

Abstract
Distinct cell types have been shown to respond to activated Ras signaling in a cell-specific manner. In contrast to its pro-tumorigenic role in some human epithelial cancers, oncogenic Ras triggers differentiation of pheochromocytoma cells and medullary thyroid carcinoma cells. Furthermore, we have previously demonstrated that in pituitary somatolactotropes, activated Ras promotes differentiation and is not sufficient to drive tumorigenesis. These findings demonstrate that lactotrope cells have the ability to evade the tumorigenic fate that is often associated with persistent activation of Ras/ERK signaling, and suggest that there may be differential expression of inhibitory signaling molecules or negative cell cycle regulators that act as a brake to prevent the tumorigenic effects of sustained Ras signaling. Here we aim to gain further insight into the mechanisms that allow GH4T2 cells to evade an oncogenic response to Ras. We show that Ral, but likely not menin, plays a key role in directing Ras-mediated differentiation of somatolactotropes, which may allow these cells to escape the tumorigenic fate that is often associated with activated Ras signaling. We also show that dominant negative Ras expression results in reduced GH4T2 cell proliferation and transformation, but does not influence differentiation. Taken together, the data presented here begin to shed light on the mechanisms by which pituitary somatolactotropes evade an oncogenic response to persistently activated Ras signaling and suggest that the architecture of the Ras signaling cascade in some endocrine cell types may be distinct from that of cells that respond to Ras in an oncogenic manner.
Key Terms: Pituitary, Lactotrope, Prolactinoma, Ras, Ral, Menin
Introduction
The Ras signaling cascade is classically linked to cell proliferation and survival in many types of malignancies, including lung, prostate, and colorectal cancers. Extracellular growth factors and mitogens bind to RTKs and G-protein coupled receptors (GPCRs), resulting in recruitment and activation of the GTPase Ras. Ras is a timed-effector that cycles between a GTP-loaded active state, and a GDP-bound inactive state. Activated Ras stimulates the protein kinase activity of Raf, resulting in the phosphorylation of MEK, and finally ERK, which ultimately catalyzes phosphorylation of a wide variety of downstream effector proteins including other kinases, phosphatases, and transcription factors. Activating mutations in proteins involved in the Ras/ERK cascade cause constitutive activation of the pathway, which can lead to uncontrolled proliferation and tumorigenesis (1, 2).
Although Ras is conventionally described to be pro-proliferative, distinct cell types have been shown to respond to activated Ras signaling in a cell-specific manner {Formatting Citation}. In contrast to its pro-tumorigenic role in some human epithelial cancers, oncogenic Ras triggers differentiation of PC12 pheochromocytoma cells and TT medullary thyroid carcinoma cells (4, 5). In PC12 cells, the differentiation outcome of activated ERK signaling is opposed by the downstream Ras effector RalGEF, a guanine nucleotide exchange factor that stimulates the GTPase Ral. Specifically, neurite outgrowth from PC12 cells is enhanced with sustained ERK signaling, but suppressed with Ral activation (6), demonstrating that Ras effectors can each have distinct inputs on the physiological events that result from activated Ras signaling.
We have previously demonstrated that in pituitary somatolactotropes, activated Ras promotes differentiation and is not sufficient to drive tumorigenesis (7). Other groups have also reported that activated Ras is insufficient for transformation of pituitary lactotropes (8–11). These findings suggest that lactotrope cells have the ability to evade the tumorigenic fate that is often associated with persistent activation of Ras/ERK signaling. However, the mechanism through which Ras/ERK signaling promotes differentiation and not proliferation or transformation in GH4T2 somatolactotropes and other endocrine cells has not been defined. Furthermore, we have shown that GH4T2 somatolactotrope cells are capable of turning off the Ras/ERK signaling pathway, even in the presence of exogenous stimulation (7).
The ability of pituitary lactotropes to evade tumorigenic responses to oncogenic V12Ras suggests that there may be differential expression of inhibitory signaling molecules or negative cell cycle regulators that act as a brake to prevent the tumorigenic effects of sustained Ras signaling. Menin, a tumor suppressor protein, is a key regulator of Ras signaling outcome in pancreatic beta cells, where V12Ras does not promote proliferation unless menin expression is lost (12). However, in non-endocrine pancreatic cells V12Ras drives proliferation, suggesting that Ras signaling outcomes are context-dependent, and that menin may function to block the proliferative response to Ras in endocrine cells. Furthermore, menin is mutated in multiple endocrine neoplasia type I (MEN1), a disease characterized by tumors of endocrine tissues including the anterior pituitary, pancreatic islet cells, and parathyroid glands (13). A better understanding of the role of menin in regulating proliferation of GH4T2 cells is needed to determine its involvement in directing Ras signaling outcome.
In the experiments presented here, we aim to gain further insight into the mechanisms that allow GH4T2 cells to evade an oncogenic response to Ras. First, we utilize V12Ras mutant constructs that selectively activate downstream effectors Raf, RalGEF, or PI3K, and find that in addition to Raf, Ral also contributes to the differentiation outcome of activated Ras signaling. We also use a doxycycline-inducible N17-HRas construct to further characterize Ras function in GH4T2 cell proliferation and differentiation. N17Ras is a dominant negative mutant that preferentially binds GDP over GTP and sequesters RasGEFs to render them inactive, resulting in inhibition of Ras activity (14). We show that dominant negative Ras expression results in reduced GH4T2 cell proliferation and transformation, but does not influence differentiation. Lastly, we knock down menin expression to determine its role in directing Ras/ERK signaling outcome, and find that loss of menin protein expression results in increased GH4T2 cell proliferation, but is not sufficient to increase transformation. Taken together, the data presented here begin to shed light on the mechanisms by which pituitary somatolactotropes evade an oncogenic response to persistently activated Ras signaling. Our findings suggest that Ral, but likely not menin, plays a role in directing Ras-mediated differentiation of somatolactotrope cells, which could allow these cells to escape the tumorigenic fate that is often associated with activated Ras signaling. Furthermore, these data suggest that the architecture of the Ras signaling cascade in some endocrine cells types may be distinct from that of cells that respond to Ras in an oncogenic manner.
Materials and Methods
Cell Lines and Culture Methods
All cells were grown in a humidified tissue culture incubator at 37°C in 5% CO2. The GH4T2 cell line was derived from GH4C1 rat somatolactotrope cells, as previously described (15, 16). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM, Gibco Life Technologies, Inc.) supplemented with 15% horse serum and 2.5% fetal bovine serum (FBS; Gibco Life Technologies, Inc.), in addition to 1X HEPES and NEAA. For serum-starved conditions, GH4T2 cells were cultured in DMEM supplemented with HEPES, and NEAA, and 0.05% FBS. For the doxycycline (dox)-inducible GH4T2 clones, 2.5% fetal bovine serum (FBS) was replaced with 2.5% tetracycline system-approved FBS (CLONTECH Laboratories Inc.). Fingerprinting methods for authentication of rat cell lines are not commercially available. However, somatolactotrope markers Pit-1, PRL, and GH are detectable by Western blot in GH4T2 and GH4C1 cell lines. 293T cells and BOSC cells are modified human embryonic kidney (HEK) 293 cells that were used to package lentivirus and retrovirus, respectively. Cells were maintained in DMEM supplemented with 10% FBS in addition to 1X HEPES and NEAA.
Doxycycline-inducible construct cloning
pCDNA3.1+ human H-Ras S17 3xHA (N-terminus) plasmid was purchased from the UMR cDNA Resource Center (University of Missouri-Rolla, Rolla, Missouri). The plasmid was cut with HindIII and XbaI to remove the Ras insert. pTRE-Tight vector was purchased from CLONTECH Laboratories Inc. and cut with HindIII, XbaI, and then calf intestinal alkaline phosphatase (Fisher Scientific). The insert and vector were ligated (3:1 ratio) and products were sequenced to confirm insertion.
Doxycycline-inducible GH4T2 clones
BS/IRES-M2 clone 13 was established by co-transfecting GH4T2 cells with BS/IRES-M2 plasmid (a kind gift from Dr. Stefania Lamartina, Istituto Di Richerche Di Biologia Molecolare, Rome, Italy) and empty pEGFP-C3 vector (Invitrogen; 10:1 ratio) by electroporation. After transfection, cells were plated in complete culture media. After 24 hours, media was changed to DMEM with 300 μg/mL G418 (Corning Cellgro) for selection. Clones were picked and maintained in DMEM with 100 μg/mL G418.
To create dox-inducible N17Ras clones, BS/IRES-M2 clone 13 was co-transfected with pTRE-H-Ras S17 3×HA plasmid and empty pQCXIP-puro plasmid (CLONTECH; 5:1 ratio). Media was changed to DMEM with 100 μg/mL G418 and 4μg/mL puromycin (Sigma) for selection. Clones were picked and maintained in DMEM with 100 μg/mL G418 and 2 μg/mL puromycin. All experiments were performed in N17Ras clones number 11 and number 37 because these clones required dox for 3HA-N17Ras expression. Here we show experimental results from clone number 11. Comparable results were obtained from clone number 37 (Supplemental Fig. 1).
Viral plasmids and transduction
pLKO shMenin and pLKO.1 shScramble constructs were purchased from Open Bio Systems. To package lentivirus, 293T cells were transfected with 4 μg lentiviral shRNA plasmid and 2 μg of packaging plasmids using Effectene, according to the manufacturer's protocol. pBABE-puro was a gift from Hartmut Land, Jay Morgenstern, and Bob Weinberg [Addgene plasmid #1764; (18)]. pBabe HcRed Hras G12V was a gift from William Hahn (Addgene plasmid #10678). pBabe puro H-Ras 12V (35S), pBabe puro H-Ras 12V (37G), and pBabe puro H-Ras 12V (40C) were gifts from Channing Der [Addgene plasmids #12588, #12589, #12590; (17)]. To package retrovirus, BOSC cells were transfected with 1.5 μg of the retroviral vector along with 1.8 μg each of pVsV-g and delta8.9 packaging plasmids using Turbofectin, according to the manufacturer's protocol. For both lentiviral and retroviral packaging, media with virus was collected after 48 hours, filtered, and snap frozen in single use aliquots for infection. For infection, GH4T2 cells were grown to ∼50% confluency and a mixture of 2 mL freshly thawed virus, 4 mL fresh media, and 6 μL polybrene (Sigma) was added directly to cells. Approximately 16 hours later, a second round of infection was completed. Cells were selected using 1 μg/mL puromycin when applicable for up to 7 days. Alternatively, cells were selected overnight with 4 μg/mL puromycin and then maintained in media containing 2 μg/mL puromycin. Selection was removed prior to plating of functional assays.
Protein Lysate Preparation, SDS-PAGE, and Western Blotting
GH4T2 cells were washed and harvested with extraction buffer (EB): 10 mM Tris, pH 7.4; 5 mM EDTA; 50 mM NaCl; 50 mM NaF; 0.1% BSA; 1% Triton X-100; 2 mM Na3VO4; 1 mM PMSF; and 1 mM DTT. Protein was quantitated using a Bio-Rad DC protein assay (Bio-Rad Laboratories). Twenty-five to 50 μg of protein sample were loaded into SDS-PAGE gels. Primary antibodies were diluted in 5% milk, or in 5% ECL milk for detection of phosphorylated proteins, and applied as follows: c-Myc (Sigma C3956; 1:1000, 1 hr), ERK1/2 p-Thr202/Tyr204 (Cell Signaling 4370; 1:1000, overnight), ERK (Cell Signaling 9107; 1:1000, overnight), GH (NIDDK AFP-411S; 1:1000, 1 hr), HA (Covance MMS-101P-200; 1:5000, overnight), Menin (Bethyl A300-105A; 1:1000, 1 hr), p21 (Sigma 029K1786; 1:7500, 1hr), p27 (Novus Biologicals NB100-1949; 1:200, 1hr), Pit-1 (BAbCo PRB-230C-500, 1:5000, 1 hr), PRL (NIDDK AFP-131581570; 1:5000, 1 hr), s6K p-Thr371 (Cell Signaling 9208; 1:1000, overnight), s6K (Cell Signaling 2708, 1:1000, overnight), α-Tubulin (Calbiochem CP06; 1:10,000, 1 hr). Secondary antibodies were diluted in TBS-T, or in 5% ECL milk for detection of phosphorylated protein, and were applied for 1 hour at the following concentrations: Goat Anti-Rabbit IgG-HRP (BioRad 170-6515; 1:5000, 1hr), Goat Anti-Mouse IgG-HRP (BioRad 170-6516; 1:5000, 1hr), Goat Anti-Monkey IGG-HRP (Cappel 55432; 1:5000, 1hr), ECL Donkey Anti-Rabbit IgG-HRP (GE Healthcare NA934V; 1:2000, 1hr). Blots were stripped using Chemicon mild reblot reagent (Chemicon, Inc.) prior to re-probing with additional primary antibodies. Film was imaged using a scanner and densitometric analysis was completed with ImageJ software (NIH).
Functional Assays
Proliferation, clonogenicity, and soft agar assays were completed as previously described (19). Briefly, for proliferation assays, cells were plated at a density of 9,000 cells/well in a 96-well plate. Cells were stained with 0.1% crystal violet (Sigma, St. Louis, MO) in 25% methanol, lysed with 10% acetic acid, and absorbance was read using a Synergy HT Microplate Reader (BioTek Instruments, Inc.). For a subset of experiments, cells were stained with trypan blue and manually counted, revealing comparable results (data not shown). Cells were plated at a density of 3,000 cells/well and 25,000 cells/well in a 6-well plate for clonogenicity and soft agar assays, respectively. For clonogenicity and soft agar assays, cells were stained with 0.1% crystal violet or Nitroblue reagent (1 mg/mL; Amresco), respectively, and colonies larger than 150 μm in diameter were counted using ImageJ software (NIH).
Luciferase Reporter Assays
GH4T2 cells were plated at a density of 45,000 cells/well in a 96-well plate and transiently co-transfected (Effectene) with a luciferase reporter construct containing the proximal 425 bp of the rPRL promoter (-425PRL-pA3-Luc) and TK-Renilla, with or without pBabe puro V12Ras, mutant V12Ras plasmids 35S/37G/40C, or pBabe puro empty vector control. Luciferase activity was measured 36-48 hours post-transfection using the Dual-Luciferase Reporter system (Promega). All results were normalized to the Renilla luciferase signal as an internal control.
Statistical Analysis
Statistical analysis was completed using unpaired t tests. A p value less than 0.05 was considered statistically significant.
Results
Different Ras effectors have been described to have opposing effects on cell proliferation and differentiation (6), and as such we first wanted to characterize the physiological effects of the Ras effectors Raf, Ral, and PI3K in GH4T2 cells. We utilized three expression plasmids each with an additional point mutation in V12Ras, resulting in selective downstream activation of only one Ras effector: the 35S mutant selectively binds Raf but binding to PI3K and RalGEF is reduced; the 37G mutant selectively binds RalGEF but binding to Raf and PI3K is impaired; the 40C mutant selectively binds PI3K but binding to Raf and RalGEF is reduced (20, 21). All mutant expression plasmids were in a pBabe retroviral backbone with puromycin selection (Addgene). Plasmids were packaged in BOSC cells, and virus-containing media was harvested and used to infect GH4T2 cells. An empty pBabe puro vector was used as an experimental control, and pBabe puro V12Ras without effector domain mutations was also included as a control.
To confirm that the effector domain mutations resulted in activation of each effector, cells were maintained in complete media with 2 μg/mL puromycin and protein lysates were harvested for Western blot. Phospho-ERK and p-s6K, a downstream effector of PI3K, are expressed in vector control cells because cells were not serum-starved (Fig. 1A). ERK was moderately increased with V12Ras expression, but p-s6K activity remained comparable to control (Fig. 1A). ERK was strongly stimulated with Raf activation, and p-s6K activity was modestly reduced compared to control (V12Ras 35S; Fig. 1A). ERK was also stimulated with RalGEF activation, but s6K activity was not changed compared to control (V12Ras 37G; Fig. 1A). ERK activity was reduced and s6K was stimulated with PI3K activation (V12Ras 40C; Fig. 1A). Taken together, these data confirm that the Raf- and PI3K-activating V12Ras mutants successfully activate their respective effectors, and that activation of V12Ras selectively stimulates ERK over PI3K/s6K signaling in GH4T2 cells. These data also show that activation of ERK signaling results in reduced PI3K activity, whereas activation of PI3K signaling results in reduced ERK signaling in GH4T2 cells.
Figure 1. Ral is responsible for one-third of Ras-mediated PRL promoter activation.
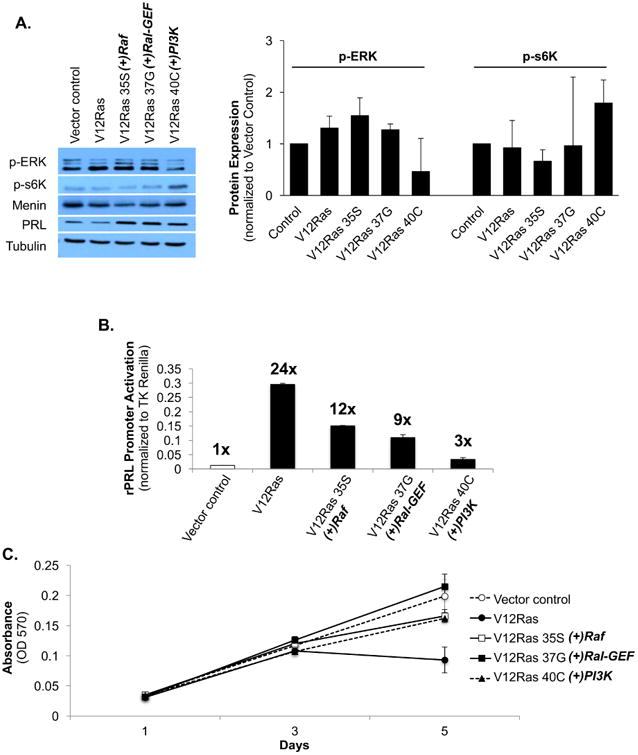
Analysis of protein expression, cell proliferation, and PRL promoter activity in GH4T2 cells following transfection or transduction with V12Ras, V12Ras effector domain mutants, or an empty vector control. A: Western blot analysis of GH4T2 cells. Cells were maintained in complete media with 2 μg/mL puromycin for selection. Whole-cell extracts were separated by SDS-PAGE and probed with the antibodies listed. For densitometric analysis protein expression was quantified, normalized to tubulin, and then normalized to vector control for each replicate. [n=2 independent experiments (right), a representative experiment is shown (left)] B: GH4T2 cells were transiently co-transfected with a luciferase reporter containing the proximal 425 bp of the rPRL promoter (-425PRL-pA3-Luc), TK-Renilla as an internal control, and the expression plasmids listed. Luciferase activity was measured 36 hours post-transfection and all results were normalized to TK-Renilla. (n=3 independent experiments) C: Analysis of GH4T2 cell proliferation with V12Ras and V12Ras effector domain mutant expression. Cells were maintained in complete media with 2 μg/mL puromycin and fresh media was added on days 2 and 4. Crystal violet staining was completed as described in methods. (n=3 independent experiments; a representative experiment is shown) Data are shown as means ±S.E.M.
To characterize the effects of Ras effector activation on GH4T2 cell differentiation, GH4T2 cells were transiently transfected with V12Ras constructs along with a PRL-Luciferase reporter construct and a TK-Renilla-Luciferase construct as an internal control. Expression of V12Ras strongly activated the PRL promoter, with a 24-fold increase in promoter activity compared to vector control (Fig. 1B). This is consistent with our previous findings that V12Ras promotes differentiation of GH4T2 cells toward a PRL-secreting, lactotrope phenotype, and also agrees with earlier data from our lab describing Ras-mediated PRL promoter activation via Ets1 and Pit-1 (7, 22, 23). Activation of Raf accounted for one-half of V12Ras-mediated PRL promoter activity, with a twelve-fold increase in activity compared to vector control (V12Ras 35S; Fig. 1B). Activation of RalGEF contributed to approximately one-third of V12Ras-mediated PRL promoter activity, with a nine-fold increase in activity compared to control (V12Ras 37G; Fig. 1B). PI3K activity only minimally contributed to V12Ras-mediated PRL promoter activation, with a three-fold increase in activity compared to control (V12Ras 40C; Fig. 1B). Together these findings demonstrate that Raf and Ral predominantly mediate Ras-driven differentiation of GH4T2 cells.
To determine the effects of Ras effector activation on GH4T2 cell proliferation, cells were maintained in complete medium with 2 μg/mL puromycin and fixed every other day for analysis. Cells expressing V12Ras without additional effector domain mutations displayed reduced cell proliferation by day 5 compared to vector control (Fig. 1C), similar to our previous report that stable expression of V12Ras without the use of an inducible system reduces cell proliferation (7). Expression of the effector domain mutants (35S, 37G, and 40C) did not significantly alter cell proliferation (Fig. 1C).
Following transduction of GH4T2 cells with V12Ras and V12Ras effector mutants, morphological differences were observed. GH4T2 cells are small, round cells that grow in clusters, as seen in cells transduced with an empty vector control (Fig. 2). Upon transduction with V12Ras, some cells were observed to produce neurite-like outgrowths (black arrows) and others became larger and appeared swollen (white arrows; Fig. 2). In cells transduced with the Raf-specific mutant of V12Ras, many cells developed neurite-like outgrowths (black arrows; Fig. 2). In cells transduced with the RalGEF-specific mutant of V12Ras, many cells displayed increased size and appeared swollen (white arrows; Fig. 2). However, cells transduced with the PI3K-specific V12Ras mutant appeared similar to control, with no neurite-like outgrowths or swelling of cells (Fig. 2).
Figure 2. Ras effectors RalGEF and ERK, but not PI3K, induce morphological changes in GH4T2 cells.
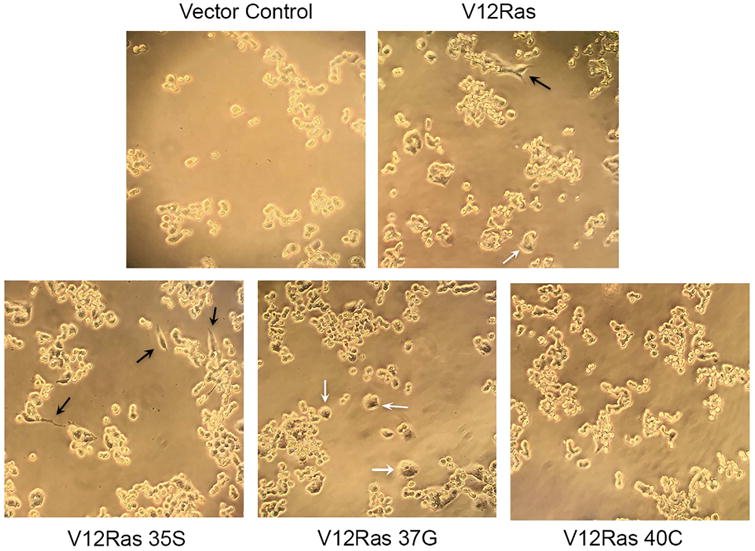
GH4T2 cell morphology with V12Ras effector mutant expression. GH4T2 cells were transduced with retrovirus encoding V12Ras, V12Ras effector mutants, or an empty vector control. Cells were selected with 2 μg/mL puromycin and images were captured 48-72 hours post-transduction. Black arrows denote neurite-like outgrowths; white arrows denote swollen cells. (n=3 independent experiments; representative images are shown)
Next, to further characterize Ras function in GH4T2 cell proliferation and differentiation, we utilized a doxycycline-inducible dominant negative N17Ras construct. GH4T2 clonal cell lines (number 11 and number 37) were established that stably expressed HA-tagged, dominant negative N17Ras under the control of a tightly regulated doxycycline-inducible promoter, as described previously (7). Comparable experimental results were obtained from both clonal cell lines and here we show data from clone number 11; data from clone 37 are shown in Supplemental Fig. 1. GH4T2 clone 11 cells were maintained in media with complete serum and treated with concentrations of doxycycline ranging from 0 to 2 μg/mL. Doxycycline addition resulted in stable, dose-regulated expression of 3HA-N17Ras within 24 hours; 3HA-N17Ras was expressed robustly at doxycycline concentrations of 1.0 μg/mL and 2.0 μg/mL, and was expressed at a low level at 0.2 and 0.4 μg/mL doxycycline (Fig. 3A).
Figure 3. Stable expression of N17Ras reduces GH4T2 cell proliferation and colony formation, and does not change the PRL:GH expression ratio.
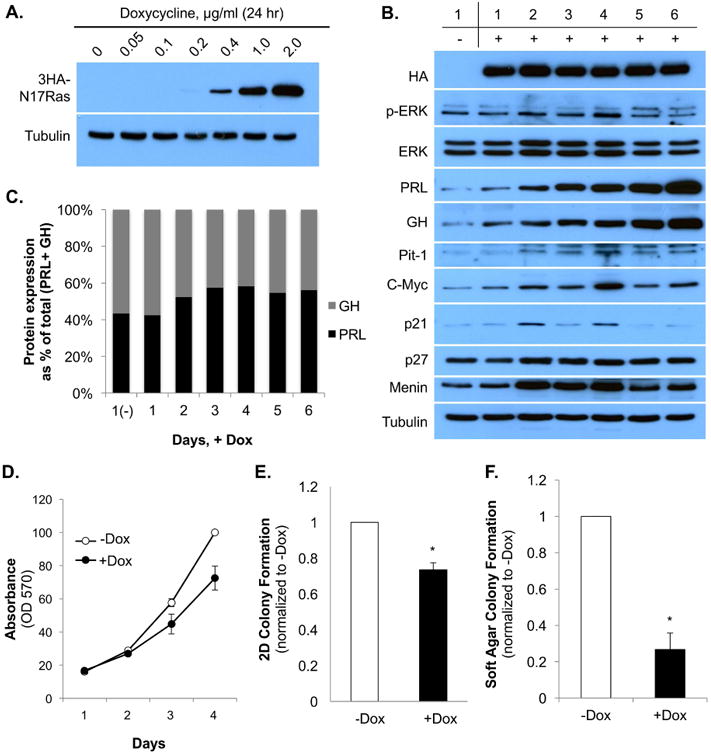
A: Western blot analysis of dox-induced 3HA-N17Ras expression. Clone 11 cells were treated ±dox with varying concentrations for 24 hours. Whole-cell extracts were separated by SDS-PAGE and probed with anti-HA and anti-tubulin antibodies. B: Western blot analysis of GH4T2 clone 11. Clone 11 cells were maintained in complete media with or without 2 μg/mL doxycycline, added on days 1, 3, and 5, and cells were harvested daily for 6 days. Day 1, (-)Dox cells were not treated with dox. Whole-cell extracts were separated by SDS-PAGE and probed with the antibodies listed. C: Quantification of PRL and GH expression ±dox. PRL (black bars) and GH (gray bars) protein expression from panel B was quantified by densitometric analysis, normalized to PRL expression on day 1, (-)Dox, and graphed as a percentage of total (PRL + GH) expression for each day. D-F: GH4T2 clone 11 cells were maintained in complete media with or without 2 μg/mL dox, and fresh media was added every other day. D: Analysis of GH4T2 clone 11 cell proliferation ±dox. Crystal violet staining was completed as described in methods. (n=3 independent experiments; a representative experiment is shown). E: Analysis of 2D GH4T2 clone 11 cell colony formation. Crystal violet staining was completed as described in methods (n=3 independent experiments). F: Analysis of GH4T2 clone 11 colony formation in soft agar. On day 17, colonies were stained overnight with Nitroblue, and were then photographed and counted with ImageJ software (n=3 independent experiments). Data are shown as means ±S.E.M. *, p <.05.
To determine the effect of dominant negative N17Ras expression on p-ERK activity in GH4T2 cells, 2 μg/mL doxycycline was added to cycling (non-starved) cells on days 1, 3, and 5, and cells were harvested daily for six days. A Western blot for HA showed stable, robust expression of 3HA-N17Ras throughout the six days of doxycycline treatment (Fig. 3B). Expression of total ERK protein was not significantly changed with doxycycline treatment (Fig. 3B). Expression of PRL was increased by day 1 of doxycycline addition, compared to day 1, (-) dox, and continued to increase for six days. GH expression showed a similar pattern with a gradual increase in protein expression throughout six days of doxycycline treatment (Fig. 3B). The PRL:GH expression ratio was not significantly changed with doxycycline addition, demonstrating that expression of dominant negative N17Ras does not promote GH4T2 cell differentiation from a bi-hormonal somatolactotrope cell towards a lactotrope or somatotrope phenotype (Fig. 3C). Pit-1 and c-Myc both showed increased protein expression beginning on day 2, with peak expression by day 4, essentially following the pattern of p-ERK activity (Fig. 3B). The cyclin-dependent kinase inhibitor p21 showed fluctuating expression, and expression of the cyclin-dependent kinase inhibitor p27 was not significantly changed with doxycycline addition (Fig. 3B). Menin displayed increased expression on days 2-4, compared to day 1, (-) dox, suggesting that expression of dominant negative Ras may have an effect on GH4T2 cell proliferation (Fig. 3B).
Therefore, we next asked if expression of dominant negative N17Ras had an effect on GH4T2 proliferation and transformation. GH4T2 clone 11 cells expressing doxycycline-inducible 3HA-N17Ras were maintained in complete media and treated with or without 2 μg/mL doxycycline on days 1 and 3, and were fixed daily for analysis. Crystal violet staining showed reduced GH4T2 cell proliferation with N17Ras expression over a 4-day period (Fig. 3D). To determine the role of N17Ras in GH4T2 transformation, clone 11 cells were maintained in complete media with or without 2 μg/mL doxycycline for colony formation assays in two-dimensions (2D) on poly-L-lysine coated tissue culture plates or in three dimensions (3D) in soft agar. Cell colonies were stained with crystal violet for 2D assays or with Nitroblue for 3D assays, and were then photographed and counted using ImageJ software. GH4T2 cells treated with doxycycline to induce expression of dominant negative N17Ras displayed a 27% reduction in 2D colony growth and a 74% reduction in 3D anchorage-independent colony growth (Fig. 3, E and F). Colony size was not noticeably different in doxycycline-treated cells (data not shown). In summary, these data indicate that dominant negative inhibition of Ras results in reduced GH4T2 cell proliferation and transformation, but does not influence the PRL:GH expression ratio.
Since menin expression was increased in GH4T2 cells with dominant negative N17Ras, and it also acts as a tumor suppressor in endocrine tissues including the pituitary gland, we next aimed to knockdown menin to further characterize its role in GH4T2 cell proliferation and transformation. A lentiviral pLKO.1 shMenin expression plasmid was packaged in 293T cells, and virus-containing media was harvested and used to infect GH4T2 cells. A lentiviral pLKO.1 vector containing a scrambled shRNA (shScr) sequence was also packaged into 293T and used as an experimental control. GH4T2 cells expressing shScr or shMenin were maintained in complete serum and protein lysate was harvested for Western blot analysis. Although menin protein expression was low in GH4T2 cells transduced with shScr control, menin was nearly undetectable in cells transduced with shMenin lentivirus, demonstrating that menin was successfully knocked down (Fig. 4A). ERK activity was modestly increased with menin knockdown, and total ERK protein expression was not changed (Fig. 4A). PRL expression was also modestly increased with menin knockdown, but GH expression did not change (Fig. 4A).
Figure 4. Menin knockdown increases GH4T2 cell proliferation but is not sufficient to increase colony formation.
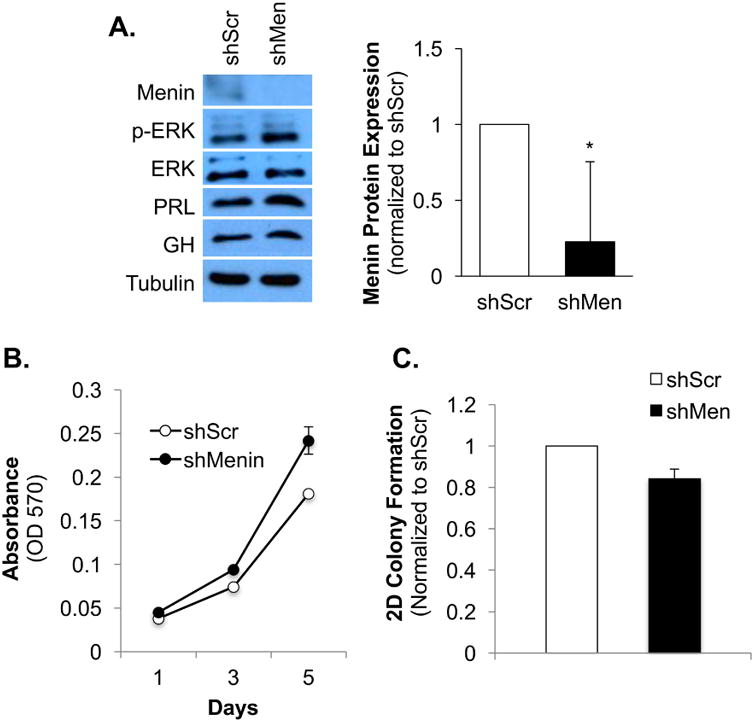
Analysis of GH4T2 protein expression, cell proliferation, and colony formation with menin knockdown. A: Western blot analysis of GH4T2 cells. GH4T2 cells were transduced with lentivirus encoding shMenin or shScr control and were maintained in complete media with 2 μg/mL puromycin for selection. Whole-cell extracts were separated by SDS-PAGE and probed with the antibodies listed. For densitometric analysis protein expression was quantified, normalized to tubulin, and then normalized to shScr control for each replicate. (n=3 independent experiments; a representative experiment is shown) B: Analysis of GH4T2 cell proliferation with menin knockdown. GH4T2 cells were maintained in complete media with 2 μg/mL puromycin and fresh media was added on days 2 and 4. Crystal violet staining was completed as described in methods. (n=3 independent experiments; a representative experiment is shown) C: Analysis of two-dimensional (2D) GH4T2 cell colony formation with menin knockdown. Cells were maintained in complete media with 2 μg/mL puromycin, fresh media was added every other day, and cells were fixed with 4% PFA after 11 days. Crystal violet staining was completed as described in methods. Data are shown as means ±S.E.M. (n=3 independent experiments).
We next sought to determine how menin knockdown affects GH4T2 cell proliferation and transformation. GH4T2 cells expressing shScr or shMenin were maintained in complete media and functional assays for cell proliferation and colony formation were completed. Menin knockdown promoted an increase in GH4T2 cell proliferation over a five-day period compared to shScr control (Fig. 4B). However, knockdown of menin was not sufficient to increase colony formation (Fig. 4C). Comparable trends for proliferation and colony formation were observed with cells maintained in serum-starved conditions (0.05% FBS; data not shown). We also characterized menin expression with activation of each Ras effector in GH4T2 cells. Expression of V12Ras resulted in a slight reduction in menin protein expression (Fig. 1A). Menin was strongly inhibited by Raf activation (V12Ras 35S; Fig. 1A), and modestly inhibited with Ral and PI3K activation (V12Ras 37G and V12Ras 40C, respectively; Fig. 1A). Thus, menin expression is not upregulated by Ras, or with selective activation of any of the Ras effectors. Taken together, these data suggest that Ral, but likely not menin, plays a key role in promoting Ras-mediated differentiation in somatolactotropes, which might allow these cells to escape the tumorigenic fate that is often associated with activated Ras signaling.
Discussion
We and others have shown that activated Ras signaling is insufficient to drive lactotrope tumorigenesis, but the precise mechanism by which lactotrope cells evade an oncogenic response to V12Ras has yet to be elucidated. We have previously shown that persistent activation of ERK with V12Ras does not increase GH4T2 cell proliferation and reduces tumorigenic characteristics (7), and here we show that inhibition of Ras activity also reduces GH4T2 proliferation and transformation (Fig. 3, D-F). Because Ras impinges upon many downstream growth factor signaling pathways, it was expected that expression of a dominant negative Ras construct would result in reduced cell growth and transformation. Our previous report shows that inducible expression of V12Ras is lost despite continual addition of dox, and that cells do not tolerate prolonged activation of ERK signaling, despite continued stimulation with either doxycycline to induce V12Ras expression, or with addition of exogenous EGF (7). However, here we show that N17Ras expression is maintained for the duration of the experiment (six days), and therefore, it is not simply that GH4T2 cells do not tolerate prolonged expression of a transgene (HA protein; Fig. 3B). Instead, GH4T2 cells appear to have a mechanism in place to prevent prolonged activation of ERK signaling. MKP-1 and MKP-2 are dual-specificity MAPK phosphatases that have been implicated in the regulation of ERK signaling in the anterior pituitary, but we have previously demonstrated that these phosphatases do not regulate the loss of p-ERK expression that is observed with prolonged Ras signaling in GH4T2 cells (7). Oncogene-induced senescence is a phenomenon frequently observed with prolonged Ras activity across many different cell types (24). However, GH4T2 cells do not stop proliferating with doxycycline-induced, low-level V12Ras expression, which would be indicative of senescence (7).
Experiments with 3HA-N17Ras-expressing clones were completed in complete media (2.5% FBS and 15% horse serum), and therefore, ERK shows baseline activation on day 1, (-) dox from stimulation by growth factors in the cell culture medium (Fig. 3B). Expression of the 3HA-N17Ras transgene is robust by day 1 of doxycycline addition and is maintained throughout the six days of analysis (HA, Fig. 3B), but p-ERK remains activated at a level comparable to day 1, (-) dox, until days 5 and 6, when ERK activity is modestly reduced (p-ERK, Fig. 3B). Although dominant negative inhibition of Ras signaling reduces GH4T2 cell proliferation and transformation (Fig. 3, D-F), sustained ERK activity with Ras inhibition suggests that ERK might be activated by a Ras-independent mechanism in these cells. ERK may be activated by other small GTPases such as Rap1 (25); further studies in GH4T2 cells are required to address this possibility.
We were surprised to find increased expression of menin with Ras inhibition (Fig. 3B), since we hypothesized that inhibition of a growth factor would either reduce or have no effect on the expression of a tumor suppressor protein. Furthermore, we show that loss of menin expression in GH4T2 cells results in increased cell proliferation, but is not sufficient to increase transformation (Fig. 4). In mouse models, Men1+/- mice develop pituitary adenomas, but very slowly, no earlier than 13 months of age (26, 27). Patients with MEN1 inherit a germline mutation in one copy of menin, but must also acquire a somatic mutation for pituitary tumorigenesis to occur (13), further confirming our findings that loss of menin expression is not sufficient for lactotrope tumorigenesis. Here we show that menin knockdown results in increased ERK activation (Fig. 4A). Previous studies in pancreatic β-cells have shown that menin functions to dictate Ras signaling outcome, with loss of menin resulting in activated ERK signaling and pancreatic tumorigenesis (12). Activin, a member of the TGFα family of proteins, has been shown to signal to menin through SMAD, resulting in inhibition of PRL expression (28). Indeed, we find that PRL expression is increased with menin knockdown (Fig. 4A), demonstrating that menin plays a role in suppressing PRL production to maintain lactotrope homeostasis.
An interesting observation from these studies was the altered morphology of GH4T2 cells with expression of the Ras effector mutants. We show that Raf activation stimulates outgrowth of neurite-like processes from GH4T2 cells, and this was also observed with expression of V12Ras (Fig. 2). Studies in PC12 pheochromocytoma cells report similar findings of neurite-like outgrowths in response to sustained ERK activation. GH4T2 and PC12 cell lines are derived from pituitary somatolactotropes and chromaffin cells of the adrenal medulla, respectively, and are both classified as neuroendocrine cells. Neurite outgrowth is a marker of neuronal differentiation (29), thus, these data suggest that Raf/ERK activation promotes neuronal differentiation of GH4T2 cells. With expression of the RalGEF-specific V12Ras mutant, cell swelling was observed, and this was also seen with V12Ras expression. Cell swelling plays an important role in hormone secretion from endocrine cells (30). As shown in Fig. 1, RalGEF is responsible for approximately one-third of Ras-mediated PRL promoter activation. We speculate that Ral activity contributes to Ras-mediated somatolactotrope differentiation not only by increasing PRL production, but also by upregulating cellular PRL secretion.
One possibility that arises from the data presented here is that Ras-driven Ral activation may be required for GH4T2 cells to evade an oncogenic response to constitutively activated ERK signaling. In PC12 cells, Ral activity has been shown to oppose ERK-mediated differentiation (6). However, here we show that Ral stimulates the PRL promoter and promotes cell swelling, which suggests that Ral is contributing to, rather than opposing, differentiation of somatolactotropes toward a lactotrope cell phenotype. RalA promotes cell motility, invasion, and is required for tumorigenesis in Ras-driven human cancer cell lines including pancreatic and bladder cancer (31, 32). However, RalB, a paralog of RalA, inhibits RalGEF-induced transformation of cells, likely by competing for RalGEF activation (31). Interestingly, in contrast to its role in epithelial cancer cells, RalA is required for exocytosis of secretory granules in neuroendocrine cells including pancreatic β-cells, where RalA contributes to insulin release (33, 34). This suggests that RalA plays a distinct role in pituitary and other neuroendocrine cells, where instead of functioning to drive tumorigenesis, it mediates hormone secretion. Further investigation into activation of RalA vs. RalB with V12Ras in GH4T2 cells would begin to address this possibility. Furthermore, inhibition of RalA or RalB following V12Ras activation may reveal a role for Ral in blocking Ras/ERK-driven proliferation and tumorigenesis of somatolactotrope cells.
Neuroendocrine cell types, including pituitary lactotropes and pancreatic β-cells, do not respond to activated Ras in a oncogenic manner (3). Here we show that Ral, but likely not menin, may play a role in directing Ras-mediated differentiation of somatolactotrope cells, which could allow these cells to escape the tumorigenic fate that is often associated with activated Ras signaling. Taken together, the data presented here begin to shed light on the mechanisms by which pituitary somatolactotropes evade an oncogenic response to persistently activated Ras signaling. Furthermore, these data suggest that the architecture of the Ras signaling cascade in some endocrine cell types may be distinct from that of cells that respond to Ras in an oncogenic manner. However, a better understanding of the mechanisms that neuroendocrine cells use to block tumorigenesis is required. If identified, the protective mechanisms that are present in neuroendocrine cells, but absent in pancreatic, colon, and lung cancer cells, may lead to the development of a novel strategy to counter the aberrant signaling events that drive malignant tumorigenesis.
Supplementary Material
A: Western blot analysis of GH4T2 clone 37. Clone 37 cells were maintained in complete media with or without 2 μg/mL doxycycline, added on days 1, 3, and 5, and cells were harvested daily for 6 days. Day 1, (-)Dox cells were not treated with dox. Whole-cell extracts were separated by SDS-PAGE and probed with the antibodies listed. B: Analysis of GH4T2 clone 37 cell proliferation ±dox. Crystal violet staining was completed as described in methods. (n=3 independent experiments; a representative experiment is shown). C: Analysis of 2D GH4T2 clone 37 cell colony formation. Crystal violet staining was completed as described in methods (n=3 independent experiments). Data are shown as means ±S.E.M. *, p <.05.
Ras-driven differentiation of GH4T2 cells is predominantly mediated by Raf and Ral.
Ras and its effectors do not upregulate menin expression in somatolactotropes.
Ral may block Ras-driven proliferation and transformation of somatolactotropes.
Acknowledgments
This research was supported by NIH R01 DK46868.
Grants supporting the writing of this paper: NIH R01 DK46868
Footnotes
Disclosure statement: The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–90. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 2.Cakir M, Grossman AB. Targeting MAPK (Ras/ERK) and PI3K/Akt pathways in pituitary tumorigenesis. Expert Opin Ther Targets. 2009;13:1121–34. doi: 10.1517/14728220903170675. [DOI] [PubMed] [Google Scholar]
- 3.Roof AK, Gutierrez-hartmann A. Consider the context : Ras / ERK and PI3K / AKT / mTOR signaling outcomes are pituitary cell type-specific. Mol Cell Endocrinol. 2017 doi: 10.1016/j.mce.2017.04.019. [DOI] [PubMed] [Google Scholar]
- 4.Nakagawa T, Mabry M, de Bustros A, Ihle JN, Nelkin BD, Baylin SB. Introduction of v-Ha-ras oncogene induces differentiation of cultured human medullary thyroid carcinoma cells. Proc Natl Acad Sci U S A. 1987;84:5923–7. doi: 10.1073/pnas.84.16.5923. Online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bar-Sagi D, Feramisco JR. Microinjection of the ras oncogene protein into PC12 cells induces morphological differentiation. [Online] Cell. 1985;42:841–8. doi: 10.1016/0092-8674(85)90280-6. [DOI] [PubMed] [Google Scholar]
- 6.Goi T, Rusanescu G, Urano T, Feig LA. Ral-specific guanine nucleotide exchange factor activity opposes other Ras effectors in PC12 cells by inhibiting neurite outgrowth. Mol Cell Biol. 1999;19:1731–41. doi: 10.1128/mcb.19.3.1731. Online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Booth A, Trudeau T, Gomez C, Lucia MS, Gutierrez-Hartmann A. Persistent ERK/MAPK activation promotes lactotrope differentiation and diminishes tumorigenic phenotype. Mol Endocrinol. 2014;28:1999–2011. doi: 10.1210/me.2014-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McAndrew J, Paterson AJ, Asa SL, McCarthy KJ, Kudlow JE. Targeting of transforming growth factor-alpha expression to pituitary lactotrophs in transgenic mice results in selective lactotroph proliferation and adenomas. Endocrinology. 1995;136:4479–88. doi: 10.1210/endo.136.10.7664668. [DOI] [PubMed] [Google Scholar]
- 9.Borrelli E, Sawchenko PE, Evans RM. Pituitary hyperplasia induced by ectopic expression of nerve growth factor. Proc Natl Acad Sci U S A. 1992;89:2764–8. doi: 10.1073/pnas.89.7.2764. Online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roh M, Paterson AJ, Asa SL, Chin E, Kudlow JE. Stage-sensitive blockade of pituitary somatomammotrope development by targeted expression of a dominant negative epidermal growth factor receptor in transgenic mice. Mol Endocrinol. 2001;15:600–13. doi: 10.1210/mend.15.4.0625. [DOI] [PubMed] [Google Scholar]
- 11.Ezzat S, Zheng L, Zhu X, Wu GE, Asa SL. Targeted expression of a human pituitary tumor – derived isoform of FGF receptor-4 recapitulates pituitary tumorigenesis. J Clin Invest. 2002;109:15–16. doi: 10.1172/JCI14036. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Chamberlain CE, Scheel DW, McGlynn K, Kim H, Miyatsuka T, Wang J, Nguyen V, Zhao S, Mavropoulos A, Abraham AG, O'Neill E, Ku GM, Cobb MH, Martin GR, German MS. Menin determines K-RAS proliferative outputs in endocrine cells. J Clin Invest. 2014;124:4093–101. doi: 10.1172/JCI69004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4) Mol Cell Endocrinol. 2014;386:2–15. doi: 10.1016/j.mce.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mosteller RD, Han J, Broek D. Identification of residues of the H-ras protein critical for functional interaction with guanine nucleotide exchange factors. Mol Cell Biol. 1994;14:1104–1112. doi: 10.1128/mcb.14.2.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conrad KE, Gutierrez-Hartmann A. The ras and protein kinase A pathways are mutually antagonistic in regulating rat prolactin promoter activity. Oncogene. 1992;7:1279–86. Online. [PubMed] [Google Scholar]
- 16.Tashjian AH, Yasumura Y, Levine L, Sato GH, Parker ML. Establishment of Clonal Strains of Rat Pituitary Tumor Cells That Secrete Growth Hormone. Endocrinology. 1968;82:342–352. doi: 10.1210/endo-82-2-342. [DOI] [PubMed] [Google Scholar]
- 17.McFall A, Ulkü A, Lambert QT, Kusa A, Rogers-Graham K, Der CJ. Oncogenic Ras blocks anoikis by activation of a novel effector pathway independent of phosphatidylinositol 3-kinase. Mol Cell Biol. 2001;21:5488–99. doi: 10.1128/MCB.21.16.5488-5499.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morgenstern JP, Land H, Cancer I, Fields I, Wca L. Advanced mammalian gene transfer : high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Booth AK, Gutierrez-Hartmann A. Signaling pathways regulating pituitary lactotrope homeostasis and tumorigenesis. Advances in experimental medicine and biology. :37–59. doi: 10.1007/978-3-319-12114-7_2. [DOI] [PubMed] [Google Scholar]
- 20.Khosravi-Far R, White Ma, Westwick JK, Solski Pa, Chrzanowska-Wodnicka M, Van Aelst L, Wigler MH, Der CJ. Oncogenic Ras activation of Raf/mitogen-activated protein kinase-independent pathways is sufficient to cause tumorigenic transformation. Mol Cell Biol. 1996;16:3923–3933. doi: 10.1128/mcb.16.7.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White MA, Nicolette C, Minden A, Polverino A, Vanaelst L, Karin M, Wigler MH. Multiple Ras Functions Can Contribute To Mammalian-Cell Transformation. Cell. 1995;80:533–541. doi: 10.1016/0092-8674(95)90507-3. [DOI] [PubMed] [Google Scholar]
- 22.Bradford AP, Conrad KE, Wasylyk C, Wasylyk B, Gutierrez-Hartmann A. Functional interaction of c-Ets-1 and GHF-1/Pit-1 mediates Ras activation of pituitary-specific gene expression: mapping of the essential c-Ets-1 domain. Mol Cell Biol. 1995;15:2849–2857. doi: 10.1128/mcb.15.5.2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bradford AP, Wasylyk C, Wasylyk B, Gutierrez-Hartmann A. Interaction of Ets-1 and the POU-homeodomain protein GHF-1/Pit-1 reconstitutes pituitary-specific gene expression. Mol Cell Biol. 1997;17:1065–1074. doi: 10.1128/mcb.17.3.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9:493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 25.Romano D, Magalon K, Ciampini A, Talet C, Enjalbert A, Gerard C. Differential involvement of the Ras and Rap1 small GTPases in vasoactive intestinal and pituitary adenylyl cyclase activating polypeptides control of the prolactin gene. J Biol Chem. 2003;278:51386–94. doi: 10.1074/jbc.M308372200. [DOI] [PubMed] [Google Scholar]
- 26.Crabtree JS, Scacheri PC, Ward JM, Garrett-Beal L, Emmert-Buck MR, Edgemon KA, Lorang D, Libutti SK, Chandrasekharappa SC, Marx SJ, Spiegel AM, Collins FS. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci U S A. 2001;98:1118–23. doi: 10.1073/pnas.98.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bertolino P, Tong WM, Galendo D, Wang ZQ, Zhang CX. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol Endocrinol. 2003;17:1880–92. doi: 10.1210/me.2003-0154. [DOI] [PubMed] [Google Scholar]
- 28.Lacerte A, Lee EH, Reynaud R, Canaff L, De Guise C, Devost D, Ali S, Hendy GN, Lebrun JJ. Activin inhibits pituitary prolactin expression and cell growth through Smads, Pit-1 and menin. Mol Endocrinol. 2004;18:1558–69. doi: 10.1210/me.2003-0470. [DOI] [PubMed] [Google Scholar]
- 29.Nikolic M, Dudek H, Kwon YT, Ramos YFM, Tsai LH. The cdk5 /p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. doi: 10.1101/gad.10.7.816. [DOI] [PubMed] [Google Scholar]
- 30.Štrbák V, Greer MA. Regulation of hormone secretion by acute cell volume changes: Ca2+-independent hormone secretion. Cell Physiol Biochem. 2000;10:393–402. doi: 10.1159/000016372. [DOI] [PubMed] [Google Scholar]
- 31.Lim KH, Baines AT, Fiordalisi JJ, Shipitsin M, Feig LA, Cox AD, Der CJ, Counter CM. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell. 2005;7:533–45. doi: 10.1016/j.ccr.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 32.Gildea JJ, Harding MA, Seraj MJ, Gulding KM, Theodorescu D. The Role of Ral A in Epidermal Growth Factor Receptor-regulated Cell Motility. Cancer Res. 2002;62:982–985. [PubMed] [Google Scholar]
- 33.Ljubicic S, Bezzi P, Vitale N, Regazzi R. The GTPase RalA regulates different steps of the secretory process in pancreatic beta-cells. PLoS One. 2009;4 doi: 10.1371/journal.pone.0007770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vitale N, Mawet J, Camonis J, Regazzi R, Bader MF, Chasserot-Golaz S. The small GTPase RalA controls exocytosis of large dense core secretory granules by interacting with ARF6-dependent phospholipase D1. J Biol Chem. 2005;280:29921–29928. doi: 10.1074/jbc.M413748200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A: Western blot analysis of GH4T2 clone 37. Clone 37 cells were maintained in complete media with or without 2 μg/mL doxycycline, added on days 1, 3, and 5, and cells were harvested daily for 6 days. Day 1, (-)Dox cells were not treated with dox. Whole-cell extracts were separated by SDS-PAGE and probed with the antibodies listed. B: Analysis of GH4T2 clone 37 cell proliferation ±dox. Crystal violet staining was completed as described in methods. (n=3 independent experiments; a representative experiment is shown). C: Analysis of 2D GH4T2 clone 37 cell colony formation. Crystal violet staining was completed as described in methods (n=3 independent experiments). Data are shown as means ±S.E.M. *, p <.05.