

Here, Erickson et al. studied whether paused RNA polymerase II (Pol II) that piles up near most human promoters is a stable or dynamic target of mechanisms that control entry into productive elongation. They demonstrate that most 5′ paused Pol II throughout the genome is turned over within 2 min and propose that Pol II occupancy near 5′ ends is governed by a cycle of ongoing assembly of preinitiated complexes that transition to pause sites followed by eviction from the DNA template.
Keywords: Spt5, Pol II pausing, transcription elongation, transcription initiation, transcriptional regulation
Abstract
Paused RNA polymerase II (Pol II) that piles up near most human promoters is the target of mechanisms that control entry into productive elongation. Whether paused Pol II is a stable or dynamic target remains unresolved. We report that most 5′ paused Pol II throughout the genome is turned over within 2 min. This process is revealed under hypertonic conditions that prevent Pol II recruitment to promoters. This turnover requires cell viability but is not prevented by inhibiting transcription elongation, suggesting that it is mediated at the level of termination. When initiation was prevented by triptolide during recovery from high salt, a novel preinitiated state of Pol II lacking the pausing factor Spt5 accumulated at transcription start sites. We propose that Pol II occupancy near 5′ ends is governed by a cycle of ongoing assembly of preinitiated complexes that transition to pause sites followed by eviction from the DNA template. This model suggests that mechanisms regulating the transition to productive elongation at pause sites operate on a dynamic population of Pol II that is turning over at rates far higher than previously suspected. We suggest that a plausible alternative to elongation control via escape from a stable pause is by escape from premature termination.
A signature feature of transcription in most metazoans is that RNA polymerase II (Pol II) piles up near start sites so that its density is often many times higher at 5′ ends than it is at positions further downstream. This unusual polymerase distribution was discovered by nuclear run-on and cross-linking analysis of the β-globin, MYC, and Hsp70 genes (Gariglio et al. 1981; Bentley and Groudine 1986; Eick and Bornkamm 1986; Gilmour and Lis 1986). Subsequently, genome-wide studies revealed that the 5′ pileup occurs at most Pol II transcribed genes (Muse et al. 2007; Zeitlinger et al. 2007; Core et al. 2008; Nechaev et al. 2010) and at enhancers (Henriques et al. 2018).
The promoter-proximal pausing model proposes that Pol II piled up near start sites has initiated transcription, elongated ∼30–60 bases, and is stably engaged on the DNA template at this position (Rougvie and Lis 1988; Krumm et al. 1995; Gilmour 2009; Jonkers and Lis 2015; Mayer et al. 2017) but is held back by the negative elongation factors DSIF and NELF (Yamaguchi et al. 1999, 2013; Wu et al. 2003; Cheng and Price 2007). In this paused state, Pol II is the target of regulators that control the transition into productive elongation. Paused Pol II is released to resume elongation by activators and coactivators that recruit the positive elongation factor PTEFb (Cdk9/CyclinT) (Marshall and Price 1995; Yang et al. 2005; Chen et al. 2015b; Yu et al. 2015) that antagonizes the negative elongation factors by phosphorylating them (Wada et al. 1998; Zhou et al. 2012). The model of regulated release of paused Pol II is consistent with many in vivo experiments (Adelman and Lis 2012; Zhou et al. 2012; Jonkers and Lis 2015; Mayer et al. 2017), although an important limitation of the model is that individual Pol II complexes have never actually been observed to pause and then resume elongation. Furthermore, in vitro, stable pauses of the type proposed to occur in vivo have not been seen in the presence of physiological nucleotide concentrations. Moreover, single-molecule studies of Pol II elongation show that pauses do not usually exceed 3 sec (Zamft et al. 2012).
An alternative fate for paused Pol II is eviction from the template by premature termination (Hay et al. 1982; Brannan and Bentley 2012; Wagschal et al. 2012; Chiu et al. 2018). Active Pol II eviction at pause sites has never been observed directly either, but several indirect lines of evidence are consistent with this possibility, including production of short nuclear capped transcripts (Nechaev et al. 2010) and the presence of termination factors at 5′ ends of genes (Brannan et al. 2012; Wagschal et al. 2012; Nilson et al. 2017). On the other hand, studies using triptolide, which blocks open complex formation by inhibiting the TFIIH-associated XPB ATPase (Titov et al. 2011; Alekseev et al. 2017), show that Pol II can persist at 5′ ends without initiation for relatively long periods (Henriques et al. 2013; Jonkers et al. 2014; Chen et al. 2015a; Shao and Zeitlinger 2017). For example, the median half-life of paused Pol II in triptolide-treated Drosophila cells was >15 min (Shao and Zeitlinger 2017), suggesting that turnover by premature termination is not very significant. However, another study, also in Drosophila, that indirectly mapped Pol II based on methyltransferase footprint size reported a more rapid average Pol II turnover rate [t(1/2) ∼ 6.5 min]. Paradoxically, this report found no difference in Pol II turnover between genes with high and low levels of pausing (Krebs et al. 2017). A caveat of all of these studies is that they assume that triptolide prevents recruitment of stable Pol II complexes at promoters—an assertion that has not been experimentally tested. Rapid turnover is also consistent with the short residence times (<1 min) of a fraction of fluorescently labeled Pol II on a reporter gene (Darzacq et al. 2007) and endogenous genes (Steurer et al. 2018), as measured by fluorescence recovery after photobleaching (FRAP) in some but not all studies (Buckley et al. 2014). Because of these conflicting results, it currently remains unresolved whether the target of post-initiation mechanisms that regulate entry of paused Pol II into productive elongation is working on relatively static polymerases or a population that is dynamically turning over.
In addition to pausing after initiation, there is evidence that Pol II can accumulate at start sites in a “poised” or “docked” state that has not initiated synthesis of the RNA chain. This model is supported by studies in Caenorhabditis elegans and quiescent B lymphocytes where Pol II is found near promoters in the absence of a nuclear run-on signal or a transcription bubble (Kouzine et al. 2013; Maxwell et al. 2014). It is not clear whether accumulation of “poised” Pol II at mammalian promoters is a general phenomenon or specific to quiescent cells with low levels of XPB ATPase (Kouzine et al. 2013) that is required for open complex formation.
Transcription initiation and elongation differ dramatically in their sensitivity to high ionic strength. Whereas preinitiation complexes are dissociated by 0.25 M KCl, after synthesis of the first 10 bases, elongation complexes become stable to 0.6 M KCl (Cai and Luse 1987). Consistent with the in vitro results, when yeast or mammalian cells are treated with 0.5 M NaCl, Pol II dissociated from promoter regions but not downstream positions within several genes (Proft and Struhl 2004; Wang et al. 2005). To investigate the dynamics of Pol II at pause sites in human cells, we revisited the question of how blocking recruitment to promoters with high salt affects Pol II occupancy genome-wide using anti-Pol II ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing). When Pol II recruitment was prevented under hypertonic conditions, a highly active turnover mechanism was revealed that caused an almost complete, yet rapidly reversible, loss of Pol II from promoter-proximal pause sites throughout the genome within 2–5 min. This phenomenon required cell viability but was unaffected by the inhibitors DRB and α-amanitin, suggesting that Pol II removal from pause sites does not require normal elongation. During recovery from hypertonic shock, Pol II associated at 5′ ends of genes even when initiation was prevented by triptolide, suggesting that it stably accumulates at start sites in a “poised” preinitiation state, which is probably an intermediate that precedes pausing. Such “poised” preinitiation complexes may account for previous reports of prolonged Pol II persistence at 5′ ends in the absence of initiation. Together, these results demonstrate active ongoing assembly of preinitiated complexes at start sites and rapid eviction from pause sites at far higher rates than previously suspected.
Results
Rapid loss of Pol II from pause sites revealed under hypertonic conditions
We investigated how high salt affects Pol II occupancy on genes in vivo by challenging HCT116 colon carcinoma cells with addition of NaCl to a final concentration of 350 mM monovalent cations followed by anti-Pol II ChIP. As determined by quantitative PCR (qPCR), high-salt treatment caused >90% loss of Pol II from the 5′ ends of representative genes within 2 min (Fig. 1A). NaCl titration showed that as little as 250 mM total salt in the medium for 10 min was sufficient to remove most of the paused Pol II from the 5′ end of the MYC gene (Fig. 1B). To determine whether this trend held genome-wide, we performed anti-Pol II ChIP-seq with a spike-in of cross-linked yeast chromatin. Yeast Pol II cross reacts with the anti-pan-Pol II C-terminal domain (CTD) antibody so that ChIP-seq signals can be normalized to the spike-in (Hu et al. 2015). Following 10 min in 500 mM total salt, there is a widespread depletion of almost 90% of Pol II from promoter-proximal pause sites, as shown in metaplots (Fig. 1C–E) and by inspection of individual genes (Fig. 1G–I). This effect of hypertonic conditions on 5′ paused Pol II is not unique to HCT116, as we observed the same effect in HAP1 myeloid leukemia cells (Supplemental Fig. S1A).
Figure 1.

Rapid removal of Pol II from promoter-proximal pauses sites under hypertonic conditions. (A) Rapid loss of Pol II from 5′ ends in high salt. NaCl was added to HCT116 cells in McCoy's medium (350 mM monovalent cations final) for 2, 5, or 10 min followed by anti-Pol II ChIP. ChIP qPCR signals were normalized to −NaCl control. The mean and SEM for three technical replicates are shown. (B) Salt concentrations ≥250 mM elicit rapid loss of Pol II from 5′ ends. NaCl was added to HCT116 cells for 10 min to the final salt concentrations shown, and Pol II was assayed after 10 min by ChIP qPCR at MYC amplicons. The mean and SEM are shown for two biological replicates. n = 5. (C) Genome-wide loss of paused Pol II from 5′ ends in high NaCl. Metaplots of anti-Pol II ChIP-seq with or without 0.5 M NaCl (final) for 10 min. Mean ChIP signals are normalized to yeast spike-in. One-hundred-base bins are shown in flanking regions −1.5 to +0.5 kb relative to the transcription start site (TSS) and −0.5 to +3.5 kb relative to the poly(A) site. Gene body regions between +500 relative to the TSS and −500 relative to the poly(A) site were divided into 20 equal bins. (D,E) Genome-wide loss of paused Pol II in high NaCl from genes with low and high pausing indices. Metaplots are as in C. The pausing index was calculated for the 9000 genes with the highest signals in control cells. One-thousand genes with the highest and lowest pausing index values were selected. (F) Genome-wide loss of paused Pol II from enhancers in high NaCl. Metaplots are as in C. Enhancer regions from Andersson et al. (2015) (red double arrow) were centered and divided into 20 equal bins. (G,H) University of California at Santa Cruz (UCSC) genome browser screenshots of Pol II ChIP signals as in C. Note the loss of Pol II from 5′ pause sites (red arrows) and transcription into 3′ flanking regions in high salt (blue arrow) (see also Supplemental Fig. S1B,C). Poly(A) sites are shown as orange lines. (I) Pol II ChIP normalized to total mapped reads in HCT116 cells treated with 350 mM NaCl for 10 and 30 min. Note Pol II “creeping” with depletion from the 5′ end (arrows).
We compared the effect of high salt on Pol II occupancy at 5′ ends of genes with low and high levels of promoter-proximal pausing as defined by the pausing index {Pol II density at start site [−500 to +500 relative to the transcription start site (TSS)]/gene body [+501 from the TSS to −500 from the poly(A) site]}. Pol II was equally effectively removed from promoter regions of genes with the lowest (Fig. 1D,G) and highest (Fig. 1E,H) pausing indices. In high salt, Pol II was also rapidly removed from enhancers, where peaks of Pol II density are commonly located close to the start sites of bidirectional transcription that gives rise to eRNAs (Fig. 1F). Pol II removal from pause sites was not peculiar to high-salt treatment, as similar results were obtained in HCT116 cells that were osmotically shocked with high sucrose, which also raises intracellular salt concentrations (Fig. 2A,B).
Figure 2.

(A) Genome-wide loss of paused Pol II from 5′ ends of genes in high sucrose. Sucrose was added to HCT116 cells to 0.63 M to exert osmotic pressure equivalent to 0.35 M NaCl for 10 min, and Pol II occupancy was assayed by ChIP-seq. (B) UCSC genome browser screenshot of Pol II ChIP on the EIF1 gene as in A. (C) UCSC genome browser screenshot of Pol II ChIP on the FSTL3 gene in HCT116 cells that lacks a 5′ peak of paused Pol II. Note Pol II resistance to high salt at genes that lack 5′ paused Pol II (see Supplemental Fig. S1D,E). (D) Pol II ChIP on a transfer RNA (tRNA) Ile gene that is resistant to eviction in high salt (see Supplemental Fig. S1F). (E) Metaplots of relative frequency (RF) of anti-histone H3 ChIP-seq with and without addition of NaCl for 10 min. Relative frequency plots are made by calculating mean read counts for each bin divided by the sum of mean counts in all bins. Note the evidence of nucleosome replacement when paused Pol II is lost in high salt (red arrow) and the maintenance of the nucleosome-free region at the TSS (black arrow). (F) Pol II eviction from 5′ ends in high salt is an active process. HCT116 cells were untreated (control) or permeabilized with 0.3 mg/mL saponin + 150 µM NaN3 for 5 min (saponin), and then NaCl was added for 10 min followed by addition of mouse M12 spike and cross-linking. Pol II occupancy at 5′ ends of five genes was assayed by ChIP normalized to mouse actin and −NaCl controls. Note that the removal of Pol II in high salt is reduced by loss of cell viability. The mean and SEM for two biological replicates and four PCR reactions are shown.
In contrast to 5′ pause sites, Pol II within gene bodies was resistant to hypertonic conditions even after treatment for 30 min (Fig. 1I), consistent with the resistance of elongation complexes to high salt in vivo and in vitro (Cai and Luse 1987; Proft and Struhl 2004; Wang et al. 2005). High-salt treatment did not eliminate Pol II occupancy from the 5′ ends of all genes, however. For the minor class of genes (such as FSTL3, ZNF395, and GRB10), where most Pol II is actively elongating and there is no 5′ pause, NaCl treatment had little effect (Fig. 2C; Supplemental Fig. S1D,E). This observation shows that Pol II engaged at the 5′ ends of genes is not inherently salt-labile. Another exception occurred at transfer RNA (tRNA) genes, where Pol II has been found previously to accumulate for unknown reasons (Raha et al. 2010). Peaks of Pol II at tRNA genes persisted or even increased in magnitude in high salt (Fig. 2D; Supplemental Fig. S1F). The latter result shows that sites of Pol II accumulation in vivo are not necessarily intrinsically sensitive to disruption by high salt and, furthermore, that high salt does not nonspecifically inhibit cross-linking of Pol II to DNA. We conclude that hypertonic stress is associated with a genome-wide net displacement of Pol II that is specific to promoter-proximal pause sites. Pol II removal does not appear to require the hypertonic stress response because it was unaffected by PD-169316, an inhibitor of the p38 MAP kinases that are required for this response (Kayali et al. 2000; Zhou et al. 2016; Supplemental Fig. S1G).
When cells were kept in high salt for 30 min, Pol II appeared to creep toward 3′ ends with depletion from the 5′ regions of gene bodies (Fig. 1I; Supplemental Fig. S1B,C). This scenario is consistent with a block to initiation, as occurs in vitro, combined with slow elongation reminiscent of that reported after oxidative stress (Nilson et al. 2017), which might result from dissociation or modification of an elongation factor. Inhibition of initiation is further suggested by the loss of most TBP from promoter regions of several mRNA-coding genes, but not a tRNA, after addition of high salt (Supplemental Fig. S1H). We also noted that in high salt, Pol II localized far downstream from poly(A) sites (Figs. 1I, 2B; Supplemental Fig. S1B,C), consistent with a termination defect and the synthesis of 3′ extended transcripts under these conditions (Vilborg et al. 2015).
We tested whether nucleosomes become rearranged following loss of paused Pol II in high salt. This possibility is suggested by experiments in Drosophila showing that paused Pol II is anti-correlated with positioned nucleosomes downstream from the TSS (Gilchrist et al. 2010). We observed that histone H3 occupancy increased downstream from start sites (Fig. 2E, red arrow) within 10 min under high-salt conditions, consistent with competition between nucleosomes and paused Pol II (Gilchrist et al. 2010). On the other hand, nucleosome-depleted regions centered over TSSs were maintained in high salt (Fig. 2E, black arrow).
Active removal of Pol II from pause sites when elongation is inhibited
Post-initiation Pol II complexes are stable to at least 0.6 M KCl in vitro (Cai and Luse 1987); however, we observed robust loss of Pol II from pause sites in vivo on exposure to only 0.25 M salt (Fig. 1B). To account for this result, we hypothesized that in high salt, formation of initiation complexes was inhibited and, in addition, that rather than passively dissociating from the template, paused Pol II was actively removed. To test this idea, we asked whether removal of paused Pol II required cell viability. Killing cells by permeabilization with saponin for 5 min in the presence of NaN3 did not reduce Pol II occupancy at pause sites and may even have slightly increased it (Supplemental Fig. S2A). Notably, inhibiting all metabolic processes by saponin/NaN3 treatment strongly stabilized Pol II at pause sites when cells were challenged with high salt (Fig. 2F). We conclude that removal of Pol II from 5′ pause sites under hypertonic conditions is an active process that requires cell viability rather than a passive displacement by high ionic strength.
There are two possible explanations for the rapid and near-complete loss of Pol II from pause sites in high salt: premature termination and release of arrested Pol II into productive elongation. To investigate whether elongation is required for Pol II removal in high salt, we asked whether it was affected by the inhibitors DRB and α-amanitin. DRB inhibits the PTEFb kinase and blocks release of paused Pol II into productive elongation (Marshall and Price 1995). We quantified Pol II occupancy at the TSS in control and DRB-treated HCT116 cells before and after NaCl treatment. As expected, Pol II was distributed as a bimodal peak around the TSS (Fig. 3A), consistent with pausing of divergent transcription complexes (Quinodoz et al. 2014). If loss of Pol II at pause sites in high salt was due to a transition to active elongation, then DRB should prevent it, but this was not the case. In the presence of 100 µM DRB for 60 min, substantial loss of Pol II from pause sites still occurred within 2 min of adding 500 mM NaCl (final) and was almost complete within 5 min (Fig. 3A,B; Supplemental Fig. S2B–E).
Figure 3.
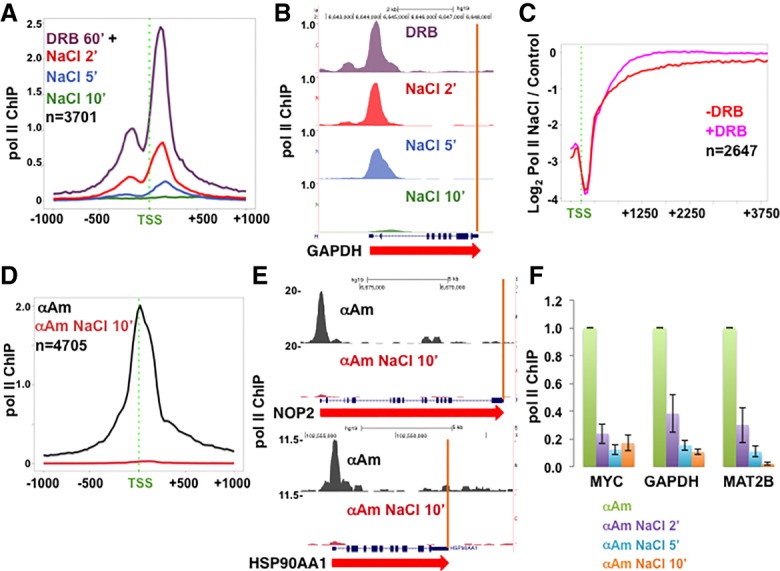
Inhibitors of elongation do not prevent turnover of promoter-proximal Pol II. (A) Turnover of DRB-arrested Pol II at 5′ ends of genes. HCT116 cells were treated with 100 µM DRB for 1 h, and then 350 mM NaCl (final monovalent cations) was added, and Pol II occupancy was assayed by ChIP-seq after 2, 5, and 10 min. Metaplots with 25-base-pair (bp) bins of mean Pol II ChIP-seq normalized to mouse M12 spike-in are shown. (B) UCSC genome browser shot of Pol II ChIP-seq signals on GAPDH as in A. Note the Pol II pileup at the 5′ end and clearance from the gene body in DRB (see also Supplemental Fig. S2B–E). (C) Ratio of Pol II occupancy in HCT116 with or without high NaCl. Pol II ChIP-signals were normalized to yeast spike-ins, and ratios with or without 350 mM NaCl for 10 min at each gene were determined for 100-bp bins. −DRB results are those in Figure 1I, and +DRB (100 µM for 60 min) results are those in Supplemental Figure S2B,C. The genes analyzed are the top one-third ranked for pausing index under control conditions. Note that loss of Pol II in high NaCl at 5′ ends is not compensated for by increased levels at downstream positions. (D,E) Turnover of α-amanitin-arrested Pol II at 5′ ends of genes. HCT116 cells were treated with 5 µg/mL α-amanitin (α-Am) for 24 h (see Supplemental Fig. S2F), and then NaCl was added for 10 min, and Pol II occupancy was assayed by ChIP-seq normalized to a mouse M12 spike-in. (D) Metaplots of mean Pol II ChIP signals. (E) Genome browser shots as in D (see also Supplemental Fig. S2G,H). (F) Rapid turnover of α-amanitin-arrested Pol II. HCT116 cells were treated with 5 µg/mL α-amanitin for 24 h, and then NaCl was added for 2, 5, or 10 min. Mouse M12 cells were spiked in, and Pol II occupancy was assayed at the 5′ ends with normalization to the mouse actin by ChIP-qPCR. Values were normalized to controls without NaCl addition. The mean and SEM for three technical replicates are shown.
To further address the possibility that loss of Pol II from 5′ ends could be caused by release of paused Pol II into gene bodies, we calculated ratios of spike-in normalized Pol II density in low and high salt throughout the 5′ ends of genes. However, even for genes with the greatest pausing, the loss of 5′ Pol II in high NaCl was not compensated for by increased Pol II density at downstream positions (Fig. 3C, red plot). Similarly, the loss of DRB-arrested Pol II at 5′ ends in high salt was not compensated for by increased density within gene bodies (Fig. 3C, pink plot). As expected, in DRB, Pol II loss is more restricted to 5′ ends because it is arrested at start sites. In summary, these results show that DRB-sensitive PTEFb-dependent Pol II release into elongation is not required for loss of paused Pol II in high salt. Moreover, there is not a detectable net displacement of Pol II from pause sites into gene bodies under these conditions.
We next tested whether α-amanitin, which binds the trigger loop and blocks translocation, affects Pol II turnover at 5′ ends in high salt. α-Amanitin (5 µg/mL for 24 h) strongly inhibited Pol II but not Pol III transcription, as determined by BrU labeling (Supplemental Fig. S2F). Pol II occupancy at 5′ ends after prolonged α-amanitin treatment is much reduced and also less dispersed away from the TSS than in control cells, presumably due to poor translocation (Fig. 3D). As with DRB, α-amanitin did not prevent the almost complete loss of Pol II from 5′ ends within 10 min in high salt, as shown by anti-Pol II ChIP-seq normalized to a mouse spike-in (Fig. 3D,E; Supplemental Fig. S2G,H). Indeed, anti-Pol II ChIP-qPCR at several promoters demonstrated substantial Pol II turnover of α-amanitin-arrested Pol II within 2 min after adding high salt (Fig. 3F). Turnover of α-amanitin-arrested complexes in high salt was inhibited by saponin permeabilization +NaN3, indicating that it is an active process rather than passive dissociation (Supplemental Fig. S2I). In summary, both DRB and α-amanitin failed to prevent the rapid removal of Pol II from 5′ ends of genes in high-salt-treated cells. These observations suggest that when elongation is impaired, an alternative mechanism, presumably a form of premature termination, is sufficient to rapidly remove most Pol II from pause sites in cells exposed to hypertonic conditions.
Rapid reversal of Pol II loss at promoters; accumulation of ‘poised’ complexes
When cells were returned to isotonic conditions after hypertonic shock, the peaks of Pol II density reappeared at thousands of 5′ pause sites within 15 min. At many genes, we observed a “rebound” effect, where, after recovery, slightly more Pol II accumulated at 5′ ends than was present prior to the osmotic shock (Fig. 4A,B). This rapid reversal of Pol II loss provides a unique opportunity to investigate how Pol II is recruited to promoter regions in vivo in real time. We took advantage of the active Pol II accumulation at unoccupied promoters during recovery from hypertonic shock to ask whether initiation is required for this process. Previously, it had been assumed that stable Pol II accumulation at 5′ ends required transcription initiation followed by pausing. As a result, it was concluded that prolonged Pol II occupancy at 5′ ends in the presence of the initiation inhibitor triptolide reflects highly stable pausing (Henriques et al. 2013; Jonkers et al. 2014; Chen et al. 2015a; Shao and Zeitlinger 2017). Pol II occupancy was assayed in cells that were treated with 10 µM triptolide for 10–60 min and exposed to 350 mM salt for 10 min followed by recovery in isotonic medium plus triptolide for 15 min. Triptolide was added before salt to allow time for XPB inactivation. As expected, triptolide did not affect the loss of Pol II at 5′ ends in high salt. Unexpectedly, however, triptolide also did not prevent Pol II rebinding at 5′ ends during recovery from high salt. This result is evident from inspection of ChIP-seq results normalized to a mouse spike-in for individual genes and in metaplots of many genes (Fig. 4C,D; Supplemental Fig. S3). Notably, the preinitiated Pol II that accumulated at 5′ ends in the presence of triptolide was confined to the TSS and did not move to upstream and downstream pause sites, in contrast to paused Pol II that accumulates during recovery from high salt in the absence of triptolide (Fig. 4F). We classified genes into those with strong and weak recruitment of Pol II preinitiation complexes, as defined by the ratio of Pol II ChIP signal around the TSS after recovery from high NaCl in triptolide/ChIP signal in triptolide before NaCl addition. The group with robust recruitment of preinitiation complexes, indicative of strong promoter activity, corresponded to those with high Pol II occupancy under normal conditions. In contrast, those genes with poor recruitment of preinitiation complexes, indicative of weak promoters, had far lower Pol II occupancy normally (Fig. 4E). In summary, these observations during recovery from hypertonic shock reveal a previously undetected process of active assembly of relatively stable “poised” preinitiation complexes operating at thousands of promoters. Furthermore, the results in Figure 4 show that it is not always correct to assume that Pol II residing at 5′ ends is in a paused state after initiation has been inhibited.
Figure 4.
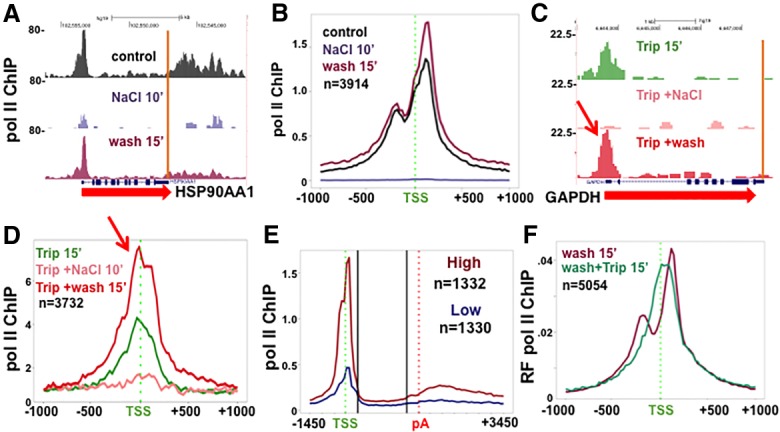
Pol II recruitment to 5′ ends during recovery from hypertonic shock does not require initiation. (A,B) Removal of Pol II from pause sites is rapidly reversed during recovery from hypertonic shock. NaCl was added to HCT116 cells for 10 min and then replaced with normal isotonic medium (wash) for 15 min. Pol II ChIP-seq was normalized to mouse M12 spike-in. (A) Pol II ChIP-seq genome browser shots. (B) Metaplots of mean Pol II ChIP signals as in A with 25-bp bins. Note the bimodal Pol II distribution following recovery from high salt. (C,D) Accumulation of “poised” preinitiation complexes at the TSS during recovery from hypertonic shock in triptolide. HCT116 was treated with 10 µM triptolide (Trip.) for 15 min, and then NaCl was added for 10 min followed by washout with medium containing 10 µM triptolide for 15 min. Pol II ChIP-seq was normalized to M12 spike-in. (C) Pol II ChIP-seq at GAPDH. (D) Metaplots of mean Pol II ChIP signals with 25-bp bins. Note the recovery of Pol II in the absence of initiation (red arrows) (see also Supplemental Fig. S3). (E) Rapid recruitment of preinitiation complexes correlates with high steady-state Pol II occupancy. Pol II ChIP-seq in HCT116 cells under normal conditions, as in Figure 1C, for genes with the highest and lowest recovery of Pol II at 5′ ends upon return to isotonic medium in triptolide. Recovery was calculated from data in Figure 3D as Pol II ChIP signal around the TSS after NaCl washout/signal before addition of NaCl. (F) “Poised” preinitiation complexes at the TSS during recovery from hypertonic shock in triptolide. Metaplots of relative frequency (RF) of Pol II ChIP 15 min after washout of high salt (25-bp bins normalized to bin 1). Results are from biological replicates of the experiments in A–E. Triptolide results are as in Supplemental Figure S3, D–F. Note that unimodal distribution of Pol II preinitiation complexes centered at the TSS after recovery from high salt.
Rapid loss of Pol II from pause sites under isotonic conditions
We asked whether the rapid turnover of Pol II at pause sites that occurs in hypertonically shocked cells also operates under isotonic conditions. If this is the case, then triptolide inhibition of initiation is predicted to deplete Pol II from pause sites, while ongoing assembly of preinitiation complexes is expected to maintain some level of Pol II occupancy at the TSS. To test this idea, we treated cells with triptolide for short periods and monitored Pol II occupancy around start sites by ChIP-seq (Fig. 5A–D; Supplemental Fig. S4A,B), as in previous studies (Henriques et al. 2013; Jonkers et al. 2014; Chen et al. 2015a; Krebs et al. 2017; Shao and Zeitlinger 2017). We mapped ChIP-seq reads at high resolution to distinguish Pol II complexes at pause sites from those at the TSS. Importantly, we found that the profile of Pol II near start sites was qualitatively altered by triptolide. Under control conditions, Pol II was distributed in the expected bimodal peak around the TSS, but, in triptolide, this profile was remodeled as Pol II shifted from upstream and downstream pause sites toward the TSS, consistent with a previous report (Chen et al. 2015a). This transition, which is easily seen in plots of the relative frequency of Pol II ChIP signals around the TSS, was first detected after 5 min in triptolide and was complete after 10 min, at which time the bimodal Pol II profile was replaced by a single peak of Pol II over the TSS both in HCT116 and HEK293 cells (Fig. 5C,D). This transition of Pol II from pause sites to the TSS is not an artifact of lower ChIP signals in triptolide, as shown in metaplots of mean ChIP signals for equivalent numbers of reads over the region from −1 to +1 kb relative to the TSS (Supplemental Fig. S4C). We interpret this result to mean that in triptolide, paused Pol II localized at peaks flanking the start site is rapidly turned over, making room for the assembly of new “poised” preinitiation complexes that localize to the TSS but are unable to transition to the pause sites. Note that the time scale of this change in Pol II positioning likely underestimates the actual rate of turnover at pause sites because it does not account for the lag in onset of XPB inhibition after adding triptolide to cells. In summary, these results show that when initiation is inhibited under isotonic conditions, a large fraction of Pol II at pause sites is turned over within 5 min, and the process is essentially complete within 10 min. This conclusion is in contrast to previous studies that did not distinguish “poised” from “paused” complexes and interpreted the persistence of 5′ peaks of Pol II in the presence of triptolide as evidence of stable pausing (Henriques et al. 2013; Chen et al. 2015a; Shao and Zeitlinger 2017).
Figure 5.
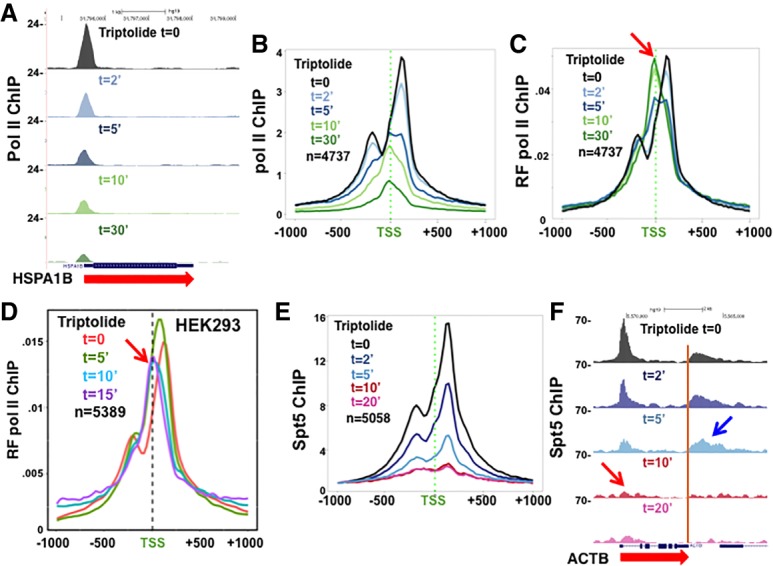
Rapid turnover of paused Pol II complexes under isotonic conditions. (A) Pol II ChIP-seq at HSPA1B at intervals after adding 10 µM triptolide to HCT116 cells normalized to mouse M12 spike-ins (see also Supplemental Figure S4A–C). (B) Metaplots of mean Pol II ChIP signals with 25-bp bins as in A. (C,D) Metaplots of relative frequency (RF) of Pol II ChIP signals in HCT116 and HEK293 cells at time points after adding 10 µM triptolide. Note that within 5 min, the Pol II distribution shifts away from pause sites and becomes centered over the TSS. (E,F) Spt5 ChIP-seq at time points after adding triptolide to HCT116 cells normalized to M12 spike-ins. (E) Metaplots of mean Spt5 ChIP signals with 25-bp bins normalized to bin 1. Note the bimodal distribution of Spt5 at pause sites, in contrast to Pol II. (F) Spt5 ChIP-seq on ACTB as in E. Note the rapid loss of Spt5 from 5′ ends (red arrow) relative to 3′ ends (blue arrow) (see also Supplemental Figure S4D–F).
The Pol II at 5′ ends when initiation is inhibited by triptolide is not pausedc
To be sure that the Pol II remaining over start sites in triptolide is really distinct from paused complexes, we asked whether it was associated with DSIF (Spt4/5), which is an integral component of paused complexes. Spt5 contacts the front and back sides of the Pol II ternary complex as well as the nascent transcript (Missra and Gilmour 2010; Bernecky et al. 2017; Ehara et al. 2017). Anti-Spt5 ChIP-seq (with a mouse spike-in for normalization) was performed at intervals after addition of triptolide. In contrast to Pol II occupancy, which shifts from pause sites to the TSS within 10 min, Spt5 was found only at pause sites upstream of and downstream from the TSS. Within 5 min of adding triptolide, Spt5 occupancy was reduced to less than half and was close to background levels within 10 min (Fig. 5E,F; Supplemental Fig. S4D–F). Rapid Spt5 loss from pause sites when initiation is blocked with triptolide occurred on both highly transcribed genes (such as ACTB and EIF1) and poorly transcribed genes that accumulate paused Pol II at their 5′ ends (such as FOS and HSPA1B) (Fig. 5F; Supplemental Fig. S4D–F).
Spt5 is associated with Pol II complexes not only at promoter-proximal pause sites but also throughout elongation and at 3′ pause sites downstream from poly(A) sites (Glover-Cutter et al. 2008; Rahl et al. 2010). In contrast to the rapid loss of Spt5 from 5′ ends in triptolide (Fig. 5E,F; Supplemental Fig. S4D–F, red arrows), it persisted for much longer periods within gene bodies and 3′ flanking regions (Fig. 5E; Supplemental Fig. S4D, blue arrows). We interpret this difference to mean that Spt5-associated paused Pol II at 5′ ends turns over rapidly relative to the rate of clearance of elongation complexes after initiation is blocked. In summary, these results show that Pol II positioned at the TSS after a few minutes in triptolide is not paused but is instead in a “poised” preinitiation state that lacks a nascent transcript required for stable Spt5 binding (Missra and Gilmour 2010; Bernecky et al. 2017; Ehara et al. 2017).
Discussion
The accumulation of paused Pol II at promoter-proximal sites is a ubiquitous feature of transcription in multicellular organisms, and these paused complexes are targeted by mechanisms that control entry into productive elongation. The stability of paused complexes has been the subject of intense investigation with conflicting results because it is difficult to distinguish Pol II entry and exit at pause sites (Henriques et al. 2013; Jonkers et al. 2014; Chen et al. 2015a; Krebs et al. 2017; Nilson et al. 2017; Shao and Zeitlinger 2017). We report the first direct observation of widespread dismantling of Pol II complexes from promoter-proximal pause sites—a phenomenon that was revealed under conditions of hypertonic shock. Eviction from pause sites under these conditions is very rapid; 80%–90% of Pol II is lost within 2 min on several genes tested after raising the salt concentration to ≥250 mM (Figs. 1A,B, 6A, left). Moreover, this is an active process that requires cell viability (Fig. 2F). In contrast, elongating Pol II situated within genes throughout the genome is resistant to hypertonic shock (Figs. 1, 2; Supplemental Fig. S1; Proft and Struhl 2004; Wang et al. 2005), and transcription often extends long distances past poly(A) sites (Fig. 6A, right), in agreement with previous work, suggesting a termination defect under hypertonic conditions (Vilborg et al. 2015).
Figure 6.
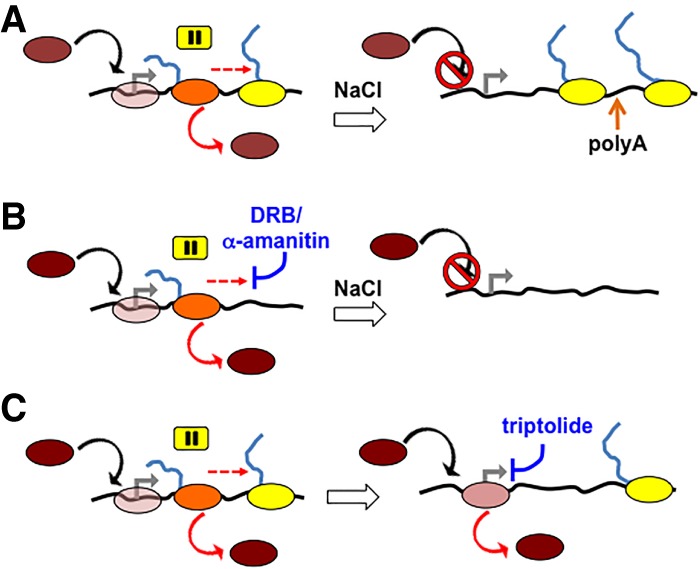
The dynamic turnover model of Pol II recruitment and eviction at human promoter regions. (A, left) Isotonic conditions with ongoing recruitment (black arrow) of free Pol II (brown) to the TSS (gray arrow; divergent transcription is not shown), where it forms a transient “poised” complex (clear pink) followed by initiation and transition to the promoter-proximal pause (orange), where eviction (solid red arrow) or release into productive elongation (dashed red arrow) occurs. (Right) Hypertonic conditions with block to recruitment and slow elongation of salt-resistant elongation complexes that transcribe far downstream from poly(A) sites. (Brown) Free Pol II; (pink) poised Pol II; (orange) paused Pol II; (yellow) elongating Pol II. (B, left) Isotonic conditions with ongoing recruitment and turnover of paused Pol II in the presence of elongation inhibitor DRB or α-amanitin. (Right) Under hypertonic conditions plus DRB or α-amanitin, recruitment is blocked, and Pol II arrested at the pause site is rapidly evicted without active elongation. (C) Triptolide blocks initiation and permits accumulation of “poised” Pol II at the TSS that turns over and remains preinitiated (solid pink).
We interpret the dramatic and specific loss of Pol II at pause sites to reflect a highly active turnover mechanism that is uncovered under hypertonic conditions when TBP binding (Supplemental Fig. S1H) and assembly of new preinitiation complexes are prevented (Cai and Luse 1987; Conaway et al. 1990). This turnover mechanism does not appear to require PTEFb-mediated pause release or normal translocation, as it is resistant to the inhibitors DRB and α-amanitin (Fig. 3; Supplemental Fig. S2). Furthermore, the loss of Pol II from pause sites in high salt is not accompanied by any detectable compensating increase in Pol II density at downstream positions within genes (Fig. 3C).
Rapid turnover of Pol II at pause sites is obvious in high salt but is also detectable under isotonic conditions when initiation is inhibited by triptolide, provided one distinguishes post-initiation complexes at pause sites from preinitiation complexes at TSSs. Within 5 min of adding triptolide, a substantial fraction of Pol II is lost from pause sites where it is associated with Spt5 and instead accumulates at the TSS in a “poised” preinitiation state lacking Spt5, presumably because there is no nascent transcript for Spt5 to contact (Fig. 5; Supplemental Fig. S4D–F). We propose that the same turnover mechanism is responsible for removing Pol II from pause sites when initiation is blocked by either high salt or triptolide even when elongation is blocked by DRB or α-amanitin. This conclusion is supported by a recent study of FRAP of GFP-tagged Pol II (Steurer et al. 2018). This report modeled the kinetics of Pol II-GFP FRAP to identify a Pol II population with an average residence time of 42 sec that has properties expected for promoter-proximal paused Pol II. Consistent with our findings, FRAP kinetics indicated that the residence time of the putative paused fraction was almost unaffected by triptolide or flavopiridol, which acts like DRB (Steurer et al. 2018).
The eviction of paused Pol II complexes reported here strongly suggests that active premature termination though the underlying mechanism is not understood. It is unclear whether this termination is similar to the torpedo mechanism that operates downstream from poly(A) sites (Proudfoot 2016), but it is notable that the latter mechanism is substantially inhibited in high salt (Fig. 1G,I; Supplemental Fig. S1B,C; Vilborg et al. 2015), whereas premature termination appears to be highly active under these conditions. An alternative possibility is that termination of paused Pol II is related to the Nrd1–Nab3–Sen1-dependent mechanism that terminates many noncoding transcripts in yeast (Steinmetz et al. 2001; Kim et al. 2006; Porrua and Libri 2013). Pausing is a common precondition for termination in prokaryotic systems, where it may promote conversion to a polymerase conformation that opens the clamp and thereby favors release from the template (Hein et al. 2014; Sekine et al. 2015).
Under steady-state conditions, we propose that a dynamic cycle operates, in which ongoing eviction of Pol II from pause sites is balanced by active recruitment of Pol II to promoters (Fig. 6). We observed this recruitment directly during recovery from hypertonic shock, which effectively purges promoters of virtually all Pol II (Fig. 4A,B). Remarkably, a robust recruitment of stable preinitiation complexes to promoters was revealed when cells were allowed to recover from hypertonic shock in the presence of triptolide (Fig. 4C,D; Supplemental Fig. S3). Furthermore, the recruitment of preinitiated complexes after return to isotonic medium correlated with steady-state levels of paused Pol II at 5′ ends under normal conditions (Fig. 4E), as expected if this process was controlled by promoter strength. One implication of these results, together with those reported previously in quiescent B cells (Kouzine et al. 2013), is that “poised” preinitiation complexes are relatively stable intermediates in a pathway leading to formation of paused post-initiation complexes (Fig. 6). Moreover, our observations in proliferating tissue culture cells suggest that accumulation of stable preinitiation complexes is not peculiar to quiescent B cells, which have limiting XPB (Kouzine et al. 2013), but is instead a general phenomenon. In summary, these results demonstrate two active ongoing processes that control Pol II accumulation at 5′ ends of genes: recruitment into preinitiated complexes and eviction of paused complexes by a mechanism that does not require active elongation (Fig. 6).
The surprisingly robust recruitment of preinitiated complexes to promoters in the presence of the initiation inhibitor triptolide means that previous studies using this approach to measure Pol II residence time at pause sites (Henriques et al. 2013; Chen et al. 2015a; Krebs et al. 2017; Shao and Zeitlinger 2017) probably underestimated rates of turnover. By taking advantage of a recruitment block under hypertonic conditions, we were able to monitor the decay of Pol II at pause sites absent the confounding recruitment of Pol II into stable preinitiated complexes. Under these circumstances, it is clear that the rate of Pol II loss from pause sites is far higher than previously suspected (e.g. Fig. 1A). We note that the formation of relatively stable preinitiation complexes in triptolide might account for the Pol II decay curves near start sites reported by Krebs et al. (2017). These investigators found rapid initial decay by 30%–40% within 2.5 min followed by a stabilization of Pol II levels at later times.
The highly dynamic nature of paused Pol II at most human promoters has implications for how the transition from pausing to active elongation is controlled. The results reported here are most consistent with the idea that the majority of polymerases at pause sites is rapidly evicted, and only a minority that avoids this fate is permitted to transition into productive elongation. This model is consistent with previous estimates based on live-cell Pol II FRAP kinetics that only ∼1% of Pol II encounters with the 5′ end of a gene result in productive elongation (Darzacq et al. 2007; Steurer et al. 2018). In the widely accepted pause–release model, control of entry into elongation is exerted by regulating escape from a stably paused state. We propose an alternative model in which control of this transition is instead exerted by regulating escape from premature termination with eviction from the template (Fig. 6). This “escape from termination” model suggests that productive elongation would be stimulated by factors that make the polymerase more resistant to termination, possibly by preventing conformational changes that cause clamp opening or swiveling (Hein et al. 2014; Sekine et al. 2015; Kang et al. 2018). In this context, it is interesting to note that Spt5, often regarded as a key “pausing” factor in metazoans, is implicated in control of termination in yeast (Baejen et al. 2017) and makes a conserved contact with the Pol II clamp domain (Martinez-Rucobo et al. 2011; Bernecky et al. 2017; Ehara et al. 2017). In the future, it will be important to elucidate the mechanisms responsible for rapid turnover and those that control the decision between eviction and elongation at promoter-proximal pause sites.
Materials and methods
Cell culture
HCT116 cells were maintained in McCoy's medium with 10% FBS and penicillin/streptomycin. NaCl shock was performed by replacing the same medium on cells after adding NaCl from a 5 M stock and mixing. Unless otherwise indicated, NaCl was added to 230 mM, making a final concentration of monovalent cations in the medium of 350 mM. Sucrose was added to 0.63 M, which has the same osmotic pressure as 0.35 M NaCl. DRB (Sigma) was used at 100 µM, triptolide (Cayman Chemical) was used at 10 µM, α-amanitin (Sigma) was used at 5 µg/mL, and PD-169316 (MedChemExpress) was used at 15 µM.
Antibodies
Rabbit anti-pan-Pol II CTD (Schroeder et al. 2000), −histone H3 C terminus (Zhang et al. 2005), and −Spt5 (Glover-Cutter et al. 2008) and B44 monoclonal anti-BrdU antibodies (Gratzner 1982) have been described. Rabbit anti-TBP was from Upstate Biotechnology (catalog no. 06-24).
ChIP
ChIP-seq has been described (Fong et al. 2017). Reads were mapped to the hg19 University of California at Santa Cruz human genome (February 2009) with Bowtie version 0.12.5 (Supplemental Table S1; Langmead et al. 2009). The pausing index was calculated as reads from −500 to +500 relative to the TSS/+501 relative to the TSS to −500 relative to the poly(A) site. ChIP-seq reads were quantified relative to spike-ins by the method of Hu et al. (2015). Cross-linked sheared yeast chromatin was spiked into human extract before immunoprecipitation. Mouse M12 cells (2 × 106) were added to human cells during cross-linking with formaldehyde. Input libraries were sequenced for each spiked extract to calculate the ratio of human:yeast or human:mouse reads. Metaplots of human genes are from a list of 5507 well-expressed genes separated from their neighbors by >2 kb (Brannan et al. 2012). Metaplots include all genes in common between the data sets for which a minimum ChIP signal was obtained. Relative frequency plots were made by calculating mean read counts for each bin divided by the sum total of mean read counts in all bins.
ChIP-qPCR primers are described in Supplemental Table S2.
qRT–PCR of BrU-labeled transcripts (Supplemental Fig. S2F)
Cells were labeled with 2 mM bromouridine for 30 min, RNA was purified with Trizol, and labeled RNA was isolated by immunoprecipitation with B44 monoclonal anti-BrdU as described (Paulsen et al. 2014). Random primed cDNA was made with SuperScript III (Invitrogen), and qPCR was performed with the primers listed in Supplemental Table S2. Fold change was calculated using the Δ/ΔCt method.
Accession numbers
ChIP-seq data sets have been deposited at Gene Expression Omnibus under accession number GSE117006.
Supplementary Material
Acknowledgments
We thank O. Rissland, S. Ramakrishnan, T. Blumenthal, M. Groudine, an anonymous reviewer, and members of our laboratory for comments on the manuscript. We thank K. Diener, B. Gao, and the University of Colorado at Denver Sequencing Facility for sequencing. M.C. and R.M.S. are scholars of the University of Colorado at Denver RNA Bioscience Initiative. This work was supported by National Institutes of Health grant R35GM118051 to D.L.B.
Author contributions: B.E., R.M.S., and M.C. performed all experiments. D.L.B., B.E., R.M.S., and M.C. designed the experiments. D.L.B. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.316810.118.
References
- Adelman K, Lis JT. 2012. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13: 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseev S, Nagy Z, Sandoz J, Weiss A, Egly JM, Le May N, Coin F. 2017. Transcription without XPB establishes a unified helicase-independent mechanism of promoter opening in eukaryotic gene expression. Mol Cell 65: 504–514.e4. [DOI] [PubMed] [Google Scholar]
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. 2015. An atlas of active enhancers across human cell types and tissues. Nature 507: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baejen C, Andreani J, Torkler P, Battaglia S, Schwalb B, Lidschreiber M, Maier KC, Boltendahl A, Rus P, Esslinger S, et al. 2017. Genome-wide analysis of RNA polymerase II termination at protein-coding genes. Mol Cell 66: 38–49.e36. [DOI] [PubMed] [Google Scholar]
- Bentley DL, Groudine M. 1986. A block to elongation is largely responsible for decreased transcription of c-myc in differentiated HL60 cells. Nature 321: 702–706. [DOI] [PubMed] [Google Scholar]
- Bernecky C, Plitzko JM, Cramer P. 2017. Structure of a transcribing RNA polymerase II–DSIF complex reveals a multidentate DNA–RNA clamp. Nat Struct Mol Biol 24: 809–815. [DOI] [PubMed] [Google Scholar]
- Brannan K, Bentley D. 2012. Control of transcriptional elongation by RNA polymerase II: a retrospective. Genet Res Int 10.1155/2012/170173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannan K, Kim H, Erickson B, Glover-Cutter K, Kim S, Fong N, Kiemele L, Hansen K, Davis R, Lykke-Andersen J, et al. 2012. mRNA decapping factors and the exonuclease Xrn2 function in widespread premature termination of RNA polymerase II transcription. Mol Cell 46: 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley MS, Kwak H, Zipfel WR, Lis JT. 2014. Kinetics of promoter Pol II on Hsp70 reveal stable pausing and key insights into its regulation. Genes Dev 28: 14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Luse DS. 1987. Transcription initiation by RNA polymerase II in vitro. Properties of preinitiation, initiation, and elongation complexes. J Biol Chem 262: 298–304. [PubMed] [Google Scholar]
- Chen F, Gao X, Shilatifard A. 2015a. Stably paused genes revealed through inhibition of transcription initiation by the TFIIH inhibitor triptolide. Genes Dev 29: 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FX, Woodfin AR, Gardini A, Rickels RA, Marshall SA, Smith ER, Shiekhattar R, Shilatifard A. 2015b. PAF1, a molecular regulator of promoter-proximal pausing by RNA polymerase II. Cell 162: 1003–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B, Price DH. 2007. Properties of RNA polymerase II elongation complexes before and after the P-TEFb-mediated transition into productive elongation. J Biol Chem 282: 21901–21912. [DOI] [PubMed] [Google Scholar]
- Chiu AC, Suzuki HI, Wu X, Mahat DB, Kriz AJ, Sharp PA. 2018. Transcriptional pause sites delineate stable nucleosome-associated premature polyadenylation suppressed by U1 snRNP. Mol Cell 69: 648–663.e647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conaway JW, Reines D, Conaway RC. 1990. Transcription initiated by RNA polymerase II and purified transcription factors from liver. Cooperative action of transcription factors τ and ε in initial complex formation. J Biol Chem 265: 7552–7558. [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. 2008. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322: 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH. 2007. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol 14: 796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehara H, Yokoyama T, Shigematsu H, Yokoyama S, Shirouzu M, Sekine S-i. 2017. Structure of the complete elongation complex of RNA polymerase II with basal factors. Science 357: 921–924. [DOI] [PubMed] [Google Scholar]
- Eick D, Bornkamm GW. 1986. Transcriptional arrest within the first exon is a fast control mechanism in c-myc gene expression. Nucleic Acids Res 14: 8331–8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Saldi T, Sheridan RM, Cortazar MA, Bentley DL. 2017. RNA Pol II dynamics modulate co-transcriptional chromatin modification, CTD phosphorylation, and transcriptional direction. Mol Cell 66: 546–557 e543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gariglio P, Bellard M, Chambon P. 1981. Clustering of RNA polymerase B molecules in the 5′ moiety of the adult β-globin gene of hen erythrocytes. Nucleic Acids Res 9: 2589–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist DA, Santos GD, Fargo DC, Xie B, Gao Y, Li L, Adelman K. 2010. Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell 143: 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour DS. 2009. Promoter proximal pausing on genes in metazoans. Chromosoma 118: 1–10. [DOI] [PubMed] [Google Scholar]
- Gilmour DS, Lis JT. 1986. RNA polymerase II interacts with the promoter region of the noninduced hsp70 gene in Drosophila melanogaster cells. Mol Cell Biol 6: 3984–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover-Cutter K, Kim S, Espinosa J, Bentley DL. 2008. RNA polymerase II pauses and associates with pre-mRNA processing factors at both ends of genes. Nat Struct Mol Biol 15: 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratzner HG. 1982. Monoclonal antibody to 5-bromo- and 5-iododeoxyuridine: a new reagent for detection of DNA replication. Science 218: 474–475. [DOI] [PubMed] [Google Scholar]
- Hay N, Skolnik-David H, Aloni Y. 1982. Attenuation in the control of SV40 gene expression. Cell 29: 183–193. [DOI] [PubMed] [Google Scholar]
- Hein PP, Kolb KE, Windgassen T, Bellecourt MJ, Darst SA, Mooney RA, Landick R. 2014. RNA polymerase pausing and nascent-RNA structure formation are linked through clamp-domain movement. Nat Struct Mol Biol 21: 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Gilchrist DA, Nechaev S, Bern M, Muse GW, Burkholder A, Fargo DC, Adelman K. 2013. Stable pausing by RNA polymerase II provides an opportunity to target and integrate regulatory signals. Mol Cell 52: 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Scruggs B, Inouye M, Muse GW, Williams L, Burkholder A, Lavender CA, Fargo DC, Adelman K. 2018. Widespread transcriptional pausing and elongation control at enhancers. Genes Dev 31: 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Petela N, Kurze A, Chan K-L, Chapard C, Nasmyth K. 2015. Biological chromodynamics: a general method for measuring protein occupancy across the genome by calibrating ChIP-seq. Nucleic Acids Res 43: e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Lis JT. 2015. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol 16: 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Kwak H, Lis JT. 2014. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. Elife 3: e02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JY, Mishanina TV, Bellecourt MJ, Mooney RA, Darst SA, Landick R. 2018. RNA polymerase accommodates a pause RNA hairpin by global conformational rearrangements that prolong pausing. Mol Cell 69: 802–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayali AG, Austin DA, Webster NJ. 2000. Stimulation of MAPK cascades by insulin and osmotic shock: lack of an involvement of p38 mitogen-activated protein kinase in glucose transport in 3T3-L1 adipocytes. Diabetes 49: 1783–1793. [DOI] [PubMed] [Google Scholar]
- Kim M, Vasiljeva L, Rando OJ, Zhelkovsky A, Moore C, Buratowski S. 2006. Distinct pathways for snoRNA and mRNA termination. Mol Cell 24: 723–734. [DOI] [PubMed] [Google Scholar]
- Kouzine F, Wojtowicz D, Yamane A, Resch W, Kieffer-Kwon K-R, Bandle R, Nelson S, Nakahashi H, Awasthi P, Feigenbaum L, et al. 2013. Global regulation of promoter melting in naive lymphocytes. Cell 153: 988–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs AR, Imanci D, Hoerner L, Gaidatzis D, Burger L, Schübeler D. 2017. Genome-wide single-molecule footprinting reveals high RNA polymerase II turnover at paused promoters. Mol Cell 67: 411–422.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm A, Hickey L, Groudine M. 1995. Promoter-proximal pausing of RNA polymerase II defines a general rate-limiting step after transcription initiation. Genes Dev 9: 559–572. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall NF, Price DH. 1995. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem 270: 12335–12338. [DOI] [PubMed] [Google Scholar]
- Martinez-Rucobo FW, Sainsbury S, Cheung ACM, Cramer P. 2011. Architecture of the RNA polymerase-Spt4/5 complex and basis of universal transcription processivity. EMBO J 30: 1302–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell CS, Kruesi WS, Core LJ, Kurhanewicz N, Waters CT, Lewarch CL, Antoshechkin I, Lis JT, Meyer BJ, Baugh LR. 2014. Pol II docking and pausing at growth and stress genes in C. elegans. Cell Rep 6: 455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, Landry HM, Churchman LS. 2017. Pause and go: from the discovery of RNA polymerase pausing to its functional implications. Curr Opin Cell Biol 46: 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missra A, Gilmour DS. 2010. Interactions between DSIF (DRB sensitivity inducing factor), NELF (negative elongation factor), and the Drosophila RNA polymerase II transcription elongation complex. Proc Natl Acad Sci 107: 11301–11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, Adelman K. 2007. RNA polymerase is poised for activation across the genome. Nat Genet 39: 1507–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechaev S, Fargo DC, dos Santos G, Liu L, Gao Y, Adelman K. 2010. Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science 327: 335–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilson KA, Lawson CK, Mullen NJ, Ball CB, Spector BM, Meier JL, Price DH. 2017. Oxidative stress rapidly stabilizes promoter-proximal paused Pol II across the human genome. Nucleic Acids Res 45: 11088–11105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Magnuson B, Wilson TE, Ljungman M. 2014. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods 67: 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrua O, Libri D. 2013. A bacterial-like mechanism for transcription termination by the Sen1p helicase in budding yeast. Nat Struct Mol Biol 20: 884–891. [DOI] [PubMed] [Google Scholar]
- Proft M, Struhl K. 2004. MAP kinase-mediated stress relief that precedes and regulates the timing of transcriptional induction. Cell 118: 351–361. [DOI] [PubMed] [Google Scholar]
- Proudfoot NJ. 2016. Transcriptional termination in mammals: stopping the RNA polymerase II juggernaut. Science 352: aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinodoz M, Gobet C, Naef F, Gustafson KB. 2014. Characteristic bimodal profiles of RNA polymerase II at thousands of active mammalian promoters. Genome Biol 15: R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha D, Wang Z, Moqtaderi Z, Wu L, Zhong G, Gerstein M, Struhl K, Snyder M. 2010. Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc Natl Acad Sci 107: 3639–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. 2010. c-Myc regulates transcriptional pause release. Cell 141: 432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougvie AE, Lis JT. 1988. The RNA polymerase II molecule at the 5′ end of the uninduced hsp70 gene of D. melanogaster is transcriptionally engaged. Cell 54: 795–804. [DOI] [PubMed] [Google Scholar]
- Schroeder SC, Schwer B, Shuman S, Bentley D. 2000. Dynamic association of capping enzymes with transcribing RNA polymerase II. Genes Dev 14: 2435–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine S-i, Murayama Y, Svetlov V, Nudler E, Yokoyama S. 2015. The ratcheted and ratchetable structural states of RNA polymerase underlie multiple transcriptional functions. Mol Cell 57: 408–421. [DOI] [PubMed] [Google Scholar]
- Shao W, Zeitlinger J. 2017. Paused RNA polymerase II inhibits new transcriptional initiation. Nat Genet 49: 1045–1051. [DOI] [PubMed] [Google Scholar]
- Steinmetz EJ, Conrad NK, Brow DA, Corden JL. 2001. RNA-binding protein Nrd1 directs poly(A)-independent 3′-end formation of RNA polymerase II transcripts. Nature 413: 327–331. [DOI] [PubMed] [Google Scholar]
- Steurer B, Janssens RC, Geverts B, Geijer ME, Wienholz F, Theil AF, Chang J, Dealy S, Pothof J, van Cappellen WA, et al. 2018. Live-cell analysis of endogenous GFP-RPB1 uncovers rapid turnover of initiating and promoter-paused RNA Polymerase II. Proc Natl Acad Sc 67: 201717920–201717929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titov DV, Gilman B, He Q-L, Bhat S, Low W-K, Dang Y, Smeaton M, Demain AL, Miller PS, Kugel JF, et al. 2011. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat Chem Biol 7: 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilborg A, Passarelli MC, Yario TA, Tycowski KT, Steitz JA. 2015. Widespread inducible transcription downstream of human genes. Mol Cell 59: 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Takagi T, Yamaguchi Y, Watanabe D, Handa H. 1998. Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. EMBO J 17: 7395–7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagschal A, Rousset E, Basavarajaiah P, Contreras X, Harwig A, Laurent-Chabalier S, Nakamura M, Chen X, Zhang K, Meziane O, et al. 2012. Microprocessor, Setx, Xrn2, and Rrp6 co-operate to induce premature termination of transcription by RNAPII. Cell 150: 1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Balamotis MA, Stevens JL, Yamaguchi Y, Handa H, Berk AJ. 2005. Mediator requirement for both recruitment and postrecruitment steps in transcription initiation. Mol Cell 17: 683–694. [DOI] [PubMed] [Google Scholar]
- Wu CH, Yamaguchi Y, Benjamin LR, Horvat-Gordon M, Washinsky J, Enerly E, Larsson J, Lambertsson A, Handa H, Gilmour D. 2003. NELF and DSIF cause promoter proximal pausing on the hsp70 promoter in Drosophila. Genes Dev 17: 1402–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Takagi T, Wada T, Yano K, Furuya A, Sugimoto S, Hasegawa J, Handa H. 1999. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 97: 41–51. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Shibata H, Handa H. 2013. Transcription elongation factors DSIF and NELF: promoter-proximal pausing and beyond. Biochim Biophys Acta 1829: 98–104. [DOI] [PubMed] [Google Scholar]
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. 2005. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 19: 535–545. [DOI] [PubMed] [Google Scholar]
- Yu M, Yang W, Ni T, Tang Z, Nakadai T, Zhu J, Roeder RG. 2015. RNA polymerase II-associated factor 1 regulates the release and phosphorylation of paused RNA polymerase II. Science 350: 1383–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamft B, Bintu L, Ishibashi T, Bustamante C. 2012. Nascent RNA structure modulates the transcriptional dynamics of RNA polymerases. Proc Natl Acad Sci 109: 8948–8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger J, Stark A, Kellis M, Hong JW, Nechaev S, Adelman K, Levine M, Young RA. 2007. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat Genet 39: 1512–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Schroeder S, Fong N, Bentley DL. 2005. Altered nucleosome occupancy and histone H3K4 methylation in response to ‘transcriptional stress’. EMBO J 24: 2379–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Li T, Price DH. 2012. RNA polymerase II elongation control. Annu Rev Biochem 81: 119–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Naguro I, Ichijo H, Watanabe K. 2016. Mitogen-activated protein kinases as key players in osmotic stress signaling. Biochim Biophys Acta 1860: 2037–2052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.