

Abstract
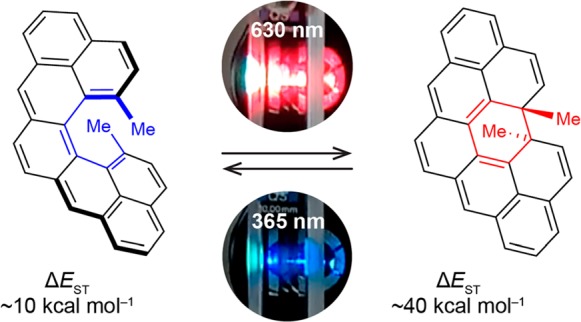
We describe the synthesis and properties of 13,14-dimethylcethrene, a prototype of a chiral diradicaloid photochemical switch that can be transformed reversibly via conrotatory electrocyclization to its more stable closed form by light (630 nm) or heat and back to its open form by light (365 nm). This system illustrates how the chemical reactivity of a diradicaloid molecule can be translated into a switching function, which alters substantially all electronic parameters, namely, the HOMO–LUMO and the singlet–triplet (ST) energy gaps, and the degree of helical twist. As a result, distinct changes in the optical and chiroptical properties of this system were observed, which allowed us to monitor the switching process by a variety of spectroscopic techniques, including NMR, UV–vis, and CD. In comparison to the previously reported parent molecule cethrene, this system benefits from two methyl substituents installed in the fjord region, which account for the stability of the closed form against oxidation and racemization. The methyl substituents increase the ST energy gap of 13,14-dimethylcethrene by ∼4 kcal mol–1 in comparison to cethrene. Our DFT calculations reveal that the larger ST gap is a result of electronic and geometric effects of the methyl substituents and show the potential of related systems to act as magnetic switches at room temperature.
Introduction
Spin-delocalized π-conjugated molecules1 that contain one or more unpaired electrons hold promise as components of materials which exhibit2 magnetic and conducting properties that are typically associated with metals. While magnetism in these systems arises from the presence3 of unpaired electrons in the ground or low-lying excited states, conductivity emerges on account of the short (∼3.1–3.2 Å) intermolecular distance4 between the molecules that are held5 together via multicentered n-electron π bonds, also referred to as “pancake bonds”. These bonds can be formed by favorable overlap of singly occupied molecular orbitals (SOMOs) in odd-electron4 or non-Kekulé6 systems or by favorable overlap of partially occupied frontier molecular orbitals (FMOs; namely, HOMO and LUMO) in the so-called “diradicaloid” Kekulé2c systems. In the latter case, the nondegenerate FMOs are close in energy and, as a consequence, a pair of electrons from the HOMO (a) partially occupies also the LUMO (b) to minimize electron repulsion. The singlet ground state of these molecules is therefore best described7 by mixing a doubly excited configuration a0b2 into the ground-state configuration a2b0. In addition to the low-lying LUMO, a common feature of diradicaloid molecules is the presence3 of a low-lying triplet excited state, which can be populated1 thermally.
In the pursuit of identifying structural features that would allow for fine tuning of key electronic parameters of diradicaloids, namely, the HOMO–LUMO and ST gaps, we turned our attention to systems that feature8 a helical π-conjugated backbone. On account of the helical structure of these systems, through-space orbital interactions arise9 within their FMOs, which can increase or decrease the FMO energies. Helical geometry is also the source of chirality10 and can create a favorable steric environment for certain intramolecular bond-rearrangement reactions, which, as we shall see, can be translated into a switching function. Recently, we developed11 the first model system of this type, a C-shaped hydrocarbon cethrene (o-1; Scheme 1, top), composed of seven fused benzenoid rings, five of which form a [5]helicene backbone. By synthesizing the diphenyl derivative o-1a, we were able to demonstrate12 its several unique features. (1) The helical geometry gives rise to through-space orbital interactions at positions 13 and 14: an antibonding interaction within the HOMO and a bonding interaction within the LUMO (see Figure 5a). These interactions decrease12c both the (a) HOMO–LUMO and (b) ST energy gaps of o-1a, in comparison to a planar diradicaloid isomer heptazethrene,13 rendering o-1a EPR active. (2) Cethrene o-1a undergoes an electrocyclic ring closure to the more stable closed form c-1a (Scheme 1, top), which proceeds in a conrotatory mode on account of steric constraints enforced by the helical geometry. Unlike most other electrocyclic reactions, however, the conrotatory ring closure of o-1a proceeds12a both (a) photochemically, which is a symmetry-allowed14 reaction, and (b) thermally with a surprisingly low activation barrier (∼14 kcal mol–1), formally a symmetry-forbidden14 reaction, the mechanism of which is not fully understood.
Scheme 1. Overview of Electrocyclic Ring Closures of Cethrene (Top, Middle) and Biphenalenylidene (Bottom) and Electrocyclic Ring Openings and Oxidations of Their Closed Forms.

Figure 5.

(a) Schematic illustration of through-space orbital interactions within the HOMO and the LUMO in o-1, o-1a, and o-1b at positions 13 and 14. (b) Thermal equilibrium between the singlet ground state and the triplet excited state of o-1 (R = H) and o-1b (R = Me). Distances between the fjord carbon atoms (dashed lines) obtained from optimized geometries (BS/U-B3LYP/6-31G(d)) are shown. (c) Relaxed potential energy surface (PES) scans of o-1 (top) and o-1b (bottom) in their singlet ground (blue) and triplet excited (red) states, performed to estimate the relative contributions of the geometric (“twist”) and electronic (“Me”) effects of the methyl substituents on the ST gap.
Because of a facile oxidation of c-1a to the planar hydrocarbon 2a, which did not allow us to isolate or even detect12a this intermediate, we were unable to perform the reverse reaction, namely, photochemical ring opening of c-1a to o-1a. Nonetheless, this process could be validated in an analogous system biphenalenylidene15 (o-3; Scheme 1, bottom) by Kubo and co-workers, who demonstrated15a that the closed form c-3 underwent an electrocyclic ring opening to give o-3 upon irradiation by UV light. In analogy to o-1a, the open form o-3 also underwent a thermal ring-closing reaction, which proceeded with a barrier (∼16 kcal mol–1) close to that of o-1a. In this case, however, irradiation of o-3 by visible light in the solid matrix did not afford c-3. Similarly to c-1a, c-3 also undergoes a facile oxidation to the planar hydrocarbon 4.
Our results and those of Kubo and co-workers indicate that chemical reactivity of cethrene and related diradicaloid molecules could be employed as a working principle in a switch that can be operated solely by light. In order to verify the reversibility of the photochemical ring-opening/-closing process and the ability of cethrene to act as a photoswitch, we designed and herein present its derivative o-1b (Scheme 1, middle), equipped with two methyl substituents (in red) in place of two hydrogen atoms in the fjord region, which are critical for suppressing the oxidation of the closed form to the flat hydrocarbon 2. In addition to improving the stability of c-1b, the methyl substituents expedite the synthesis of the [5]helicene core as well as increase its configurational stability.16 Our present results demonstrate that 13,14-dimethylcethrene can be switched reversibly between an open (o-1b) and a closed (c-1b) form by light, and we introduce this system as a prototype of a diradicaloid photoswitch as well as the first example of a carbohelicene-based chiroptical photoswitch.17 During the switching process, the (1) HOMO–LUMO and (2) ST gaps as well as (3) the degree of helical twist are altered simultaneously, leading to significant changes in the optical and chiroptical properties. Furthermore, our DFT calculations suggest that additional decrease of the ST gap of the open form, which could be achieved by suitable structural modification, can make this system act also as a switch between two singlet-ground-state forms, one of which displays magnetic properties on account of the thermally accessible triplet excited state.
Results
Synthesis
With 5 as the starting point,16 diester intermediate 6 was prepared (Scheme 2) through a palladium-catalyzed Heck cross-coupling reaction with methyl acrylate followed by a reduction in a yield of 73% over the two steps. Diester 6 was subsequently transformed first into a diacid and then its bis(acyl chloride), which afforded the key intermediate 7 in a Friedel–Crafts acylation mediated by AlCl3 in a 43% yield over the three steps. Compound 7 contains all seven six-membered rings of the cethrene core, and its structure was confirmed by 1D/2D NMR spectroscopy (sections S7 and S8 in the Supporting Information) as well as X-ray crystallographic analysis (section S5). Finally, a reduction of 7 followed by a dehydration provided the dihydro precursor 8 (61% over two steps), which upon oxidation with p-chloranil afforded the target compound c-1b in 91% yield. In the final step, the oxidant p-chloranil first generates the open form o-1b, which undergoes an in situ thermal electrocyclic ring closure to yield the closed form c-1b that can be isolated by column chromatography as a stable compound.
Scheme 2. Synthesis of the Closed Form of 13,14-Dimethylcethrene.

Reaction conditions: (a) methyl acrylate (MA), Pd(OAc)2, PPh3, K2CO3, Bu4NBr, DMF, 110 °C, 20 h; (b) H2, Pd/C, CH2Cl2/EtOH, room temperature, 3 h; (c) LiI, 2,4,6-collidine, 185 °C, 3 h; (d) (COCl)2, 65 °C, 2.5 h; (e) AlCl3, CH2Cl2, −78 to −10 °C, 5 h; (f) NaBH4, CH2Cl2/EtOH, room temperature, 1.5 h; (g) p-TSA, toluene, 90 °C, 5 min; (h) p-chloranil, C6H6, room temperature, 16 h.
Structural Characterization
To validate the structures, the proton and carbon resonances for both c-1b and o-1b were fully assigned by means of COSY, NOESY, HMQC/HSQC, and HMBC NMR techniques (sections S7 and S8). In the 1H NMR spectrum of c-1b (Figure 1, bottom), the resonances for protons H-1 and H-2 are in the region typical for a double bond (6.2–6.7 ppm), in agreement with the crystal structure (vide infra; Figure 2). Resonances for protons H-3, H-4, H-5, H-6, and H-7 of the pentaphene moiety are in the aromatic region (6.9–7.8 ppm), the highest chemical shift being displayed by H-6 (∼7.8 ppm).
Figure 1.
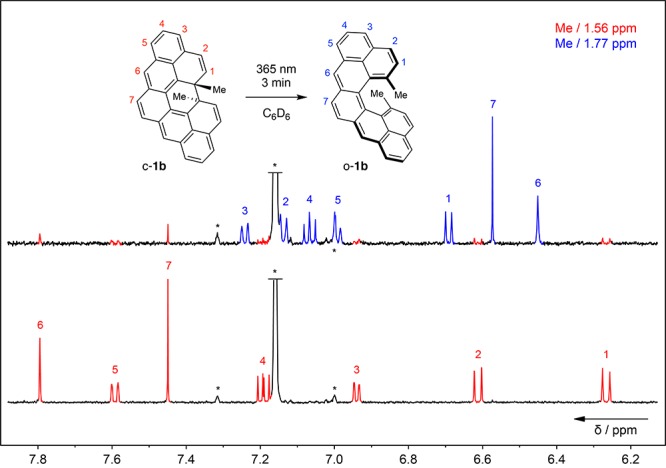
1H NMR spectra (600 MHz, C6D6, 25 °C) of c-1b (red) recorded before (bottom) and after (top) irradiation at 365 nm for 3 min, which generates o-1b (blue). The assignment of the proton resonances is shown (for 2D spectra, see the Supporting Information). Black asterisks denote the residual solvent and its satellite signals.
Figure 2.
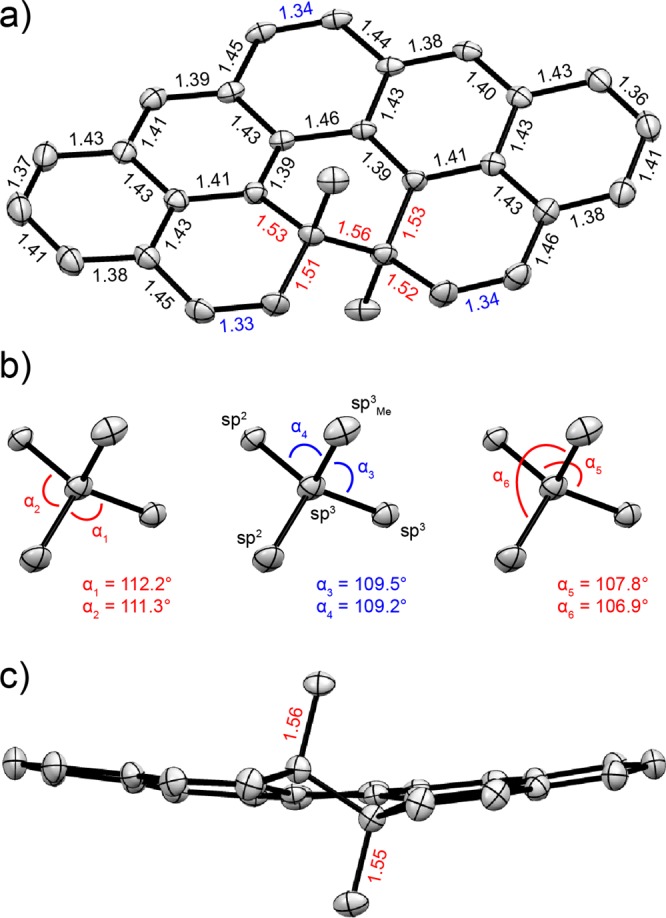
(a, c) Top (a) and side (c) views of the racemic solid-state structure of c-1b obtained from a single-crystal X-ray diffraction analysis. The thermal ellipsoids are shown at the 50% probability level, and the bond lengths are given in Å. The bond-length values typical for single and double bonds are highlighted in red and blue, respectively. (b) Tetrahedral angles (α) of the sp3 carbon atom bearing the methyl group. Angles with values close to an ideal tetrahedral angle value (∼109.5°) are highlighted in blue, and those that have higher (left) or lower (right) values are highlighted in red.
The structure of the open form o-1b was confirmed with a sample generated by irradiating a solution of c-1b (∼10–4 M in C6D6) at 365 nm for 3 min (Figure 1). Upon irradiation, the proton resonances of c-1b (in red) almost completely disappeared and the resonances that belong to o-1b (in blue) became visible (o-1b:c-1b ≈ 7:1). In the 1H NMR spectrum of o-1b, the resonances for protons H-6 and H-7 are shifted upfield to 6.4–6.6 ppm, in accord with the quinoidal structure (highlighted in blue in Figure 5b). The remaining proton resonances in the region 6.9–7.3 ppm reflect the aromatic character of the naphthalene subunits, except for the resonance of H-1 protons (6.68 ppm), which are shielded from the opposing terminal benzenoid ring.
The structure of compound c-1b was also validated by X-ray crystallographic analysis (Figure 2 and section S5) of a single crystal grown from a hexane solution by slow evaporation of the solvent. The solid-state structure reveals an almost flat, slightly bent geometry of c-1b, with the two skeletal quaternary sp3 carbon atoms each bearing one methyl substituent protruding above and below the skeleton plane. The structure appears to be only partially strained, judging from (1) slightly elongated C(sp3)–C(sp3) (1.55–1.56 Å) and C(sp2)–C(sp3) (1.51–1.53 Å) bonds (Figure 2a,c), (2) small bond-length alteration of the benzenoid rings (Figure 2a), and (3) minor distortion of some tetrahedral angles of the quaternary sp3 carbon atoms from an ideal value of ∼109.5° (Figure 2b).
UV–Vis Kinetic Measurements
The kinetic parameters of the thermal conrotatory electrocyclic ring closure of o-1b to c-1b, namely, the rate constants (k) and the activation energies (Ea), were determined through UV–vis spectroscopic measurements of samples dissolved in toluene or CH2Cl2. Compounds c-1b and o-1b have distinct UV–vis spectra (Figure 3a), where the former shows absorption mostly in the UV region (red trace), while the latter has a characteristic absorption band (λmax = 626 nm in toluene) in the visible region (blue trace). In the kinetic studies, a solution of c-1b (∼10–4 M) was irradiated by light (365 nm), which provided o-1b via a photochemical electrocyclic ring-opening reaction. Subsequently, the decrease of the absorbance (A) of o-1b at λmax = 626 nm in toluene and 624 nm in CH2Cl2 was followed with time (t) at various temperatures (Figures S1 and S2). A plot of ln(At/A0) against t was used for determination of the k values at specific temperatures (T), and then the plot of ln k against 1/T was used to determine Ea values (Figure 3b and Figures S3 and S4). In toluene, the ring closure proceeds with an Ea = 20.3 ± 0.26 kcal mol–1, while Ea = 17.1 ± 1.3 kcal mol–1 in CH2Cl2.
Figure 3.
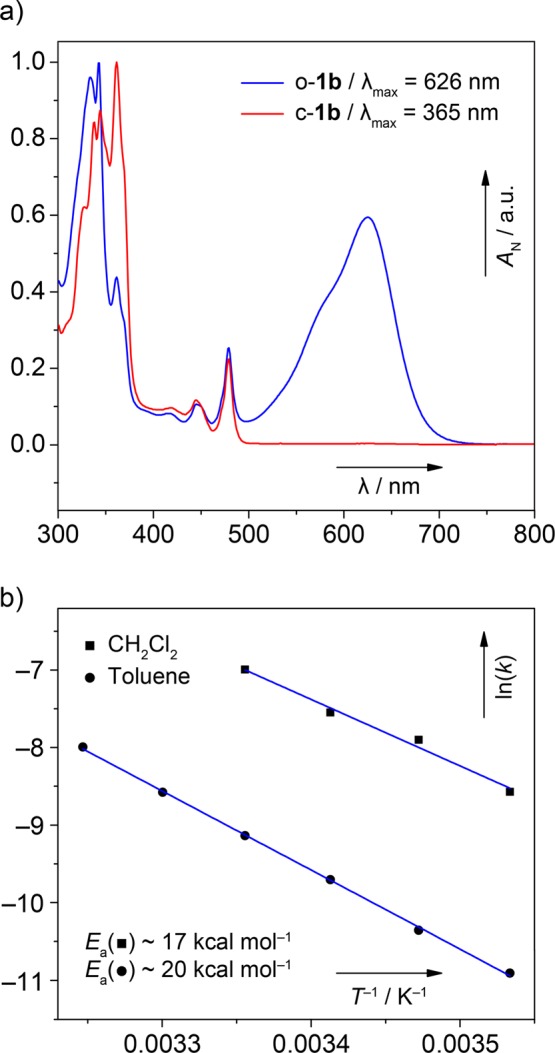
(a) Normalized UV–vis spectra of a solution of c-1b in toluene (∼10–4 M, 298 K) before (red trace) and after (blue trace) irradiation (365 nm, 1 min), which generates o-1b. The absorption bands with low intensity at 450 and 480 nm belong to an unknown impurity. (b) Arrhenius plots of ln k against 1/T used for determination of Ea in CH2Cl2 and toluene.
Circular Dichroism (CD) Spectroscopy
The R,R and S,S enantiomers of c-1b were separated by HPLC employing a chiral stationary phase (Figure S8), and the absolute configuration of each enantiomer was assigned with the aid of TD-DFT calculations (Figure S9). The complementary CD spectra for both enantiomers are shown in Figure 4 (red traces). Irradiation (365 nm, 1 min) of a solution of (R,R)-c-1b generated the corresponding M enantiomer of o-1b, while the P enantiomer was obtained from (S,S)-c-1b. These enantiomers also displayed mirror-image Cotton effects in their CD spectra (blue traces).
Figure 4.
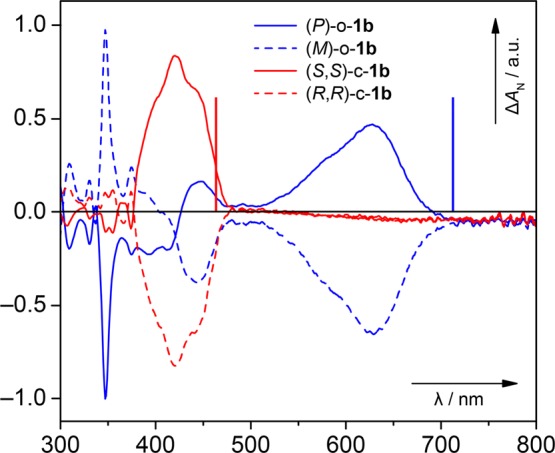
CD spectra of the enantiomers of c-1b (red traces), separated by HPLC employing a chiral stationary phase, and o-1b (blue traces), generated from the enantiomers of c-1b upon irradiation (365 nm, 1 min). The spectra of toluene samples were recorded at 283 K, and the absolute configurations were assigned with the aid of TD-DFT calculations (vertical lines; Figure S9).
DFT Calculations
The electronic structures of both o-1b and c-1b in their singlet ground and lowest triplet excited states and the thermally activated conrotatory electrocyclic ring closure of o-1b to c-1b were studied with the help of DFT calculations. Among the set of functionals that we employed in our calculations, only the CAM-B3LYP method required the use of a broken-symmetry formalism to obtain the structure of o-1b in its singlet ground state. Naturally, the optimization of the o-1b electrocyclization transition-state structures relied on broken-symmetry wave functions because the ring closure follows a reaction coordinate of a formally forbidden pericyclic process with an avoided crossing on its symmetric pathway from o-1b to c-1b. The corresponding activation barriers from the set of DFT methods used are similar, in the range of ∼22–24 kcal mol–1, and are summarized in Table S3. The calculated singlet–triplet (ST) energy gaps for o-1b and c-1b are shown in Table S2. The HOMO–LUMO energy gap calculated with the same method (B3LYP/6-311+G(d)) as that used12c previously for parent cethrene (o-1) was found to be 1.86 eV (Table S4). The ground-state energy of the closed form c-1b is lower by 6.3 kcal mol–1 (B3LYP/cc-pVTZ) in comparison to that of the open form o-1b.
Discussion
Diradicaloid π-conjugated molecules have recently regained1 considerable interest, although current research has mainly been focused18 on the physical properties of these systems to understand the relationship between their diradicaloid character and structure. The chemical reactivity that is associated with the diradicaloid character in these polycyclic aromatic hydrocarbons is without a doubt of comparable significance, but it remains largely unexplored. Reactions with oxygen as well as concerted or nonconcerted dimer and polymer formations are well-known1a examples from the early studies of parent systems, o- and p-quinodimethanes,19,20 or even extended structures such as longer acenes.21 These reactions typically represent, however, an undesired feature that impedes the synthesis and isolation of diradicaloid species, and significant efforts have therefore been made1,18,22 to develop strategies for decreasing the reactivity, and thus increasing the stability, of these molecules.
It has been demonstrated on several occasions that some of the concerted reactions can be reversed, for example by light irradiation, which offers an opportunity to steer the undesired chemical reactivity of diradicaloids to a useful function. These reactions include thermal dimerization of pleiadene or electrocyclic ring closure of its dimethyl derivative, both of which can be reversed photochemically, as demonstrated23 by Michl and co-workers. What links these two examples with cethrene12a (o-1) and its analogue biphenalenylidene15a (o-3) is the low activation energy of their formally symmetry-forbidden thermal transformations, a feature that arises on account of the diradicaloid character. In the present study, we took advantage of such unusual reactivity and translated it into a switching function that allows us to turn on and off the two most common characteristics of diradicaloids, namely, small HOMO–LUMO and singlet–triplet (ST) energy gaps, using 13,14-dimethylcethrene as a model system. In addition, the helically twisted backbone of cethrene brings an additional element, that of chirality, and this system therefore acts also as a chiroptical switch.
Synthesis and Properties
The closed form of 13,14-dimethylcethrene c-1b was synthesized (Scheme 2) in eight steps and an overall 17% yield starting from the [5]helicene precursor 5, which has the two desired methyl substituents already installed in the fjord region and is equipped with two bromo substituents at positions that allow for a construction of the two remaining six-membered rings of the cethrene core. The methyl substituents expedite the synthesis of 5, which can be accessed in only two steps from easily accessible precursors via a series of reactions that we described16 previously. Moreover, the methyl groups push the configurational stability of the [5]helicene core to the limit set by [9]helicene (the enantiomerization barrier of 10,11-dimethyl[5]helicene is ∼44 kcal mol–1 at 500 K16), making enantioenriched o-1b resistant toward racemization even at elevated temperatures. The solid-state structure of c-1b (Figure 2) unambiguously confirms the anti orientation of the methyl groups, which experimentally justifies the conrotatory mode of the electrocyclization of o-1b that we could previously support12a,24 only indirectly.
The open form o-1b was generated by irradiation (365 nm, 3 min) of a solution of a pure sample of c-1b, during which the NMR resonances that belong to c-1b almost completely disappeared and those of o-1b became visible. When this solution was left standing at room temperature, all resonances of o-1b gradually disappeared and those of c-1b appeared again, as the thermal electrocyclic ring closure of o-1b to c-1b took place.
The open and closed forms have considerably different electronic parameters, namely, the HOMO–LUMO and ST energy gaps. The former is clearly reflected by distinct UV–vis spectra (Figure 3a). While c-1b shows absorption mostly in the UV region (red trace), o-1b has a characteristic absorption band (λmax = 626 nm; S0–S1 transition) in the visible region (blue trace). With regard to the ST gap (ΔEST), the closed form c-1b is computed to have a very high (ΔEST ≈ 40 kcal mol–1, Table S2) and the open form o-1a a relatively low (ΔEST ≈ 10 kcal mol–1) triplet energy. EPR spectroscopy was used to probe whether the thermally populated triplet excited state of o-1b (Figure 5b) could be detected, as in the case of o-1a (ST gap of ∼6 kcal mol–1, EPR/DFT12c). EPR measurements performed on the samples of o-1b revealed, however, that this compound is EPR silent at room temperature, which supports that the ST gap of o-1b is larger than that of o-1a. The DFT-predicted ST gap for o-1b is even larger than that of EPR-silent planar heptazethrene (∼9 kcal mol–1, DFT12c). The most obvious reason for its increase is a higher degree of helical twist, caused by the steric effect of methyl substituents, which results in a longer distance (3.37 Å) between the fjord carbon atoms in comparison with that (3.03 Å) of o-1a (Figure 5b). The increased distance results in a less efficient through-space orbital overlap within the FMOs (Figure 5a), which is crucial for decreasing the ST gap in o-1a relative to heptazethrene.
To assess the geometric effect of the methyl substituents on the ST gap in o-1b more quantitatively, we performed relaxed potential energy surface (PES) scans for o-1 and o-1b in their singlet ground and triplet excited states along the coordinate defining the distance (d) between the fjord carbon atoms from ∼2.8 to 3.9 Å (Figure 5c). In the case of o-1, the increase of d from 3.03 Å, its minimum-energy value, to 3.37 Å, the minimum-energy value for o-1b, resulted in an increase in the ST gap by only 1.2 kcal mol–1. In the case of o-1b, the ST gap increased comparably by 1.8 kcal mol–1. The results of these calculations show that the sole geometric effect of the methyl substituents accounts only for about 30–45% of the ∼4 kcal mol–1 energy difference between the ST gaps of o-1 and o-1b. The remaining portion can be therefore attributed to the apparently stronger electronic effect of the methyl substituents, which destabilize the LUMO to a higher extent than the HOMO. Dissecting the effect of the methyl groups into electronic and geometric components nicely illustrates the large effect of subtle structural changes on the electronic parameters of helical diradicaloids, which can be employed as a useful tool to fine tune the properties of this class of materials.
Reactivity and Switching
The kinetics of the thermal ring closure was studied by UV–vis measurements (Figure 3). It was found that, in toluene, the reaction proceeds with an Ea of ∼20 kcal mol–1, a value that is larger by ∼6 kcal mol–1 in comparison to that (∼14 kcal mol–1) of o-1a reported12a previously, yet this value is quite small considering the conrotatory ring closure is formally a forbidden process. The results of our previous studies indicate12a that the low energies of the first singly and doubly excited states contribute to the lowering of the barrier. The increased Ea in the case of o-1b therefore does not come as a surprise, as the HOMO–LUMO gap of o-1b (1.86 eV, DFT; 1.80 eV, onset of absorption) is larger than that of o-1a (1.68 eV, DFT; 1.70 eV, onset of absorption12c), as discussed above.
In the more polar solvent CH2Cl2, the thermal ring closure of o-1b proceeds more quickly (Ea ≈ 17 kcal mol–1) in comparison to the reaction in toluene. Our DFT calculations show, however, that the electrocyclization barrier for o-1b via a C2-symmetric transition state (Ea ≈ 22–24 kcal mol–1) is insensitive to solvation (Table S3) and compares very well to that calculated previously for o-1a (Ea ≈ 23 kcal mol–1), despite the geometric and electronic changes brought about by the two methyl substituents, which is in agreement with our recent mechanistic hypothesis.12a In addition, an alternate mechanism that could involve a radical cation intermediate can now be also safely dismissed, as no oxidant was present in the sample of o-1b generated from a pure sample of c-1b solely by light.
The chirality and the distinct electronic properties (HOMO–LUMO and ST energy gaps) of o-1b and c-1b attracted our attention to investigate 13,14-dimethylcethrene as a conceptually new model for the design of chiroptical magnetic switches that could be operated solely by light. The photochemical processes, namely, ring opening of c-1b and the reverse process, ring closure of o-1b, were therefore studied by means of NMR (Figure 1), UV–vis (Figure 3), and CD (Figure 4) spectroscopy. In accord with the Woodward–Hoffmann rules,14 both conrotatory reactions proceed readily, with irradiation times of 1 min being sufficient to achieve a full conversion at concentrations of ∼10–4 M. This allows for efficient switching from the closed to the open form by UV light (365 nm) and from the open to the closed form by visible light (630 nm). The full irradiation cycle is shown in Figure 6a, where a colorless solution of c-1b is irradiated at 365 nm, affording a blue solution of o-1b, which upon irradiation at 630 nm turns into a colorless solution of c-1b (see also the video in the Supporting Information). The robustness of this system was demonstrated by repeating the full irradiation cycle multiple times in an aerated solution (Figure 6b). Under these conditions, the system showed only a moderate decomposition (∼1.5% loss of absorbance per cycle). The same degree of decomposition was observed when the irradiation cycles were performed under an inert atmosphere (Figures S5–S7), which indicates that the decomposition pathway of photoexcited o-1b or c-1b does not involve reactions with oxygen.
Figure 6.
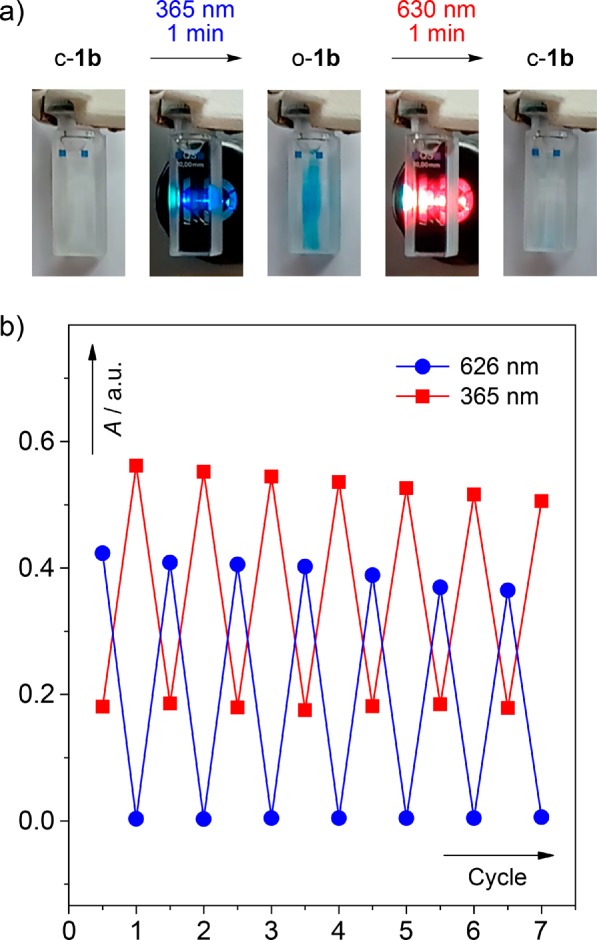
(a) Photographs documenting one full irradiation cycle. (b) Graph showing the changes in absorbance at 365 and 626 nm of a solution of c-1b in toluene (∼10–4 M, 298 K) under aerated conditions over seven irradiation cycles.
Although o-1b possesses an ST gap that is too large for detection of an EPR signal, the electronic effect of the methyl substituents on the ST gap, demonstrated clearly by our DFT calculations, indicates that it should also be possible to decrease the ST gap in a derivative of o-1b. Our preliminary calculations reveal that such a decrease can be achieved by installment of donor and acceptor substituents at relevant positions of the cethrene core, where the orbital coefficients in the HOMO and the LUMO differ markedly. Work on such a system, which can be switched between two singlet ground-state forms, one of which would display thermally accessible and magnetically active triplet state, is in progress in our laboratories.
Conclusion
We demonstrated that installment of two methyl substituents in the fjord region of cethrene leads to a robust system where the chemical reactivity of the diradicaloid core can be translated into a switching function. The methyl substituents improve not only the stability of this system against oxidation but also racemization and expedite the synthesis. Photochemical studies reveal that, in accord with the Woodward–Hoffmann rules, the conrotatory electrocyclic ring closure and ring opening can be efficiently mediated by visible and UV light, respectively. On account of the distinct electronic and geometric parameters of the open and closed forms, namely, the HOMO–LUMO and ST energy gaps and the degree of helical twist, the switching process can be monitored by a variety of spectroscopic techniques, which illustrate that this system functions as a chiroptical diradicaloid photoswitch. Moreover, substantial alteration of the ST gap upon switching suggests that systems structurally related to 13,14-dimethylcethrene can be employed in a conceptually new design of all-organic chiral magnetic switches that can be operated solely by light.
Experimental Section
Synthesis and Characterization
Experimental procedures and characterization data for all new compounds described in this work are compiled in the Supporting Information (sections S1 and S7–S9). Compound 5 was described16 previously and was prepared according to the published protocols. All chemicals and solvents were purchased from commercial sources and were used without further purification unless stated otherwise. The reactions and experiments that are sensitive to oxygen were performed using Schlenk techniques and argon-saturated solvents. The solvents were saturated with argon by either passing argon gas through the solvent or using the freeze–pump–thaw technique in three cycles. The NMR experiments were performed on instruments operating at 400, 500, and 600 MHz proton frequencies. The instruments were equipped with a direct-observe 5 mm BBFO smart probe (400 and 600 MHz), an indirect-detection 5 mm BBI probe (500 MHz), or a five-channel cryogenic 5 mm QCI probe (600 MHz). All probes were equipped with actively shielded z gradients (10 A). The experiments were performed at 295 or 298 K unless indicated otherwise, and the temperatures were calibrated using a methanol standard showing accuracy within ±0.2 K. Standard pulse sequences were used, and the data were processed using 2-fold zero-filling in the indirect dimension for all 2D experiments. Highly deuterium enriched benzene (C6D6, >99.96% D) was used in NMR experiments. Chemical shifts (δ) are reported25 in parts per million (ppm) relative to the solvent residual peak (1H and 13C NMR, respectively): C6D6 (δ = 7.16 and 128.06 ppm), CD2Cl2 (δ = 5.32 and 53.84 ppm), and CD3SOCD3 (δ = 2.50 and 39.52 ppm). The UV–vis spectra of toluene or CH2Cl2 samples were recorded at room temperature. The HPLC separation of enantiomers of c-1b was performed on an HPLC instrument equipped with a diode array UV–vis detector (λ = 200–600 nm) and a chiral-stationary-phase column (Chiralpak IA, 0.46 × 25 cm). Conditions: sample injection, 25 μL of a solution of c-1b in n-heptane/t-BuOMe (1/1, ∼1 mg in 1 mL); separation, n-heptane/t-BuOMe (98/2); flow rate, 1.0 mL min–1; 293 K. The CD spectra of toluene samples were recorded at 283 K in a 1 cm quartz glass cuvette. All solutions were argon-saturated unless stated otherwise.
Single-Crystal X-ray Diffraction (XRD)
Single crystals of compound 7 suitable for X-ray diffraction analysis were grown from the corresponding CH2Cl2 solution by slow evaporation of the solvent. Diffraction data were collected at 123 K using Cu Kα radiation on a Bruker APEX II diffractometer. Integration of the frames and data reduction were carried out using26 the APEX2 software. The structure was solved by the charge-flipping method using27 Superflip. All non-hydrogen atoms were refined anisotropically by full-matrix least squares on F2 using28 CRYSTALS. Single crystals of compound c-1b suitable for X-ray diffraction analysis were grown from the corresponding hexane solution by slow evaporation of the solvent in the dark. Diffraction data were collected at 160(1) K on a Rigaku OD XtaLAB Synergy, Dualflex, Pilatus 200 K diffractometer using a single-wavelength X-ray source (Mo Kα radiation: λ = 0.71073 Å)29 from a microfocus sealed X-ray tube and an Oxford liquid-nitrogen Cryostream cooler. The selected suitable single crystal was mounted using polybutene oil on a flexible loop fixed on a goniometer head and immediately transferred to the diffractometer. Pre-experiment, data collection, data reduction, and analytical absorption correction30 were performed31 with the program suite CrysAlisPro. Using32 Olex2, the structure was solved33 with the SHELXT small-molecule structure-solution program and refined34 with the SHELXL2016/6 program package by full-matrix least-squares minimization on F2. PLATON was used35 to check the result of the X-ray analysis. For more details about the data collection and refinement parameters of both compounds, see the corresponding CIF files in the Supporting Information. Both structures were analyzed using36 Mercury. The crystallographic views of their solid-state structures are shown in Figures S10 (7) and S11 (c-1b). The crystal parameters and structure refinements are summarized below and in Table S1. The crystallographic parameters were deposited with the Cambridge Crystallographic Data Centre (CCDC).
Crystal parameters for compound 7:
C30H22O2; 0.04 × 0.08 × 0.08 mm; monoclinic, C2/c (No. 15); a = 9.2601(11), b = 14.9860(11), and c = 14.9657(14) Å; α = 90, β = 107.491(4), and γ = 90°; V = 1980.8(3) Å3; Z = 4; T = 123 K; ρcalc = 1.390 g cm–3; μ = 0.670 mm–1. CCDC no. 1563973.
Crystal parameters for compound c-1b:
C30H20; 0.09 × 0.14 × 0.21 mm; monoclinic, P21/c (No. 14); a = 7.82299(16), b = 17.3405(4), and c = 13.9697(3) Å; α = 90, β = 96.824(2), and γ = 90°; V = 1881.62(7) Å3; Z = 4; T = 160(1) K; ρcalc = 1.343 g cm–3; μ = 0.076 mm–1. CCDC no. 1563974.
UV–Vis Kinetic Measurements
The activation energy (Ea) for the thermal transformation of o-1b to c-1b was determined through kinetic measurements. A CH2Cl2 or toluene solution of c-1b (∼10–4 M) was irradiated at 365 nm for 2.5 min at room temperature, whereupon a majority of c-1b was converted to o-1b. The UV–vis spectra before and after irradiation are shown in Figures S1a (CH2Cl2) and S2a (toluene). The absorption band with a maximum at 624 nm (CH2Cl2) and 626 nm (toluene) corresponds to o-1b (S0–S1 transition). As this band does not have any overlap with absorption bands of c-1b, the decrease of this band’s maximum intensity (Figures S1b and S2b) was used for determination of the rate constants (k) at various temperatures (Figures S3a and S4a). Because the electrocyclization of o-1b to give c-1b is a unimolecular process, it was assumed that this transformation follows first-order kinetics (ln(At/A0) = −kt). The ln k values were plotted against 1/T (Figures S3b and S4b), and the Arrhenius equation (ln k = ln A – Ea/(RT)) was then used to determine the values of Ea in CH2Cl2 and toluene.
DFT Calculations
The DFT calculations were performed37 with the Gaussian 09 (Revision D.01) suite of electronic structure programs. The gas-phase geometry optimizations were done with the respective functional (B3LYP, CAM-B3LYP, BMK, or M06-2X) and the 6-31G(d) or 6-311+G(d) basis set and ultrafine integration grid (Integral = Ultrafine keyword in Gaussian). Frequency analysis was performed to test the character of the stationary points and to provide zero-point vibrational energy corrections (ZPVEs), which were used unscaled. The geometries obtained with the latter basis set were used for calculations of the optical properties. The restricted (R-prefix) formalism was used to model the singlet states, and the unrestricted (U-prefix) formalism was used in the modeling of the triplet states. The solution of the SCF equations for the restricted singlet wave functions was tested for stability and optimized to obtain the lowest-energy solution if an RHF → UHF instability was found (Stable = Opt keyword in Gaussian). The broken-symmetry (BS) singlet wave function obtained this way was used to reoptimize the geometries of the molecules. The final energies were calculated with the cc-pVTZ basis set either employing the polarizable continuum model (SMD38 with toluene or CH2Cl2 as the solvent) to account for the collective solvation effects in calculations of light-absorption properties or using the gas phase otherwise. The relaxed potential energy surface scans (Figure 5) along the coordinate, which describes the distance between the carbon atoms at positions 13 and 14 in o-1 and o-1b, were performed for both the singlet ground state and the lowest triplet excited states at the B3LYP/6-31G(d) level of theory with no solvation and ZPVE corrections. The TD-DFT/cc-pVDZ calculations served to predict the absorption properties of o-1b (Table S4), and TD-DFT calculations (B3LYP/cc-pVDZ/PCM(toluene)) on R-B3LYP/6-31G(d) geometries served to model the circular dichroism (CD) spectra of the enantiomers of o-1b and c-1b (Figure 4 and Figure S9).
Acknowledgments
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (Grant Agreement No. 716139), the Swiss National Science Foundation (SNSF, T.Š. PZ00P2_174175, M.J. PZ00P2_148043 and PP00P2_170534), the Novartis University of Basel Excellence Scholarship (P.R. and M.J.), and the Experientia Foundation (T.Š.). We thank Prof. Dr. Marcel Mayor for a generous support of our research at the University of Basel (UB) and Dr. Alessandro Prescimone (UB) for X-ray crystallographic analysis of 7 and gratefully acknowledge the computational facilities of the University of Fribourg.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b05465.
Synthetic procedures and characterization data for all new compounds, UV–vis, HPLC, CD, and crystallographic data, results of DFT calculations, assignment of 1H and 13C NMR resonances, NMR and HRMS spectra, and Cartesian coordinates (PDF)
Crystallographic data for 7 (CIF)
Crystallographic data for c-1b (CIF)
Video documentation of the photochemical switching (MPG)
The authors declare no competing financial interest.
Supplementary Material
References
- a Konishi A.; Kubo T. Top. Curr. Chem. 2017, 375, 83. 10.1007/s41061-017-0171-2. [DOI] [PubMed] [Google Scholar]; b Konishi A.; Kubo T. In Chemical Science of π-Electron Systems; Akasaka T., Osuka A., Fukuzumi S., Kandori H., Aso Y., Eds.; Springer: Tokyo, Japan, 2015; pp 337–360. [Google Scholar]; c Kubo T. Chem. Rec. 2015, 15, 218–232. 10.1002/tcr.201402065. [DOI] [PubMed] [Google Scholar]; d Sun Z.; Zeng Z.; Wu J. Acc. Chem. Res. 2014, 47, 2582–2591. 10.1021/ar5001692. [DOI] [PubMed] [Google Scholar]; e Abe M. Chem. Rev. 2013, 113, 7011–7088. 10.1021/cr400056a. [DOI] [PubMed] [Google Scholar]; f Morita Y.; Suzuki S.; Sato K.; Takui T. Nat. Chem. 2011, 3, 197–204. 10.1038/nchem.985. [DOI] [PubMed] [Google Scholar]; g Morita Y.; Nishida S. In Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds; Hicks R. G., Ed.; Wiley: Wiltshire, U.K., 2010; pp 81–145. [Google Scholar]
- a Ravat P.; Marszalek T.; Pisula W.; Müllen K.; Baumgarten M. J. Am. Chem. Soc. 2014, 136, 12860–12863. 10.1021/ja507421x. [DOI] [PubMed] [Google Scholar]; b Shimizu A.; Kubo T.; Uruichi M.; Yakushi K.; Nakano M.; Shiomi D.; Sato K.; Takui T.; Hirao Y.; Matsumoto K.; Kurata H.; Morita Y.; Nakasuji K. J. Am. Chem. Soc. 2010, 132, 14421–14428. 10.1021/ja1037287. [DOI] [PubMed] [Google Scholar]; c Pal S. K.; Itkis M. E.; Tham F. S.; Reed R. W.; Oakley R. T.; Haddon R. C. Science 2005, 309, 281–284. 10.1126/science.1112446. [DOI] [PubMed] [Google Scholar]; d Kubo T.; Shimizu A.; Sakamoto M.; Uruichi M.; Yakushi K.; Nakano M.; Shiomi D.; Sato K.; Takui T.; Morita Y.; Nakasuji K. Angew. Chem., Int. Ed. 2005, 44, 6564–6568. 10.1002/anie.200502303. [DOI] [PubMed] [Google Scholar]; e Itkis M. E.; Chi X.; Cordes A. W.; Haddon R. C. Science 2002, 296, 1443–1445. 10.1126/science.1071372. [DOI] [PubMed] [Google Scholar]
- a Michl J.; Bonačić-Koutecký V. Tetrahedron 1988, 44, 7559–7585. 10.1016/S0040-4020(01)86250-5. [DOI] [Google Scholar]; b Salem L.; Rowland C. Angew. Chem., Int. Ed. Engl. 1972, 11, 92–111. 10.1002/anie.197200921. [DOI] [Google Scholar]
- a Kubo T.; Katada Y.; Shimizu A.; Hirao Y.; Sato K.; Takui T.; Uruichi M.; Yakushi K.; Haddon R. C. J. Am. Chem. Soc. 2011, 133, 14240–14243. 10.1021/ja2065768. [DOI] [PubMed] [Google Scholar]; b Goto K.; Kubo T.; Yamamoto K.; Nakasuji K.; Sato K.; Shiomi D.; Takui T.; Kubota M.; Kobayashi T.; Yakusi K.; Ouyang J. J. Am. Chem. Soc. 1999, 121, 1619–1620. 10.1021/ja9836242. [DOI] [Google Scholar]
- a Cui Z.-H.; Lischka H.; Beneberu H. Z.; Kertesz M. J. Am. Chem. Soc. 2014, 136, 5539–5542. 10.1021/ja412862n. [DOI] [PubMed] [Google Scholar]; b Mou Z.; Uchida K.; Kubo T.; Kertesz M. J. Am. Chem. Soc. 2014, 136, 18009–18022. 10.1021/ja509243p. [DOI] [PubMed] [Google Scholar]; c Cui Z.-H.; Lischka H.; Beneberu H. Z.; Kertesz M. J. Am. Chem. Soc. 2014, 136, 12958–12965. 10.1021/ja505624y. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tian Y.-H.; Huang J.; Kertesz M. Phys. Chem. Chem. Phys. 2010, 12, 5084–5093. 10.1039/b925259b. [DOI] [PubMed] [Google Scholar]; e Miller J. S.; Novoa J. J. Acc. Chem. Res. 2007, 40, 189–196. 10.1021/ar068175m. [DOI] [PubMed] [Google Scholar]; f Suzuki S.; Morita Y.; Fukui K.; Sato K.; Shiomi D.; Takui T.; Nakasuji K. J. Am. Chem. Soc. 2006, 128, 2530–2531. 10.1021/ja058387z. [DOI] [PubMed] [Google Scholar]; g Takano Y.; Taniguchi T.; Isobe H.; Kubo T.; Morita Y.; Yamamoto K.; Nakasuji K.; Takui T.; Yamaguchi K. J. Am. Chem. Soc. 2002, 124, 11122–11130. 10.1021/ja0177197. [DOI] [PubMed] [Google Scholar]
- Mou Z.; Kertesz M. Angew. Chem., Int. Ed. 2017, 56, 10188–10191. 10.1002/anie.201704941. [DOI] [PubMed] [Google Scholar]
- Das A.; Müller T.; Plasser F.; Lischka H. J. Phys. Chem. A 2016, 120, 1625–1636. 10.1021/acs.jpca.5b12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravat P.; Ribar P.; Rickhaus M.; Häussinger D.; Neuburger M.; Juríček M. J. Org. Chem. 2016, 81, 12303–12317. 10.1021/acs.joc.6b02246. [DOI] [PubMed] [Google Scholar]
- a Takamuku S.; Nakano M.; Kertesz M. Chem. - Eur. J. 2017, 23, 7474–7482. 10.1002/chem.201700999. [DOI] [PubMed] [Google Scholar]; b Beaujean P.; Kertesz M. Theor. Chem. Acc. 2015, 134, 147. 10.1007/s00214-015-1750-3. [DOI] [Google Scholar]
- Rickhaus M.; Mayor M.; Juríček M. Chem. Soc. Rev. 2016, 45, 1542–1556. 10.1039/C5CS00620A. [DOI] [PubMed] [Google Scholar]
- Juríček M. Chimia 2018, 72, 322–327. 10.2533/chimia.2018.322. [DOI] [PubMed] [Google Scholar]
- a Šolomek T.; Ravat P.; Mou Z.; Kertesz M.; Juríček M. J. Org. Chem. 2018, 83, 4769–4774. 10.1021/acs.joc.8b00656. [DOI] [PubMed] [Google Scholar]; b Ravat P.; Šolomek T.; Ribar P.; Juríček M. Synlett 2016, 27, 1613–1617. 10.1055/s-0035-1561447. [DOI] [Google Scholar]; c Ravat P.; Šolomek T.; Rickhaus M.; Häussinger D.; Neuburger M.; Baumgarten M.; Juríček M. Angew. Chem., Int. Ed. 2016, 55, 1183–1186. 10.1002/anie.201507961. [DOI] [PubMed] [Google Scholar]
- a Li Y.; Heng W.-K.; Lee B. S.; Aratani N.; Zafra J. L.; Bao N.; Lee R.; Sung Y. M.; Sun Z.; Huang K.-W.; Webster R. D.; López Navarrete J. T.; Kim D.; Osuka A.; Casado J.; Ding J.; Wu J. J. Am. Chem. Soc. 2012, 134, 14913–14922. 10.1021/ja304618v. [DOI] [PubMed] [Google Scholar]; b Clar E.; Macpherson I. A. Tetrahedron 1962, 18, 1411–1416. 10.1016/S0040-4020(01)99296-8. [DOI] [Google Scholar]
- a Woodward R. B.; Hoffmann R.. The Conservation of Orbital Symmetry; Verlag Chemie: Weinheim, Germany, 1970. [Google Scholar]; b Fleming I.Molecular Orbitals and Organic Chemical Reactions; Wiley: Chichester, U.K., 2010; pp 253–368. [Google Scholar]; c Woodward R. B.; Hoffmann R. Angew. Chem., Int. Ed. Engl. 1969, 8, 781–932. 10.1002/anie.196907811. [DOI] [Google Scholar]; d Woodward R.; Hoffmann R. J. Am. Chem. Soc. 1965, 87, 395–397. 10.1021/ja01080a054. [DOI] [Google Scholar]
- a Uchida K.; Ito S.; Nakano M.; Abe M.; Kubo T. J. Am. Chem. Soc. 2016, 138, 2399–2410. 10.1021/jacs.5b13033. [DOI] [PubMed] [Google Scholar]; b Pogodin S.; Agranat I. J. Am. Chem. Soc. 2003, 125, 12829–12835. 10.1021/ja035968k. [DOI] [PubMed] [Google Scholar]
- Ravat P.; Hinkelmann R.; Steinebrunner D.; Prescimone A.; Bodoky I.; Juríček M. Org. Lett. 2017, 19, 3707–3710. 10.1021/acs.orglett.7b01461. [DOI] [PubMed] [Google Scholar]
- A few examples of heterohelicene-based chiroptical photoswitches have been reported:; a Isla H.; Crassous J. C. R. Chim. 2016, 19, 39–49. 10.1016/j.crci.2015.06.014. [DOI] [Google Scholar]; b Li W.; Li X.; Xie Y.; Wu Y.; Li M.; Wu X.-Y.; Zhu W.-H.; Tian H. Sci. Rep. 2015, 5, 9186. 10.1038/srep09186. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Irie M.; Fukaminato T.; Matsuda K.; Kobatake S. Chem. Rev. 2014, 114, 12174–12277. 10.1021/cr500249p. [DOI] [PubMed] [Google Scholar]; d Walko M.; Feringa B. L. Chem. Commun. 2007, 1745–1747. 10.1039/b702264f. [DOI] [PubMed] [Google Scholar]; e Okuyama T.; Tani Y.; Miyake K.; Yokoyama Y. J. Org. Chem. 2007, 72, 1634–1638. 10.1021/jo0620213. [DOI] [PubMed] [Google Scholar]; f Tani Y.; Ubukata T.; Yokoyama Y.; Yokoyama Y. J. Org. Chem. 2007, 72, 1639–1644. 10.1021/jo062022v. [DOI] [PubMed] [Google Scholar]; g Wigglesworth T. J.; Sud D.; Norsten T. B.; Lekhi V. S.; Branda N. R. J. Am. Chem. Soc. 2005, 127, 7272–7273. 10.1021/ja050190j. [DOI] [PubMed] [Google Scholar]
- a Wang Q.; Hu P.; Tanaka T.; Gopalakrishna T. Y.; Herng T. S.; Phan H.; Zeng W.; Ding J.; Osuka A.; Chi C.; Siegel J. S.; Wu J. Chem. Sci. 2018, 9, 5100–5105. 10.1039/C8SC01388H. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lukman S.; Richter J. M.; Yang L.; Hu P.; Wu J.; Greenham N. C.; Musser A. J. J. Am. Chem. Soc. 2017, 139, 18376–18385. 10.1021/jacs.7b10762. [DOI] [PubMed] [Google Scholar]; c Li G.; Phan H.; Herng T. S.; Gopalakrishna T. Y.; Liu C.; Zeng W.; Ding J.; Wu J. Angew. Chem., Int. Ed. 2017, 56, 5012–5016. 10.1002/anie.201700441. [DOI] [PubMed] [Google Scholar]; d Zeng W.; Phan H.; Herng T. S.; Gopalakrishna T. Y.; Aratani N.; Zeng Z.; Yamada H.; Ding J.; Wu J. Chem. 2017, 2, 81–92. 10.1016/j.chempr.2016.12.001. [DOI] [Google Scholar]; e Liu J.; Ravat P.; Wagner M.; Baumgarten M.; Feng X.; Müllen K. Angew. Chem., Int. Ed. 2015, 54, 12442–12446. 10.1002/anie.201502657. [DOI] [PubMed] [Google Scholar]; f Sun Z.; Lee S.; Park K. H.; Zhu X.; Zhang W.; Zheng B.; Hu P.; Zeng Z.; Das S.; Li Y.; Chi C.; Li R.-W.; Huang K.-W.; Ding J.; Kim D.; Wu J. J. Am. Chem. Soc. 2013, 135, 18229–18236. 10.1021/ja410279j. [DOI] [PubMed] [Google Scholar]; g Shimizu A.; Hirao Y.; Matsumoto K.; Kurata H.; Kubo T.; Uruichi M.; Yakushi K. Chem. Commun. 2012, 48, 5629–5631. 10.1039/c2cc31955a. [DOI] [PubMed] [Google Scholar]; h Kubo T.; Shimizu A.; Uruichi M.; Yakushi K.; Nakano M.; Shiomi D.; Sato K.; Takui T.; Morita Y.; Nakasuji K. Org. Lett. 2007, 9, 81–84. 10.1021/ol062604z. [DOI] [PubMed] [Google Scholar]
- a Segura J. L.; Martín N. Chem. Rev. 1999, 99, 3199–3246. 10.1021/cr990011e. [DOI] [PubMed] [Google Scholar]; b Charlton J. L.; Alauddin M. M. Tetrahedron 1987, 43, 2873–2889. 10.1016/S0040-4020(01)86825-3. [DOI] [Google Scholar]
- Brown C. J.; Farthing A. C. Nature 1949, 164, 915–916. 10.1038/164915b0.15407665 [DOI] [Google Scholar]
- Zade S. S.; Bendikov M. J. Phys. Org. Chem. 2012, 25, 452–461. 10.1002/poc.1941. [DOI] [Google Scholar]
- Anthony J. E. Chem. Rev. 2006, 106, 5028–5048. 10.1021/cr050966z. [DOI] [PubMed] [Google Scholar]
- a Steiner R. P.; Michl J. J. Am. Chem. Soc. 1978, 100, 6413–6415. 10.1021/ja00488a023. [DOI] [Google Scholar]; b Kolc J.; Michl J. J. Am. Chem. Soc. 1970, 92, 4147–4148. 10.1021/ja00716a075. [DOI] [Google Scholar]
- Although there was some structural ambiguity regarding the sp3 carbon atoms in the solid-state structure of c-3 obtained from crystallographic analysis on account of disordered alignment of the R,R and S,S enantiomers, the planar molecular backbone clearly indicated the anti configuration of c-3 (see ref (15a)).
- Fulmer G. R.; Miller A. J. M.; Sherden N. H.; Gottlieb H. E.; Nudelman A.; Stoltz B. M.; Bercaw J. E.; Goldberg K. I. Organometallics 2010, 29, 2176–2179. 10.1021/om100106e. [DOI] [Google Scholar]
- APEX2, Version 2 User Manual, M86–E01078; Bruker Analytical X-ray Systems, Inc.: Madison, WI, 2006. [Google Scholar]
- Palatinus L.; Chapuis G. J. Appl. Crystallogr. 2007, 40, 786–790. 10.1107/S0021889807029238. [DOI] [Google Scholar]
- Betteridge P. W.; Carruthers J. R.; Cooper R. I.; Prout K.; Watkin D. J. J. Appl. Crystallogr. 2003, 36, 1487. 10.1107/S0021889803021800. [DOI] [Google Scholar]
- Rigaku Oxford Diffraction, 2015.
- Clark R. C.; Reid J. S. Acta Crystallogr., Sect. A: Found. Crystallogr. 1995, 51, 887–897. 10.1107/S0108767395007367. [DOI] [Google Scholar]
- CrysAlisPro (version 1.171.39.13a); Rigaku Oxford Diffraction, 2016.
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spek A. L. J. Appl. Crystallogr. 2003, 36, 7–13. 10.1107/S0021889802022112. [DOI] [Google Scholar]
- a Macrae C. F.; Bruno I. J.; Chisholm J. A.; Edgington P. R.; McCabe P.; Pidcock E.; Rodriguez-Monge L.; Taylor R.; van de Streek J.; Wood P. A. J. Appl. Crystallogr. 2008, 41, 466–470. 10.1107/S0021889807067908. [DOI] [Google Scholar]; b Bruno I. J.; Cole J. C.; Edgington P. R.; Kessler M.; Macrae C. F.; McCabe P.; Pearson J.; Taylor R. Acta Crystallogr., Sect. B: Struct. Sci. 2002, 58, 389–397. 10.1107/S0108768102003324. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 09, Revision D.01; Gaussian, Inc., Wallingford, CT, 2009.
- Marenich A. V.; Cramer C. J.; Truhlar D. G. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.