

Abstract

Understanding the spontaneous organization of atoms on well-defined surfaces promises to enable control over the shape and size of supported nanostructures. Atomic layer deposition (ALD) boasts atomic-scale control in the synthesis of thin films and nanoparticles. Yet, the possibility to control the shape of ALD-grown nanostructures remains mostly unexplored. Here, we report on the bottom-up formation of both linear and V-shaped anatase TiO2 nanorods (NRs) on graphene nanoplatelets during TiCl4/H2O ALD carried out at 300 °C. NRs as large as 200 nm form after only five ALD cycles, indicating that diffusional processes rather than layer-by-layer growth are behind the NR formation. In particular, high-resolution transmission electron microscopy reveals that the TiO2 NRs and graphene nanoplatelets are in rotational alignment as a result of lattice matching. Crucially, we also show that individual nanocrystals can undergo in-plane oriented attachment.
Introduction
Atomic layer deposition (ALD) is a gas-phase bottom-up technique based on cyclic sequences of self-limiting reactions that bring about the deposition of less than a monolayer per cycle.1−4 It has already established itself as the method of choice for the layer-by-layer deposition of conformal thin films in applications that require atomic-level precision.4 Although mostly developed for flat substrates, ALD is also scalable to high-surface-area substrates, which are relevant for a variety of applications spanning catalysis, energy storage and conversion, and medicine.2,5−16 Given its unparalleled precision and scale-up potential, considerable research effort has been put in expanding its capability to the deposition of nanostructures other than thin films such as nanoparticles (NPs).2,6,7,17−21 However, the formation and growth of NPs are mediated not only by ALD surface chemistry but also by nonequilibrium phenomena such as adatom and NP diffusion and aggregation.17,22 Because such mechanisms are a strong function of reaction conditions and adlayer–support interactions, control over the NP size can be achieved only under certain system-dependent conditions.17,20,21 Nonetheless, if properly understood, unconventional growth pathways can expand the range of nanostructures that can be synthesized by ALD.22,23 After achieving control over the NP size, the natural next step in the advancement of ALD of NPs is the synthesis of shape-controlled NPs and, in particular, of one-dimensional (1D) nanocrystals such as nanorods (NRs).
Inspired by a recent finding,23 Wen and He24 posed the question: “Can oriented attachment be an efficient growth mechanism for the synthesis of 1D nanocrystals via ALD?” Oriented attachment is a nonclassical growth mechanism, mostly observed in the liquid phase, that involves the self-organization of particles or crystallites that migrate, align, and then fuse along a preferential crystallographic orientation, such that the resulting crystal grows in an asymmetric fashion.25−27 Although still poorly understood, a growing body of evidence suggests that oriented attachment dominates the solution-based growth not only of important nanomaterials but also of minerals in biogenic and geological environments.25,28−30 For this reason, De Yoreo et al.28 argued that a rational exploitation of oriented attachment can bring about significant advances in the design and synthesis of nanomaterials. Along these lines, Shi et al.23 have shown that high-temperature (≥600 °C) ALD of TiO2 can indeed be used to grow NRs via a mechanism that the authors argued to be a vapor-phase variant of oriented attachment. In fact, in contrast to the oriented attachment of individual crystals that align and fuse, they proposed that the NRs grow at the expense of an amorphous layer encapsulating the lateral facets, which migrates and attaches onto the facets exposed to the gas phase. Although in principle oriented attachment can be exploited to synthesize 1D nanocrystals via ALD, several questions of both practical and fundamental nature remain unanswered: (1) because oriented attachment transcends the layer-by-layer model of ALD, what is the interplay between the self-limiting behavior of ALD reactions and the atomistic processes behind the formation of NRs? In particular, can the NR size still be controlled by the number of cycles? (2) Can the substrate promote oriented attachment during ALD? For example, a variety of nanostructures are known to self-align on two-dimensional (2D) materials in an attempt to minimize the interfacial energy.31−34
Here, we report on the bottom-up formation of anatase NRs on gram-scale batches of graphene nanoplatelets at temperatures as low as 300 °C via TiCl4/H2O ALD. We show that NRs as large as 200 nm can form even after only five cycles, indicating that the growth is dominated by diffusional processes. The number of cycles affects mostly the number rather than the size of the NRs. Statistical analysis of the shape of the nanostructures at different reactant dosings reveals a competitive process between growth pathways, leading to either symmetric growth, and thus NPs, or asymmetric growth, and thus NRs. In particular, the population of the different observed nanostructures is a nonlinear function of the exposure time of TiCl4 and H2O even at saturation conditions. Finally, transmission electron microscopy (TEM) provides direct evidence not only of oriented attachment of individual TiO2 nanocrystals but also of an in-plane lattice alignment between TiO2 and graphene nanoplatelets.
Methods
Materials
Graphene nanoplatelets (6–8 nm thick and 15 m wide, and a surface area of about 150 m2 g–1) and the Ti precursor, titanium tetrachloride (TiCl4), were obtained from Strem Chemicals and used as received. Demineralized water was used as the coreactant. Both precursors were contained in stainless steel bubblers.
ALD Experiments
The ALD experiments were carried out in a home-built fluidized bed reactor operated at atmospheric pressure already described elsewhere.17,35 The reactor consists of a glass column (2.6 cm in internal diameter and 50 cm in height), placed on top of a double-motor vibration table (Paja PTL 40/40-24) to assist the fluidization process. The titanium precursor (TiCl4) was kept either at 30 °C or at 0 °C by means of an ice bath to investigate the influence of the vapor pressure on the Ti loading and the morphology of the ALD-grown TiO2 nanostructures. The coreactant (H2O) was kept either at 30 °C or at 80 °C by means of a heating tape wrapped around the bubbler. The precursors were delivered to the reactor by flowing N2—the carrier gas through the bubblers. The reactor was connected to three separate gas lines: two for the precursor delivery and one for the purging gas. The reactor was heated by an infrared lamp parallel to the glass column with a proportional–integral–derivative controller to maintain the desired temperature, that is, 100, 150, 200, or 300 °C. An ALD cycle consisted of sequential exposures of TiCl4 (5 s to 5 min) and H2O (5 s to 5 min), separated by N2 (99.999 vol %) purging steps (5–10 min). In each experiment, 0.75 g of graphene nanopowder was loaded in the reactor. Prior to the deposition, the nanopowder was dried overnight at 120 °C. An optimized gas flow of 0.8 NL min–1 was used to achieve uniform fluidization of the nanopowder at all times. To remove adventitious carbon and activate the inherently defective surface of the graphene nanoplatelets, the nanopowder was pretreated in situ with ozone-enriched air obtained by flowing synthetic air (20 wt % O2) through an ozone generator (Certizon Ozonizer C200, O3 output of 200 mg/h) prior to each ALD experiment. After the pretreatment and before the ALD experiment, the reactor was purged with N2 for 30 min.
Material Characterization
The morphology of the TiO2/graphene composites was investigated
via TEM, scanning electron
microscopy (SEM), and high-resolution TEM (HRTEM). The as-synthesized
TiO2/graphene composites were suspended in ethanol and
transferred to regular TEM grids of 3.05 mm in diameter. Such grids
were then used for all the microscopes. TEM images were taken using
a JEOL JEM1400 transmission electron microscope operating at a voltage
of 120 kV. The images were then analyzed by using the software ImageJ
to determine the size and shape of the TiO2 nanostructures.
For each sample, 900–1000 nanostructures were characterized
by measuring the following descriptors: perimeter (P), area (A), aspect ratio (AR) of the fitted ellipse,
and circularity: 4πA/P2 (C). On account of these parameters, the
analyzed structures were grouped into three different categories:
NPs, asymmetric NPs (ANPs), and NRs. NPs are defined as nanostructures
with C ≥ π/4 ≃ 0.785 (an object
with P and A such that C ≥ π/4 cannot
be approximated by a rectangle). The NP size was then determined by
calculating the equivalent projected diameter: 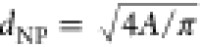 . NRs are defined as nanostructures
having
AR ≥ 3. The length and the width of the NRs are then defined
as the height and the base of the equivalent rectangle having the
same P and A. ANPs are structures
that fall in neither of the previous groups, that is, structures characterized
by either AR ≤ 3 and C ≤ π/4
or AR ≥ 3 and C ≥ π/4. SEM images were obtained
by using a JEOL JSM-840 scanning electron microscope with a LaB6 source at a voltage of 40 kV. HRTEM micrographs were taken
using a FEI Cs-corrected cubed Titan operating at 300 kV. Crystallographic
analysis (determination of lattice spacing, crystal orientation, exposed
facets, etc.) was carried out by analyzing the fast Fourier transform
(FFT) of the HRTEM images with the aid of the software CrysTBox36,37 and jems V4.
. NRs are defined as nanostructures
having
AR ≥ 3. The length and the width of the NRs are then defined
as the height and the base of the equivalent rectangle having the
same P and A. ANPs are structures
that fall in neither of the previous groups, that is, structures characterized
by either AR ≤ 3 and C ≤ π/4
or AR ≥ 3 and C ≥ π/4. SEM images were obtained
by using a JEOL JSM-840 scanning electron microscope with a LaB6 source at a voltage of 40 kV. HRTEM micrographs were taken
using a FEI Cs-corrected cubed Titan operating at 300 kV. Crystallographic
analysis (determination of lattice spacing, crystal orientation, exposed
facets, etc.) was carried out by analyzing the fast Fourier transform
(FFT) of the HRTEM images with the aid of the software CrysTBox36,37 and jems V4.
The crystal structure of the composites was analyzed by X-ray powder diffraction (XRD). The composites were transferred onto a Si wafer coated with 300 nm of SiO2 to remove the influence of the Si signal in the XRD patterns. The diffractograms were obtained by a PANalytical X-pert Pro diffractometer with Cu Kα radiation, a secondary flat crystal monochromator, and an X’celerator RTMS detector system. The angle of interest 2θ was measured from 10° to 80° with steps of 0.001°.
The chemical state and the composition of the composites were investigated by X-ray photoelectron (XPS) carried out with a ThermoFisher K-Alpha system using Al Kα radiation with a photon energy of 1486.7 eV. The composites were immobilized on a carbon tape placed on a Si wafer. XPS scans were acquired using a 200 μm spot size, a 55 eV pass energy, and a 0.1 eV/step with charge neutralization. The XPS spectra were fitted with the CasaXPS software.
Elemental analysis was carried out by means of inductively coupled plasma optical emission spectrometry (ICP–OES) and instrumental neutron activation analysis (INAA). The two techniques gave comparable Ti loadings. For ICP–OES, approximately 25 mg of powder was dissolved in a solution containing 4.5 mL of HCl (30%), 1.5 mL of HNO3 (65%), and 1 mL of HF (40%) using a microwave (Multiwave PRO). The destruction time in the microwave was 180 min at maximum power. After destruction, the samples were diluted with 50 mL of Milli-Q water and then analyzed with a PerkinElmer Optima 4300 DV optical emission spectrometer. For INAA, around 25 mg of powder was loaded into high-purity polyethylene capsules. The samples as well as a reference sample and an empty capsule were then sealed together in a polyethylene foil, packed in an irradiation container, and irradiated by a constant neutron flux. INAA has a detection limit in the range of 10–100 ng. The measurements were performed for 1 h after an appropriate time in order to let the nuclides decay. Then, the results were interpreted by a UNIX-based computer system, which converts all the information, for example, peak area and energies, in terms of Ti weight fraction (wt %). The Ti loading (L) was expressed in terms of at nm–2 by using the following equation:1,17
where x is the wt % of Ti in the powder sample, NA is Avogadro’s number, MWTi is the molecular weight of titanium, and S is the surface area of the graphene nanoplatelets.
Results and Discussion
Self-Limiting Behavior
The hallmark of ALD is its self-limiting nature.1,2 We tested whether TiO2 can be grown in a self-limiting fashion on gram-scale batches of graphene nanoplatelets by following the amount of deposited Ti as a function of the exposure time and the partial pressure of the precursors: TiCl4 and H2O (see Figure 1). For information regarding the experimental method and materials, the interested reader is referred to the Methods section. The evolution of the Ti loading (at nm–2) as a function of precursor exposure time shows that the surface reactions saturate after an exposure of about 30 s of TiCl4 and 15 s of H2O. This is true at different temperatures (200 and 300 °C), number of cycles (i.e., 5 and 10), and precursor partial pressures (see Figure 1b–e). In particular, past the saturation point, the Ti loading remains fairly constant within a time window of several minutes. In addition, the value of the Ti loading at saturation after 10 cycles is ∼2.3 at nm–2, which is about two times the saturation value after 5 cycles. In fact, exposure times of 30 s for the exposure of both precursors resulted in a linear increase of the Ti loading with the number of cycles in the cycle range 1–10 and in the temperature range 100–300 °C (see Figure 1a). If we consider that a monolayer of anatase exposing {100} facets has a Ti density of ∼16.7 at nm–2, a growth per cycle (GPC) of ∼0.23 Ti at nm–2 translates into ∼1.4% of a monolayer per cycle. This GPC is about 10 times lower than the steady-state GPC (i.e., in the limit of a large number of cycles) typically reported for TiCl4/H2O ALD, which is in the range of 10–15% of a monolayer per cycle.38−40 Such low GPC is consistent with the low reactivity of graphitic surfaces, which, lacking dangling bonds, induce what is referred to as substrate-inhibited ALD growth.1 Nonetheless, ALD growth can still start from the very first cycle because, in contrast to ideal graphene or perfect graphitic surfaces, the surface of graphene nanoplatelets is inherently defective, and as such it can be activated by the incorporation of the oxygen-containing species during the ozone pretreatment step.17,41,42
Figure 1.

Self-limiting behavior of the surface chemistry of the ALD reactions. (a) Evolution of the amount of titanium deposited (i.e., the loading) with the number of ALD cycles at different temperatures. Evolution of the Ti loading with the exposure time of TiCl4 (b) and H2O (c) at 300 °C after 10 cycles. The loading indicated by the red open squares and black circles of (b,c) was obtained by having the TiCl4 bubbler at 0 and 30 °C, respectively. Evolution of the Ti loading with the exposure time of TiCl4 (d) and H2O (e) at 200 °C after five cycles. The loading indicated by the red open square of (e) was obtained by having the H2O bubbler at 80 °C. Unless otherwise specified, all the experiments were carried out with the TiCl4 and H2O bubbler kept at 30 °C. The vapor pressure of TiCl4 is ∼2.5 Torr at 0 °C and ∼14.6 Torr at 30 °C, whereas H2O has a vapor pressure of ∼31.8 Torr at 30 °Cand of ∼355.1 Torr at 80 °C.43 The error bars indicate 95% confidence intervals.
Effect of Deposition Temperature on Crystallinity, Morphology, and Chemical State
Although the deposition temperature had virtually no effect on the amount of deposited Ti, it did affect the crystallinity, the morphology, and the chemical state of the ALD-grown TiO2. The XPS spectra of the composites obtained after 10 cycles in the temperature range 100–300 °C reveal that the Ti atoms are mostly in the Ti4+ state, with a small fraction being in the Ti3+ state (see Figure 2b). In particular, the fraction of the latter decreases from about 10% at 100 °C to less than 4% at 300 °C. The Ti3+ contribution to the Ti 2p spectra is probably due to the presence of nonstoichiometric TiO2. This is corroborated by the shift toward lower binding energies of the peak associated with the Ti–O bonds in the O 1s region with increasing deposition temperature (see Figure 2d), which is known to be correlated with a reduction in the concentration of oxygen vacancies.44,45 The deposition of nonstoichiometric TiO2 at low temperature is also consistent with the absence of the signature of crystallinity in the XRD patterns of the composites obtained at temperatures below 200 °C (see Figure 2a). On the other hand, a clear signature of the presence of anatase TiO2 is evident for the composites obtained at 200 and 300 °C. In particular, the peaks associated with the anatase structure were narrower for the composites obtained at 300 °C, indicating the formation of larger crystallites at higher temperature.
Figure 2.

XRD spectra (a) and XPS spectra (b–d) of the TiO2 graphene composites obtained after 10 cycles at different temperatures. In particular, (b) shows the XPS spectra of the Ti 2p region, (c) shows the spectra of the C 1s region, and (d) shows the spectra of the O 1s regions. All the experiments were carried out by following the pulsing sequence: 30 s TiCl4-5 min N2 purge-30 s H2O-5 min N2 purge.
The transition from amorphous to anatase TiO2 with increasing temperature is also reflected in the evolution of the morphology of the composites as revealed by TEM analysis (see Figure 3). At 100 °C, the ALD-grown TiO2 appears in the form of irregularly shaped 2D flakes (see Figure 3a). On the other hand, at 200 °C, while 2D flakes are still visible, a fraction of the ALD-grown TiO2 forms into distinct crystallites with sharp corners (see Figure 3b). Crucially, at 300 °C, the 2D flakes are no longer present; instead, the same amount of TiO2 is concentrated in nanostructures ranging from individual and agglomerated NPs to linear, curved, and v-shaped NRs (see Figures 3c and 6).
Figure 3.

Effect of the deposition temperature on the morphology of the ALD-grown TiO2. Representative TEM images of the TiO2/graphene composites obtained after 10 cycles at 100, 200, and 300 °C. All the experiments were carried out by following the pulsing sequence: 30 s TiCl4—5 min N2 purge—30 s H2O—5 min N2 purge.
Figure 6.

(a) TEM micrograph of a graphene nanoplatelet after 10 ALD cycles carried out at 300 °C using the pulsing sequence: 2 min TiCl4—5 min N2—30 s H2O—5 min N2. (b) TEM micrograph of the tip of a NR encapsulated by an amorphous layer. Field emission SEM micrograph of the TiO2/graphene composites obtained after 10 cycles at 300 °C (c) using the same pulsing sequence as in (a). Selection of TEM micrographs of NRs of peculiar morphology: curved NR (d), NR aligned with the edge of the nanoplatelet (e), and v-shaped NRs (f).
To test whether the formation of NRs is simply triggered by an increase in temperature, we annealed at 300 °C the composites obtained after 10 cycles at 100 and 200 °C for 2 h (≃the average duration of 10 cycles) in synthetic air and in N2 + H2O, respectively. In particular, we annealed the composites in the same reactor where all the ALD experiments were performed. After annealing, the morphology did change in that 2D flakes were no longer present and most of the TiO2 formed into percolated NPs; however, no NRs were observed (see Figure S1). This suggests that the sequential nature of ALD, and thus the timing of each precursor exposure, is crucial for the formation of NRs.
Effect of Precursor Exposure Time on the Morphology and Unfolding of Asymmetric Growth
The precursor exposure time had a dramatic effect on the morphology of the ALD-grown TiO2. Figure 4 shows the evolution of the morphology of the TiO2 nanostructures deposited after 10 cycles at 300 °C with varying TiCl4 exposure times in the range 30–180 s. Strikingly, although the morphology varies considerably within such a range of exposures, the Ti loading remains virtually the same (see Figures 1b and 4). This means that prolonged TiCl4 exposures affect the growth process by exacerbating the effect of diffusion phenomena rather than by the addition of more TiO2.
Figure 4.
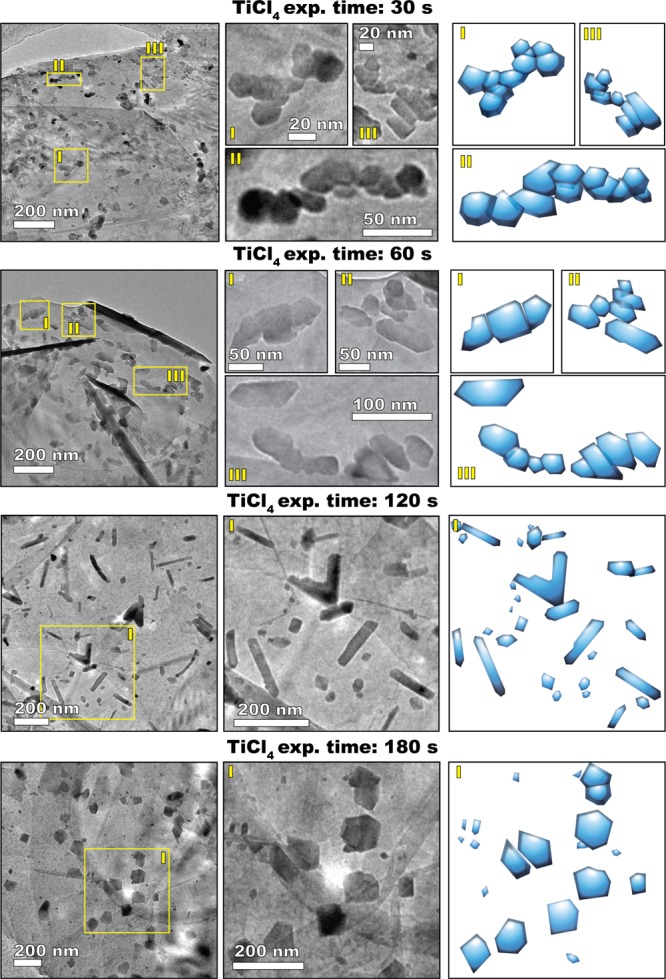
Effect of the TiCl4 exposure time on the morphology of the TiO2 nanostructures grown at 300 °C after 10 ALD cycles (the water exposure time was of 30 s in all cases). The images in the first two columns are TEM micrographs, whereas the third column contains schematic representations of the morphology of the observed nanostructures.
TiCl4 exposures of 30 s resulted in the formation of a large number of both round and ANPs of about 20 nm in size that are mostly clustered in elongated chainlike agglomerates. Sporadically, also NRs with sharp facets were observed (see also Figure 3c). Image analysis shows that about 50% of the nanostructures have an aspect ratio (AR) ranging from 1.5 to 6 and that the NRs (AR ≥ 3 & circularity ≤ π/4) account for about 2.5% of the total population (see Figure 5a,c).
Figure 5.

Summary of the statistical analysis of the shape of the nanostructures obtained with ALD at 300 °C. Box plots and data overlap of the evolution of the AR as a function of the TiCl4 and H2O exposure time (a,b). The boxes indicate the 10th, 25th, 75th, and 90th percentiles of the population, the full circle indicates the maximum, the white diamonds indicate the average, and the horizontal line indicates the median. Evolution of the average fraction of NRs, that is, the number of NRs divided by the total number of observed nanostructures, as a function of TiCl4 (c) and H2O (d) exposure time, and of the number of cycles (e). The asterisks indicate that the experiments were carried out with the TiCl4 bubbler kept at 30 °C, otherwise all the data were obtained with experiments in which the TiCl4 and H2O bubbler were kept at 0 and 30 °C respectively. Box plots and data overlap of the evolution of the (f) length and (g) width of the NRs and of the (h) size of the NPs with the number of cycles (pulse sequence: 2–5 min-30 s-5 min). The boxes indicate the 10th, 25th, 75th, and 90th percentiles of the population, the full circle indicates the average, and the horizontal line indicates the median.
Increasing the TiCl4 exposure time to 60 s resulted in a larger fraction of elongated structures having an AR between 1.5 and 3. In this case, the nanostructures are less irregular and more symmetric with respect to their major axis. TEM images clearly show that such structures mostly consist of few individual NPs that are partially aligned and fused. In fact, the average size of the ANPs doubled compared to the previous case (see Figure S2). This indicates that longer exposures promoted the diffusion and coalescence of the individual NPs. Furthermore, although the fraction of NRs was comparable to the previous case, the number of structures having an AR of about 2 and sharp lateral edges increased.
Crucially, TiCl4 exposures of 120 s resulted in the formation of a significantly higher number of both linear and V-shaped NRs presenting sharp lateral facets (see also Figure 6). This was true for two different TiCl4 partial pressures (in the precursor bubbler): ∼2.5 and ∼14.6 Torr43 (see Figures 5d and S7). In particular, the length of the NRs was distributed in the range 50–500 nm, whereas the width ranged from ∼10 to 60 nm (see Figure S2). Interestingly, the average NR width is always about equal to the average NP size (∼20 nm), which is consistent with the NPs being the building blocks of the NRs (see Figure 5g,h). In this case, the fraction of NRs accounted for ∼7–8% of the total population (see Figure 5c). It is worth noting that the fraction of NRs is calculated on a number basis: the total number of NRs divided by the total number of observed nanostructures. On a weight basis, however, NRs would account for a fraction of TiO2 much larger than 8%. In the first approximation, if we assume that all TiO2 is distributed in NPs and NRs and that the average NR weight is equal to average NP weight times the average NR AR (∼5), then the NRs account for about 30% of the total TiO2 mass.
TiCl4 exposures of 180 s suppressed the formation of NRs and, instead, promoted the growth of large and nearly symmetric NPs. Therefore, there exists an optimum in the TiCl4 exposure time that promotes asymmetric growth and thus maximizes the fraction of NRs. Interestingly, varying the H2O exposure time affected the morphology in a similar fashion, with 30 s of H2O exposure being the optimal time when using TiCl4 exposures of 120 s (see Figures S3, 5b and d). In addition, varying the number of cycles also resulted in an evolution of the relative fraction of NRs, exhibiting an optimum (see Figure 5e). In particular, an increase in the number of cycles from 5 to 10 resulted in an increase in the NR fraction, whereas the NR and NP size remained virtually unaffected (see Figure 5e–h). On the other hand, increasing the number of cycles from 10 to 20 not only decreased the fraction of NRs but also broadened the size distribution of both NRs and ANPs (see Figures 5f and S2). As we will point out later, the existence of an optimum and the evolution of the size of the different nanostructures can be explained in terms of a competition between diffusion phenomena, leading to either symmetric or asymmetric growth, and NP formation resulting from deposition.
Analogies with Liquid-Phase Oriented Attachment
The transition from individual NPs to NRs with varying TiCl4 exposure times presented in Figure 4 bears a striking resemblance to the solution growth of Pt3Fe NRs from NP building blocks via oriented attachment observed by Liao et al.30 By using real-time TEM, they could monitor the spontaneous formation of NRs in a solution containing Pt and Fe precursors that undergo reduction upon electron beam illumination. In particular, they observed three distinct stages. In the first stage, a large number of small NPs nucleate upon precursor reduction. These NPs grow by parallel atom attachment and NP coalescence until they reach a critical size. In particular, in this stage, coalescing NPs relax in spherical shapes. In the second stage, colliding NPs form into NP chains that do not coalesce into spherical NPs. When most NPs form into NP chains, these can in turn undergo end-to-end attachment and form longer chains. In the third stage, polycrystalline chains can straighten and form into single-crystal NRs via the alignment and mass redistribution between the individual NPs within the chain.
The similarities between the evolution of the morphology with TiCl4 exposure time revealed by ex situ TEM (Figure 4) and the mechanism observed by Liao et al.30 in real time suggest that an analogous three-stage process is likely to be behind the formation of TiO2 NRs during ALD on graphene nanoplatelets. Yet, it must be noted that the latter is considerably more complex than the growth of Pt3 Fe NRs in solution in that: the nucleation of TiO2 NPs is heterogeneous; the diffusion of TiO2 species and of NPs takes place over a surface, that is, in 2D; new material is added cyclically into the system, rather than being added all at once at the beginning of the process; TiO2 is deposited in each cycle on both the substrate and the pre-existing nanostructures; and both the reaction environment and the surface chemistry change periodically as a result of the pulsing sequence and of ALD surface reactions.
Although oriented attachment is typically observed in liquid-phase processes,25−27 the same three-step process described by Liao et al.30 (i.e., NP nucleation, collision of NPs followed by oriented attachment, and formation of NRs by mass redistribution) can also apply to the formation of NRs in vapor-solid processes. In principle, oriented attachment can take place in solid-phase processes provided that the individual crystals can diffuse and collide and if there exists particle–particle interactions that mediate the alignment of crystals that come in close contact with each other. To the best of our knowledge, only Shi et al.23 have so far reported on the experimental realization of oriented attachment in a vapor–solid deposition process. Furthermore, molecular dynamics simulations have shown that, while in vacuum TiO2 crystals simply merge along the direction of their collision, the presence of water vapor triggers the alignment of colliding TiO2 and thus their oriented attachment.46 In particular, water molecules mediate the interaction between colliding crystals without being consumed by the overall process. In fact, water molecules do adsorb on the surface of the TiO2 crystals, but these are then released as a result of the merging of aligned crystals.46 Finally, although such a role in mediating particle–particle interactions has so far been described only for water, similar mechanisms might also hold for other chemical species such as TiCl4.
To further substantiate the role of TiCl4 in mediating the growth of NRs, we also carried out preliminary ALD experiments using titanium isopropoxide (TTIP) as a titanium source. Using a pulsing sequence analogous to the one that gave an optimum fraction of NRs with TiCl4 and H2O: 2 min TTIP-5 min N2 purge-30 s H2O-5 min N2 purge and a temperature of 300 °C we only obtained a high density of rounded NPs of a few nanometers in size and no NRs after 10 cycles (see Figure S4). By using the same precursor for TiO2 ALD on graphene, also Zhang et al.47 could only obtain small NPs of about 2 nm. Therefore, we suggest that both TiCl4 and water vapors can promote the formation of NRs by mediating the interactions between individual TiO2 nanocrystals.
The analogy between liquid-phase and solid-state oriented attachment also requires that individual TiO2 nanocrystals can diffuse over the surface of graphene nanoplatelets. Although the diffusion of three-dimensional atomic clusters has not been extensively studied, there is considerable experimental and theoretical research that suggests that whenever the cluster is not in epitaxy with the substrate, cluster diffusion is commonplace.17,48−51 The greater the degree of epitaxial misalignment, the greater the mobility, which is minimum when the cluster approaches epitaxy with the substrate.48−51 As a result, adsorbed clusters that are not in epitaxy can experience transient mobility until rotational diffusion brings them in epitaxy, after which they are virtually immobile. Interestingly, as we will discuss in the next section, we do find that the lattice of the NRs is in rotational alignment with the underlying substrate lattice. Finally, chemical species in the vapor phase can alter the surface properties of supported NPs as well as their interaction with the substrate, thereby promoting or inhibiting mobility.17,52,53 Water vapor, in particular, has long been known to promote the sintering of several oxides by increasing the surface diffusion.54 Therefore, it is indeed possible that TiO2 nanocrystals can diffuse over the surface of graphene nanoplatelets and that their mobility as well as the rate of merging of two or more nanocrystals is affected by the presence of TiCl4 and water vapor.
Solid-State Oriented Attachment, Heteroepitaxial Alignment, and the Effect of the Substrate
Figure 6 shows some examples of the different morphologies that the ALD-grown NRs can display: straight NRs (Figure 6b), NRs aligned with the edge of a nanoplatelet (Figure 6e), curved NRs (Figure 6c,d), and V-shaped NRs (Figure 6f). Such morphologies offer a clue into the growth mechanism. In particular, the presence of curved NRs is an indirect evidence of oriented attachment. In fact, Zhang et al.55 have shown that NRs forming in solutions via oriented attachment of small anatase NPs exposing {011} facets, such as the ones formed during ALD as shown in Figure 7b, bend in order to lower their energy. We could indeed find direct evidence of oriented attachment via HRTEM analysis. For example, Figure 7a shows a TEM micrograph of two distinct single-crystal NRs that are fused at one extremity while retaining a perfect alignment along {011} facets as shown by the FFT analysis. Another example of oriented attachment is given in Figure S5l, which shows a small TiO2 NP attached to one end of a NR with a perfect lattice alignment. More evidence is also provided by Figure 7f, where a single-crystal straight NR is shown to be fused with a NP, which exhibits a perfect alignment with the NR along the {011} facets, as highlighted in the inset (I). The NR fused to an NP of Figure 7f is consistent with the mechanism revealed by Liao et al.,30 where individual NPs, NP chains, and NRs first undergo end-to-end oriented attachment and then form into straight single-crystal NRs via mass redistribution. The fact that the NRs are bound by {011} is no surprise as they exhibit the lowest surface energy among the anatase crystal facets.56 This is also consistent with the findings of Shi et al.,23 who also observe NRs bound by {011} facets. The driving force for oriented attachment is in fact the reduction of the total surface energy.24
Figure 7.

(a) HRTEM micrograph of two NRs undergoing oriented attachment together with FFT patterns of the indicated areas and inverse FFT (IFFT) images obtained by singling out the frequencies associated with the {011} facets. (b) Representative HRTEM micrograph of a TiO2 NP exhibiting the tetragonal dipyramidal structure, characteristic of anatase crystals, exposing {011} facets. (c) HRTEM micrograph of a supported NR, FFT patterns of the indicated areas, and IFFT images obtained by singling out the frequencies associated with the lattice of graphene (I) and of TiO2 (II). (d) Analysis of the FFT pattern of the area indicated as (II), highlighting the heteroepitaxial alignment between TiO2 and graphene nanoplatelets. (e) HRTEM image and corresponding IFFT of a V-shaped NR, and FFT patterns of the corresponding indicated areas. The IFFT image was obtained by singling out the FFT patterns associated with the lattice of both TiO2 and graphene nanoplatelets. (f) HRTEM image and corresponding IFFT of a NR with an irregular bottom tip. The inset highlights the incorporation by the oriented attachment of a distinct crystallite at the bottom of the NR. The IFFT was obtained as in (e).
The lattice mismatch and the poor chemical affinity between graphitic surfaces and oxides such as TiO2 entail high interfacial energies.31,57 The minimization of the latter can drive not only the transition from 2D flakes to NPs and NRs but also the in-plane alignment between the crystal lattices of TiO2 and graphene. Indeed, HRTEM images and their Fourier transforms clearly show that the lattices of the TiO2 NRs and graphene nanoplatelets are rotationally aligned (see Figures 7c–e and S5a–h). For example, the {013} and {011} facets of the NR in Figure 7c are perfectly aligned with the zigzag and armchair directions of the graphitic lattice, respectively (see insets (I) and (II) of Figure 7c,d). Heteroepitaxial alignment is known to be driven by lattice matching.31,58−61 In fact, we find that TiO2 that is aligned with carbon hexagon minimizes the lattice mismatch. For the NR in Figure 7c, the interplanar spacing of {033} and {017} facets is about the length of one carbon hexagon (lattice mismatch ≃0.8% and ≃0.4%, respectively), whereas the spacing between {013} and {020} is about the distance between two carbon atoms in the carbon hexagon (lattice mismatch ≃12 and ≃10%, respectively).
An atomic-scale rotational alignment between adsorbates and the substrate often translates into a preferential alignment between the adsorbates, especially if they are 1D nanostructures such as NRs.31,32 Indeed, we found that the orientation of the NRs exhibits a 12-fold symmetry; in other words, the angle between the major axis of two NRs lying on the same graphene nanoplatelet is a multiple of 30° (see Figure S6). This is a clear indication that the substrate has an active role in mediating the directional growth of the deposited TiO2 into NRs. We conclude that TiO2 species and individual NPs diffuse and aggregate in an asymmetric fashion so that the TiO2 nanocrystals grow in the direction that minimizes the interfacial energy. A diffusion field that exhibits a 12-fold symmetry can explain, among other things, the formation of V-shaped NRs with an internal angle of about 60° or 30° as the ones shown in Figures 6f and 7e. Finally, also the edges of the graphene nanoplatelets can impart directionality to the growth process, as they act as impassable boundaries for diffusing species as well as for growing nanocrystals. For example, Figure S5i shows a TiO2 crystalline NP whose lattice sharply terminates in correspondence of the edge of the graphene nanoplatelet. Analogously, edges can act as a template for the directional growth of NRs, as shown in Figure 6e.
On the Growth Mechanism
In light of the results of our analysis and, in particular, of the following two observations: (i) nonlinear dependence of the NR fraction with the exposure time of both precursors and the number of cycles and (ii) rotational alignment between the lattices of TiO2 and graphene nanoplatelets driven by lattice matching, we propose the following growth mechanism. In each cycle, TiO2 species form on both the graphene nanoplatelets and the already-deposited TiO2 as a result of ALD surface reactions. During the first cycles, TiO2 species diffuse and form into anatase NPs to minimize the interfacial energy. Once formed, the NPs can grow by capturing diffusing TiO2 species or by undergoing diffusion and coalescence with neighboring NPs. In particular, the NPs will tend to diffuse and grow in accord with the symmetry of the lattice of the underlying graphene. Before reaching a critical size, two colliding NPs can coalesce into a symmetric NP. With increasing number of cycles, the deposition of more TiO2 leads to an increase in the number of NPs, which in turn increases the chances of two NPs undergoing diffusive aggregation. Upon reaching a certain size, the coalescence step slows down (the characteristic time for the coalescence of two spherical NPs scales with R4).62 As the NPs grow in size and number, they can come in contact without coalescing, thereby forming NP chains. In particular, NPs in close proximity can align and undergo oriented attachment or form irregular polycrystalline chains. As previously discussed, in the presence of chemical atmospheres that promote surface diffusion and oriented attachment and given enough time, NP chains can form into single-crystal NRs via alignment and mass redistribution between the individual NPs within the chain. Furthermore, the oriented attachment of NPs can still lead to steps or gaps along the lateral sides of the forming NR, if the NPs are of different sizes or if their centers of mass are not aligned. In this case, a straight NR can form via the preferential attachment of migrating TiO2 species to the steps in the lateral facets. In the limit of long times, however, NP chains and NRs will inevitably tend to relax into the most stable shape, that is, the tetragonal bipyramidal shape24,26,27 (see Figure 7b). There are, therefore, three processes in series that are all mediated by TiO2 mobility: NP aggregation into NP chains, NP relaxation into NRs, and relaxation of NRs into symmetric bipyramidal NPs. It follows that if both TiCl4 and H2O promote the mobility of TiO2, there will be an optimal exposure time that maximizes the formation of NRs. Also, with the increasing number of cycles, large and irregular structures can form as new NPs attach to pre-existing nanostructures and growing neighboring nanostructures come into contact with each other. Because the time scale associated with the relaxation of a polycrystalline chains into NRs will likely increase with chain length and complexity, the fraction of NRs is bound to decrease after a critical number of cycles. Finally, this picture implies that the optimal exposure time varies from cycle to cycle and on the extent of saturation of the ALD surface reactions because the time scales associated with the diffusive process leading to the formation of NRs depend on the TiO2 coverage and the size of the nanostructures. This particular insight paves the way for an optimization of the process aimed at maximizing the NR fraction.
Conclusions
In conclusion, we have demonstrated the use of ALD as a route for the bottom-up synthesis of TiO2 NRs on gram-scale batches of graphene nanoplatelets at temperatures as low as 300 °C. NRs >200 nm in length could be obtained even after only five cycles, indicating that the growth is dominated by diffusional processes rather than by a layer-by-layer mechanism. In particular, complex structures such as V-shaped and curved NRs were observed. The formation of NRs was induced by modulating the exposure time of the precursors: TiCl4 and H2O. Prolonged annealing experiments suggest that the sequential nature of the ALD process is crucial for the NR formation. Statistical analysis of the evolution of the shape and the relative number of different nanostructures formed at different exposure times reveals a competition between growth pathways, leading to either asymmetric or symmetric growth. In particular, the number of NRs is a nonlinear function of the exposure time of the precursors and of the number of cycles: there exists an optimum. In particular, we show that the morphology of the ALD-grown TiO2 nanostructures evolves within the saturation window of the ALD surface reactions. In other words, we obtain different morphologies while retaining the same amount of the material being deposited. The number of cycles mostly affects the relative number of the NRs rather their size. HRTEM reveals that TiO2 nanocrystals can undergo oriented attachment during ALD. Crucially, analysis of the relative lattice orientation clearly shows an in-plane rotational alignment between the lattices of the TiO2 nanocrystals and graphene nanoplatelets. Hence, the minimization of the interfacial energy by lattice matching is considered to be among the major driving forces for the diffusional processes underlying the NR formation and growth. The insights presented here are also relevant to ALD of other oxides that are known to form NRs such as vanadium oxide and zinc oxide.63,64 We hope that our work will motivate future studies on the exploitation of nonclassical growth pathways for the synthesis nanostructures with controlled size and shape via ALD. We believe that fundamental studies on the role of the substrate in mediating the self-organization of forming nanocrystals can expand the capabilities of ALD and other synthesis methods, especially with regard to nanostructures supported on 2D materials.
Acknowledgments
The research leading to these results has received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP/2007–2013)/ERC Grant, Agreement no. 279632.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcc.8b05572.
TEM and HRTEM images and extended size distributions (PDF)
Author Contributions
† F.G. and D.L.Z. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Puurunen R. L. Surface Chemistry of Atomic Layer Deposition: A Case Study for the Trimethylaluminum/Water Process. J. Appl. Phys. 2005, 97, 121301. 10.1063/1.1940727. [DOI] [Google Scholar]
- Van Bui H.; Grillo F.; van Ommen J. R. Atomic and Molecular Layer Deposition: Off the Beaten Track. Chem. Commun. 2017, 53, 45–71. 10.1039/c6cc05568k. [DOI] [PubMed] [Google Scholar]
- George S. M. Atomic Layer Deposition: An Overview. Chem. Rev. 2010, 110, 111–131. 10.1021/cr900056b. [DOI] [PubMed] [Google Scholar]
- Ahvenniemi E.; Akbashev A. R.; Ali S.; Bechelany M.; Berdova M.; Boyadjiev S.; Cameron D. C.; Chen R.; Chubarov M.; Cremers V.; et al. Review Article: Recommended reading list of early publications on atomic layer deposition-Outcome of the “Virtual Project on the History of ALD”. J. Vac. Sci. Technol., A 2017, 35, 010801. 10.1116/1.4971389. [DOI] [Google Scholar]
- Grillo F.; Kreutzer M. T.; van Ommen J. R. Modeling the Precursor Utilization in Atomic Layer Deposition on Nanostructured Materials in Fluidized Bed Reactors. Chem. Eng. J. 2015, 268, 384–398. 10.1016/j.cej.2015.01.067. [DOI] [Google Scholar]
- Lu J.; Elam J. W.; Stair P. C. Atomic layer deposition-Sequential self-limiting surface reactions for advanced catalyst “bottom-up” synthesis. Surf. Sci. Rep. 2016, 71, 410–472. 10.1016/j.surfrep.2016.03.003. [DOI] [Google Scholar]
- O’Neill B. J.; Jackson D. H. K.; Lee J.; Canlas C.; Stair P. C.; Marshall C. L.; Elam J. W.; Kuech T. F.; Dumesic J. A.; Huber G. W. Catalyst Design with Atomic Layer Deposition. ACS Catal. 2015, 5, 1804–1825. 10.1021/cs501862h. [DOI] [Google Scholar]
- Marichy C.; Bechelany M.; Pinna N. Atomic Layer Deposition of Nanostructured Materials for Energy and Environmental Applications. Adv. Mater. 2012, 24, 1017–1032. 10.1002/adma.201104129. [DOI] [PubMed] [Google Scholar]
- Longrie D.; Deduytsche D.; Detavernier C. Reactor Concepts for Atomic Layer Deposition on Agitated Particles: A Review. J. Vac. Sci. Technol., A 2014, 32, 010802. 10.1116/1.4851676. [DOI] [Google Scholar]
- King D. M.; Liang X.; Weimer A. W. Functionalization of Fine Particles Using Atomic and Molecular Layer Deposition. Powder Technol. 2012, 221, 13–25. 10.1016/j.powtec.2011.12.020. [DOI] [Google Scholar]
- King D. M.; Spencer J. A.; Liang X.; Hakim L. F.; Weimer A. W. Atomic Layer Deposition on Particles Using a Fluidized Bed Reactor with in Situ Mass Spectrometry. Surf. Coat. Technol. 2007, 201, 9163–9171. 10.1016/j.surfcoat.2007.05.002. [DOI] [Google Scholar]
- Hakim L. F.; Blackson J.; George S. M.; Weimer A. W. Nanocoating Individual Silica Nanoparticles by Atomic Layer Deposition in a Fluidized Bed Reactor. Chem. Vap. Deposition 2005, 11, 420–425. 10.1002/cvde.200506392. [DOI] [PubMed] [Google Scholar]
- van Ommen J. R.; Kooijman D.; de Niet M.; Talebi A.; Goulas A. Continuous Production of Nanostructured Particles Using Spatial Atomic Layer Deposition. J. Vac. Sci. Technol., A 2015, 33, 021513. 10.1116/1.4905725. [DOI] [Google Scholar]
- McCormick J. A.; Cloutier B. L.; Weimer A. W.; George S. M. Rotary Reactor for Atomic Layer Deposition on Large Quantities of Nanoparticles. J. Vac. Sci. Technol., A 2007, 25, 67–74. 10.1116/1.2393299. [DOI] [Google Scholar]
- Duan C.-L.; Liu X.; Shan B.; Chen R. Fluidized Bed Coupled Rotary Reactor for Nanoparticles Coating Via Atomic Layer Deposition. Rev. Sci. Instrum. 2015, 86, 075101. 10.1063/1.4926349. [DOI] [PubMed] [Google Scholar]
- Kääriäinen T. O.; Kemell M.; Vehkamäki M.; Kääriäinen M.-L.; Correia A.; Santos H. A.; Bimbo L. M.; Hirvonen J.; Hoppu P.; George S. M.; et al. Surface Modification of Acetaminophen Particles by Atomic Layer Deposition. Int. J. Pharm. 2017, 525, 160–174. 10.1016/j.ijpharm.2017.04.031. [DOI] [PubMed] [Google Scholar]
- Grillo F.; Van Bui H.; Moulijn J. A.; Kreutzer M. T.; van Ommen J. R. Understanding and Controlling the Aggregative Growth of Platinum Nanoparticles in Atomic Layer Deposition: An Avenue to Size Selection. J. Phys. Chem. Lett. 2017, 8, 975–983. 10.1021/acs.jpclett.6b02978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulas A.; Ruud van Ommen J. Atomic Layer Deposition of Platinum Clusters on Titania Nanoparticles at Atmospheric Pressure. J. Mater. Chem. A 2013, 1, 4647. 10.1039/c3ta01665j. [DOI] [Google Scholar]
- Lubers A. M.; Muhich C. L.; Anderson K. M.; Weimer A. W. Mechanistic Studies for Depositing Highly Dispersed Pt Nanoparticles on Carbon by Use of Trimethyl(methylcyclopentadienyl)platinum(iv) Reactions with O2 and H2. J. Nanopart. Res. 2015, 17, 179. 10.1007/s11051-015-2982-9. [DOI] [Google Scholar]
- Mackus A. J. M.; Weber M. J.; Thissen N. F. W.; Garcia-Alonso D.; Vervuurt R. H. J.; Assali S.; Bol A. A.; Verheijen M. A.; Kessels W. M. M. Atomic Layer Deposition of Pd and Pt Nanoparticles for Catalysis: On the Mechanisms of Nanoparticle Formation. Nanotechnology 2016, 27, 034001. 10.1088/0957-4484/27/3/034001. [DOI] [PubMed] [Google Scholar]
- Dendooven J.; Ramachandran R. K.; Solano E.; Kurttepeli M.; Geerts L.; Heremans G.; Rongé J.; Minjauw M. M.; Dobbelaere T.; Devloo-Casier K.; et al. Independent Tuning of Size and Coverage of Supported Pt Nanoparticles Using Atomic Layer Deposition. Nat. Commun. 2017, 8, 1074. 10.1038/s41467-017-01140-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knez M. Diffusion Phenomena in Atomic Layer Deposition. Semicond. Sci. Technol. 2012, 27, 074001. 10.1088/0268-1242/27/7/074001. [DOI] [Google Scholar]
- Shi J.; Li Z.; Kvit A.; Krylyuk S.; Davydov A. V.; Wang X. Electron Microscopy Observation of TiO2 Nanocrystal Evolution in High-Temperature Atomic Layer Deposition. Nano Lett. 2013, 13, 5727–5734. 10.1021/nl403566u. [DOI] [PubMed] [Google Scholar]
- Wen K.; He W. Can Oriented-Attachment Be an Efficient Growth Mechanism for the Synthesis of 1D Nanocrystals via Atomic Layer Deposition?. Nanotechnology 2015, 26, 382001. 10.1088/0957-4484/26/38/382001. [DOI] [PubMed] [Google Scholar]
- Penn R. L. Imperfect Oriented Attachment: Dislocation Generation in Defect-Free Nanocrystals. Science 1998, 281, 969–971. 10.1126/science.281.5379.969. [DOI] [PubMed] [Google Scholar]
- Ivanov V. K.; Fedorov P. P.; Baranchikov A. Y.; Osiko V. V. Oriented Attachment of Particles: 100 Years of Investigations of Non-Classical Crystal Growth. Russ. Chem. Rev. 2014, 83, 1204–1222. 10.1070/rcr4453. [DOI] [Google Scholar]
- He W.; Wen K.; Niu Y.. Nanocrystals from Oriented-Attachment for Energy Applications; Springer International Publishing: Cham, 2018; pp 1–13. [Google Scholar]
- De Yoreo J. J.; Gilbert P. U. P. A.; Sommerdijk N. A. J. M.; Penn R. L.; Whitelam S.; Joester D.; Zhang H.; Rimer J. D.; Navrotsky A.; Banfield J. F.; et al. Crystallization by Particle Attachment in Synthetic, Biogenic, and Geologic Environments. Science 2015, 349, aaa6760. 10.1126/science.aaa6760. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Huang F.; Lin Z. Progress of Nanocrystalline Growth Kinetics Based on Oriented Attachment. Nanoscale 2010, 2, 18–34. 10.1039/b9nr00047j. [DOI] [PubMed] [Google Scholar]
- Liao H.-G.; Cui L.; Whitelam S.; Zheng H. Real-Time Imaging of Pt3Fe Nanorod Growth in Solution. Science 2012, 336, 1011–1014. 10.1126/science.1219185. [DOI] [PubMed] [Google Scholar]
- Yang J.; Kim K.; Lee Y.; Kim K.; Lee W. C.; Park J. Self-Organized Growth and Self-Assembly of Nanostructures on 2D Materials. FlatChem 2017, 5, 50–68. 10.1016/j.flatc.2017.07.004. [DOI] [Google Scholar]
- Lee W. C.; Kim K.; Park J.; Koo J.; Jeong H. Y.; Lee H.; Weitz D. A.; Zettl A.; Takeuchi S. Graphene-Templated Directional Growth of an Inorganic Nanowire. Nat. Nanotechnol. 2015, 10, 423–428. 10.1038/nnano.2015.36. [DOI] [PubMed] [Google Scholar]
- Luo Z.; Somers L. A.; Dan Y.; Ly T.; Kybert N. J.; Mele E. J.; Johnson A. T. C. Size-Selective Nanoparticle Growth on Few-Layer Graphene Films. Nano Lett. 2010, 10, 777–781. 10.1021/nl9026605. [DOI] [PubMed] [Google Scholar]
- Evans E. L.; Bahl O. P.; Thomas J. M. The Decoration Of, and Epitaxial Growth of Gold On, Graphite Surfaces. Carbon 1967, 5, 587–589. 10.1016/0008-6223(67)90038-3. [DOI] [Google Scholar]
- Van Bui H.; Grillo F.; Kulkarni S. S.; Bevaart R.; Van Thang N.; van der Linden B.; Moulijn J. A.; Makkee M.; Kreutzer M. T.; van Ommen J. R. Low-temperature Atomic Layer Deposition Delivers More Active and Stable Pt-Based Catalysts. Nanoscale 2017, 9, 10802–10810. 10.1039/c7nr02984e. [DOI] [PubMed] [Google Scholar]
- Klinger M.; Jäger A. Crystallographic Tool Box(CrysTBox): automated tools for transmission electron microscopists and crystallographers. J. Appl. Crystallogr. 2015, 48, 2012–2018. 10.1107/s1600576715017252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger M. More features, more tools, more CrysTBox. J. Appl. Crystallogr. 2017, 50, 1226–1234. 10.1107/s1600576717006793. [DOI] [Google Scholar]
- Ritala M.; Leskelä M.; Nykänen E.; Soininen P.; Niinistö L. Growth of Titanium Dioxide Thin Films by Atomic Layer Epitaxy. Thin Solid Films 1993, 225, 288–295. 10.1016/0040-6090(93)90172-l. [DOI] [Google Scholar]
- Ritala M.; Leskelä M.; Johansson L.-S.; Niinistö L. Atomic Force Microscopy Study of Titanium Dioxide Thin Films Grown by Atomic Layer Epitaxy. Thin Solid Films 1993, 228, 32–35. 10.1016/0040-6090(93)90557-6. [DOI] [Google Scholar]
- Puurunen R. L.; Sajavaara T.; Santala E.; Miikkulainen V.; Saukkonen T.; Laitinen M.; Leskelä M. Controlling the Crystallinity and Roughness of Atomic Layer Deposited Titanium Dioxide Films. J. Nanosci. Nanotechnol. 2011, 11, 8101–8107. 10.1166/jnn.2011.5060. [DOI] [PubMed] [Google Scholar]
- Sun S.; Zhang G.; Gauquelin N.; Chen N.; Zhou J.; Yang S.; Chen W.; Meng X.; Geng D.; Banis M. N.; et al. Single-Atom Catalysis Using Pt/Graphene Achieved through Atomic Layer Deposition. Sci. Rep. 2013, 3, 13053. 10.1166/jnn.2011.5060. [DOI] [Google Scholar]
- Karasulu B.; Vervuurt R. H. J.; Kessels W. M. M.; Bol A. A. Continuous and Ultrathin Platinum Films on Graphene Using Atomic Layer Deposition: A Combined Computational and Experimental Study. Nanoscale 2016, 8, 19829–19845. 10.1039/c6nr07483a. [DOI] [PubMed] [Google Scholar]
- Yaws C. L.; Satyro M. A.. The Yaws Handbook of Vapor Pressure; Gulf Professional Publishing, 2015; pp 1–314. [Google Scholar]
- Rumaiz A. K.; Ali B.; Ceylan A.; Boggs M.; Beebe T.; Ismat Shah S. Experimental studies on vacancy induced ferromagnetism in undoped TiO2. Solid State Commun. 2007, 144, 334–338. 10.1016/j.ssc.2007.08.034. [DOI] [Google Scholar]
- Santara B.; Giri P. K.; Imakita K.; Fujii M. Evidence of Oxygen Vacancy Induced Room Temperature Ferromagnetism in Solvothermally Synthesized Undoped TiO2 Nanoribbons. Nanoscale 2013, 5, 5476. 10.1039/c3nr00799e. [DOI] [PubMed] [Google Scholar]
- Raju M.; van Duin A. C. T.; Fichthorn K. A. Mechanisms of Oriented Attachment of TiO2 Nanocrystals in Vacuum and Humid Environments: Reactive Molecular Dynamics. Nano Lett. 2014, 14, 1836–1842. 10.1021/nl404533k. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Guerra-Nuñez C.; Utke I.; Michler J.; Agrawal P.; Rossell M. D.; Erni R. Atomic Layer Deposition of Titanium Oxide on Single-Layer Graphene: An Atomic-scale Study toward Understanding Nucleation and Growth. Chem. Mater. 2017, 29, 2232–2238. 10.1021/acs.chemmater.6b05143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen P. Growth of Nanostructures by Cluster Deposition: Experiments and Simple Models. Rev. Mod. Phys. 1999, 71, 1695–1735. 10.1103/revmodphys.71.1695. [DOI] [Google Scholar]
- Guerra R.; Tosatti E.; Vanossi A. Slider Thickness Promotes Lubricity: From 2D Islands to 3D Clusters. Nanoscale 2016, 8, 11108–11113. 10.1039/c6nr00520a. [DOI] [PubMed] [Google Scholar]
- Celardo G. L.; Archetti D.; Ferrini G.; Gavioli L.; Pingue P.; Cavaliere E. Evidence of diffusive fractal aggregation of TiO2 nanoparticles by femtosecond laser ablation at ambient conditions. Mater. Res. Express 2017, 4, 015013. 10.1088/2053-1591/aa50e9. [DOI] [Google Scholar]
- Dietzel D.; de Wijn A. S.; Vorholze M.; Schirmeisen A. Friction Fluctuations of Gold Nanoparticles in the Superlubric Regime. Nanotechnology 2018, 29, 155702. 10.1088/1361-6528/aaac21. [DOI] [PubMed] [Google Scholar]
- Grillo F.; Moulijn J. A.; Kreutzer M. T.; van Ommen J. R. Nanoparticle Sintering in Atomic Layer Deposition of Supported Catalysts: Kinetic Modeling of the Size Distribution. Catal. Today 2018, 10.1016/j.cattod.2018.02.020. [DOI] [Google Scholar]
- Grillo F.; Van Bui H.; La Zara D.; Aarnink A. A. I.; Kovalgin A. Y.; Kooyman P.; Kreutzer M. T.; van Ommen J. R. From Single Atoms to Nanoparticles: Autocatalysis and Metal Aggregation in Atomic Layer Deposition of Pt on TiO2 Nanopowder. Small 2018, 14, 1800765. 10.1002/smll.201800765. [DOI] [PubMed] [Google Scholar]
- Varela J. A.; Whittemore O. J.; Longo E. Pore Size Evolution during Sintering of Ceramic Oxides. Ceram. Int. 1990, 16, 177–189. 10.1016/0272-8842(90)90053-i. [DOI] [Google Scholar]
- Zhang H.; Finnegan M. P.; Banfield J. F. Titania Nanorods Curve to Lower Their Energy. Nanoscale 2013, 5, 6742–6746. 10.1039/c3nr02616g. [DOI] [PubMed] [Google Scholar]
- Chen W.; Kuang Q.; Wang Q.; Xie Z. Engineering a High Energy Surface of Anatase Tio2 Crystals Towards Enhanced Performance for Energy Conversion and Environmental Applications. RSC Adv. 2015, 5, 20396–20409. 10.1039/c5ra00344j. [DOI] [Google Scholar]
- Li W.; Wang F.; Liu Y.; Wang J.; Yang J.; Zhang L.; Elzatahry A. A.; Al-Dahyan D.; Xia Y.; Zhao D. General Strategy to Synthesize Uniform Mesoporous TiO2/Graphene/Mesoporous TiO2 Sandwich-Like Nanosheets for Highly Reversible Lithium Storage. Nano Lett. 2015, 15, 2186–2193. 10.1021/acs.nanolett.5b00291. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Zhou W.; Lu A.-Y.; Fang W.; Lee Y.-H.; Hsu A. L.; Kim S. M.; Kim K. K.; Yang H. Y.; Li L.-J.; et al. van der Waals Epitaxy of MoS2 Layers Using Graphene As Growth Templates. Nano Lett. 2012, 12, 2784–2791. 10.1021/nl204562j. [DOI] [PubMed] [Google Scholar]
- Hong Y. J.; Yang J. W.; Lee W. H.; Ruoff R. S.; Kim K. S.; Fukui T. van der Waals Epitaxial Double Heterostructure: InAs/Single-Layer Graphene/InAs. Adv. Mater. 2013, 25, 6847–6853. 10.1002/adma.201302312. [DOI] [PubMed] [Google Scholar]
- Dang W.; Peng H.; Li H.; Wang P.; Liu Z. Epitaxial Heterostructures of Ultrathin Topological Insulator Nanoplate and Graphene. Nano Lett. 2010, 10, 2870–2876. 10.1021/nl100938e. [DOI] [PubMed] [Google Scholar]
- Lee W. C.; Kim K.; Park J.; Koo J.; Jeong H. Y.; Lee H.; Weitz D. A.; Zettl A.; Takeuchi S. Graphene-Templated Directional Growth of an Inorganic Nanowire. Nat. Nanotechnol. 2015, 10, 423–428. 10.1038/nnano.2015.36. [DOI] [PubMed] [Google Scholar]
- Wynblatt P.; Gjostein N. A. Supported Metal Crystallites. Prog. Solid State Chem. 1975, 9, 21–58. 10.1016/0079-6786(75)90013-8. [DOI] [Google Scholar]
- Li Z.; Wang F.; Wang X. Hierarchical Branched Vanadium Oxide Nanorod@Si Nanowire Architecture for High Performance Supercapacitors. Small 2017, 13, 1603076. 10.1002/smll.201603076. [DOI] [PubMed] [Google Scholar]
- Claudia P.; Andreas K.; Horst W. Self-assembly of ZnO: From Nanodots to Nanorods. Angew. Chem., Int. Ed. 2002, 41, 1188–1191. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.