns and signaling networks in human cancers that do not identify the JAK/STAT3 pathway as causal in epithelial carcinogenesis (Kandoth et al. 2013; Hoadley et al. 2014). The human TMA results suggested that STAT3 was linked to normal pancreatic tissue and differentiated pancreatic carcinomas (Fig. 1; Supplemental Fig. S1C), leading us to hypothesize that STAT3 may play a fundamental role in cancer through the maintenance of epithelial identity.
To experimentally test this premise, we used a genetically tractable murine model system and investigated the contribution of STAT3 to KRAS-driven pancreatic and lung carcinomas. There are several significant findings in our study. First, STAT3 is not required for KRAS-driven tumorigenesis. CRISPR/Cas9-mediated deletion of STAT3 in pretumor cells from the lung or pancreatic epithelium does not inhibit tumor formation. Conversely, activation of STAT3 by pY705 increases the latency of tumor formation. Our results uncover a negative interaction between STAT3 pY705 and KRAS mutations, which may reflect the fact that gain-of-function STAT3 mutations are almost completely absent from human cancers carrying mutant KRAS (http://www.cbioportal.org).
Second, we highlight the fundamental role of STAT3 in the maintenance of epithelial identity and differentiation in KRAS-induced tumor cells. STAT3 pY705 maintains an epithelial phenotype in both lung and pancreatic tumors. In contrast, it is the loss of STAT3 that leads to the acquisition of a mesenchymal-like tumor phenotype, a change known to associate with metastasis and resistance to drug therapeutics. The modification of STAT3 by S727 phosphorylation has been reported to cooperate with KRAS in tumorigenesis (Gough et al. 2009). For this reason, we tested the effect of a phosphomimetic, STAT3 S727E, and found this modification (particularly in the absence of pY705) to have an effect similar to that of the loss of STAT3 to foster mesenchymal transition. Together, our results define a previously unrecognized function of activated STAT3 as a general tumor modifier (rather than strictly a tumor promoter or suppressor) that has an important regulatory role in directing epithelial identity.
To understand the mechanistic impact of STAT3 in this capacity, we compared the transcriptome profile of mutant KRAS lung tumors formed by STAT3 knockout or hyperactive STAT3 Y640F cells. The differences in gene expression unequivocally confirm the increase in regulators and effectors of epithelial identity in STAT3 pY705 tumors, matching the histology of the tumors (Fig. 5B,C; Supplemental Fig. S6A–E). The increase in TP63 transcription factor expression is particularly significant, since it is often designated a master regulator of epithelial cell fate (Pellegrini et al. 2001; Crum and McKeon 2010; Melino et al. 2015; Yoh and Prywes 2015). Genes that are responsive to TP63, particularly the ΔNP63 isoform, show increased expression in tumors with persistently active STAT3 (Fig. 5; Supplemental Fig. S6; Truong et al. 2006). These results indicate that the tumor microenvironment has an important influence on the status of tumor cell STAT3 tyrosine phosphorylation to promote TP63 expression and maintain epithelial identity. A noticeable feature of the epithelial lung tumors expressing wild-type STAT3 or STAT3 Y640F was an increase in immune cell infiltration (CD45), frequently accompanied by areas of necrosis (CD45) (Fig. 4E; Supplemental Fig. S5). This may have contributed to the longer latency needed for the formation of these tumors (Fig. 4C). As RNA sequencing (RNA-seq) was performed on RNA extracted from whole lung tumors, stromal tissue was also included in the preparations. Stromal cells from tumors expressing STAT3 Y640F may be responsible for the apparent increase in immune cell markers such as CD5; immune cell regulators IL-10, MST1, CXCL5, CXCL9, and CXCR5; mast cell proteases; and interferon-induced genes GTPase1, IFITM6, and 2′5′OASE.
KRASG12D lung tumors with activated STAT3 showed clear modulation of genes involved in the TP63 pathway, identifying TP63 as a potential downstream effector of STAT3. The ΔNP63 isoform, in particular, has been shown to be a direct target of STAT3 (Chu et al. 2008), and several studies demonstrated a role of STAT3 with TP63 in differentiation (Ma et al. 2010; Ripamonti et al. 2013; Tadokoro et al. 2014). To evaluate the genetic interaction between TP63 and STAT3 in tumor differentiation, we analyzed a lung tumor phenotype with loss of TP63 and/or STAT3 (Fig. 5G). The mutant KRAS tumors that lacked either STAT3 or TP63 lost their epithelial characteristics and were undifferentiated anaplastic carcinomas. Genetic complementation of the STAT3 knockout tumor cells with ectopic TP63, particularly ΔNP63, was able to promote epithelial differentiation. Importantly, ectopic expression of wild-type STAT3 in TP63 knockout cells could not promote differentiation, and the hyperactive STAT3 Y640F was severely limited in promoting epithelial differentiation in the absence of TP63. Together, the result implicates TP63 as a critical downstream effector of STAT3 in KRAS lung tumors with a squamous phenotype (Fig. 5G,H). Genetic complementation was also evaluated in the KRAS pancreatic tumors (Supplemental Fig. S7). Although TP63 expression is low in these pancreatic cells and apparently not induced by STAT3 activity, the action of both factors is required for the tumor cell epithelial identity of PDAC.
The switch from an epithelial tumor identity to a partial mesenchymal phenotype has been proposed to promote malignancy and resistance to cancer drug therapeutics. Our studies are the first to identify a function of STAT3 in the regulation of the epithelial cell identity of tumors, dependent on the action of TP63. The influence of the STAT3 transcription factor on cancer development has been investigated for nearly two decades, and studies have provided conflicting results. Studies in mice suggest the existence of context-dependent (i.e., tissue- and oncogene-dependent) mechanisms that underlie the role of STAT3 in tumorigenesis. Our experimental findings and cancer genomics data suggest that it is essential to thoroughly understand the full spectrum of context-dependent roles for STAT3 in tumor progression prior to therapeutic intervention.
Materials and methods
Mammalian cells and reagents
Premalignant and tumor-derived KRASG12D p53 knockout pancreatic and lung epithelial cell lines were described previously (Ischenko et al. 2013, 2014a). These cells were grown on gelatinized plates in CnT-PR epithelial medium (Zenbio) supplemented with 1× penicillin/streptomycin (Gibco). For standard proliferation assays, cells were seeded into six-well plates (4 × 105 cells per well) and counted with a Coulter counter for each time point. 3D Matrigel and 3D suspension cultures were analyzed as described before (Ischenko et al. 2013, 2015). For flow cytometry, cells were lifted with Accutase (Sigma); stained with FITCI-, PE-, or APC-conjugated antibodies to CD326 (EpCAM), CD133, SCA1, CD44, and CD24 (eBioscience or BD Pharmingen); and analyzed using FACSCalibur (BD) with CellQuest software. Cell viability was measured using propidium iodide (PI) staining. Lentiviral vectors (adapted from the pWPXL/pEF1a backbone) encoding mutant STAT3 alleles were derived using site-directed mutagenesis. The final plasmids were sequence-confirmed. For CRISPR/Cas9-mediated knockouts, guide RNA (gRNA) oligos were cloned into LentiCRISPRv2 (Sanjana et al. 2014). The gRNA sequences were as follows: CACCGGCAGCTGGACACACGCTACC (mouse STAT3 knockout 1), CACCGGTACAGCGACAGCTTCCCCA (mouse STAT3 knockout 2), CACCGCCACGCACAGAATAGCGTGA (mouse TP63 knockout), CACCGTGTACAGCACCGGCCGATGC (human STAT3 knockout 1), and CACCGACGCCGGTCTTGATGACGAG (human STAT3 knockout 2). Recombinant lentiviruses were produced by transient transfection of 293T cells according to standard protocols.
Tumorigenicity in mice
All animal studies were approved by the Institutional Animal Care and Use Committee at Stony Brook University. Cells (104) in 100 µL of Matrigel (diluted 1:10 with Opti-MEM) were transplanted subcutaneously in nude mice or introduced by tail vein injection. Orthotopic pancreatic injections were performed as described (Kim et al. 2009). Tumor latency was defined as the period between injection of tumorigenic cells into mice and the appearance of tumors of ≥1 mm in diameter. The end point was a tumor diameter of 1 cm. Statistical analyses was performed using Student's t-test. P ≤ 0.05 was considered statistically significant. Mouse tissue was harvested and processed as described (Ischenko et al. 2014a). Two pathologists read and scored the histology slides independently.
IHC
Paraffin-embedded 3-µm sections were processed and stained with H&E for histology as described (Ischenko et al. 2014a). The following antibodies were used for immunohistochemical analysis: anti KRT17/19 (Cell Signaling, D3209), SOX9 (Cell Signaling, D8G8H), STAT3 (Cell Signaling, 4904), STAT3 pY705 (Cell Signaling, 9131), CDH1 (Cell Signaling, 3195), ACTA2, CD45, ΔNP63 (Biolegend), KRT5, and KRT14 (Biorbyt). Histology was performed by HistoWiz, Inc. (http://www.histowiz.com) using standard operating procedures and fully automated workflow and by the Research Histology Core Laboratory, Department Pathology, Stony Brook Medicine.
Protein expression analyses
Western blot analyses were performed using antibodies against STAT3 (BD, 610190), CTNNB1 (Cell Signaling, 6B3), ERK1/2 (Cell Signaling, 9102), HES1 (Cell Signaling, D6P24), KRT17/19 (Cell Signaling, D3209), STAT3 pY705 (Cell Signaling, 9131), STAT pS727 (Cell Signaling, 9134), SNAI1 (Cell Signaling, C15D3), SNAI2 (Cell Signaling, C19G7), SOX9 (Cell Signaling, D8G8H), MYC-T58/S62 (Millipore, 04-217), P-ERK1/2 (Millipore, 04-797); MYC (Santa Cruz Biotechnology, N-262), P63 (Santa Cruz Biotechnology, 4A4), SMA (Santa Cruz Biotechnology, 53142), SOCS3 (Santa Cruz Biotechnology, 6A463), ΔNP63 (Biolegend, 619001), KRT5 (Biorbyt, orb128270), and KRT14 (GeneTex, GTX104124). Whole-cell extracts were prepared by lysing cells in buffer containing 10 mM TrisHCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Triton X-100, 40 mM NaVO4, 0.1% SDS, and 1× protease inhibitors (Roche). Nuclear extracts were prepared using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Scientific). Western blots were imaged using Image Studio software (LI-COR). RPPA analysis was performed by the MD Anderson Cancer Center Reverse Phase Protein Array Core Facility. RPPA data analysis was carried out using publicly available data sets (http://www.cbioportal.org) and the existing literature (Akbani et al. 2014). The TCGA RPPA data were downloaded as z-scores from the cBioPortal. The heat maps were generated using Heatmapper software with Pearson correlation and centroid linkage.
RNA expression analyses
Total RNA was isolated with the PureLink RNA minikit (ThermoFisher) according to the manufacturer's specifications. First strand cDNA synthesis was performed using the iScript cDNA synthesis kit (Bio-Rad) followed by quantitative PCR using the SsoAdvanced Universal and CFX96 Touch real-time PCR detection system (Bio-Rad). Primers were designed through the Massachusetts General Hospital PrimerBank. Primer sequences are available on request. Statistical differences were detected using a two-tailed t-test. RNA-seq of tumor samples with basic bioinformatics and statistical analyses were performed by Novogene Corp. (http://en.novogene.com).
Human pancreatic TMAs
TMAs containing a wide range of normal pancreas, PanIN, and PDAC were constructed using paraffin tissue blocks. Five-micrometer sections from five TMA slides originally containing 179 normal, PanIN, PDAC, and metastasis samples were cut and stained for STAT3 and pY705 STAT3 using a Ventana Discovery Ultra automated stainer (Roche Diagnostics). Slides were scored blindly for relative staining intensity (negative, weak, moderate, or strong), and the degree of progression for each sample was confirmed. In cases where staining intensity was heterogeneous, samples were categorized by the highest level of epithelial intensity.
Zebrafish xenograft assays
All zebrafish experiments were approved by the Stony Brook University Institutional Animal Use and Care Committee. Two-day-old kdrl:RFP transgenic embryos were anesthetized with 0.016% tricaine and transferred to embryo medium containing 4% Ficoll. Fifty to 100 cells (control, STAT3 knockout, STAT3 Y640F, and STAT3 S727E) were injected into the common cardinal vein of the embryos using a CellTram Vario (Eppendorf) and transferred to a 33°C incubator. Embryos were imaged every 2 d after injection using a Leica DMI6000B inverted microscope and, on the eighth day after injection, with a spinning disc confocal microscope (Yokogawa, CSU-10; Carl Zeiss,AxioImager; Hamamatsu Photonics, EM-CCD camera).
Supplementary Material
Acknowledgments
We thank Alice Nemajerova, Laura Parrilla Monge, and Nithya Sivaram for their assistance with rodent surgery and IHC of tumor samples, and Alkiviadis Pierrakeas for technical support (Stony Brook University). This work was supported by National Institutes of Health grants RO1AI105114 and R03AI094054 and the Carol Baldwin Breast Cancer Research Award to N.C.R., and Damon Runyon Cancer Research Foundation grant DRR4714 to B.L.M.
Author contributions: O.P. and N.C.R. designed the research. S.D., J.S., B.L.M., and O.P. performed the research. S.D., H.C.C., B.L.M., and O.P. contributed new reagents/analytic tools. O.P., H.C.C., and N.C.R. analyzed the data. O.P. and N.C.R. wrote the paper.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.311852.118.
References
- Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V, et al. 2009. A mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 137: 87–98. [DOI] [PubMed] [Google Scholar]
- Akbani R, Ng PK, Werner HM, Shahmoradgoli M, Zhang F, Ju Z, Liu W, Yang JY, Yoshihara K, Li J, et al. 2014. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat Commun 5: 3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, Rueger MA, Bae SK, Kittappa R, McKay RD. 2006. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 442: 823–826. [DOI] [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. 2016. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531: 47–52. [DOI] [PubMed] [Google Scholar]
- Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, Nebelsiek T, Lundgren-May T, Canli O, Schwitalla S, et al. 2009. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 15: 91–102. [DOI] [PubMed] [Google Scholar]
- Brabletz T, Kalluri R, Nieto MA, Weinberg RA. 2018. EMT in cancer. Nat Rev Cancer 18: 128–134. [DOI] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE Jr. 1999. Stat3 as an oncogene. Cell 98: 295–303. [DOI] [PubMed] [Google Scholar]
- Buchert M, Burns CJ, Ernst M. 2016. Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene 35: 939–951. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2012. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489: 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2014. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511: 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KS, Sano S, Kiguchi K, Anders J, Komazawa N, Takeda J, DiGiovanni J. 2004. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J Clin Invest 114: 720–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu WK, Dai PM, Li HL, Chen JK. 2008. Transcriptional activity of the ΔNp63 promoter is regulated by STAT3. J Biol Chem 283: 7328–7337. [DOI] [PubMed] [Google Scholar]
- Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA, Bardeesy N. 2011. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res 71: 5020–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couto JP, Daly L, Almeida A, Knauf JA, Fagin JA, Sobrinho-Simoes M, Lima J, Maximo V, Soares P, Lyden D, et al. 2012. STAT3 negatively regulates thyroid tumorigenesis. Proc Natl Acad Sci 109: E2361–E2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crescenzo R, Abate F, Lasorsa E, Tabbo F, Gaudiano M, Chiesa N, Di Giacomo F, Spaccarotella E, Barbarossa L, Ercole E, et al. 2015. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 27: 516–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crum CP, McKeon FD. 2010. p63 in epithelial survival, germ cell surveillance, and neoplasia. Annu Rev Pathol 5: 349–371. [DOI] [PubMed] [Google Scholar]
- Dang CV. 2012. MYC on the path to cancer. Cell 149: 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cock JM, Shibue T, Dongre A, Keckesova Z, Reinhardt F, Weinberg RA. 2016. Inflammation triggers Zeb1-dependent escape from tumor latency. Cancer Res 76: 6778–6784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Iglesia N, Konopka G, Puram SV, Chan JA, Bachoo RM, You MJ, Levy DE, Depinho RA, Bonni A. 2008. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev 22: 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotto GP, Rustgi AK. 2016. Squamous cell cancers: a unified perspective on biology and genetics. Cancer Cell 29: 622–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst M, Najdovska M, Grail D, Lundgren-May T, Buchert M, Tye H, Matthews VB, Armes J, Bhathal PS, Hughes NR, et al. 2008. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J Clin Invest 118: 1727–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foshay KM, Gallicano GI. 2008. Regulation of Sox2 by STAT3 initiates commitment to the neural precursor cell fate. Stem Cells Dev 17: 269–278. [DOI] [PubMed] [Google Scholar]
- Fukuda A, Wang SC, Morris JP, Folias AE, Liou A, Kim GE, Akira S, Boucher KM, Firpo MA, Mulvihill SJ, et al. 2011. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 19: 441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, Vasile E, DePinho RA, Jacks T. 2009. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 16: 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. 2009. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324: 1713–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabner B, Schramek D, Mueller KM, Moll HP, Svinka J, Hoffmann T, Bauer E, Blaas L, Hruschka N, Zboray K, et al. 2015. Disruption of STAT3 signalling promotes KRAS-induced lung tumorigenesis. Nat Commun 6: 6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, et al. 2009. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15: 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M. 2007. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11: 291–302. [DOI] [PubMed] [Google Scholar]
- He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, Sieghart W, Peck-Radosavljevic M, Leffert HL, Karin M. 2010. Hepatocyte IKKβ/NF-κB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 17: 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. 2005. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7: 469–483. [DOI] [PubMed] [Google Scholar]
- Ho PL, Lay EJ, Jian W, Parra D, Chan KS. 2012. Stat3 activation in urothelial stem cells leads to direct progression to invasive bladder cancer. Cancer Res 72: 3135–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MDM, Niu B, McLellan MD, Uzunangelov V, et al. 2014. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158: 929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischenko I, Zhi J, Moll UM, Nemajerova A, Petrenko O. 2013. Direct reprogramming by oncogenic Ras and Myc. Proc Natl Acad Sci 110: 3937–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischenko I, Liu J, Petrenko O, Hayman MJ. 2014a. Transforming growth factor-β signaling network regulates plasticity and lineage commitment of lung cancer cells. Cell Death Differ 21: 1218–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischenko I, Petrenko O, Hayman MJ. 2014b. Analysis of the tumor-initiating and metastatic capacity of PDX1-positive cells from the adult pancreas. Proc Natl Acad Sci 111: 3466–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischenko I, Petrenko O, Hayman MJ. 2015. A MEK/PI3K/HDAC inhibitor combination therapy for KRAS mutant pancreatic cancer cells. Oncotarget 6: 15814–15827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhan JR, Andrechek ER. 2016. Stat3 accelerates Myc induced tumor formation while reducing growth rate in a mouse model of breast cancer. Oncotarget 7: 65797–65807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. 2013. Mutational landscape and significance across 12 major cancer types. Nature 502: 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T. 2005. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121: 823–835. [DOI] [PubMed] [Google Scholar]
- Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE. 2009. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc 4: 1670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AW, Pattabiraman DR, Weinberg RA. 2017. Emerging biological principles of metastasis. Cell 168: 670–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim JC, Lee SE, Quinley C, Kim H, Herdman S, Corr M, Raz E. 2012. Signal transducer and activator of transcription 3 (STAT3) protein suppresses adenoma-to-carcinoma transition in Apcmin/+ mice via regulation of Snail-1 (SNAI) protein stability. J Biol Chem 287: 18182–18189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, Yoshimura A, Reindl W, Sipos B, Akira S, et al. 2011. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 19: 456–469. [DOI] [PubMed] [Google Scholar]
- Liu K, Jiang M, Lu Y, Chen H, Sun J, Wu S, Ku WY, Nakagawa H, Kita Y, Natsugoe S, et al. 2013. Sox2 cooperates with inflammation-mediated Stat3 activation in the malignant transformation of foregut basal progenitor cells. Cell Stem Cell 12: 304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Meng Y, Kwiatkowski DJ, Chen X, Peng H, Sun Q, Zha X, Wang F, Wang Y, Jing Y, et al. 2010. Mammalian target of rapamycin regulates murine and human cell differentiation through STAT3/p63/Jagged/Notch cascade. J Clin Invest 120: 103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques IJ, Weiss FU, Vlecken DH, Nitsche C, Bakkers J, Lagendijk AK, Partecke LI, Heidecke CD, Lerch MM, Bagowski CP. 2009. Metastatic behaviour of primary human tumours in a zebrafish xenotransplantation model. BMC Cancer 9: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald OG, Maitra A, Hruban RH. 2012. Human correlates of provocative questions in pancreatic pathology. Adv Anat Pathol 19: 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melino G, Memmi EM, Pelicci PG, Bernassola F. 2015. Maintaining epithelial stemness with p63. Sci Signal 8: re9. [DOI] [PubMed] [Google Scholar]
- Musteanu M, Blaas L, Mair M, Schlederer M, Bilban M, Tauber S, Esterbauer H, Mueller M, Casanova E, Kenner L et al. 2010. Stat3 is a negative regulator of intestinal tumor progression in Apc(Min) mice. Gastroenterology 138: 1003–1011.e5. [DOI] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA, Thiery JP. 2016. Emt: 2016. Cell 166: 21–45. [DOI] [PubMed] [Google Scholar]
- Pellegrini G, Dellambra E, Golisano O, Martinelli E, Fantozzi I, Bondanza S, Ponzin D, McKeon F, De Luca M. 2001. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci 98: 3156–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pencik J, Schlederer M, Gruber W, Unger C, Walker SM, Chalaris A, Marie IJ, Hassler MR, Javaheri T, Aksoy O, et al. 2015. STAT3 regulated ARF expression suppresses prostate cancer metastasis. Nat Commun 6: 7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Mancera PA, Guerra C, Barbacid M, Tuveson DA. 2012. What we have learned about pancreatic cancer from mouse models. Gastroenterology 142: 1079–1092. [DOI] [PubMed] [Google Scholar]
- Pilati C, Amessou M, Bihl MP, Balabaud C, Nhieu JT, Paradis V, Nault JC, Izard T, Bioulac-Sage P, Couchy G, et al. 2011. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J Exp Med 208: 1359–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putoczki TL, Thiem S, Loving A, Busuttil RA, Wilson NJ, Ziegler PK, Nguyen PM, Preaudet A, Farid R, Edwards KM, et al. 2013. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell 24: 257–271. [DOI] [PubMed] [Google Scholar]
- Qin HR, Kim HJ, Kim JY, Hurt EM, Klarmann GJ, Kawasaki BT, Duhagon Serrat MA, Farrar WL. 2008. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res 68: 7736–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripamonti F, Albano L, Rossini A, Borrelli S, Fabris S, Mantovani R, Neri A, Balsari A, Magnifico A, Tagliabue E. 2013. EGFR through STAT3 modulates ΔN63α expression to sustain tumor-initiating cell proliferation in squamous cell carcinomas. J Cell Physiol 228: 871–878. [DOI] [PubMed] [Google Scholar]
- Roy N, Hebrok M. 2015. Regulation of cellular identity in cancer. Dev Cell 35: 674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy N, Malik S, Villanueva KE, Urano A, Lu X, Von Figura G, Seeley ES, Dawson DW, Collisson EA, Hebrok M. 2015. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev 29: 658–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11: 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler C, Levy DE, Decker T. 2007. JAK–STAT signaling: from interferons to cytokines. J Biol Chem 282: 20059–20063. [DOI] [PubMed] [Google Scholar]
- Shen Y, Schlessinger K, Zhu X, Meffre E, Quimby F, Levy DE, Darnell JE Jr. 2004. Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol Cell Biol 24: 407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark GR, Darnell JE Jr. 2012. The JAK–STAT pathway at twenty. Immunity 36: 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadokoro T, Wang Y, Barak LS, Bai Y, Randell SH, Hogan BL. 2014. IL-6/STAT3 promotes regeneration of airway ciliated cells from basal stem cells. Proc Natl Acad Sci 111: E3641–E3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan EH, Morton JP, Timpson P, Tucci P, Melino G, Flores ER, Sansom OJ, Vousden KH, Muller PA. 2014. Functions of TAp63 and p53 in restraining the development of metastatic cancer. Oncogene 33: 3325–3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong AB, Kretz M, Ridky TW, Kimmel R, Khavari PA. 2006. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev 20: 3185–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varon D, Shai E. 2015. Platelets and their microparticles as key players in pathophysiological responses. J Thromb Haemost 13: S40–S46. [DOI] [PubMed] [Google Scholar]
- Wakefield LM, Hill CS. 2013. Beyond TGFβ: roles of other TGFβ superfamily members in cancer. Nat Rev Cancer 13: 328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Yu Y, Sun C, Liu T, Liang T, Zhan L, Lin X, Feng XH. 2016. STAT3 selectively interacts with Smad3 to antagonize TGF-β signalling. Oncogene 35: 4422. [DOI] [PubMed] [Google Scholar]
- Wen Z, Zhong Z, Darnell JE Jr. 1995. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82: 241–250. [DOI] [PubMed] [Google Scholar]
- Winslow MM, Dayton TL, Verhaak RG, Kim-Kiselak C, Snyder EL, Feldser DM, Hubbard DD, DuPage MJ, Whittaker CA, Hoersch S, et al. 2011. Suppression of lung adenocarcinoma progression by Nkx2-1. Nature 473: 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Weinberg RA. 2008. Epithelial–mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 14: 818–829. [DOI] [PubMed] [Google Scholar]
- Yoh K, Prywes R. 2015. Pathway regulation of p63, a director of epithelial cell fate. Front Endocrinol 6: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Lee H, Herrmann A, Buettner R, Jove R. 2014. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 14: 736–746. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Raje V, Yakovlev VA, Yacoub A, Szczepanek K, Meier J, Derecka M, Chen Q, Hu Y, Sisler J, et al. 2013. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J Biol Chem 288: 31280–31288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Shi G, Schmidt CM, Hruban RH, Konieczny SF. 2007. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am J Pathol 171: 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.