

In this study, Smith et al. investigated how cells modulate chromosome mobility in response to DNA damage. They show that global chromosome mobility is regulated by the Rad51 recombinase and its mediator, Rad52, and their findings indicate that interplay between recombination factors and the checkpoint restricts increased mobility until recombination proteins are assembled at damaged sites.
Keywords: DNA repair, homologous recombination, chromosome mobility, homology search, DNA damage checkpoint signaling
Abstract
During homologous recombination, cells must coordinate repair, DNA damage checkpoint signaling, and movement of chromosomal loci to facilitate homology search. In Saccharomyces cerevisiae, increased movement of damaged loci (local mobility) and undamaged loci (global mobility) precedes homolog pairing in mitotic cells. How cells modulate chromosome mobility in response to DNA damage remains unclear. Here, we demonstrate that global chromosome mobility is regulated by the Rad51 recombinase and its mediator, Rad52. Surprisingly, rad51Δ rad52Δ cells display checkpoint-dependent constitutively increased mobility, indicating that a regulatory circuit exists between recombination and checkpoint machineries to govern chromosomal mobility. We found that the requirement for Rad51 in this circuit is distinct from its role in recombination and that interaction with Rad52 is necessary to alleviate inhibition imposed by mediator recruitment to ssDNA. Thus, interplay between recombination factors and the checkpoint restricts increased mobility until recombination proteins are assembled at damaged sites.
DNA damage-induced chromosomal mobility is a well-conserved and important facet of the cellular response to DNA damage that is positioned at the intersection of repair and checkpoint pathways (Seeber and Gasser 2017; Smith and Rothstein 2017). In Saccharomyces cerevisiae, both damaged and undamaged chromosomal loci expand their exploration of nuclear space in response to the formation of DNA double-strand breaks (DSBs) (Dion et al. 2012; Mine-Hattab and Rothstein 2012; Seeber et al. 2013). Sites close to the location of a DSB undergo a large increase in nuclear exploration, termed local mobility (an ∼10-fold increase in explored volume), while undamaged sites elsewhere in the nucleus (on both homologous and nonhomologous chromosomes) undergo a smaller increase, termed global mobility (an approximately fourfold increase). We propose that local mobility primarily drives homology search, while global mobility is also induced to promote efficient pairing in the crowded nucleus (Mine-Hattab and Rothstein 2013).
The ability of cells to modulate chromosomal mobility in response to DNA damage is conserved throughout evolution from yeast to humans. In S. cerevisiae and Schizosaccharomyces pombe, dynamic telomere-led movements may serve to promote stringent pairing of homologs during meiosis (Yamamoto and Hiraoka 2001; Koszul and Kleckner 2009). Also, in S. pombe, persistent DSBs migrate to the nuclear periphery and become associated with microtubule-linked KASH domain proteins (Swartz et al. 2014). In mammalian cells, expansions in chromosome mobility occur after the formation of DSBs (Aten et al. 2004) or the uncapping of telomeres (Dimitrova et al. 2008). 53BP1-mediated mobility is important for both DNA damage repair and the avoidance of ectopic recombination (Lottersberger et al. 2015). Furthermore, the directional movement of damaged telomeres has also been observed in human ALT (alternative lengthening of telomeres) cells (Cho et al. 2014). In Drosophila and human cells, damaged heterochromatic sites move into subdomains of the nucleus where recombination is permitted (Chiolo et al. 2011; Tsouroula et al. 2016; Amaral et al. 2017).
What are the factors that control chromosomal mobility? The checkpoint factors Mec1, Rad9, and Rad53 are required for both local and global mobility (Dion et al. 2012; Seeber et al. 2013). Sae2 generates ssDNA substrates that are necessary for checkpoint protein loading and contributes to the timing of local mobility following a DSB (Mine-Hattab and Rothstein 2012). Several models have been proposed for how the DNA damage checkpoint regulates the physical motion of chromosomes. Checkpoint-induced chromatin remodeling has been suggested as a possible mechanism for local as well as global mobility (Seeber et al. 2013; Hauer et al. 2017), as have changes in cytoskeletal elements such as microtubules and nuclear actin (Spichal et al. 2016; Strecker et al. 2016; Lawrimore et al. 2017). Recent evidence suggests that changes to the DNA polymer underlie altered chromosome mobility both locally and globally (Amitai et al. 2017; Herbert et al. 2017; Mine-Hattab et al. 2017). It has also been observed that the recombination machinery is critical for increased local mobility after DNA damage. Both RAD51 (Mine-Hattab and Rothstein 2012) and RAD54 (Dion et al. 2012) are required; however, the mechanism for this requirement remains unclear.
Although recent work has focused on how the DNA damage checkpoint and its downstream effectors control chromosomal mobility, it is unknown how recombination factors such as Rad51 and Rad52 cooperate with the checkpoint to transduce a damage signal into a global nuclear reorganization. In this study, we show how the recombination machinery controls this reorganization. We found that the Rad51 recombinase is required for global mobility after DNA damage in diploid cells. Moreover, the recombination mediator Rad52 is also required for global mobility. Interestingly, a rad51Δ rad52Δ double-mutant strain displays constitutively increased global mobility driven by an activated DNA damage checkpoint, indicating that the proper recruitment of these two proteins impinges on checkpoint signaling and mobility. The recruitment of Rad51 to DNA, but not its recombination functions, is required for global mobility. Furthermore, the interaction between Rad51 and Rad52 at ssDNA is required to signal competence for global mobility. Our results demonstrate that a regulatory circuit between the recombination machinery and the DNA damage checkpoint ensures that globally increased chromosomal mobility occurs only after the proper assembly of recombination proteins at sites of DNA damage.
Results
Rad51 is required for global mobility
To determine how the recombination machinery affects global mobility, we used a previously reported system for visualizing chromosome movements (Mine-Hattab and Rothstein 2012). Briefly, as shown in Figure 1A, this system includes a multiple tandem array of tetO sequences inserted at the URA3 locus on chromosome V, which is bound by an RFP-tagged TetR protein. A YFP-tagged component of the spindle pole body, Spc110, acts as a reference for cellular motion. A repair protein that colocalizes to DSBs (Rad52 or Ddc1) is tagged with CFP to detect breaks (Lisby et al. 2001). We imaged early S-phase cells every 10 sec using three-dimensional (3D) epifluorescence microscopy to measure the positions of the tetO array and the spindle pole body. As shown previously, the mean square displacement (MSD) curves of the tracked chromosomal array plateau, which is indicative of confined Brownian diffusion (Marshall et al. 1997; Heun et al. 2001). The radius of that confinement (Rc) can then be inferred from the plateau of the fitted MSD curve.
Figure 1.
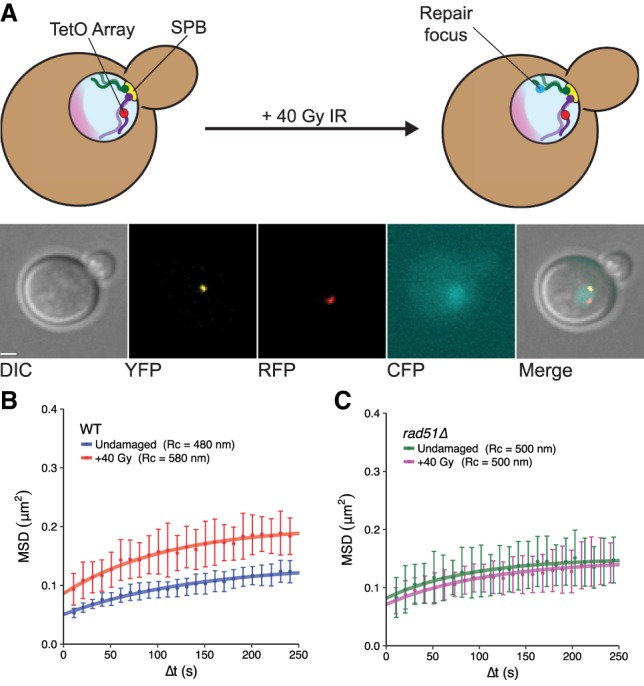
Rad51 is required for DNA damage-induced global mobility. (A) Diagram of the chromosomal locus tracking system. The tetO array is situated at the URA3 locus on one copy of chromosome V in diploid yeast. The other homolog is unlabeled. RFP-tagged TetR associates with the tetO array, forming foci. Spc110 is tagged with YFP to serve as a reference point for cellular and nuclear motion, and Rad52 or Ddc1 is tagged with CFP to assess whether DSBs have formed in a given cell. The nucleolus is shaded pink. Following irradiation with ionizing radiation (IR), random DSBs form and are visualized by the assembly of repair foci. Images below the schematic show z-series projections of a representative undamaged cell of this system (with each channel indicated) following deconvolution. Bar, 1 µm. (B) MSDs for the tagged URA3 locus in wild-type strains either undamaged (blue) or irradiated with 40 Gy of γ-radiation (red) show global mobility. (C) As in B, in a rad51Δ strain, undamaged cells (green) are plotted alongside irradiated cells (magenta). All mobility experiments were performed in Rad52-CFP-tagged strains. Error bars of MSD plots represent the 95% confidence interval (CI).
To induce global mobility, we produced random DSBs by treating cells with ionizing radiation (IR) (Mine-Hattab and Rothstein 2012). Approximately four DSBs per cell are created after 40 Gy of γ-radiation (Ma et al. 2008). Because chromosome V is small (∼2% of the genome), IR-induced DSBs form there infrequently. Therefore, any observed increase in Rc reflects global mobility. As expected, 40 Gy of IR produces a global mobility response at the URA3 locus (undamaged: Rc = 480 nm ± 30 nm; +40 Gy: Rc = 580 nm ± 40 nm; P-value = 0.01) (Fig. 1B). Previous studies have indicated that the S. cerevisiae RecA homolog RAD51 is critical for local chromosome mobility after DNA damage (Dion et al. 2012, 2013; Mine-Hattab and Rothstein 2012). To assess the contribution of Rad51 to global mobility, we subjected diploid rad51Δ cells to 40 Gy of radiation (Fig. 1C). In contrast to wild type, rad51Δ cells do not display global mobility (undamaged: Rc = 500 nm ± 30 nm; +40 Gy: Rc = 500 nm ± 50 nm; P-value = 0.89). Thus, in diploid cells, Rad51 is required for the mobility of undamaged sites in the context of damage elsewhere in the nucleus.
Rad52 is also required to regulate global mobility
Since Rad51 is important for global mobility, we tested whether deletion of the recombination mediator RAD52, which is required for the recruitment of Rad51 to sites of DNA damage (Fig. 2A), affects this process. To evaluate mobility, we constructed rad52Δ strains and detected DSB formation with CFP-tagged Ddc1. Global mobility does not occur in the absence of Rad52 (undamaged: Rc = 480 nm ± 30 nm; +40 Gy: Rc = 490 nm ± 20 nm; P-value = 0.24) (Fig. 2B). Since global mobility cannot occur in the absence of Rad52 or Rad51, we next tested whether this increase in movement in wild-type cells was a direct consequence of Rad52 recruiting Rad51 to DSBs by constructing double-mutant strains and assaying for mobility. If mobility was controlled purely by such recruitment, rad51Δ rad52Δ cells should not display global mobility following irradiation. Surprisingly, however, we observed that the double mutant displayed elevated mobility in not only damaged cells but also undamaged cells (undamaged: Rc = 580 nm ± 50 nm; damaged: Rc = 600 nm ± 40 nm; P-value = 0.93) (Fig. 2C), suggesting that the regulation of increased mobility provided by these recombination factors is more complex than anticipated. We next investigated the mechanisms underlying this genetic interaction and the roles of both Rad51 and Rad52 during the initiation of global mobility.
Figure 2.
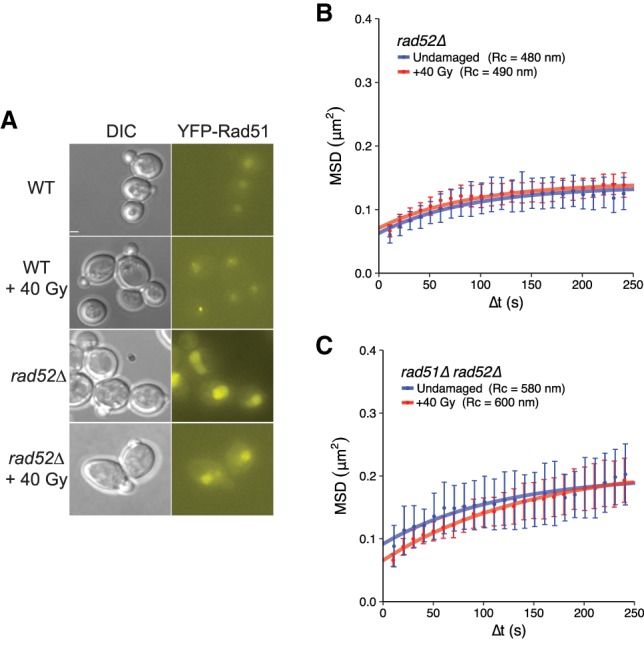
Effects of rad52Δ in single-mutant and rad51Δ rad52Δ double-mutant backgrounds. (A) rad52Δ cells do not form YFP-Rad51 foci effectively after irradiation. The genotypes of the strains depicted are as follows: wild type (RAD52/RAD52 YFP-RAD51/RAD51) and rad52Δ (rad52Δ/rad52Δ YFP-RAD51/RAD51). Bar, 1.8 µm. Wild type, 0 Gy: 6% foci, 152 cells; wild type, +40 Gy: 89.1% foci, 148 cells; rad52Δ, 0 Gy: 3% foci, 270 cells; rad52Δ, +40 Gy: 5% foci, 79 cells. (B) rad52Δ cells display no global mobility response after γ-irradiation (compare undamaged [blue] with irradiated [red]). (C) rad51Δ rad52Δ cells—either undamaged (blue) or irradiated (red)—show elevated mobility in both cases. All mobility experiments were performed in Ddc1-CFP-tagged strains. Error bars of MSD plots represent the 95% CI.
The absence of both Rad51 and Rad52 induces constitutive mobility through elevated checkpoint activation
Previous studies indicated that the DNA damage checkpoint is important for local and global mobility in haploids (Dion et al. 2012; Seeber et al. 2013; Strecker et al. 2016; Hauer et al. 2017). We suspected that the increase in mobility observed in rad51Δ rad52Δ cells might be due to increased checkpoint activation. First, to assess the importance of the DNA damage checkpoint in wild-type diploids, we treated cells with the checkpoint kinase inhibitor caffeine (Gentner and Werner 1975; Hall-Jackson et al. 1999; Heffernan et al. 2002) and found that global mobility does not occur after inhibition (undamaged: Rc = 500 nm ± 50 nm; +40 Gy: Rc = 450 nm ± 40 nm; P-value = 0.68) (Fig. 3A). Moreover, artificial checkpoint induction driven by galactose-inducible colocalization of LacI-tagged Ddc1 and Ddc2 in undamaged cells (Bonilla et al. 2008) is sufficient to trigger global mobility (untreated: Rc = 460 nm ± 40 nm; +galactose: Rc = 640 nm ± 90 nm; P-value = 0.02) (Fig. 3B). This induction of mobility is also sensitive to caffeine (+galactose, +caffeine: Rc = 400 nm ± 20 nm; P-value [compared with +galactose] < 0.001) (Fig. 3B) and requires a lacO array for colocalization (Supplemental Fig. S1B). Therefore, the DNA damage checkpoint is necessary and sufficient for global mobility in wild-type diploid cells.
Figure 3.
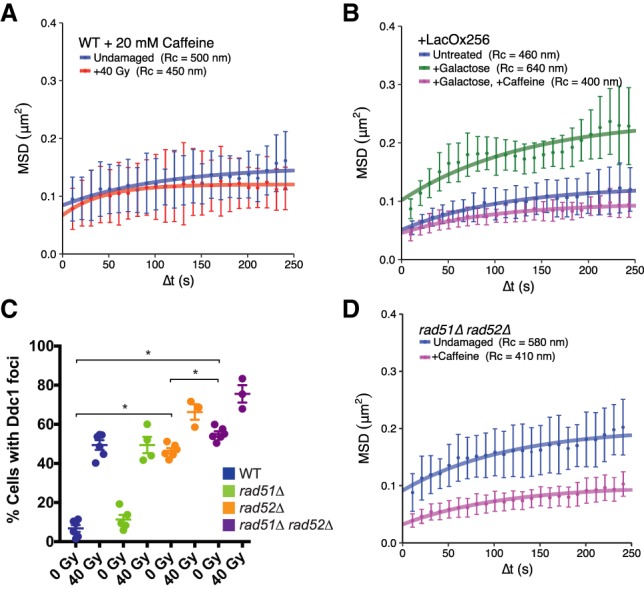
rad51Δ rad52Δ cells display elevated mobility as a result of an activated DNA damage checkpoint. (A) Both undamaged (blue) and irradiated (red) cells treated with 20 mM caffeine show similar confinement radii, demonstrating that a checkpoint response is required for global mobility. (B) Colocalization of Ddc1-LacI and Ddc2-LacI (expressed after their induction by galactose) leads to artificial checkpoint activation and global mobility in the absence of DNA damage (see the Materials and Methods; Supplemental Fig. S1A). Strains containing a lacO array for colocalization of these sensors (either uninduced [blue], induced [green], or induced in the presence of caffeine [magenta]) are shown. A control strain in which the lacO array has not been included does not undergo global mobility after galactose induction (Supplemental Fig. S1B). (C) Counts of Ddc1 foci in undamaged and damaged wild-type, rad51Δ, rad52Δ, and rad51Δ rad52Δ cells. Each point represents the percentage of cells with Ddc1 foci in a given experiment. Error bars are mean ± SEM. (*) P-value < 0.05, as calculated by unpaired t-test. (D) Undamaged rad51Δ rad52Δ cells treated with 20 mM caffeine (magenta) and untreated (blue) show that constitutive mobility in the double mutant is due to an activated checkpoint. Mobility experiments in A and B were performed in Rad52-CFP-tagged cells, while those in D were performed in Ddc1-CFP-tagged cells. Error bars of MSD plots represent the 95% CI.
Based on the fact that the rad51Δ rad52Δ double mutant exhibits constitutive mobility, we hypothesized that the DNA damage checkpoint is activated in these cells. To test this idea, we examined how deletion of RAD51 and RAD52 affects checkpoint activation. We assessed both phosphorylation of the checkpoint effector kinase Rad53 (Sun et al. 1996) and recruitment of the checkpoint adaptor Ddc1 (Melo et al. 2001) to sites of DNA damage in wild-type, rad51Δ, rad52Δ, and rad51Δ rad52Δ double-mutant cells. While we did not observe high levels of phosphorylated Rad53 in double-mutant cells (Supplemental Fig. S1C), spontaneous Ddc1 foci were significantly increased in these backgrounds, indicating that in rad52Δ and rad51Δ rad52Δ cells, the engagement of checkpoint complexes is highly elevated compared with wild type (wild type, undamaged: 7.4%; rad52Δ: 46% [P-value {compared with wild type} < 0.001]; rad51Δ rad52Δ: 56% [P-value {compared with wild type} < 0.001]) (Fig. 3C). Notably, there is a small but significant increase in the levels of Ddc1 foci formed in rad51Δ rad52Δ cells compared with rad52Δ (P-value = 0.001), suggesting that the loss of both repair proteins leads to an additive increase in checkpoint factor recruitment.
Since the DNA damage checkpoint is critical for global mobility and since rad51Δ rad52Δ cells activate the checkpoint, we measured mobility in rad51Δ rad52Δ mutants treated with caffeine to determine whether the aberrant checkpoint activation in these cells drives constitutive increased global mobility. Caffeine treatment completely blocks increased mobility in undamaged rad51Δ rad52Δ cells (Rc = 410 nm ± 40 nm; P-value [compared with untreated] [Fig. 2C] = 0.01) (Fig. 3D). Therefore, the constitutive mobility caused by loss of both Rad51 and Rad52 is a consequence of an activated DNA damage checkpoint. This result, along with the results outlined above, argues that the recombination machinery defines a regulatory circuit that controls the activation of increased mobility following damage and checkpoint activation. We next investigated the molecular mechanisms of this regulation.
The interaction between Rad51 and Rad52 modulates the activation of global mobility
Because the absence of Rad51 and Rad52 leads to increased checkpoint engagement and mobility, we reasoned that these two repair proteins establish an inhibitory circuit that restricts global mobility to contexts in which the recombination machinery is properly assembled. To test this hypothesis, we constructed separation-of-function mutant strains of Rad51 and Rad52. First, we examined rad51-II3A mutant strains, in which Rad51 can form filaments on resected ssDNA but cannot catalyze strand exchange (Cloud et al. 2012), in order to determine whether the regulation of global mobility is a product of recombination per se or a consequence of repair factor binding to ssDNA. We observed that the mobility of the URA3 locus in undamaged rad51-II3A cells is slightly elevated compared with wild type (although not statistically significant [rad51-II3A undamaged: Rc = 510 nm ± 40 nm] compared with wild-type undamaged [data from Figs. 1B, 4A]; P-value = 0.71) and that mobility is significantly increased after irradiation when compared with undamaged wild-type cells (rad51-II3A, +40 Gy: Rc = 580 nm ± 60 nm; P-value for wild-type undamaged [data from Fig. 1B] compared with rad51-II3A, +40 Gy: 0.03) (Fig. 4A). Thus, the Rad51-II3A protein allows global mobility despite its inability to allow recombination, suggesting that the association of Rad51 with DNA, rather than its recombination functions, controls global chromosomal mobility.
Figure 4.
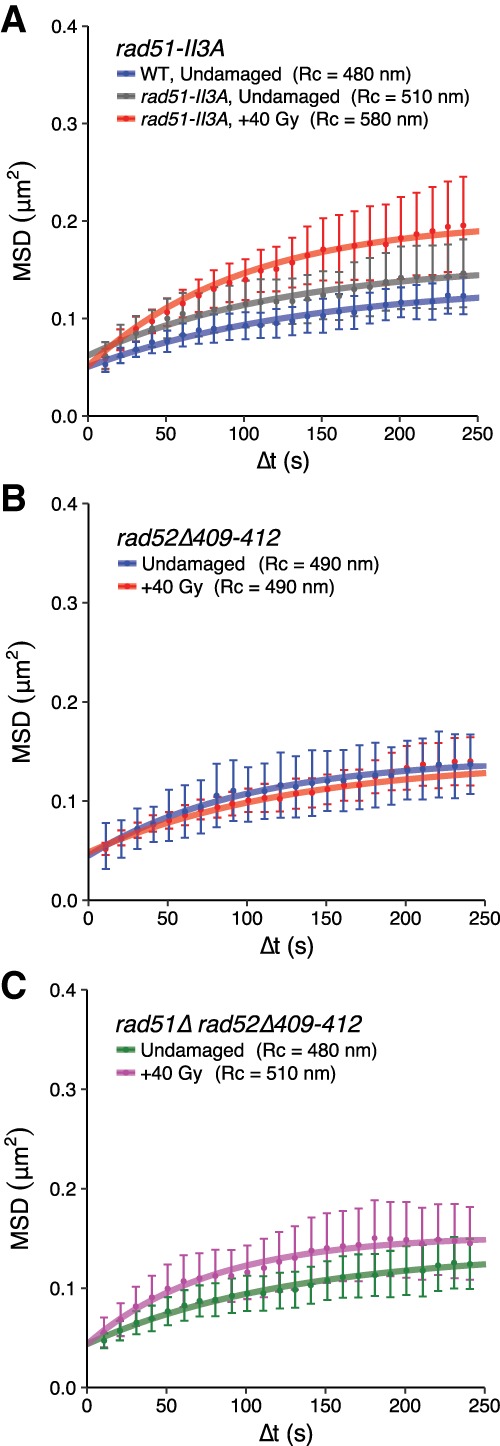
The interaction of Rad51 and Rad52 is required for global mobility. (A) Strand exchange functions are dispensable for global mobility after DNA damage. rad51-II3A cells display slightly elevated mobility (gray) compared with wild type (blue) and undergo an increase in mobility when damaged (red). (B) rad52Δ409–412 cells (either undamaged [blue] or irradiated [red]) show no increase in mobility. (C) No global mobility is observed in undamaged (green) or irradiated (magenta) rad51Δ rad52Δ409–412 cells. The MSD experiments in A were performed in Rad52-CFP-tagged cells, while those in B and C were performed in Ddc1-CFP-tagged cells. Error bars of MSD plots represent the 95% CI.
The association of Rad51 with DNA requires an interaction with Rad52, and this interaction may be critical for global mobility. To directly assess whether this interaction is required for global mobility, we examined rad52Δ409–412 strains in which the Rad51 interaction domain within Rad52 has been deleted. This mutant Rad52 protein can bind to ssDNA and catalyze annealing reactions but cannot interact with Rad51 and has defects in homologous recombination (Krejci et al. 2002). We found that these mutant cells are completely deficient for global mobility (undamaged: Rc = 490 nm ± 40 nm; damaged: Rc = 490 nm ± 30 nm; P-value = 0.63) (Fig. 4B). Thus, the interaction between Rad51 and Rad52 is required for global mobility. Since a rad51Δ rad52Δ double mutant has constitutive global mobility, we examined the effect of this mutant protein in combination with a rad51Δ. Unlike the rad51Δ rad52Δ double-deletion strain, the rad51Δ rad52Δ409–412 double mutant shows a total loss of global mobility (undamaged: Rc = 480 nm ± 30 nm; damaged: Rc = 510 nm ± 40 nm; P-value = 0.51) (Fig. 4C). These results indicate that the association of the mutant Rad52 protein with ssDNA in a background lacking Rad51 is sufficient to prevent the constitutive mobility observed in a rad51Δ rad52Δ double mutant.
These results suggest that the inhibitory circuit established by Rad51 and Rad52 to control global mobility is mediated through their interaction. We hypothesized, based on the observation that global mobility does not occur in rad51Δ rad52Δ409–412 (unlike rad51Δ rad52Δ), that the recruitment of Rad52 to ssDNA blocks global mobility until Rad51 can be recruited. Following an interaction between these two proteins, Rad52 would be displaced, thus alleviating the inhibition. We suspected that mobility is blocked in rad52Δ409–412 cells because the mutant Rad52 protein cannot be evicted from ssDNA due to loss of its interaction with Rad51. This hypothesis predicts that in rad52Δ409–412 cells, mutant Rad52 protein will colocalize with Rad51 more frequently than in wild type.
To test the hypothesis, we measured colocalization of Rad51 and Rad52 on ssDNA by visualizing tagged wild-type Rad51 alongside either wild-type Rad52-CFP or mutant Rad52Δ409–412-CFP (Fig. 5A). In rad52Δ409–412-CFP/rad52Δ409–412 cells, we observed a dramatic decrease in the intensity of Rad51 foci (mean focus intensity ± SEM is 120,000 AU ± 17,000 AU in wild type [n = 68 foci] and 7600 AU ± 1100 AU in rad52Δ409–412-CFP/rad52Δ409–412 [n = 45 foci]; P-value < 0.001, unpaired t-test) (Fig. 5A), indicating that the Rad51 interaction domain in Rad52 is required for proper Rad51 focus formation. In irradiated wild-type cells, similar to previous reports (Lisby et al. 2004; Burgess et al. 2009), there are more Rad51 foci than Rad52 foci in the population (67% and 35% of cells, respectively) (Fig. 5B). A minority of Rad51 foci (32%) colocalizes with Rad52, while 61% of Rad52 foci colocalize with Rad51 (Fig. 5C). Consistent with the notion that the absence of the interaction domain leads to defects in Rad52 removal, we observed an increase in the overall levels of Rad52Δ409–412 foci after irradiation compared with wild-type Rad52 (35% for wild type and 60% for rad52Δ409–412-CFP/rad52Δ409–412; P-value = 0.007) (Fig. 5B) as well as a twofold increase in the proportion of Rad51 foci that colocalizes with Rad52Δ409–412-CFP (32% for wild type and 65% for rad52Δ409–412-CFP/rad52Δ409–412; P-value < 0.001) (Fig. 5C). There is also a concomitant decrease in the number of Rad52Δ409–412-CFP foci that colocalize with Rad51 (61% for wild-type and 41% for rad52Δ409–412-CFP/rad52Δ409–412; P-value = 0.03) (Fig. 5C), likely due to the inability of Rad52Δ409–412 to adequately recruit Rad51. These results support the hypothesis that in the absence of the Rad51 interaction domain, recruitment of the recombinase is defective, and removal of Rad52 from ssDNA is impaired.
Figure 5.
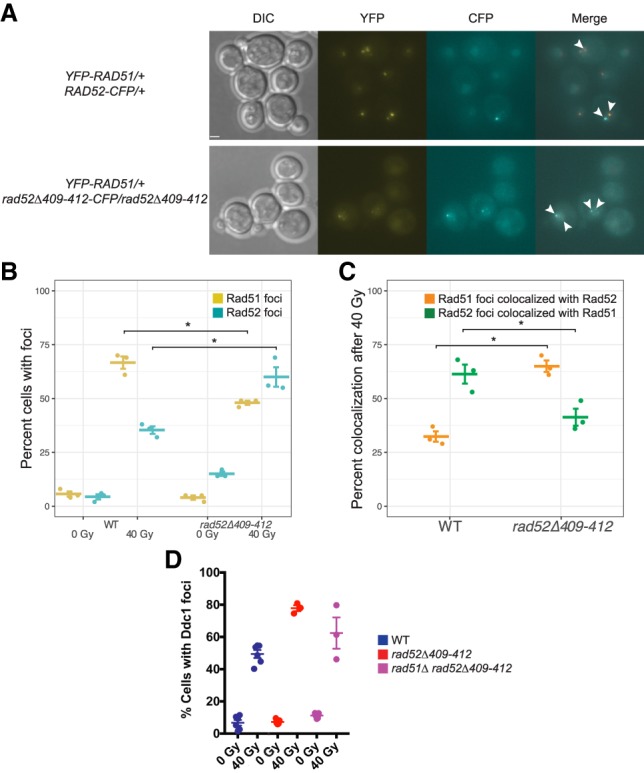
The Rad52Δ409–412 protein affects recombinase and checkpoint factor recruitment. (A) Images of either YFP-RAD51/+ RAD52-CFP/+ or YFP-RAD51/+ rad52Δ409–412-CFP/rad52Δ409–412 cells after treatment with 40 Gy of IR. Rad51 foci are significantly weaker in rad52Δ409–412 cells. Mean focus intensity ± SEM is 120,000 AU ± 17,000 AU in wild type (n = 68 foci) and 7600 AU ± 1100 AU in rad52Δ409–412-CFP/rad52Δ409–412 (n = 45 foci). P-value < 0.001, unpaired t-test. Colocalized Rad51 and Rad52 foci are marked with arrowheads. Bar, 1.7 µm. (B) The percentage of cells with Rad51 (yellow) or Rad52 (cyan) foci before and after irradiation in wild-type (left) and rad52Δ409–412-CFP/rad52Δ409–412 (right) cells. Each point represents a biological replicate with mean and SEM plotted. (C) The percentage of total Rad51 foci that colocalize with Rad52 foci (orange) or Rad52 foci that colocalize with Rad51 (green) in irradiated wild-type or rad52Δ409–412-CFP/rad52Δ409–412 cells, as in B. (D) Ddc1 focus counts, performed as in Figure 3C. Wild-type data are from Figure 3C. (*) P-values < 0.05, as calculated by unpaired t-test.
We next asked whether the mutant Rad52 protein retained at ssDNA affects checkpoint factor recruitment and activation. We examined checkpoint activation in rad52Δ409–412 and rad51Δ rad52Δ409–412 cells by measuring Rad53 phosphorylation and Ddc1 focus formation. In the absence of irradiation, there are no major differences in Rad53 phosphorylation between rad52Δ409–412 and rad52Δ as well as between rad51Δ rad52Δ409–412 and rad51Δ rad52Δ (Supplemental Fig. S1C). On the other hand, we observed that both rad52Δ409–412 and rad51Δ rad52Δ409–412 cells have reduced levels of spontaneous Ddc1 foci compared with rad52Δ and rad51Δ rad52Δ, respectively (47% for rad52Δ and 7% for rad52Δ409–412 [P-value < 0.001]; 55% for rad51Δ rad52Δ and 11% for rad51Δ rad52Δ409–412 [P-value < 0.001]) (cf. Figs. 3C and 5D). Thus, the Rad52Δ409–412 protein, which can bind to DNA but not properly interact with Rad51 (Krejci et al. 2002), suppresses the spontaneous checkpoint activation as well as the constitutive global mobility observed in the rad51Δ rad52Δ double mutant. These results are consistent with a model in which retention of Rad52 at ssDNA in the absence of an interaction with Rad51 negatively regulates both checkpoint activation and downstream global mobility. Altogether, our results demonstrate that global mobility is regulated by the interplay between recombination factors and the DNA damage checkpoint machinery and suggest that the cell integrates the signals from these two pathways to coordinate appropriate changes in nuclear organization in response to DSBs.
Discussion
Our group and others have proposed that DNA damage-induced increased chromosomal mobility is a mechanism for homology search and assists broken DNA ends in efficiently navigating through the complexity of nuclear space to locate homology (for review, see Smith and Rothstein 2017). The results presented here demonstrate that a regulatory circuit between checkpoint and recombination factors governs the signaling events that produce global mobility. Figure 6 depicts a schematic describing the molecular interactions that initiate the global mobility response. In wild-type cells, after a DSB, resection of 5′ ends reveals stretches of ssDNA that rapidly accumulate RPA complexes (Symington et al. 2014). The Ddc2–Mec1 complex as well as the 9-1-1 sliding clamp are then recruited to these resected ends, leading to the phosphorylation of downstream checkpoint factors (Gobbini et al. 2013). While resection proceeds and Rad51 is being recruited, Rad52 inhibits mobility until a proper presynaptic filament structure can be formed, after which Rad51 interacts with Rad52, and RPA and Rad52 are displaced from ssDNA (Sugiyama and Kowalczykowski 2002; Gibb et al. 2014). These transactions between Rad51, Rad52, and RPA—as well as other proteins bound to RPA—serve as a switch with both activating and inhibiting components. The cell uses this cross-talk between the homologous recombination machinery and the DNA damage checkpoint to determine when it is appropriate to increase mobility throughout the nucleus.
Figure 6.
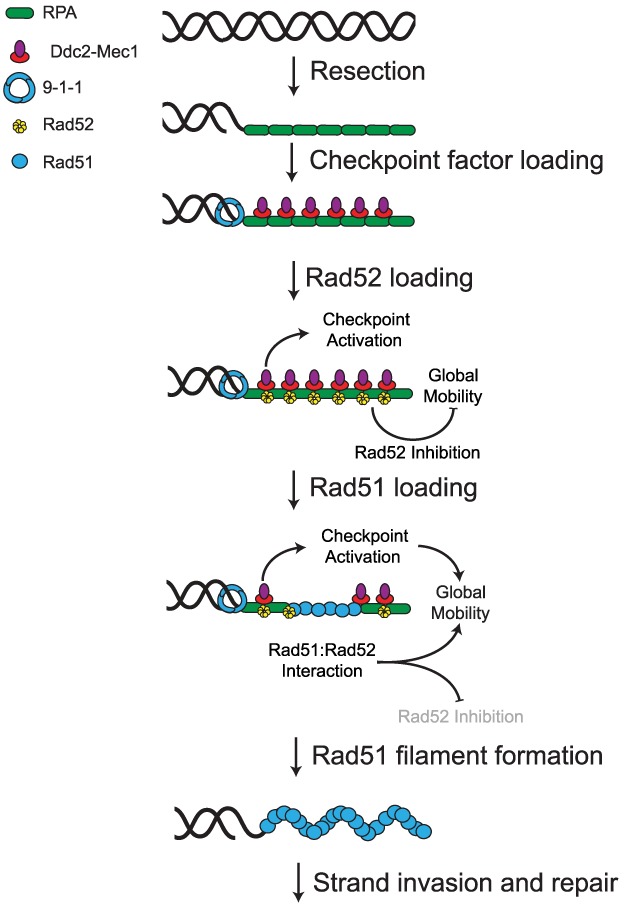
A model for the role of the recombination machinery in regulating global mobility. After DSB formation, end resection yields RPA-bound 3′ ssDNA tracts that serve as platforms for both checkpoint signaling and repair factor loading. At this time, the DNA damage checkpoint becomes active, but the recruitment of Rad52 to the ssDNA inhibits the initiation of global mobility until Rad51 is sufficiently recruited. The interaction of Rad51 with Rad52 alleviates this inhibition (shown by grayed-out Rad52 inhibition), allowing the checkpoint to promote global mobility. Thus, proper assembly of the recombination machinery to sites of damage offers an additional layer of regulatory input to the checkpoint apparatus that restricts the activation of mobility to cells that are committed to homologous recombination.
The mutant strains used in this study reveal important underlying features of this pathway (see Supplemental Fig. S2). First, experiments in rad51-II3A strains reveal that global mobility is regulated at the level of the processed DSB and not during strand exchange or homology search. The notion that global mobility is regulated prior to recombination is reasonable because both the repair proteins and the participants in the Mec1/ATR checkpoint cascade share resected ssDNA as a substrate. This close physical proximity likely facilitates necessary regulatory interactions. Furthermore, results from studies of the rad52Δ409–412 mutant suggest that the Rad52 protein blocks global mobility unless Rad51 triggers its removal (Figs. 4, 5). The simplest explanation for these results is that Rad52 acts as a repressor of global mobility when bound to ssDNA. However, this view is not consistent with the observation that rad52Δ cells are defective in mobility following irradiation (Fig. 2B). Instead, Rad51 may have an activating role beyond that of simply displacing Rad52, suggesting that an interaction between the recombinase and mediator per se is required to initiate mobility. This hypothesis explains the independent requirement for each factor at damage sites. It is important to note that this regulatory circuit can be bypassed, since rad51Δ rad52Δ cells are constitutive for mobility, which we attribute to an increased level of checkpoint engagement (increased spontaneous Ddc1 foci) (Fig. 3C). In fact, the capacity of cells to bypass the requirement for the recombination machinery after a certain level of checkpoint activation has been reached may explain the observation that Rad51 is dispensable for global mobility in zeocin-treated haploid cells (Seeber et al. 2013). Our results clearly demonstrate that checkpoint activation is not the sole determinant for global mobility, since it is not observed in multiple genetic backgrounds where the checkpoint has been activated (Supplemental Fig. S1C). Thus, the interactions between the repair proteins and the checkpoint machinery are important for the appropriate control of global mobility.
How might Rad52 inhibit global mobility? It is possible that Rad52 recruitment to resected ends directly influences checkpoint activation. The Rad52Δ409–412 mutant protein prevents spontaneous checkpoint focus formation singly or in combination with rad51Δ (Figs. 3C, 5D). Reduced checkpoint activation may reflect the strand-annealing activity of the mutant protein, which allows it to properly resolve more lesions than a rad52Δ mutant strain. However, we favor the hypothesis that the association of this mediator with ssDNA prevents checkpoint factor recruitment and activation. In this scenario, the reduction in checkpoint focus formation may occur by Rad52 sterically hindering the binding of checkpoint factors (e.g., Ddc2–Mec1 and the 9-1-1 complex) on the RPA-bound ssDNA substrate. Alternatively, the reduction may occur through a direct protein–protein interaction between Rad52 and checkpoint factors that prevents their activation or accumulation.
How do Rad51 and Rad52 cooperate to drive global mobility? In vitro, RPA is removed from ssDNA following the formation of a tripartite complex between itself, Rad51, and Rad52 (Sugiyama and Kowalczykowski 2002). Therefore, their interaction and the displacement of RPA and associated factors may be the trigger for global mobility to begin. In support of this view, Rad52Δ409–412-CFP foci more often colocalize with Rad51 foci than in wild type, suggesting a defect in the displacement of Rad52 when the interaction between mediator and recombinase is impaired (Fig. 5C). The interaction between Rad51 and Rad52 may also facilitate downstream interactions with repair factors such as Rad54 (Dion et al. 2012) or with checkpoint complexes. For example, Flott et al. (2011) have reported that Rad51 is phosphorylated by Mec1 within its Walker A ATPase domain, and we suspected that this interaction might affect mobility. To investigate this connection, we examined a phosphorylation-dead mutant at this site (rad51-S192A) as well as a phosphomimetic mutant (rad51-S192E) and found that both are defective for global mobility (Supplemental Fig. S1D,E). However, since both mutations cause a strong ssDNA-binding defect (Flott et al. 2011), it is possible that the lack of mobility observed in these mutants is due to their inability to displace Rad52. The importance of the Rad51–Rad52 interaction has been observed previously in studies of checkpoint adaptation (Lee et al. 2003) and may underlie other mechanisms by which the cell senses the progression of DSB end processing and repair.
Studies in haploid cells have implicated the checkpoint in the control of global mobility and proposed various mechanisms by which mobility is generated. Seeber et al. (2013) reported that checkpoint activation targets members of the INO80 chromatin remodeling complex to produce global mobility, and a later study by Hauer et al. (2017) reported that changes in histone occupancy on DNA following damage may be consequential for chromatin confinement and mobility. Strecker et al. (2016) reported that the activation of the DNA damage checkpoint leads to phosphorylation of the kinetochore protein Cep3 and suggested that modulation of centromere to kinetochore attachments alters chromosome tethering to promote both local and global mobility. In addition, DNA damage causes a release of telomeric sequences as well as expansion of pericentric chromatin domains (Lawrimore et al. 2017). Here, we found in diploid cells that the DNA damage checkpoint is necessary and sufficient for the induction of global mobility (Fig. 3A,B) and that the activity of Rad51 and Rad52 affects checkpoint factor engagement and downstream mobility. Our discovery of this relationship not only reinforces the importance of the DNA damage checkpoint but also uncovers a novel role for the recombination machinery in regulating global mobility.
Why might the cell link the initiation of mobility to the recruitment of recombination factors? A simple explanation is that it is advantageous for the cell to restrict the activation of mobility to only those contexts in which it has committed to homologous recombination (i.e., following resection) and has properly assembled the mechanical complexes, such as the Rad51 presynaptic filament. Thus, tying mobility to homologous recombination progression and checkpoint activation adds a layer of stringency that prevents increased mobility at inappropriate times or after an inappropriate damage stimulus. For example, mistimed mobility could result in translocations or chromosome loss. The notion that mobility is restricted to contexts in which homologous recombination is favorable may explain the observations that diploids, which can use the homolog for recombination, undergo mobility more readily than haploids, which are limited to sister chromatid exchange (Dion et al. 2012; Mine-Hattab and Rothstein 2012). Alternatively, increased mobility could disrupt nonhomologous contacts through mechanical force during the homology search, serving as a stringency mechanism (Lottersberger et al. 2015). In either case, the linkage between recombination progression and checkpoint activation is undoubtedly critical for the control of mobility.
Materials and methods
Strains
Unless noted otherwise, all strains are RAD5+ derivatives of W303 (Thomas and Rothstein 1989; Zhao et al. 1998; Reid et al. 2016). Genetic constructs were created as listed in Supplemental Table S1 (McDonald and Rothstein 1994; Reid et al. 2002; Lisby et al. 2003, 2004; Muller et al. 2005; Bonilla et al. 2008; Cloud et al. 2012; Mine-Hattab and Rothstein 2012).
Galactose induction
For DNA damage checkpoint induction experiments, cells were grown overnight in 3- to 5-mL cultures of SC + 2% raffinose at 23°C. In the morning, 2% galactose was added, and strains were induced for 90 min. Strains were then prepared for microscopy.
Caffeine treatment
Caffeine treatment was performed as described previously (Barlow and Rothstein 2009). Briefly, fresh 100 mM stock solutions of caffeine were prepared each experimental day. Cells were treated with 20 mM caffeine for 30 min. Cells were irradiated and imaged in the presence of caffeine.
Checkpoint induction
The DNA damage checkpoint was induced in undamaged cells as described previously (Bonilla et al. 2008). Briefly, Ddc1-GFP-LacI and Ddc2-GFP-LacI constructs were placed under the control of galactose-inducible promoters. A lacO array was inserted on chromosome IV. Following galactose induction, the colocalization of these two damage sensors on the array was sufficient to trigger Mec1 activation and a checkpoint response.
γ-Irradiation
Overnight cultures of strains designated for irradiation were diluted slightly in fresh medium and allowed to grow for 1 h at 23° C. Aliquots of these cultures were then exposed to defined doses of irradiation using a Nordion 220 60Co irradiator and then immediately prepared for imaging.
Immunoblotting
Protein was harvested from cells via precipitation in 5% trichloroacetic acid followed by washing in acetone. Protein preparations were separated by SDS-PAGE (Laemmli 1970) using 10% gels and transferred to nitrocellulose membranes with a Trans-Blot Turbo transfer system (Bio-Rad). Membranes were blotted overnight with either mouse EL7.E1, α-Rad53 monoclonal antibody (1:1000; Abcam), or α-PGK1 22C5D8 monoclonal antibody (1:20,000; Thermo Fisher) as a loading control.
Microscopy
Microscopy was performed as described previously (Lisby et al. 2004; Mine-Hattab and Rothstein 2012). Cells were pelleted from treated or untreated cultures and resuspended at higher density before being placed on a 1.4% agarose slab for visualization. Images were acquired on a Leica DM5500B upright microscope (Leica Microsystems) illuminated with a 100-W mercury arc lamp. High-efficiency filter cubes were used for fluorophore imaging (Chroma 41028, Chroma 31044v2, and Chroma 41002C for YFP, CFP, and RFP, respectively). Images were captured with a Hamamatsu Orca AG cooled digital CCD (charge-coupled device), and analysis of image data was performed with Volocity software (Perkin-Elmer). For mobility experiments, we captured 15 z-stacks spaced by 300 nm every 10 sec for 70 time points. Exposure times were as follows: 30 msec for differential interference contrast (DIC), 100 msec for YFP, 100 msec for RFP, 800 msec for Rad52-CFP, and 2 sec for Ddc1-CFP. CFP images were taken as part of a complete stack of all colors performed before time-lapse imaging began. For Rad51 and Rad52 focus experiments, we captured 21 z-stacks spaced as for mobility experiments. DIC exposure time was 30 msec, and the YFP and CFP exposure time was 800 msec.
Image analysis
Analyses were performed as described previously (Mine-Hattab and Rothstein 2012). Briefly, positions of the tetO array and the spindle pole body were measured every 10 sec. The positions of the spindle pole body were subtracted from the tetO array in order to correct for nuclear motion. Rc values were obtained by averaging individual MSD plots for each cell in an experiment into a mean MSD, fitting the resultant curve, and extracting the plateau value from that curve. For these mean MSD curves, only the first 25%–30% of the MSD values were used to generate a fit to avoid the imprecise MSD values at high Δt. Plateau values were extracted from the fit and used to determine Rc values, which we report to the nearest 10 nm. Error bars on MSD plots represent the 95% confidence interval (CI) at each Δt value. The Y intercept of an MSD curve reflects the accuracy with which the position of the locus can be determined and includes the error of this measurement as well as how much the tracked locus moves during acquisition (for review, see Michalet 2010). Although MSD plots are often depicted passing through the origin, it is worth noting that an MSD curve would only do so in an ideal experiment in which there is no localization error. In addition, Rc values were determined for individual cells and used to calculate one SEM for the Rc values displayed in Supplemental Table S2. Individual Rc values were also used to detect cells with Rc values of >1.5 times the interquartile range for a given experiment, which were excluded from analysis. Individual Rc values were also used for further statistical analysis.
Intensity measurements (Fig. 5A) were also performed using Volocity. Sum intensity values were background-subtracted by multiplying the mean intensity of a non-focus-containing nuclear region by the voxel size of the measured Rad51 foci.
Statistics
We found that the distribution of Rc values for individual undamaged cells is approximately normally distributed with a peak centered around 450 nm. However, damaged cells display distribution with ∼25% less mobile cells and 75% cells with increased Rc (Supplemental Fig. S3). Since this distribution does not meet assumptions of normality, we used the nonparametric two-sided Wilcoxon rank sum test (Mann and Whitney 1947) to estimate the shift in Rc distribution relative to control and determine a P-value for each experiment. Supplemental Table S2 lists all test results. Statistical analyses were performed in R (http://www.R-project.org).
Supplementary Material
Acknowledgments
We thank the entire Rothstein laboratory for experimental feedback and suggestions, and Fred Chang, Roger Greenberg, Ignacio Izeddin, Gaelle Legube, Judith Mine-Hattab, and Eric Greene for critically reading the manuscript. We also thank Luke Berchowitz for fruitful discussion, assistance with protein blotting, and critical reading of the manuscript. We also thank Doug Bishop, Steve Jackson, and David Toczyski for providing strains and reagents. This work was supported by National Institutes of Health grants T32 GM007088 (to M.J.S.), T32 GM008798 (to E.E.B.), T32 CA009503 (to E.E.B.), TL1TR001875 (to E.E.B.), and R35 GM118180 (to R.R.).
Author contributions: M.J.S. and R.R. conceived the study. M.J.S. and E.E.B. performed the methodology, validated the results, and performed the formal analysis. M.J.S. performed the investigation. R.R. acquired the resources. M.J.S. wrote the original draft of the manuscript. M.J.S., E.E.B., and R.R. reviewed and edited the manuscript. M.J.S. visualized the study. R.R. supervised the study, was the project administrator, and acquired the funding.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.317966.118.
References
- Amaral N, Ryu T, Li X, Chiolo I. 2017. Nuclear dynamics of heterochromatin repair. Trends Genet 33: 86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitai A, Seeber A, Gasser SM, Holcman D. 2017. Visualization of chromatin decompaction and break site extrusion as predicted by statistical polymer modeling of single-locus trajectories. Cell Rep 18: 1200–1214. [DOI] [PubMed] [Google Scholar]
- Aten JA, Stap J, Krawczyk PM, van Oven CH, Hoebe RA, Essers J, Kanaar R. 2004. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science 303: 92–95. [DOI] [PubMed] [Google Scholar]
- Barlow JH, Rothstein R. 2009. Rad52 recruitment is DNA replication independent and regulated by Cdc28 and the Mec1 kinase. EMBO J 28: 1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla CY, Melo JA, Toczyski DP. 2008. Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol Cell 30: 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RC, Lisby M, Altmannova V, Krejci L, Sung P, Rothstein R. 2009. Localization of recombination proteins and Srs2 reveals anti-recombinase function in vivo. J Cell Biol 185: 969–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiolo I, Minoda A, Colmenares SU, Polyzos A, Costes SV, Karpen GH. 2011. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144: 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho NW, Dilley RL, Lampson MA, Greenberg RA. 2014. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 159: 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloud V, Chan YL, Grubb J, Budke B, Bishop DK. 2012. Rad51 is an accessory factor for Dmc1-mediated joint molecule formation during meiosis. Science 337: 1222–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, Chen YC, Spector DL, de Lange T. 2008. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456: 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion V, Kalck V, Horigome C, Towbin BD, Gasser SM. 2012. Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat Cell Biol 14: 502–509. [DOI] [PubMed] [Google Scholar]
- Dion V, Kalck V, Seeber A, Schleker T, Gasser SM. 2013. Cohesin and the nucleolus constrain the mobility of spontaneous repair foci. EMBO Rep 14: 984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flott S, Kwon Y, Pigli YZ, Rice PA, Sung P, Jackson SP. 2011. Regulation of Rad51 function by phosphorylation. EMBO Rep 12: 833–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentner NE, Werner MM. 1975. Repair in Schizosaccharomyces pombe as measured by recovery from caffeine enhancement of radiation-induced lethality. Mol Gen Genet 142: 171–183. [DOI] [PubMed] [Google Scholar]
- Gibb B, Ye LF, Kwon Y, Niu H, Sung P, Greene EC. 2014. Protein dynamics during presynaptic-complex assembly on individual single-stranded DNA molecules. Nat Struct Mol Biol 21: 893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobbini E, Cesena D, Galbiati A, Lockhart A, Longhese MP. 2013. Interplays between ATM/Tel1 and ATR/Mec1 in sensing and signaling DNA double-strand breaks. DNA Repair 12: 791–799. [DOI] [PubMed] [Google Scholar]
- Hall-Jackson CA, Cross DA, Morrice N, Smythe C. 1999. ATR is a caffeine-sensitive, DNA-activated protein kinase with a substrate specificity distinct from DNA-PK. Oncogene 18: 6707–6713. [DOI] [PubMed] [Google Scholar]
- Hauer MH, Seeber A, Singh V, Thierry R, Sack R, Amitai A, Kryzhanovska M, Eglinger J, Holcman D, Owen-Hughes T, et al. 2017. Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat Struct Mol Biol 24: 99–107. [DOI] [PubMed] [Google Scholar]
- Heffernan TP, Simpson DA, Frank AR, Heinloth AN, Paules RS, Cordeiro-Stone M, Kaufmann WK. 2002. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol Cell Biol 22: 8552–8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert S, Brion A, Arbona JM, Lelek M, Veillet A, Lelandais B, Parmar J, Fernandez FG, Almayrac E, Khalil Y, et al. 2017. Chromatin stiffening underlies enhanced locus mobility after DNA damage in budding yeast. EMBO J 36: 2595–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heun P, Laroche T, Shimada K, Furrer P, Gasser SM. 2001. Chromosome dynamics in the yeast interphase nucleus. Science 294: 2181–2186. [DOI] [PubMed] [Google Scholar]
- Koszul R, Kleckner N. 2009. Dynamic chromosome movements during meiosis: a way to eliminate unwanted connections? Trends Cell Biol 19: 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci L, Song B, Bussen W, Rothstein R, Mortensen UH, Sung P. 2002. Interaction with Rad51 is indispensable for recombination mediator function of Rad52. J Biol Chem 277: 40132–40141. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- Lawrimore J, Barry TM, Barry RM, York AC, Cook DM, Akialis K, Tyler J, Vasquez P, Yeh E, Bloom K. 2017. Microtubule dynamics drive enhanced chromatin motion and mobilize telomeres in response to DNA damage. Mol Biol Cell 28: 1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Pellicioli A, Vaze MB, Sugawara N, Malkova A, Foiani M, Haber JE. 2003. Yeast Rad52 and Rad51 recombination proteins define a second pathway of DNA damage assessment in response to a single double-strand break. Mol Cell Biol 23: 8913–8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Rothstein R, Mortensen UH. 2001. Rad52 forms DNA repair and recombination centers during S phase. Proc Natl Acad Sci 98: 8276–8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Mortensen UH, Rothstein R. 2003. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat Cell Biol 5: 572–577. [DOI] [PubMed] [Google Scholar]
- Lisby M, Barlow JH, Burgess RC, Rothstein R. 2004. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118: 699–713. [DOI] [PubMed] [Google Scholar]
- Lottersberger F, Karssemeijer RA, Dimitrova N, de Lange T. 2015. 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair. Cell 163: 880–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Resnick MA, Gordenin DA. 2008. Apn1 and Apn2 endonucleases prevent accumulation of repair-associated DNA breaks in budding yeast as revealed by direct chromosomal analysis. Nucleic Acids Res 36: 1836–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann HB, Whitney DR. 1947. On a test of whether one of two random variables is stochastically larger than the other. Ann Math Stat 18: 50–60. [Google Scholar]
- Marshall WF, Straight A, Marko JF, Swedlow J, Dernburg A, Belmont A, Murray AW, Agard DA, Sedat JW. 1997. Interphase chromosomes undergo constrained diffusional motion in living cells. Curr Biol 7: 930–939. [DOI] [PubMed] [Google Scholar]
- McDonald JP, Rothstein R. 1994. Unrepaired heteroduplex DNA in Saccharomyces cerevisiae is decreased in RAD1 RAD52-independent recombination. Genetics 137: 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo JA, Cohen J, Toczyski DP. 2001. Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev 15: 2809–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalet X. 2010. Mean square displacement analysis of single-particle trajectories with localization error: Brownian motion in an isotropic medium. Phys Rev E Stat Nonlin Soft Matter Phys 82: 041914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mine-Hattab J, Rothstein R. 2012. Increased chromosome mobility facilitates homology search during recombination. Nat Cell Biol 14: 510–517. [DOI] [PubMed] [Google Scholar]
- Mine-Hattab J, Rothstein R. 2013. DNA in motion during double-strand break repair. Trends Cell Biol 23: 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mine-Hattab J, Recamier V, Izeddin I, Rothstein R, Darzacq X. 2017. Multi-scale tracking reveals scale-dependent chromatin dynamics after DNA damage. Mol Biol Cell 28: 3133–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller EG, Snydsman BE, Novik I, Hailey DW, Gestaut DR, Niemann CA, O'Toole ET, Giddings TH Jr., Sundin BA, Davis TN. 2005. The organization of the core proteins of the yeast spindle pole body. Mol Biol Cell 16: 3341–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid RJ, Lisby M, Rothstein R. 2002. Cloning-free genome alterations in Saccharomyces cerevisiae using adaptamer-mediated PCR. Methods Enzymol 350: 258–277. [DOI] [PubMed] [Google Scholar]
- Reid RJ, Du X, Sunjevaric I, Rayannavar V, Dittmar J, Bryant E, Maurer M, Rothstein R. 2016. A synthetic dosage lethal genetic interaction between CKS1B and PLK1 is conserved in yeast and human cancer cells. Genetics 204: 807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeber A, Gasser SM. 2017. Chromatin organization and dynamics in double-strand break repair. Curr Opin Genet Dev 43: 9–16. [DOI] [PubMed] [Google Scholar]
- Seeber A, Dion V, Gasser SM. 2013. Checkpoint kinases and the INO80 nucleosome remodeling complex enhance global chromatin mobility in response to DNA damage. Genes Dev 27: 1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MJ, Rothstein R. 2017. Poetry in motion: increased chromosomal mobility after DNA damage. DNA Repair 56: 102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spichal M, Brion A, Herbert S, Cournac A, Marbouty M, Zimmer C, Koszul R, Fabre E. 2016. Evidence for a dual role of actin in regulating chromosome organization and dynamics in yeast. J Cell Sci 129: 681–692. [DOI] [PubMed] [Google Scholar]
- Strecker J, Gupta GD, Zhang W, Bashkurov M, Landry MC, Pelletier L, Durocher D. 2016. DNA damage signalling targets the kinetochore to promote chromatin mobility. Nat Cell Biol 18: 281–290. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Kowalczykowski SC. 2002. Rad52 protein associates with replication protein A (RPA)–single-stranded DNA to accelerate Rad51-mediated displacement of RPA and presynaptic complex formation. J Biol Chem 277: 31663–31672. [DOI] [PubMed] [Google Scholar]
- Sun Z, Fay DS, Marini F, Foiani M, Stern DF. 1996. Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes Dev 10: 395–406. [DOI] [PubMed] [Google Scholar]
- Swartz RK, Rodriguez EC, King MC. 2014. A role for nuclear envelope-bridging complexes in homology-directed repair. Mol Biol Cell 25: 2461–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Rothstein R, Lisby M. 2014. Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics 198: 795–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BJ, Rothstein R. 1989. The genetic control of direct-repeat recombination in Saccharomyces: the effect of rad52 and rad1 on mitotic recombination at GAL10, a transcriptionally regulated gene. Genetics 123: 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsouroula K, Furst A, Rogier M, Heyer V, Maglott-Roth A, Ferrand A, Reina-San-Martin B, Soutoglou E. 2016. Temporal and spatial uncoupling of DNA double strand break repair pathways within mammalian heterochromatin. Mol Cell 63: 293–305. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Hiraoka Y. 2001. How do meiotic chromosomes meet their homologous partners?: lessons from fission yeast. Bioessays 23: 526–533. [DOI] [PubMed] [Google Scholar]
- Zhao X, Muller EG, Rothstein R. 1998. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 2: 329–340. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.