

Abstract
Background & Aims
Defects in lysosome function and autophagy contribute to pathogenesis of alcoholic liver disease. We investigated the mechanisms by which alcohol consumption affects these processes, evaluating the functions transcription factor EB (TFEB), which regulates lysosomal biogenesis.
Methods
We performed studies with GFP-LC3 mice, mice with liver-specific deletion of transcription factor EB (TFEB), mice with disruption of the transcription factor E3 gene (TFE3-knockout mice), mice with disruption of the Tefb and Tfe3 genes (TFEB, TFE3 double-knockout mice), and Tfebflox/floxalbumin cre-negative mice (controls). TFEB was overexpressed from adenoviral vectors or knocked down with small interfering RNAs in mouse livers. Mice were placed on diets of chronic ethanol feeding plus an acute binge to induce liver damage (ethanol diet); some mice were also given injections of torin1, an inhibitor of the kinase activity of the mechanistic target of rapamycin (mTOR). Liver tissues were collected and analyzed by immunohistochemistry, immunoblots, and quantitative real-time PCR to monitor lysosome biogenesis. We analyzed levels of TFEB in liver tissues from patients with alcoholic hepatitis and from healthy donors (controls) by immunohistochemistry.
Results
Liver tissues from mice on the ethanol diet had lower levels of total and nuclear TFEB, compared with control mice, and hepatocytes had reduced lysosome biogenesis and autophagy. Hepatocytes from mice on the ethanol diet had increased translocation of mTOR into lysosomes, resulting increased mTOR activation. Administration of torin1 increased liver levels of TFEB and reduced steatosis and liver injury induced by ethanol. Mice that overexpressed TFEB in liver developed less-severe ethanol-induced liver injury and had increased lysosomal biogenesis and mitochondrial bioenergetics compared to mice carrying a control vector. Mice with knockdown of TFEB, as well as TFEB, TFE3 double-knockout mice, developed more severe liver injury in response to the ethanol diet than control mice. Liver tissues from patients with alcohol-induced hepatitis had lower nuclear levels of TFEB than control tissues
Conclusions
We found chronic ethanol feeding plus an acute binge to reduce hepatic expression of the transcription factor TFEB, which is required for lysosomal biogenesis and autophagy. Strategies to block mTOR activity or increase levels of TFEB might be developed to protect liver from ethanol-induced damage.
Keywords: fatty liver, gene regulation, hepatic protection, mouse model
Graphical abstract
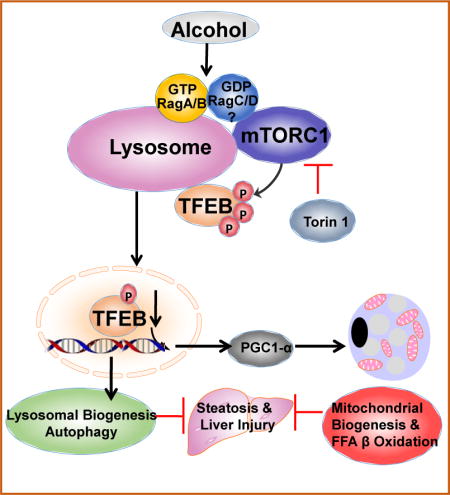
Introduction
Alcoholic liver disease (ALD) is a major cause of chronic liver disease worldwide, claiming 3.3 million deaths globally in 20121. The pathogenesis of alcohol-induced liver injury is characterized by hepatic steatosis, inflammation, and fibrosis, which can progress to cirrhosis and liver cancer2–4. Despite major progress in understanding mechanisms for ALD, no successful treatment for ALD is available.
Cells can adapt and protect themselves in response to stress by activating cellular protective mechanisms including autophagy and lysosomal biogenesis. Autophagy is a catabolic process that degrades cellular proteins and subcellular organelles such as mitochondria, which is critical for prevention and recovery from alcohol-induced liver injury5, 6. However, it seems that acute and chronic alcohol exposure may differentially regulate hepatic autophagy7, 8. We previously demonstrated that autophagy is activated in mouse livers and primary cultured hepatocytes in response to acute alcohol5, 9. While two independent groups reported that chronic ethanol exposure enhanced autophagic flux in mice and increased autophagosome numbers in rats10, 11, earlier studies indicated that chronic ethanol exposure disrupts lysosome function12. Moreover, animals with chronic ethanol exposure and heavy drinkers develop hepatomegaly with increased hepatic protein accumulation13. These observations suggest a defect in hepatic autophagy in chronic alcohol conditions. However, the mechanisms by which alcohol impairs lysosomal functions in the liver are largely unknown.
The lysosome contains more than 50 acid hydrolases and is the terminal component of autophagy. The transcription factor EB (TFEB) is a master regulator of lysosome biogenesis and autophagy-related gene transcription14, 15, which is mainly regulated by the mechanistic target of rapamycin complex 1 (mTORC1). mTORC1 is a conserved serine-threonine kinase and acts as a nutrient and energy sensor to regulate cell growth by coordinating signals from nutrients and growth factors as well as cellular energy levels16. The tumor suppressor proteins tuberous sclerosis complex 1 (TSC1) and TSC2 form a complex and negatively regulate the GTP-loading state of Rheb, a Ras-related GTP-binding protein. Rheb interacts with and activates mTORC1 when it binds with GTP16, 17. However, amino acids activate mTORC1 by activating the Ragulator-Rag complex independent of the TSC1/TSC2 complex18. There are four Rag proteins in mammals: Rag A, B, C and D proteins, in which Rag A and Rag B are functionally redundant, whereas Rag C and Rag D are also functionally redundant18, 19. Increasing evidence indicates that mTORC1 translocates to lysosomes and becomes activated by Rag GTPases16, 20, which results in the inactivation of TFEB via phosphorylation14, 21. TFEB overexpression enhances both lysosomal and mitochondrial functions, which markedly attenuates high fat diet-induced steatosis in mice22. A previous descriptive study showed that mice fed with ethanol diet for 35-62 days decreased hepatic nuclear TFEB levels, which seemed to be associated with deceased autophagy7. However, the causal role and mechanisms by which TFEB regulates alcohol-induced liver injury was not investigated in this correlative study8. Chronic feeding plus acute binge alcohol (hereafter referred to as “Gao-binge”) in mice mimics consumption patterns of human alcoholics, and it causes greater liver injury and inflammation than other chronic Lieber-DeCarli alcohol mouse models4, 23. However, autophagy status in this Gao-binge model in mouse livers has not been characterized. Moreover, whether Gao-binge alcohol affects TFEB and lysosomal biogenesis in the liver has not been studied. The aim of this study was to determine autophagic flux and TFEB-mediated lysosomal biogenesis in the liver in addition to the contributions of TFEB and its underlying mechanisms in Gao-binge-induced liver injury. We found that Gao-binge impaired TFEB-mediated lysosomal biogenesis due to activation of mTORC1 resulting in insufficient autophagy in mouse livers. Overexpression or ablation of TFEB in mouse livers attenuated or exacerbated alcohol-induced liver injury in mice.
Materials and Methods
Animal Experiments
GFP-LC3 transgenic mice were generated by Dr. N. Mizushima and were purchased from RIKEN (Japan). TFEB flox mice were generated as described previously22 and were crossed with Albumin Cre (The Jackson Laboratory) to generate liver-specific TFEB knockout (KO) mice. TFE3 KO (C57BL/6N) mice were generated as described previously24 and obtained from Dr. David Fisher (Harvard Medical School). All animals received humane care. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Kansas Medical Center. Age matched litter mates from TFEB flox, Albumin Cre negative and wild type (WT) TFE3 (C57BL/6N) mice were used as control WT mice for all the KO mice. Eight to twelve week old male mice were treated with Gao-binge alcohol model4, 23. Briefly, mice were acclimated to the Lieber-DeCarli liquid control diet (F1259SP, Bio-Serv) for 5 days followed by further feeding with the liquid control or ethanol diet (F1258SP, Bio-Serv, 5% ethanol) for 10 days. The volume of control diet given to mice was matched to the volume of ethanol diet consumed. On the last day of feeding, mice were further given 5 g/kg ethanol or 9 g/kg maltose dextran and sacrificed 8 hours later. In some experiments, one dose of leupeptin (40 mg/kg, i.p.) or torin 1 (2 mg/kg, i.p.) was given right before the gavage on the final day. For knockdown or overexpression of TFEB, one dose of Ad-U6-shRNA-negative control (Neg, 1×109 PFU/mouse, i.v.) or Ad-U6-shRNA-TFEB (1×109 PFU/mouse, i.v.), or Ad-Null (5 ×108 PFU/mouse, i.v.) or Ad-TFEB (5 ×108 PFU/mouse, i.v.) was given on the day prior to ethanol feeding. On the last day of feeding, mice were euthanized 8 hours after the gavage. Blood and liver tissues were collected. Liver injury was determined by measuring serum alanine aminotransferase (ALT). Liver cryosections and H & E staining as well as confocal microscopy were performed as described previously25.
Cellular Fractionation and Western Blot Analysis
Total liver lysates were prepared using radioimmunoprecipitation assay (RIPA) buffer (1% NP40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl (lauryl) sulfate). Nuclear and cytoplasmic fractions of liver tissue were prepared by using a commercial kit (Thermo scientific, 78835). Lysosomal and cytosolic fractions were prepared as described previously26. Briefly, liver tissues were homogenized in HIM buffer (200 mM mannitol, 70 mM sucrose, 5 mM Hepes, 0.5 mM EGTA (pH 7.5)) containing protease inhibitors using a dounce homogenizer. Homogenates were centrifuged at 1,000 × g to remove debris and nuclei, and the supernatant was centrifuged at 10,000 × g for 10 minutes to separate lysosomal and cytosolic fractions. The supernatant was kept as the cytosolic fraction, and the pellet containing the lysosomal fraction was further washed by centrifugation and re-suspended in HIM buffer. Protein (30 g) was separated by a 12% SDS-PAGE gel before transfer to a PVDF membrane. Membranes were probed using appropriate primary and secondary antibodies and developed with SuperSignal West Pico chemiluminescent substrate (Life Technologies, 34080).
Antibodies
The following antibodies were used for western blot analysis: TFEB (Bethyl, A303-673A), β-Actin (Sigma, A5441), GAPDH (Cell Signaling, 2118), p62 (Abnova, H00008878-M01), GFP (Santa Cruz), phos-s6-ribosomal protein (Cell Signaling, 4858), phos-4EBP1 (Cell Signaling, 9451), s6-ribosomal protein (Cell Signaling, 2217), 4EBP1 (Cell Signaling, 9452), phos-ERK (Cell Signaling, 9101), ERK (Cell Signaling, 9102), anti-lymphocyte antigen B superfamily (Ly6B, AbD Serotec, MCA771G), Lamin A/C (Cell Signaling, 2032), Lamp1 (DSHB, 1D4B), TFE3 (Sigma, HPA023881), PGC1α (Abnova, PAB12061), mTOR (Cell Signaling, 2972). Antibodies for ATPase H+ Transporting V1 Subunit 1A (VATP6V1a) and ATPase H+ Transporting V1 Subunit 1B2 (VATP6V1b2) are gifts from Dr. Dennis Brown from Harvard Medical School. The Anti-LC3 antibody was generated as previously described5. HRP-conjugated, FITC-conjugated and Cy3-conjugated secondary antibodies were from Jackson ImmunoResearch.
Statistical Analysis
All experimental data were expressed as means ± S.E. and subjected to one-way ANOVA analysis with Bonferroni post hoc test or Student’s t-test where appropriate. A p<0.05 was considered significant.
For additional materials and methods, please refer to the Supporting Material.
Results
Gao-binge alcohol induces autophagic flux but is insufficient for p62 degradation in mouse liver
To determine autophagic flux in mouse livers, we treated GFP-LC3 transgenic mice with Gao-binge model with or without leupeptin (Leu), a lysosomal inhibitor. As shown in Fig. 1A, GFP-LC3 has a diffuse pattern in the cytosol of hepatocytes in control mice. The number of GFP-LC3 puncta increased in the mouse hepatocytes of alcohol feeding plus binge group (EtOH) due to their targeting to autophagosomal membranes. As expected, blocking lysosomal degradation by Leu increased the number of GFP-LC3 puncta compared with control group (Ctrl). The number of GFP-LC3 puncta was much higher in mice treated with alcohol together with Leu (EtOH+Leu) than either EtOH or Leu alone (Ctrl+Leu). Consistent with the confocal imaging data, EtOH treatment increased endogenous LC3-II levels, which further increased in the presence of Leu (Fig. 1B & C). Leu alone treatment increased the levels of LC3-II and p62, suggesting the successful blocking of basal autophagy in the liver. We also found that the levels of GFP-LC3-II markedly increased in EtOH+Leu group compared to either EtOH or Ctrl+Leu groups. Based on the guidelines of autophagy research27, these data suggest that Gao-binge increases autophagic flux in mouse livers. However, we found that hepatic protein and mRNA levels of p62 did not change after alcohol (Fig. 1B & D & data not shown). The failure to efficiently degrade hepatic p62 suggests that Gao-binge-induced autophagy may not be sufficient, which could be due to either impaired lysosomal/autolysosomal functions or decreased lysosome numbers.
Figure 1. Gao-binge induces autophagic flux in mouse livers.
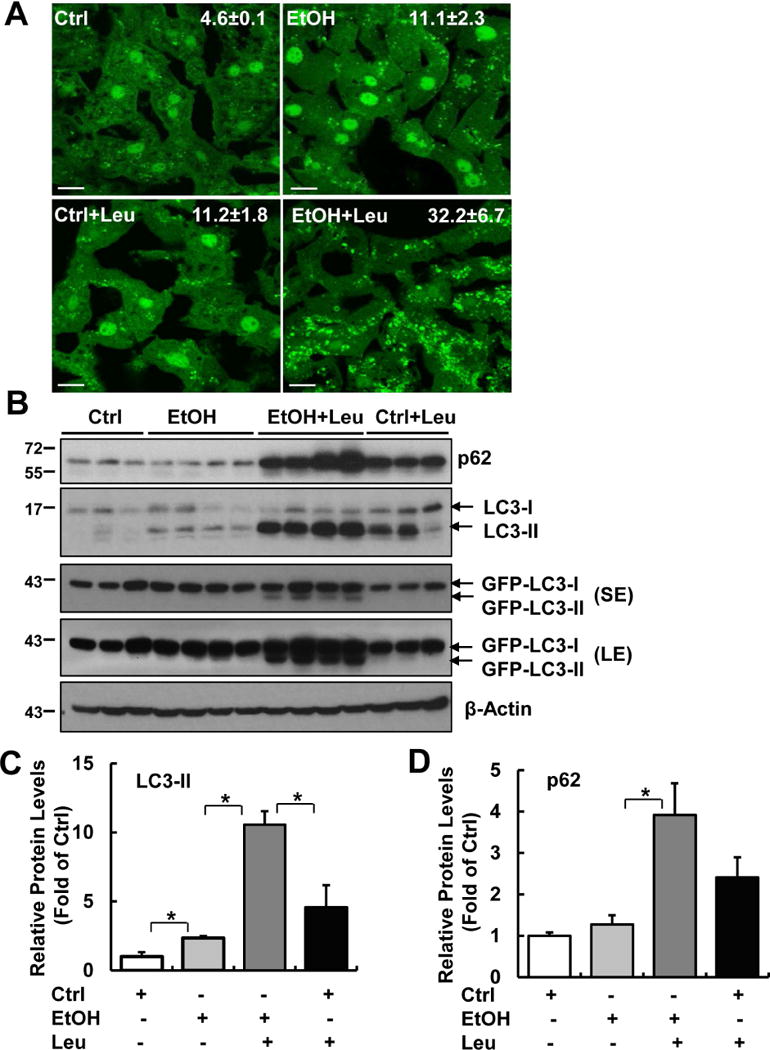
Male GFP-LC3 transgenic mice were treated with Gao-binge model. One dose of Leupeptin (40 mg/kg, i.p) was given to mice prior to the gavage. Mice were sacrificed 8 hours later after the gavage. (A) Representative confocal microscopy images from liver cryosections are shown. GFP-LC3 puncta per cell in each group were quantified (n=3-4). Ctrl: control diet+maltose gavage; EtOH: ethanol diet+ ethanol gavage. Data are means ± SE. More than 60 cells were counted in each mouse. Scale Bar: 10 μm. (B) Total liver lysates were subjected to western blot analysis. SE: short exposure; LE: long exposure. (C & D) Densitometry analysis of (B). The levels of p62 and LC3-II were normalized to the loading control (β-Actin). Data represent means ± SE (n= 3-4). *p<0.05; one-way ANOVA analysis.
Impaired TFEB in Gao-binge alcohol mouse livers and is associated with human alcoholic hepatitis
Lysosomes are the terminal degradative organelles to ensure the completion of autophagic flux by fusing with incoming autophagosomes. Thus, the lack of sufficient lysosome numbers to meet the autophagy demand or impaired lysosomal function would compromise autophagic flux. We found that EtOH caused a 30% reduction of lysosome numbers in hepatocytes as judged by Lamp1 positive vesicles/puncta (labelled as red, Fig. 2A & B). Analysis of the colocalization of GFP-LC3 puncta with Lamp1 revealed that the percentage of green-only GFP-LC3 puncta (which are not colocalized with Lamp1) was significantly higher in alcohol-treated mouse livers than that of Ctrl mice (Fig. 2C & D). These results suggest that there is a lack of sufficient number of lysosomes to fuse with autophagosomes induced by alcohol, which probably contributes to the insufficient hepatic autophagy induced by Gao-binge alcohol.
Figure 2. Gao-binge decreases the number of lysosomes with increased accumulation of autophagosomes and inhibits TFEB in mouse livers.
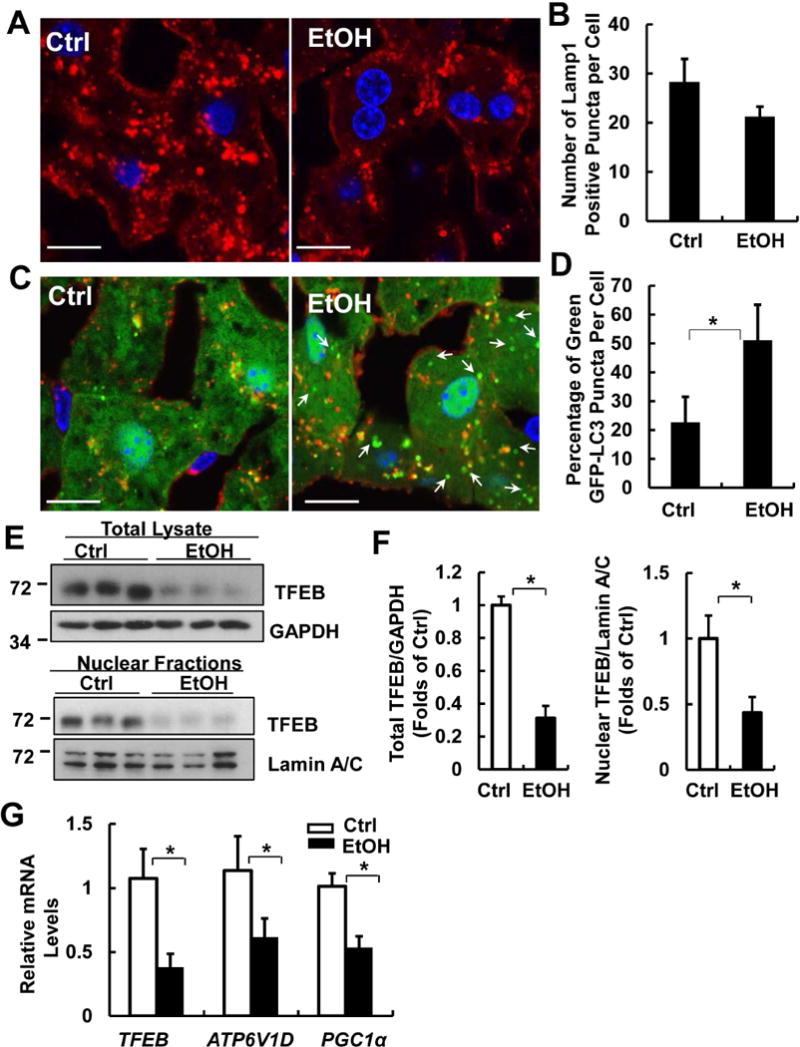
Male GFP-LC3 transgenic mice were treated with Gao-binge model and cryosections of mouse livers were subjected to immunostaining for Lamp1 followed by confocal microscopy. Representative images of immunostaining of Lamp1 (A) and quantified number of Lamp1 positive vesicles are shown (B). Scale Bar: 10 μm. Data are means ± SE (n=3-4). More than 50 cells were counted in each mouse. (C) Representative images of the colocalization of Lamp1 with GFP-LC3 puncta are shown. Arrows denote the green-only GFP-LC3 puncta. (D) Percentage of GFP-LC3 puncta that are not colocalized with Lamp1 positive vesicles. Data are means ± SE (n=3-4). More than 50 cells were counted in each mouse. *p<0.05; Student t test. (E) Male C57BL/6J WT mice were treated with Gao-binge model. Total lysates and nuclear fractions from mouse livers were subjected to western blot analysis. (F) Densitometry analysis of (E). (G) mRNA from mouse livers was used for qPCR. Results were normalized to 18s and expressed as fold change compared to Ctrl group. Data shown are means ± SE (n =4). *p<0.05; Student t test.
Since TFEB is a master regulator for lysosomal biogenesis, we wondered whether decreased lysosome numbers in EtOH-treated mouse livers could be due to impaired hepatic TFEB function. We found that EtOH markedly decreased the total and nuclear TFEB protein levels in mouse livers compared to control mice (Fig. 2E & F). Moreover, EtOH significantly decreased the mRNA levels of hepatic TFEB, ATP6V1D (ATPase H+ Transporting V1 Subunit D) and PGC1α (peroxisome proliferator-activated receptor gamma coactivator 1-alpha), three known TFEB target genes (TFEB is its own target gene), compared to Ctrl mice (Fig. 2G). These data indicate that Gao-binge alcohol not only decreases TFEB proteins but also reduces its transcription activity in mouse livers.
Immunostaining for TFEB also revealed that TFEB mainly localized in the cytosol and nuclei but EtOH significantly decreased the number of hepatocytes with nuclear TFEB (Fig. 3A&B, arrows). Decreased hepatic TFEB was also observed in mice that fed with Lieber-DeCarli alcohol diet for 32 days compared with their pair-fed control mice (Supplemental Fig. 1A&B). In contrast, neither mice fed with a Lieber-DeCarli alcohol diet for 10 days nor mice received an acute alcohol gavage altered the total or nuclear TFEB levels in the mouse livers (Supplemental Fig. 1C&F), suggesting decreased hepatic TFEB is specific to Gao-binge or prolonged alcohol exposure.
Figure 3. Decreased nuclear TFEB proteins in Gao-binge alcohol mouse livers and human alcoholic hepatitis.
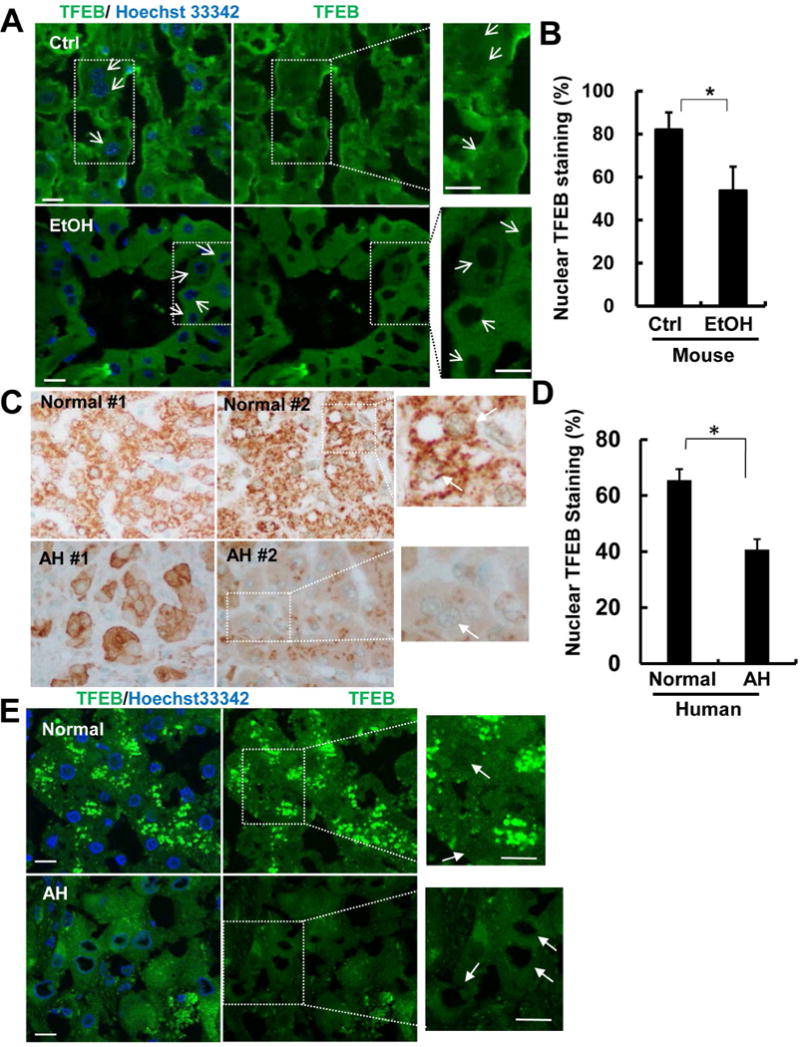
Male C57BL/6J WT mice were treated with Gao-binge model. Cryosections of mouse livers were subjected to immunostaining for TFEB and nuclei were stained with Hoechst33342 followed by confocal microscopy. Representative images are shown (A). Arrows denote the nuclear staining of TFEB. Right panels are enlarged photographs from the boxed areas. Scale Bar: 10 μm. (B) The percentage of hepatocytes with nuclear TFEB staining were quantified from 5 fields of 40X magnification from 3 mice of each group. Data are means ± SE. *p<0.05; Student t test. (C) Immunohistochemical staining of liver TFEB of alcoholic hepatitis (AH) patients or healthy donors (normal liver), and representative images are shown. (D) The percentage of hepatocytes with nuclear TFEB staining were quantified from 4 fields of 40X magnification (n=6 healthy controls; and n=6 AH). Data are means ± SE. *p<0.05; Student t test. (E) Cryo-liver sections of AH patient and healthy donor were subjected immunostaining of TFEB and nuclear were stained with Hoechst33342 followed by confocal microscopy. Images in right panels were enlarged photographs from the boxed areas in the left panels. Arrows denote the nuclear TFEB staining.
To further determine whether impaired TFEB signaling pathway would also be associated with human ALD, we performed immunohistochemistry and fluorescence immunostaining for hepatic TFEB in livers from healthy human donors and alcoholic hepatitis (AH) patients obtained from the Clinical Resources for Alcoholic Hepatitis Investigator of Johns Hopkins University. The detail demographic, clinical and biological characterization information of the AH patients and healthy controls has been recently described by Khanova E et al.28. Consistent with the TFEB changes in EtOH-treated mice, we found that TFEB mainly localized in the cytosol and nuclei in normal healthy livers but the nuclear TFEB staining markedly decreased in AH livers (Fig. 3C&E, arrows). These data suggest that impaired hepatic TFEB is associated not only with EtOH-treated mice but also with human AH.
Gao-binge alcohol increases mTOR lysosomal translocation and activates mTORC1 in mouse livers
Since mTOR negatively regulates TFEB by directly phosphorylating TFEB and promoting its degradation by proteasome29, 30, we next determined the mTOR activation in EtOH-treated mouse livers. We found that mTOR displayed a diffuse pattern in hepatocytes in Ctrl mice. In contrast, mTOR showed a punctated pattern in EtOH-treated mouse livers that colocalized with Lamp1 staining (Fig. 4A arrows). EtOH increased mTOR and RagA but decreased Lamp1 protein levels in isolated lysosomal fractions compared to Ctrl mice (Fig. 4B & C). Decreased Lamp1 levels in the lysosomal fractions from EtOH-treated mice could be due to decreased lysosomal biogenesis as a result of TFEB inactivation. These data indicate that EtOH increases mTOR lysosomal translocation in hepatocytes. We further found that EtOH markedly increased mTORC1 activity as shown by increased levels of phosphorylated S6 and 4EBP1, two well-known substrate proteins that are phosphorylated by mTORC1 (Fig. 4D). In contrast, EtOH did not change the levels of phosphorylated extracellular signal-regulated kinase 1/2 (ERK1/2) compared to Ctrl mice, although the levels of phosphorylated ERK1/2 varied among different mice (Fig. 4D & E). While EtOH decreased the levels of phosphorylated TFEB (p-TFEB), EtOH increased the ratio of p-TFEB vs total TFEB (Fig. 4D & E). Taken together, these data indicate that Gao-binge alcohol increases lysosomal mTOR translocation and activates mTORC1 but not ERK in mouse livers.
Figure 4. Gao-binge activates mTOR in mouse livers.
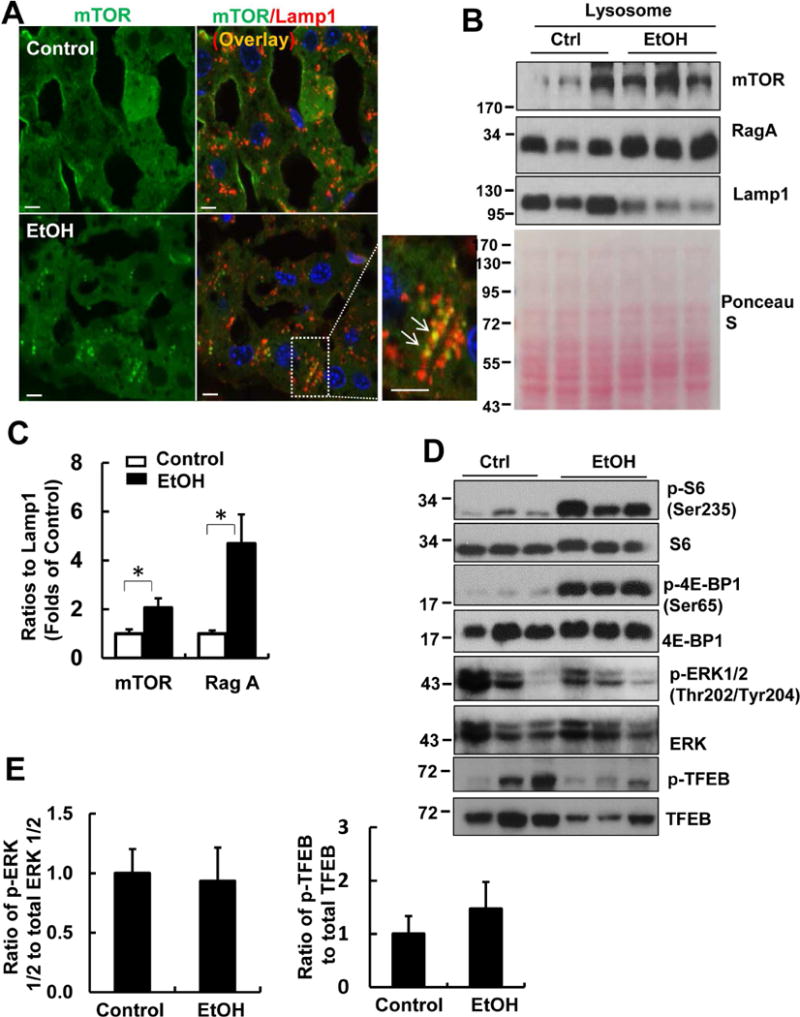
Male C57BL/6J WT mice were treated with Gao-binge model. (A) Cryosections of mouse livers were subjected to immunostaining for mTOR (green) and Lamp1 (red) followed by confocal microscopy. Representative images are shown. Right panel is an enlarged photograph from the boxed area. Arrows denote the colocalization of mTOR with Lamp1. Scale Bar: 10 μm. (B) Lysosomal fractions from mouse livers were subjected to western blot analysis. Membrane was pre-stained with Ponceau S as a loading control. (C) Densitometry analysis of (B) are shown (means ± SE, n=3). *p<0.05; Student t test. (D) Total liver lysates were subjected to western blot analysis. (E) Densitometry analysis of (D) are shown (means ± SE, n=3).
Pharmacological inhibition of mTOR attenuates whereas inhibition of lysosomal degradation exacerbates Gao-binge-induced liver injury
Since EtOH activated mTOR in mouse livers, we wondered whether inhibition of mTOR would attenuate mTOR-mediated TFEB inactivation and subsequent liver injury induced by EtOH. We found that Torin 1 (Tor), a potent mTORC1 inhibitor, improved EtOH-induced insufficient autophagy as demonstrated by further increased numbers of GFP-LC3 puncta and levels of GFP-LC3-II and endogenous LC3-II (Supplemental Fig. 2A & B). Decreased hepatic TFEB levels and expression of TFEB target genes induced by EtOH were partially recovered by Tor (Supplemental Fig. 2B & C). As a result, Tor markedly attenuated EtOH-induced liver injury and steatosis as demonstrated by decreased serum levels of ALT and hepatic TG as well as numbers of lipid droplets (LDs) as shown by H & E and LipidTOX™ staining (Supplemental Fig. 3A-D). In contrast, we found that inhibition of lysosome by Leu exacerbated EtOH increased serum levels of ALT and hepatic TG (Supplemental Fig. 4A & B). Leu also further increased EtOH-induced LD as revealed by H & E staining (Supplemental Fig. 4C) and LipidTOX™ staining for neutral lipid (Supplemental Fig. 4D). These data indicate that pharmacological inhibition of mTOR improves TFEB-mediated lysosomal pathway and attenuates EtOH-induced liver injury whereas inhibition of lysosomal degradation exacerbates EtOH-induced liver injury.
Hepatocyte-specific TFEB KO mice and TFE3 whole body KO mice do not exacerbate Ga-binge-induced steatosis and liver injury
To further determine the role of TFEB in EtOH-induced liver injury, we generated hepatocyte-specific TFEB KO mice and whole body TFE3 KO mice and subjected these mice and their matched WT mice to Gao-binge model. To our surprise, we found that levels of serum ALT and hepatic TG were almost identical between hepatocyte-specific TFEB KO mice and their matched WT littermates (TFEB f/f, Alb Cre-) after Gao-binge (Supplemental Fig. 5A & B). TFEB belongs to microphthalmia-associated transcription factor (MITF) family proteins, which include MITF, TFEB, TFE3 and TFEC in mammals31. Indeed, we found that TFE3 was also expressed in both hepatocyte-specific TFEB KO and WT mouse livers (Supplemental Fig. 5C). Moreover, the levels of VATP6V1a and VATP6V1b2, two subunits of the lysosomal VATPase, did not decrease in hepatocyte-specific TFEB KO mice compared to WT mice. These data suggest that TFE3 may compensate for the hepatic loss of TFEB in mouse livers. To determine the role of TFE3 in liver injury, we treated TFE3 whole body KO mice and their matched WT mice with Gao-binge. We found no differences in serum ALT and hepatic TG levels after EtOH between TFE3 KO and their matched WT mice with or without EtOH treatment (Supplemental Fig. 5D & E). We also found similar levels of TFEB, VATP6V1a and VATP6V1b2 between TFE3 KO and WT mouse livers (Supplemental Fig. 5F). Taken together, these data suggest that single chronic deletion of either TFEB or TFE3 is not sufficient to impair lysosomal biogenesis in the liver likely due to the compensatory effects of the other, and these single KO mice do not exacerbate alcohol-induced liver injury.
Acute knockdown of TFEB or deletion of both TFEB and TFE3 in mouse livers impairs hepatic lysosomal biogenesis and exacerbates Gao-binge-induced liver injury
To overcome the compensatory effects of the chronic deletion of TFEB in hepatocyte-specific TFEB KO mice, we acutely knocked down TFEB in mouse livers using an adenovirus (Ad)-shRNA approach. We found that Ad-shRNA TFEB markedly decreased hepatic TFEB at both mRNA and protein levels. Knockdown of TFEB also led to significantly decreased expression of PGC1α and ATP6V1H, two well-known TFEB target genes (Fig. 5A & B). Mice that received Ad-shRNA TFEB had significantly elevated serum ALT levels that were approximately 10 fold higher than the mice that received Ad-shRNA control after EtOH treatment (Fig. 5C). Mice with knockdown of TFEB also had more severe steatosis than mice that received control viruses after EtOH treatment, as demonstrated by elevated liver TG and increased numbers of large-sized LDs by H & E staining (Fig. 5D & E) as well as Oil Red O and LipidTOX™ staining (Supplemental Fig. 6A). We found that the diameters of some of the LDs reached around 20~30 μm, which squeezed the nuclei to the edge of the hepatocytes (Supplemental Fig. 6B arrow). Moreover, mice with knockdown of TFEB increased hepatic infiltration of neutrophils compared with mice received control shRNA after EtOH (Supplemental Fig. 7A&B). In addition to acute knockdown of TFEB, we also generated mice with double knockout (DKO) of both TFEB and TFE3 in the liver and treated these mice with EtOH. We found that DKO mice had decreased protein levels of PGC1α, VATP6V1a and VATP6V1b and increased serum ALT levels as well increased hepatic infiltration of neutrophils compared with matched WT littermates after EtOH (Fig. 6A-C & Supplemental Fig. 7 C&D). TFEB and TFE3 DKO mice had slightly higher levels of basal hepatic TG compared with WT mice, but the levels of hepatic TG did not further increase after EtOH (Fig. 6D). These data suggest that TFE3 may compensate for the chronic loss of TFEB in mouse livers, and TFEB-mediated lysosomal biogenesis is a critical protective mechanism against alcohol-induced liver injury.
Figure 5. Knockdown of hepatic TFEB exacerbates Gao-binge-induced liver injury and steatosis.
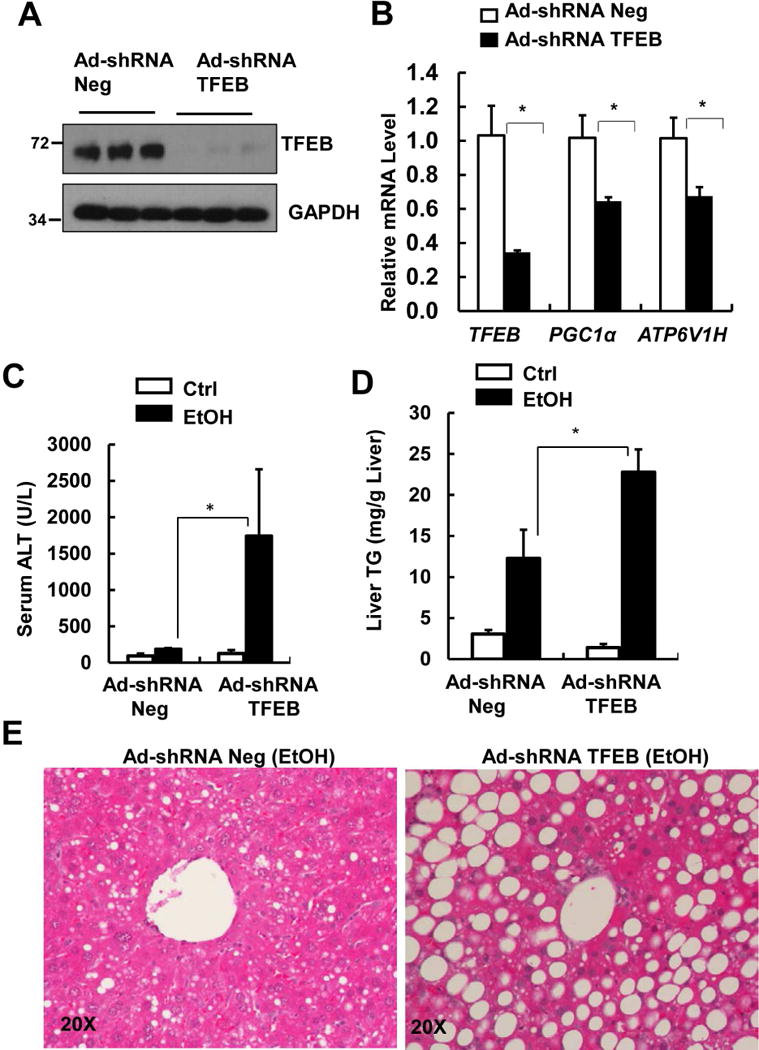
Male C57BL/6J WT mice were injected with Ad-shRNA negative and Ad-shRNA TFEB (1×109 PFU/mouse via tail vein) followed by Gao-binge model. (A) Total liver lysates were subjected to western blot analysis. (B) Hepatic mRNA was extracted followed by qPCR (n=4-6). Data are means ± SE. *p<0.05; Student t test. (C) Serum ALT levels and (D) hepatic TG levels were quantified. Data are means ± SE (n=4-6). * p<0.05; One-way ANOVA analysis. (E) Representative images of H& E staining are shown.
Figure 6. TFEB/TFE3 DKO mice are more susceptible to Gao-binge-induced liver injury.
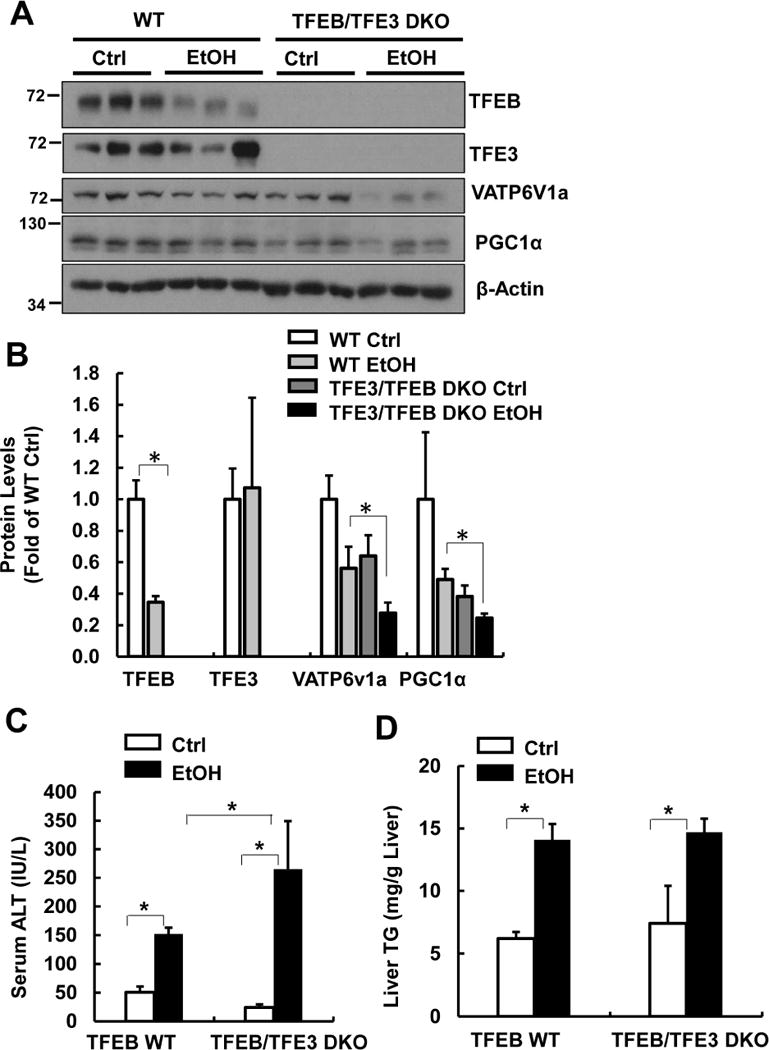
Male TFEB/TFE3 DKO and matched WT mice were treated with Gao-binge model. (A) Total liver lysates were subjected to western blot analysis and (B) densitometry analysis of (A). Serum ALT (C) and liver TG levels (D) were measured. Data are means ± SE (n=3-10). *p<0.05; One-way ANOVA analysis.
Overexpression of TFEB increases lysosomal biogenesis, improves mitochondrial bioenergetics and protects against Gao-binge-induced liver injury
We found that administration of Ad-TFEB dramatically increased TFEB protein levels in the liver but not in muscle or pancreas (Fig. 7A). Moreover, hepatic expression of several lysosomal genes such as ATP6V1D, ATP6V0E1 and ATP6V1H as well as PGC1α significantly increased in Ad-TFEB-treated mice compared to Ad-null treated mice (Fig. 7B). In addition to the mRNA changes, overexpression of TFEB also led to marked increase of TFEB, PGC1α, VATP6V1a and VATP6V1b2 proteins in mouse livers (Fig. 7C). More importantly, overexpression of TFEB significantly inhibited EtOH-induced liver injury and steatosis as demonstrated by markedly decreased levels of serum ALT and hepatic TG (Fig. 7D & E) as well as hepatic Oil Red O staining and hepatic infiltration of neutrophils (Supplemental Fig. 8 & 9). Moreover, EtOH impaired hepatic mitochondrial bioenergetics and State 3 respiration, which were significantly improved by overexpression of TFEB (Supplemental Fig. 10). Consistently, EtOH also significantly decreased hepatic mRNA levels of fatty acid beta-oxidation genes, which were significantly recovered by overexpression of TFEB or further decreased by knockdown of TFEB, respectively (Supplemental Figure 11). These results indicate that overexpression of hepatic TFEB protects against alcohol-induced steatosis and liver injury likely via induction of lysosomal biogenesis coupled with improved mitochondria bioenergetics.
Figure 7. Overexpression of TFEB protects against alcohol-induced steatosis and liver injury.
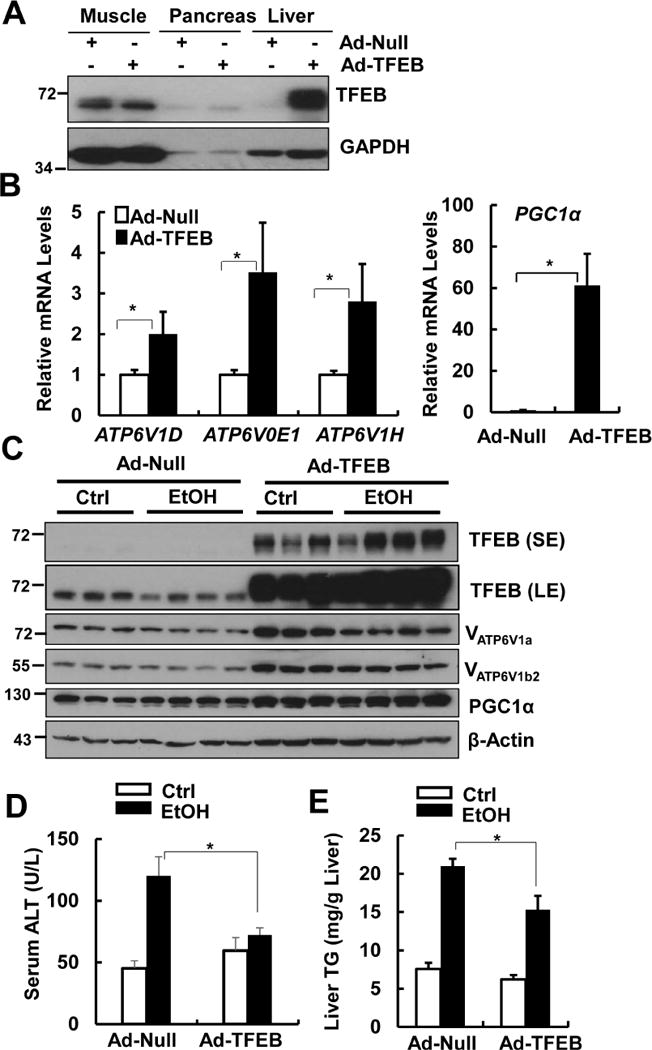
Male WT C57BL/6J mice were injected with Ad-Null and Ad-TFEB (5×108 PFU/mouse via tail vein) followed by Gao-binge model. (A) Total muscle, pancreas and liver tissue lysates from mice that were treated with control diet+maltose gavage were subjected to western blot analysis. (B) Hepatic mRNA was extracted followed by qPCR (n=4-6). Data are means ± SE (n=4-6). * p<0.05; Student t test. (C) Total liver lysates were subjected to western blot analysis. SE: short exposure; LE: long exposure. (D) Serum ALT levels and (E) hepatic TG levels were quantified. Data are means ± SE (n=4-6). * p<0.05; one-way ANOVA analysis.
Discussion
We used the protocol of chronic feeding plus acute binge alcohol (“Gao-binge, EtOH” model)4, 23 to identify steps in lysosomal biogenesis in the autophagy protective pathway that are impaired by alcohol in the liver. We found that EtOH activated mTORC1 and decreased TFEB proteins and TFEB-mediated lysosomal biogenesis, resulting in insufficient autophagy in mouse livers. More importantly, we also found decreased TFEB in human ALD samples compared with healthy human donor liver samples. Pharmacological or genetic inhibition of lysosomal functions greatly exacerbated EtOH-induced liver injury. In contrast, overexpression of TFEB increased lysosomal biogenesis and protected against EOH-induced steatosis and liver injury.
TFEB binds specifically to a 10-bp (GTCACGTGAC) motif found in the promoter regions of many genes encoding for lysosomal biogenesis and autophagy induction15. Under conditions that have high autophagy demand, TFEB coordinates an efficient transcriptional program to upregulate genes that are responsible for both early (autophagosome formation) and late (lysosome biogenesis) steps of autophagy. TFEB is mainly regulated at the posttranslational level via phosphorylation. At least three kinases, including ERK2, mTORC1 and protein kinase Cβ (PKCβ), have been identified to phosphorylate TFEB21. ERK2 phosphorylates TFEB at Ser142 whereas mTORC1 phosphorylates TFEB at Ser142, Ser211 and Ser122, leading to the retention of TFEB in the cytosol and subsequent proteasome-mediated degradation and inactivation of TFEB15, 21, 30, 32. In contrast, PKCβ induces phosphorylation of Ser461, Ser466, and Ser468, which stabilizes TFEB and increases its activity33. Moreover, lysosomal Ca2+ release via mucolipin 1 activates the phosphatase calcineurin, which dephosphorylates TFEB at Ser142 and Ser211 and promotes TFEB nuclear translocation34. We found that EtOH increased lysosomal mTOR translocation and mTORC1 activation in mouse livers. Consistent with the known negative regulatory role of mTORC1 on TFEB, EtOH increased ratio of p-TFEB/total TFEB but decreased nuclear TFEB translocation and TFEB-mediated transcription of lysosomal biogenesis genes. Torin1, a specific mTORC1 inhibitor, partially reversed decreased hepatic TFEB proteins induced by EtOH, further supporting a critical role of mTORC1 in regulating EtOH-induced inactivation of TFEB. However, the partial recovery of TFEB levels by Torin 1 suggests that other pathways in addition to mTORC1 may also be involved in regulating TFEB inactivation after EtOH. ERK1/2 may not play an important role in EtOH-Induced inactivation of TFEB because EtOH did not activate ERK1/2. It was reported that acute ethanol decreased activity of classic PKC isoforms in rat hepatocytes35. Whether EtOH would also affect PKC activity and in turn affect TFEB activation needs to be further investigated. Acute ethanol also increased intracellular Ca2+ levels in hepatocytes, so it will be interesting to further evaluate whether calcium-dependent calcineurin would also be involved in EtOH-induced inactivation of TFEB.
The four Rag family proteins (Rag A, B, C and D) are tethered on the surface of lysosomes by the protein complex Ragulator in close proximity to Rheb. In response to amino acids, distinct heterodimeric complexes (RagA/B GTP-bound and RagC/D GDP-bound) are formed, which bind directly to Raptor, a subunit of the mTORC1 complex, thereby recruiting the mTORC1 complex to the lysosome36. The finding that EtOH increased hepatic lysosomal RagA levels suggests that EtOH may activate mTORC1 via the Rag GTPases and Ragulator complexes. Ethanol metabolism in the liver results in changing cellular redox status by increasing NADH/NAD+ ratio2. In response to oxidative stress, the Nemo-like kinase (NLK), a distinct member of the MAP kinase (MAPK) subfamily, inhibits mTORC1 activation through phosphorylating Raptor and disrupting its interaction with the Rag complex on the lysosome37. Whether ethanol metabolites such as acetaldehyde or the change of NAD+ levels may regulate NLK-mediated mTOR lysosomal translocation and activation remain to be studied.
Our initial conclusion that Gao-binge induced autophagic flux was based on the recommended autophagic flux assay, which relies on changes in LC3-II levels in the presence or absence of a lysosomal inhibitor27. However, this assay has its own limitations and may not accurately reflect all autophagy scenarios such as the Gao-binge model. Decreased lysosomal biogenesis will cause an accumulation of early autophagosomes and a decreased number of matured autolysosomes due to the lack of lysosomes to fuse with autophagosomes. However, the insufficient numbers of mature autolysosomes may still be able to degrade some autophagic contents, although it will not reach the maximal capacity of autophagic degradation. Accordingly, Leu still enhanced EtOH-induced LC3-II levels by blocking all lysosomal degradation. Our data thus revealed a novel autophagic flux scenario that might have been neglected by using the traditional autophagic flux assay. To better assess autophagy status under certain conditions, additional assays to determine TFEB activity and number of lysosomes should also be considered in conjunction with the traditional autophagic flux assay.
We found that hepatocyte-specific TFEB KO mice and TFE3 KO mice were not more sensitive to EtOH-induced liver injury compared to their matched WT control mice. However, this is likely due to the complementary compensation for each other due to the chronic deletion of either TFEB or TFE3. We previously reported that acute knockdown of Parkin in mouse livers could overcome compensatory protective effects in Parkin KO mice against acetaminophen-induced liver injury38. Our observation that acute knockdown of hepatic TFEB and TFEB/TFE3 DKO mice markedly exacerbated EtOH-induced liver injury further underscores the notion that compensatory effects are common in KO mice due to the chronic loss of a gene. Therefore, caution needs to be emphasized for the interpretation of data obtained from KO mice.
Multiple mechanisms may account for the protective effects induced by TFEB activation against EtOH-induced steatosis and liver injury. It is likely that enhanced lysosomal degradation by overexpression of TFEB might help to remove the excess LDs or damaged mitochondria via lipophagy or mitophagy, respectively. Moreover, TFEB also markedly increased expression of PGC1α, which is an important factor for mitochondrial biogenesis and subsequent mitochondrial fatty acid beta oxidation. Indeed, we found that overexpression of TFEB markedly improved mitochondrial State3 respiration and mitochondrial reserve oxidative capacity as well as the expression of fatty acid beta oxidation genes following Gao-binge alcohol. It is known that fatty acids generated from lipophagy are potentially toxic to the liver, and timely removal of these fatty acids are important to reduce hepatic lipotoxicity39, 40. The findings that activation of TFEB improves both autophagy and mitochondrial functions strongly support a beneficial role of activation of TFEB in preventing or treating ALD.
In conclusion, we found that EtOH activated mTORC1 and impaired hepatic TFEB-mediated lysosomal biogenesis resulting in insufficient autophagy in mice. It has long been known that chronic alcohol consumption can cause hepatomegaly due to the accumulation of proteins and lipids in the liver. Impaired lysosomal functions has been suggested to be critical for the decreased protein turnover for decades in ALD, but the mechanisms are unknown. Our present study has identified that impaired TFEB may be the missing key factor that contribute to the hepatic lipid accumulation of liver injury in ALD. Indeed, pharmacological inhibition of mTOR or overexpression of TFEB protected against EtOH-induced steatosis and liver injury. Targeting TFEB-mediated lysosomal biogenesis may thus be a novel avenue for preventing and treating ALD. The possible cellular events that lead to the impaired TFEB-mediated lysosomal biogenesis and liver injury by EtOH and the possible protective effects by activating TFEB against EtOH-induced steatosis and liver injury were summarized in Supplemental Fig. 12.
Supplementary Material
Acknowledgments
We thank Drs. Dennis Brown and David Fisher from Harvard Medical School for providing VATP6V1a and VATP6V1b2 antibodies and TFE3 KO mice. Dr. Zhaoli Sun of Johns Hopkins University for providing us the paraffin-embedded and cryo-liver sections from healthy liver donors and alcoholic hepatitis patients via the Clinical Resources for Alcoholic Hepatitis Investigators (1R24AA025017). We thank Mr. Brian Bridges from KUMC Liver Center for collecting the healthy donor livers and alcoholic human cirrhotic liver samples as well as Barbara Fegley for the EM studies.
The research was supported in part by the NIAAA R01 AA020518, U01 AA024733, National Institute of General Medical Sciences P20GM103549 & P30GM118247. The electron microscopy core facility was supported in part by NIH COBRE grant 9P20GM104936.
Abbreviations
- ALD
Alcoholic Liver Disease
- ALT
alanine aminotransferase
- ATP6V1D
ATPase H+ Transporting V1 Subunit D
- ATP6V1H
ATPase H+ Transporting V1 Subunit H
- ATP6V0E1
ATPase H+ Transporting V0 Subunit E1
- Ctrl
control diet plus maltose binge
- EtOH
ethanol diet plus ethanol binge
- EM
electron microscopy
- H & E
hematoxylin and eosin
- KO
knockout
- LAMP1
Lysosomal-associated membrane protein 1
- LC3
Microtubule-associated protein light chain 3
- LDs
lipid droplets
- Leu
leupeptin
- mTORC1
mechanistic target of rapamycin complex 1
- PGC1α
peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- RIPA
radioimmunoprecipitation
- TG
triglycerides
- Tor
Torin 1
- TSC1
tuberous sclerosis complex 1
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions: W.X.D. & H.M.N. conceived and designed the study; X.C., S.W., K.Z., Y.L., J.A.W., T.L., H.C., P.K., X.C.H., L.L. & H.M.N. did acquisition and analysis of data; X.C. did statistical analysis; A.B. provided technical and material support. W.X.D supervised the study; W.X.D, X.C. & H. M.H. drafted the manuscript.
Conflict of Interest: These authors declare that they have nothing to disclose.
References
- 1.Liangpunsakul S, Haber P, McCaughan GW. Alcoholic Liver Disease in Asia, Europe, and North America. Gastroenterology. 2016;150:1786–97. doi: 10.1053/j.gastro.2016.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nagy LE, Ding WX, Cresci G, et al. Linking Pathogenic Mechanisms of Alcoholic Liver Disease With Clinical Phenotypes. Gastroenterology. 2016;150:1756–68. doi: 10.1053/j.gastro.2016.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572–85. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams JA, Manley S, Ding WX. New advances in molecular mechanisms and emerging therapeutic targets in alcoholic liver diseases. World J Gastroenterol. 2014;20:12908–33. doi: 10.3748/wjg.v20.i36.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding WX, Li M, Chen X, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–52. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding WX, Manley S, Ni HM. The emerging role of autophagy in alcoholic liver disease. Exp Biol Med (Maywood) 2011;236:546–56. doi: 10.1258/ebm.2011.010360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomes PG, Trambly CS, Fox HS, et al. Acute and Chronic Ethanol Administration Differentially Modulate Hepatic Autophagy and Transcription Factor EB. Alcohol Clin Exp Res. 2015;39:2354–63. doi: 10.1111/acer.12904. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Ding WX. A Gene Transcription Program Decides the Differential Regulation of Autophagy by Acute Versus Chronic Ethanol? Alcohol Clin Exp Res. 2016;40:47–9. doi: 10.1111/acer.12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ni HM, Du K, You M, et al. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am J Pathol. 2013;183:1815–25. doi: 10.1016/j.ajpath.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin CW, Zhang H, Li M, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol. 2013;58:993–9. doi: 10.1016/j.jhep.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eid N, Ito Y, Maemura K, et al. Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: an immunohistochemical and electron microscopic study. J Mol Histol. 2013;44:311–26. doi: 10.1007/s10735-013-9483-x. [DOI] [PubMed] [Google Scholar]
- 12.Kharbanda KK, McVicker DL, Zetterman RK, et al. Ethanol consumption reduces the proteolytic capacity and protease activities of hepatic lysosomes. Biochim Biophys Acta. 1995;1245:421–9. doi: 10.1016/0304-4165(95)00121-2. [DOI] [PubMed] [Google Scholar]
- 13.Baraona E, Leo MA, Borowsky SA, et al. Alcoholic hepatomegaly: accumulation of protein in the liver. Science. 1975;190:794–5. doi: 10.1126/science.1198096. [DOI] [PubMed] [Google Scholar]
- 14.Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. Embo j. 2012;31:1095–108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–33. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang J, Dibble CC, Matsuzaki M, et al. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol. 2008;28:4104–15. doi: 10.1128/MCB.00289-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sancak Y, Bar-Peled L, Zoncu R, et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zoncu R, Bar-Peled L, Efeyan A, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–83. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol. 2013;14:133–9. doi: 10.1038/nrm3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Settembre C, Fraldi A, Medina DL, et al. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14:283–96. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Settembre C, De Cegli R, Mansueto G, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15:647–58. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bertola A, Mathews S, Ki SH, et al. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nat Protoc. 2013;8:627–37. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steingrimsson E, Tessarollo L, Pathak B, et al. Mitf and Tfe3, two members of the Mitf-Tfe family of bHLH-Zip transcription factors, have important but functionally redundant roles in osteoclast development. Proc Natl Acad Sci U S A. 2002;99:4477–82. doi: 10.1073/pnas.072071099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ni HM, Bockus A, Boggess N, et al. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55:222–32. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding WX, Ni HM, DiFrancesca D, et al. Bid-dependent generation of oxygen radicals promotes death receptor activation-induced apoptosis in murine hepatocytes. Hepatology. 2004;40:403–13. doi: 10.1002/hep.20310. [DOI] [PubMed] [Google Scholar]
- 27.Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khanova E, Wu R, Wang W, et al. Pyroptosis by Caspase11/4-Gasdermin-D Pathway in Alcoholic Hepatitis. Hepatology. 2017 doi: 10.1002/hep.29645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sha Y, Rao L, Settembre C, et al. STUB1 regulates TFEB-induced autophagy-lysosome pathway. EMBO J. 2017;36:2544–2552. doi: 10.15252/embj.201796699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martina JA, Diab HI, Li H, et al. Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Cell Mol Life Sci. 2014;71:2483–97. doi: 10.1007/s00018-014-1565-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vega-Rubin-de-Celis S, Pena-Llopis S, Konda M, et al. Multistep regulation of TFEB by MTORC1. Autophagy. 2017:0. doi: 10.1080/15548627.2016.1271514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferron M, Settembre C, Shimazu J, et al. A RANKL-PKCbeta-TFEB signaling cascade is necessary for lysosomal biogenesis in osteoclasts. Genes Dev. 2013;27:955–69. doi: 10.1101/gad.213827.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288–99. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Domenicotti C, Paola D, Vitali A, et al. Mechanisms of inactivation of hepatocyte protein kinase C isoforms following acute ethanol treatment. Free Radic Biol Med. 1998;25:529–35. doi: 10.1016/s0891-5849(98)00079-3. [DOI] [PubMed] [Google Scholar]
- 36.Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014;24:400–6. doi: 10.1016/j.tcb.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuan HX, Wang Z, Yu FX, et al. NLK phosphorylates Raptor to mediate stress-induced mTORC1 inhibition. Genes Dev. 2015;29:2362–76. doi: 10.1101/gad.265116.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams JA, Ni HM, Haynes A, et al. Chronic Deletion and Acute Knockdown of Parkin Have Differential Responses to Acetaminophen-induced Mitophagy and Liver Injury in Mice. J Biol Chem. 2015;290:10934–46. doi: 10.1074/jbc.M114.602284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Czaja MJ, Ding WX, Donohue TM, Jr, et al. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131–58. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, Zong WX, Ding WX. Recycling the danger via lipid droplet biogenesis after autophagy. Autophagy. 2017:1–3. doi: 10.1080/15548627.2017.1371394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.