

Abstract
Epigenetic mechanisms (i.e., histone post-translational modification and DNA methylation) play a role in regulation of gene expression. The pedunculopontine nucleus (PPN), part of the reticular activating system, manifests intrinsic gamma oscillations generated by voltage-dependent, high threshold N- and P/Q-type Ca2+ channels. We studied whether PPN intrinsic gamma oscillations are affected by inhibition of histone deacetylation. We showed that, a) acute in vitro exposure to the histone deacetylation Class I and II inhibitor trichostatin A (TSA, 1 μM) eliminated oscillations in the gamma range, but not lower frequencies, b) pre-incubation with TSA (1 μM, 90–120 min) also decreased gamma oscillations, c) Ca2+ currents (ICa) were reduced by TSA, especially on cells with P/Q-type channels, d) a HDAC Class I inhibitor MS275 (500 nM), and a Class IIb inhibitor Tubastatin A (150–500 nM), failed to affect gamma oscillations, e) MC1568, a HDAC Class IIa inhibitor (1 μM), blocked gamma oscillations, and f) the effects of both TSA and MC1568 were blunted by blockade of CaMKII with KN-93 (1 μM). These results suggest a cell type specific effect on gamma oscillations when histone deacetylation is blocked, suggesting that gamma oscillations through P/Q-type channels modulated by CaMKII may be linked to processes related to gene transcription.
Introduction
Epigenetic mechanisms (i.e., histone post-translational modification and DNA methylation) play a role in regulation of gene expression in response to a wide range of environmental stimuli, such as learning, stress, or drugs of abuse1. In particular, acetylation/deacetylation of histones is associated with the modulation of transcription in multiple ways, by relaxing chromatin structure that would increase the accessibility of transcription factors to their target genes, and also by direct acetylation of transcription factors2. Histone acetylation/deacetylation is a dynamic process controlled by the antagonistic actions of two large families of enzymes: the histone acetyltransferases (HATs) and the histone deacetylases (HDACs). The balance between the actions of these enzymes serves as a key regulatory mechanism for many cellular processes and disease states. The HDACs have been classified into four classes (Class I-IV), based on localization and amino acid sequence similarities2,3. While Class I HDACs (HDACI) are localized in the nucleus, Class IIs (HDACII) shuttle between the nucleus and the cytoplasm where they can modify non-histone proteins.
HDACI/II knockdown using lentiviral transfection of cultured hippocampal neurons altered excitatory synaptic transmission and the formation of dendritic spines4. These authors showed that HDACI/II had different synaptic roles during early developmental stages compared to what was observed in neuronal cultures kept in vitro over 16 days. Synaptic effects of Class III (HDACIII) knockdown were mimicked by 18–24 hour pre-incubation of cultures with the histone deacetylation inhibitor trichostatin A (TSA). Other authors have failed to observe similar results after chronic treatment with TSA of hippocampal organotypic cultures, suggesting the existence of molecular compensations associated to the permanent knockdown of HDACs5. Knockdown of many of the individual HDAC isoforms are lethal, emphasizing the importance of performing acute experiments using HDAC inhibitors in order to assess their role in the modulation intrinsic neuronal properties.
The epigenetic profile of numerous HDAC inhibitors is being explored for translational applications6. HDACs and their inhibitors are promising candidates in treating cancer and several neurodegenerative disorders7. Many of these inhibitors have proven to have synergistic effects with other drugs8. However, the mechanisms of action of HDAC inhibitors need to be elucidated. Indeed, there are potential neurological side effects by modulating HDAC functions, and more research is needed to design more specific HDAC inhibitors with the lowest range of neurological side effects7. The neurological sequelae induced by HDAC inhibitors include fatigue, status epilepticus, somnolence, and gait problems8. That is, some side effects interfere with arousal and might be linked to altered PPN physiology since this nucleus modulates states of arousal.
The present study was designed to determine if modulation of HDACs affects the manifestation of gamma band oscillations in PPN neurons. The PPN is the only reticular activating system (RAS) nucleus that is active during high frequency (gamma range) EEG activity in both waking and rapid eye movement (REM) sleep9,10. The locus coeruleus and raphe nuclei are active during waking and slow wave sleep, but not during REM sleep. The PPN is the only cell group in the RAS in which every neuron manifests intrinsic gamma oscillations9,10. These oscillations are mediated only by voltage-dependent, high threshold P/Q-type and N-type Ca2+ channels9–12. We addressed the question; does blockade of HDACs affect the manifestation of gamma oscillations generated by one or both of these types of channels? If so, what are the intracellular signaling pathways involved in these effects?
Results
Recordings of gamma band oscillations in PPN neurons (total number of cells studied n = 150) were performed using PPN slices at 36 °C in the presence of synaptic blockers (SB, namely, the selective NMDA receptor antagonist 2-amino-5-phosphonovaleric acid (APV, 40 μM), the competitive AMPA/kainate glutamate receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μM), the glycine receptor antagonist strychnine (STR, 10 μM), the specific GABAA receptor antagonist gabazine (GBZ, 10 μM), and the nicotinic receptor antagonist mecamylamine (MEC, 10 μM) and the sodium channel blocker tetrodotoxin (TTX). Whole cell current clamp recordings of PPN cells showed that no significant rundown of oscillatory activity PPN neurons was evident during the recording period. Only one neuron per slice was used during acute application of HDAC inhibitors in order to prevent uncontrolled effects by their long lasting pre-incubation. Recordings from cells after pre-incubation were compared to those acquired in control slices. Please see the Methods section for the choice of concentration of each agent based on affinity and IC50.
Inhibitory effects on intrinsic gamma oscillations by acute and prolonged application of HDAC Class I and II inhibitors
We started by testing a widely used inhibitor of HDAC Class I and II, trichostatin A (TSA)4,5. No membrane potential changes were observed after acute TSA (1 μM) application (n = 8 PPN cells, SB + TTX: −51.9 ± 1.3 mV; +TSA: −50.3 ± 0.9 mV; paired Student’s t-test, df = 7; t = −1.52; p = 0.172). Bath application of TSA significantly increased the input resistance of PPN neurons (Fig. 1A), by 63 ± 23% (n = 7 PPN cells, paired Student’s t-test, df = 6; t = −3.19; p = 0.019), while blocking PPN membrane oscillations (Fig. 1B), reaching its maximum blocking effect after 20 min (Fig. 1C). No statistically significant decreases were observed in the power of lower frequency (10–15 Hz, 15–20 Hz, or 20–30 Hz) membrane oscillations (Table 1). However, the reduction in the gamma band range (30–100 Hz) was statistically significant (Table 1). Lower concentrations of TSA (250 nM) did not affect gamma band power (Paired Student’s t-test, n = 5; df = 4; t = 1.03; p = 0.36).
Figure 1.

Effects of acute TSA. (A) Voltage records in response to hyperpolarizing steps after SB + TTX (black record) and following TSA (20 min; 1 μM, red record) showing the increase in resistance induced by acute TSA exposure. (B) Ramp-induced oscillations after SB + TTX (black record) as previously reported for PPN neurons and effects of acute TSA exposure (20 min; 1 μM, red record) showing elimination of fast oscillations and decreased amplitude of oscillations. (C) Representative plots of power spectrum over time for acute TSA (1 μM) bath application. (D) Ramp-induced oscillations in a representative PPN cell showing low amplitude, low frequency oscillations in the presence of SB + TTX after prolonged exposure to TSA (120 min; left side record).
Table 1.
Mean power changes (% of control) mediated by Trichostatin-A (TSA, 1 μM).
| 10–15 Hz | 15–20 Hz | 20–30 Hz | 30–100 Hz | |
|---|---|---|---|---|
| Acute effect (n = 7) | 76.6 ± 19.9% | 53.2 ± 20.1% | 49.7 ± 14.7% | 36.4 ± 14.5%# |
| Pre-incubation effect (n = 19) | 99.0 ± 11.3% | 75.7 ± 23.2% | 95.9 ± 12.6% | 45 ± 3.4%* |
#p < 0.05; paired Student’s t-test (df = 6, t = −3.53; p = 0.012). *p < 0.05; Student’s t-test (df = 18; t = −2.6, p = 0.02).
Class I and II HDACs have been described to act through their association with other proteins at nuclear and/or cytoplasmic sites2. Therefore, TSA might require longer periods of incubation prior to reaching its maximal effects. With this in mind, we pre-incubated PPN slices with 1 μM TSA for 90–120 min at 36 °C in the presence of SB + TTX. Membrane oscillations were recorded from PPN neurons (n = 8) in slices without pre-incubation and compared to those from slices following TSA pre-incubation, allowing us to compare changes in power spectrum with and without long-term TSA exposure. Figure 1D shows a representative PPN cell with no gamma band frequency oscillations after 120 min pre-incubation with TSA. On average, low frequency band power was unaffected, while only gamma band power was significantly reduced by TSA pre-incubation (Table 1). Neither membrane potential (without TSA: −53.1 ± 1.7 mV; n = 9; after TSA pre-incubation: −50.5 ± 1.2 mV; n = 12; One-way ANOVA; F(1, 19) = 1.6; p = 0.22), nor input resistance (without TSA: 135 ± 9 MΩ; n = 8; after TSA pre-incubation: 147 ± 8 MΩ; n = 12; One-way ANOVA; F(1, 18) = 0.8; p = 0.39), were altered by TSA pre-incubation.
Effects of HDAC Class I and II inhibitors on PPN cells expressing different calcium channels
We then tested whether the TSA-mediated reduction in gamma oscillations might be a consequence of its blocking effect on Ca2+ currents (ICa) mediated by high-threshold, voltage-dependent channels. ICas were recorded after gaining access to the neuronal intracellular space and series resistance was compensated and stable. We used square voltage steps with high Cs+ intracellular pipette solution and synaptic receptor blockers (SB) plus TTX (see Methods). Recordings of ICa were carried out for up to 30 min without significant rundown, similarly to previously reports13,14. The holding potential was at first set at −50 mV and then depolarized using square pulses up to 40 mV. Bath application of TSA (1 μM) was found to reduce ICa (Fig. 2A), without affecting their kinetics (Table 2). In a group of PPN neurons (n = 8), the ICa blocking effects of TSA were observed throughout a wide range of test potentials (Fig. 2B). There were significant reductions in ICa between −20 mV and +40 mV. Importantly, recorded PPN cells expressing P/Q type Ca2+ channels showed greater TSA blocking effects on ICa compared to those expressing only N-type channels (Fig. 2C,D). N-type only PPN neurons manifested an 18.5 ± 2.8% TSA-mediated reduction of ICa (Paired Student’s t-test; n = 7; df = 6; t = −3.62; p = 0.015). PPN cells expressing P/Q-type channels (i.e., P/Q-only plus N + P/Q cell types) showed a 47.8 ± 8.8% reduction after acute TSA (1 μM) application (Paired Student’s t-test; n = 10; df = 9; t = −2.74; p = 0.029), a significantly greater effect (One-way ANOVA, F(1,15) = 7.2; p < 0.05; Bonferroni’s post-hoc t-test, t = 2.68, p = 0.02).
Figure 2.

Effects of acute TSA (1 μM) on voltage-dependent Ca2+ currents (ICa) in different PPN cell types. (A) Representative ICa recorded using a 100 msec long depolarizing square step from a holding potential of −50 mV to 0 mV before (black record) and after (red record) acute bath application of TSA (1 μM). (B) Standard current-voltage (I-V) relationship of ICa before (black circles; n = 8 PPN neurons), and after TSA (20 min; 1 μM, red circles, n = 8 PPN neurons). *p < 0.05, **p < 0.01 One-way ANOVA, Bonferroni post-hoc test comparing SB + TTX vs acute TSA (1 μM). Note significant decreases in ICa after TSA from −20 mV to 40 mV. (C) Representative ICa recorded from a N-only PPN and a N + P/Q PPN neuron using a 100 msec long depolarizing square steps from a holding potential of −50 mV to 0 mV in the presence of SB, TTX and TEA-Cl (black record), after acute bath application TSA (15 min., 1 μM; red record), and after ω-conotoxin GVIA (20 min., CgTx, 2.5 μM, green record) to block N-type channels. Note marked effect on the N-only cells, but partial effect on the N + P/Q cell. (D) Percent reduction in ICa amplitude by TSA (1 μM) in a group of PPN cells expressing only N-type (n = 7; dashed red bar), or P/Q-type (n = 10; solid red bar) Ca2+ channels. *p < 0.05 significant reduction of ICa by acute bath application of TSA for N-only PPN neurons (Paired Student’s t-test; n = 7; df = 6; t = −3.62; p = 0.015), or PPN cells expressing P/Q-type channels (i.e., P/Q-only plus N + P/Q types) (paired Student’s t-test; n = 10; df = 9; t = −2.74; p = 0.029). One-way ANOVA comparison of both groups showed significant differences (F(1,15) = 7.2; p < 0.05; Bonferroni’s post-hoc t-test, t = 2.68, p = 0.02).
Table 2.
Time course of activation (τON) and deactivation (τOFF) of calcium currents before and after acute application of HDAC inhibitors.
| SB + TTX + TEA-Cl | +TSA (1 μM) | SB + TTX + TEA-Cl | +MC1568 (1 μM) | SB + TTX + TEA-Cl + KN93 (1 μM) | +TSA (1 μM) | |
|---|---|---|---|---|---|---|
| τON (ms) | 3.2 ± 0.9 (n = 7) | 8.7 ± 2.8 (n = 7; p > 0.05) | 3.1 ± 1.4 (n = 8) | 8.7 ± 3.4 (n = 8; p > 0.05) | 2.8 ± 0.8 (n = 7) | 3.6 ± 1.0 (n = 7; p > 0.05) |
| τOFF (ms) | 2.2 ± 0.6 (n = 7) | 5.1 ± 1.3 (n = 7; p > 0.05) | 2.6 ± 0.9 (n = 8) | 6.2 ± 1.7 (n = 8; p > 0.05) | 1.1 ± 0.3 (n = 7) | 1.7 ± 0.3 (n = 7; p > 0.05) |
Paired Student’s t-test comparisons showed no significant differences:
Acute TSA (1 μM): τON; t = 2.3, df = 6, p = 0.06, τOFF; t = 2.4, df = 6, p = 0.055.
Acute MC1568 (1 μM): τON; t = 2.2, df = 7, p = 0.06, τOFF; t = 2.3, df = 7, p = 0.06.
Acute KN93 (1 μM) + TSA (1 μM): τON; t = 2.2, df = 6, p = 0.07, τOFF; t = 1.3, df = 6, p = 0.23.
Effects of specific HDAC Class I and Class II inhibitors
We then tested a specific HDAC Class I inhibitor, MS275 (500 nM)2. Bath application of MS275 did not affect the input resistance of PPN neurons (n = 6 PPN cells, paired Student’s t-test, df = 5; t = −2.26; p = 0.074). No effect on PPN membrane oscillations was observed following acute MS275 application (Fig. 3A). Gamma band power in the power spectra showed that mean values were not affected by MS275 (n = 6 PPN cells, Mann-Whitney Rank Sum Test, p = 0.9).
Figure 3.
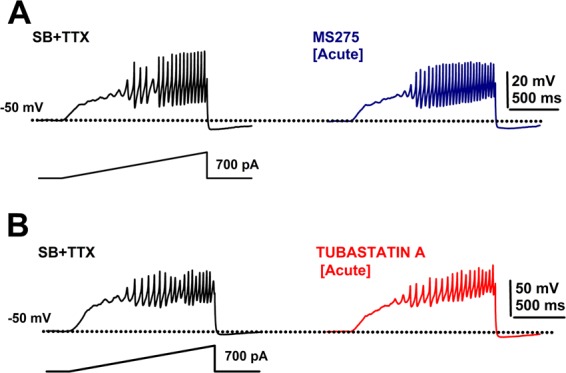
Effects of acute MS275 and tubastatin A. (A) Ramp-induced oscillations after SB + TTX (black record) as previously reported for PPN neurons, and effects of acute MS275 exposure (20 min; 500 nM, blue record) showing no effect on gamma oscillations. (B) Ramp-induced oscillations before (SB + TTX; black record) and after acute tubastatin A exposure (20 min; 150 nM, blue record) showing no effect on gamma oscillation amplitude or frequency.
We continued by testing the specific HDAC Class II inhibitors tubastatin A (150–500 nM; Class IIb, HDAC 6 inhibitor)15, and MC1568 (1 μM; Class IIa inhibitor)2,16–19. Acute tubastatin A (150 nM) bath application did not affect gamma oscillations in PPN neurons (n = 5) (Fig. 3B). No change was observed on either membrane potential (n = 5 PPN cells, SB + TTX: −53 ± 2 mV; + tubastatin A 150 nM: −51 ± 2 mV; paired Student’s t-test, df = 4; t = 0.8; p = 0.47), or input resistance of PPN neurons (n = 7 PPN cells, SB + TTX: 175 ± 14 MΩ; + tubastatin A150 nM: 230 ± 42 MΩ; paired Student’s t-test, df = 4; t = 1.88; p = 0.13). Furthermore, tubastatin A (500 nM) did not change the mean power of gamma oscillations in the power spectrum (Paired Student’s t-test, df = 4; t = −0.58; p = 0.59).
Acute application of the HDAC Class IIa inhibitor MC1568 did not change the membrane potential (n = 8 PPN cells, SB + TTX: −49 ± 1.0 mV; +MC1568: −50 ± 1.5 mV; paired Student’s t-test, df = 7; t = −0.47; p = 0.65), although it increased the input resistance of PPN neurons by 12 ± 4% (n = 7 PPN cells, SB + TTX: 193 ± 8 MΩ; + MC1568: 217 ± 28 MΩ; paired Student’s t-test, df = 6; t = 2.73; p = 0.034). PPN membrane gamma band oscillations were drastically reduced (Fig. 4A). In addition, pre-incubated PPN slices with MC1568 (1 μM) (90–120 min at 36 °C in the presence of SB + TTX) also reduced the presence of gamma oscillations in these neurons during ramp-induced depolarization (Fig. 4B). The mean reduction of power in the spectra of membrane oscillations before and following acute and pre-incubation with MC1568 in different PPN neurons is shown on Table 3. No statistically significant decreases were observed in the power of lower frequency membrane oscillations after either acute or pre-incubation application of MC1568 (10–15 Hz, 15–20 Hz, or 20–30 Hz; Student’s t-test, p > 0.05) (Table 3). However, under both experimental conditions gamma band (30–100 Hz) power reduction was statistically significant (Table 3).
Figure 4.
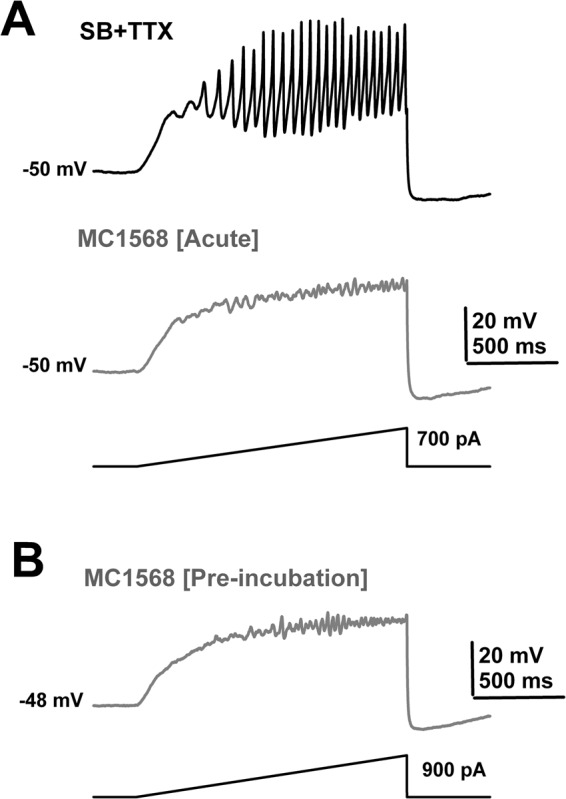
Effects of acute MC1568. (A) Ramp-induced oscillations recorded from a PPN neuron before (SB + TTX; black record), and after acute MC1568 exposure (20 min; 1 μM, grey record) showing elimination of fast oscillations and decreased amplitude of oscillations. (B) Ramp-induced oscillations in a representative PPN cell showing low amplitude, low frequency oscillations in the presence of SB + TTX after prolonged (pre-incubation) exposure to MC1568 (125 min; 1 μM, pre-incubation).
Table 3.
Mean power changes (% of control) mediated by MC1568 (1 μM).
| 10–15 Hz | 15–20 Hz | 20–30 Hz | 30–100 Hz | |
|---|---|---|---|---|
| Acute effect (n = 8) | 62.7 ± 14.9% | 80.3 ± 26.6% | 69.2 ± 38.4% | 37.7 ± 13.5%# |
| Pre-incubation effect | 108.7 ± 38.4% | 59.3 ± 11.4% | 48.4 ± 33.8% | 2.4 ± 10.9%* |
| (n = 25) |
#p < 0.05, paired Student’s t-test (df = 7, t = 4.311; p = 0.004). *p < 0.05; One-way ANOVA, (F(1,23) = 7.43; Bonferroni’s post-hoc t-test, t = 2.73, p = 0.012).
Effects of HDAC Class I and II inhibitors on CaMKII modulation
We further studied whether the TSA- and MC1568-mediated reductions of ICa required intracellular Ca2+-calmodulin‐dependent kinase type II (CaMKII) activation. Pre-incubating PPN slices with KN-93 (1 μM; >30 minutes; in SB + TTX at 36 °C) recorded ICa showed substantial mean current density and voltage dependence (Fig. 5A). Acute TSA (1 μM) exposure was less effective in reducing ICa in the presence of KN-93 (Fig. 5B). Indeed, the mean ICa reduction by TSA decreased from 56.0 ± 9.6% (Fig. 5C, black bar; n = 8; paired Student’s t-test; t = −3.77; df = 7; p = 0.007) to 26.3 ± 6.3% in the presence of KN-93 (Fig. 5C, green bar; n = 7; paired Student’s t-test; t = −3.96; df = 6; p = 0.007). No change on ICa kinetics was observed during KN-93, KN-93 + TSA, or MC1568 acute application (Table 2). In conclusion, the acute effects of TSA on ICa were blunted by KN-93 (Fig. 5C; One-way ANOVA; F(1, 13) = 6.3; p < 0.05; Bonferroni’s post-hoc t-test, t = 2.51, p = 0.03). We also observed similar effects of KN-93 on the MC1568-mediated reduction of ICa. The mean reduction of ICa by MC1568 was blunted by KN-93 (Fig. 5D; One-way ANOVA; F(1, 10) = 9.24; p = 0.013; post-hoc Bonferroni t-test; t = 3.04; p < 0.05). The mean reduction of ICa by MC1568 decreased from 63.7 ± 11.7% in the absence of KN-93 (n = 6 PPN cells; Student’s t-test, t = 3.46; df = 5, p = 0.018) to 23.0 ± 7.0% (n = 6 PPN cells; Student’s t-test, t = −3.75; df = 5, p = 0.013) after KN-93.
Figure 5.

Effects of pre-incubation with the Ca2+-calmodulin‐dependent kinase type II (CaMKII) inhibitor KN-93 (1 μM) on the TSA- (1 μM) and MC1568- (1 μM) induced reduction of ICa. (A) Standard current-voltage (I-V) relationship of ICa after 30 min incubation with KN-93 (n = 5 PPN neurons). (B) Representative ICa recorded using 100 msec long depolarizing square steps from a holding potential of −50 mV to 0 mV after 30 min incubation with KN-93 (1 μM; black record) and after acute bath application TSA (20 min, 1 μM; green record). (B) Percent reduction in ICa amplitude in a group of PPN cells (n = 8) recorded after acute bath application TSA (20 min, 1 μM; black bar), and following pre-incubation with KN-93 (1 μM; n = 7; green bar). **p < 0.01 significant reduction of ICa by acute bath application of TSA (Paired Student’s t-test; t = −3.77; df = 7; p = 0.007), or KN93 + TSA (paired Student’s t-test; t = −3.96; df = 6; p = 0.007). Note the statistically significant KN-93-dependent blunting effect of the TSA-induced reduction of ICa (One-way ANOVA; F(1,13) = 6.3; p < 0.05; post-hoc Bonferroni’s t-test, t = 2.51, p = 0.03). (C) Percent reduction in ICa amplitude in a group of PPN cells (n = 6) recorded after acute bath application MC1568 (20 min, 1 μM; solid grey bar), and following pre-incubation with KN-93 (1 μM; n = 6; dashed grey bar). *p < 0.05 significant reduction of ICa by acute bath application of MC1568 (n = 6 PPN cells; paired Student’s t-test, t = 3.46; df = 5, p = 0.018), or KN93 + MC1568 (n = 6 PPN cells; paired Student’s t-test, t = −3.75; df = 5, p = 0.013). Note the statistically significant blunting effect of KN-93 on the mean ICa reduction by MC1568 (One-way ANOVA; F(1, 10) = 9.24; p = 0.013; post-hoc Bonferroni t-test; t = 3.04; p < 0.05).
Discussion
Gamma oscillations in the PPN
In order to better appreciate the importance of the relationship between HDACs and gamma band oscillations in PPN neurons, we need to understand the role of gamma frequency oscillations in this region. During waking and REM sleep, the EEG manifests low amplitude, high frequency activity in the beta/gamma frequency range (~20–30/30–90 Hz)20. The PPN is most active during the two states of high frequency activity, waking and REM sleep21. The PPN is the arm of the RAS that modulates ascending projections through the thalamus to modulate arousal, and descending projections through the pons and medulla to modulate posture and locomotion21. The PPN is composed of different populations of cholinergic, glutamatergic, and GABAergic neurons22. PPN neurons exhibit beta/gamma frequencies in vivo during active waking and REM sleep, but not during slow wave sleep23–28. Moreover, the presence of gamma band activity has been confirmed in the cortical EEG and PPN of the cat in vivo when the animal is active;24 and in the region of the PPN in humans during stepping, but not at rest29. PPN neurons were found to fire at low frequencies ~10 Hz at rest, but the same neurons increased firing to gamma band frequencies when the animal woke up, or when the animal began walking on a treadmill30. That is, the same cells were involved in both arousal and motor control. Thus, there is convincing evidence for the presence of gamma band activity during active waking and movement in the PPN in vitro, in vivo, and in a number of species.
We described the mechanisms behind gamma band activity in the PPN9–13,21, and they will not be reiterated. Briefly, intrinsic gamma oscillations are mediated by voltage-dependent, high threshold N- and P/Q-type Ca2+ channels that are present in every PPN neuron, regardless of cell or transmitter type. The two Ca2+ channel subtypes are modulated by different intracellular pathways, specifically, N-type and P/Q-type channels are modulated by the cAMP/PK pathway and the CaMKII pathway, respectively. Moreover, there are three cell types in the PPN, those bearing only N-type Ca2+ channels (N-only cells), those with both N- and P/Q-type (N + P/Q cells), and those with only P/Q-type Ca2+ channels (P/Q-only cells)9,31–33. These results suggest that there is a “waking” pathway mediated by CaMKII and P/Q-type channels and a “REM sleep” pathway mediated by cAMP/PKA and N-type channels. These findings also suggest that different PPN cells may fire during waking (those with N + P/Q or “Wake-REM on”, and P/Q-only, or “Wake-on” cells) vs REM sleep (those with N + P/Q, or “Wake-REM on”, and only N-type, or “REM on” cells)10.
New Findings
The present results showed that, (a) acute in vitro exposure to the histone deacetylation Class I and II inhibitor TSA led to the elimination of high threshold, voltage-dependent Ca2+ channel-mediated oscillations, specifically in the gamma band range, but not lower frequencies, (b) pre-incubation with TSA led to a similar decrease specifically in gamma band oscillations, (c) a significant reduction in ICa was elicited by TSA, especially on cells with P/Q-type channels and less so on N-only cells, (d) a HDAC Class I inhibitor MS275, and a Class IIb inhibitor tubastatin A, both failed to affect intrinsic gamma oscillations in PPN neurons, (e) MC1568, a HDAC Class IIa inhibitor, blocked gamma oscillations, and f) the effects of both TSA and MC1568 were blunted by blockade of CaMKII with KN-93. Figure 6 provides a summary of the findings described.
Figure 6.
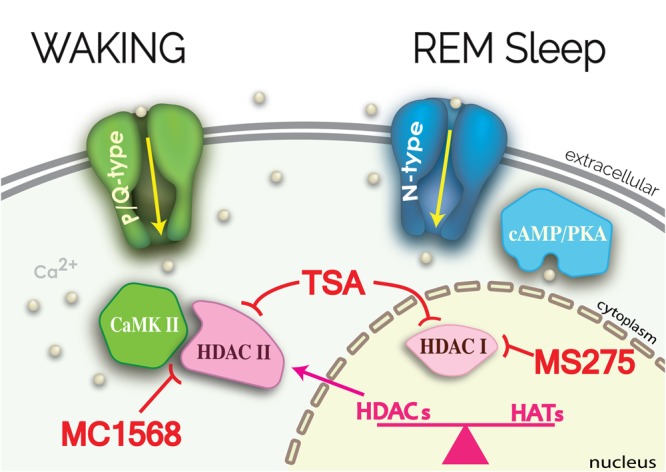
Summary of results. P/Q-type channels are modulated by CaMKII to generate gamma band oscillations (green structures). TSA blocked these oscillations and reduced ICa in PPN cells by inhibiting HDAC I and II (pink structures). KN-93 blunted the effects of TSA, suggesting that HDAC II action on CaMKII may be required for potential transcription following P/Q-type channel-mediated gamma oscillations. Inhibition of HDAC I by MS275, or of HDAC IIb by tubastatin A, in the nucleus had no effects on ICa or oscillations. However, MC1568 inhibition of HDAC IIa had the same effect as TSA in reducing ICa and oscillations. These effects were evident in cells with P/Q-type channels (N + P/Q and P/Q-only cells) but there were no effects on N-only cells that are modulated by cAMP/PKA (blue structures).
The studies described have certain limitations that should be noted, especially since the results shown are among the first to address histone acetylation/deacetylation in this part of the brain. The PPN is composed of three types of neurons previously described to express different neurotransmitters and have different dendritic branching22 that would result in different input resistance ranges. Furthermore, input resistance values were consistent with our previous reports13. Moreover, TSA has a host of nuclear and cytoplasmic actions affecting a number of mechanisms. Biochemical studies showed a direct inhibitory effect of TSA on HDAC classes I and II34. Acute treatment with TSA blunted seizure-triggered down-regulation of glutamate receptor expression in hippocampal CA3 neurons;35 and one hour of acute incubation with TSA (1 μM) occluded synaptic activity-mediated nuclear transcription factor activation of hippocampal neurons in culture36. Moreover, chronic treatment with TSA showed neuroprotective effects against losing mitochondrial Ca2+ homeostasis in striatal neurons in culture37. Finally, TSA was initially used to prevent cochlear cell death, followed by a result showing that specific inhibitors that HDAC6 (i.e., a HDAC class II) mediated the TSA effects38. Our methods tried to limit those actions by using synaptic blockers to eliminate fast synaptic transmission, as well as TTX to prevent action potential generation and thus circuit effects. The remaining activity, therefore, was mostly that of intrinsic membrane oscillations, while most synaptic/receptor interactions were negated. Moreover, these oscillations were induced only on the recorded cell by using ramp stimuli, limiting the manifestation of the effects of TSA to those oscillations. These effects were not unspecific since TSA produced frequency- and cell type-specific effects, i.e. TSA reduced oscillations in the gamma range but not lower frequency oscillations, and TSA reduced oscillations mainly in cells with P/Q-type channels but not in cells with N-type channels. TSA did not affect low frequency oscillations, having a statistically significant effect only on >30 Hz oscillations. TSA also showed cell specificity since it did not significantly affect N-only PPN neurons, which displayed normal gamma oscillations. Its effects were mainly on N + P/Q and P/Q-only cells.
Another issue is that of timing. Transcription is generally thought to occur in the range of hours to days, therefore, could changes in gene transcription by TSA and MC1568 occur during the time frame studied here? We know that agents like TSA and MC1568 can act within 15–30 min to modulate immediate early genes like c-fos and c-jun, at least in other cell types39,40. Thus, we can assume that at least some transcription is taking place during the recording period. It remains to be determined through genomic and proteomic analyses which genes and proteins may be affected by the manifestation of gamma oscillations. Such analysis is ongoing and beyond the current scope. Such studies are notoriously difficult on the brain because of the cellular heterogeneity and the multitude of receptors and channels involved. However, the methods employed here limited activity to intrinsic membrane oscillations in a nucleus in which every cell manifests gamma oscillations. As such, the functional homogeneity of the PPN makes it an ideal site for genomic and proteomic study.
We are careful not to conclude that any form of gene transcription takes place as a result of the manifestation of gamma band oscillations, or change as a result of HDAC inhibition. Given that proteomic and genomic analyses are not included herein, such conclusions would be premature. The results, however, are of exceptional value in demonstrating that gamma oscillations are linked to processes that may be involved in gene transcription.
Clinical Implications
Upon waking, sensory information onto PPN neurons induces arousal, representing a continuous flow of afferent input21. The role of gamma band activity in PPN neurons has been proposed to stabilize coherence related to arousal, providing a stable state during waking13,41,42. This constant process was suggested to help evaluate the world around us on a continuing basis, that is, to bestow preconscious awareness9–12. The sustained flux of afferent information was termed “bottom-up gamma”43,44, which may provide a remodeling of neurons in this part of the RAS, a daily update of sensory experience. The results described above suggest that the continuous flow of information may modulate at least some processes involved in transcription, which could represent one mechanism behind such remodeling. In addition, the PPN relays gamma band activity to cells in the intralaminar thalamus, which also express N- and P/Q-type Ca2+ channels45,46. The intralaminar thalamus projects to the cortex to elicit arousal, but its effects on the cortex differ between waking and REM sleep. Coherence in the EEG between distant sites on the cortex is present during waking, but is absent during REM sleep47,48. That is, the brainstem appears to dictate cortical EEG activity but does so differently during waking vs REM sleep. Our results suggest that the action of TSA was restricted mainly to cells with P/Q-type channels, which are thought to be active during waking but not REM sleep9–12. Neuroepigenetic modulation of these PPN neurons through these particular HDAC mechanisms may occur specifically during waking.
Clinically, we see at least two possible new directions of research to test the involvement of this mechanism in certain disorders. First, blocking histone deacetylation with TSA may have deleterious effects on intrinsic gamma oscillations in the PPN, inducing an inhibition of arousal, which accounts for some of the side effects of this medication. Importantly, the modulation of processes related to transcription during waking could have additional consequences. Second, we previously proposed that insomnia represents not too little sleep but too much waking10,21,43. We proposed that insomnia, in at least some cases, may be due to over expression of P/Q-type Ca2+ channels, since those were proposed to be preferentially active during waking. A potential process by which this could occur is through transcription leading to over expression of P/Q-type channels. Moreover, we know that gene regulation can be mediated by voltage-dependent Ca2+ channels49, and that a number of human disorders result when P/Q-type channels mutate or become dysfunctional50. These findings point to a larger number of potentially fruitful areas of research that need to be pursued.
Methods
Slice preparation
Pups aged 9–13 days from adult timed-pregnant Sprague-Dawley rats (280–350 g) were anesthetized with ketamine (70 mg/kg, I.M.) until tail pinch reflex was absent. This age range was selected due to the developmental decrease in REM sleep of the rat that occurs between 10 and 30 days51. This period of investigation enabled sampling from a baseline period (9–13 days), as determined by our previous body of work on the PPN52. The PPN is the only cell group in the RAS in which every neuron manifests intrinsic gamma oscillations9,10. Basically, the rat undergoes a developmental decrease in REM sleep from about 10 to 30 days of life, with the most significant changes occurring after 14–15 days. The time period studied is a stable epoch during which an exuberant, intrinsic gamma oscillations in vitro correlates with higher REM sleep in vivo during development that then decreases to the adult level by 30 days. Pups were decapitated and the brain was rapidly removed then cooled in oxygenated sucrose-artificial cerebrospinal fluid (sucrose-aCSF). The sucrose-aCSF consisted of (in mM): 233.7 sucrose, 26 NaHCO3, 3 KCl, 8 MgCl2, 0.5 CaCl2, 20 glucose, 0.4 ascorbic acid, and 2 sodium pyruvate. Sagittal sections (400 μm) containing the PPN were cut and slices were allowed to equilibrate in normal aCSF at room temperature. The aCSF was composed of (in mM): 117 NaCl, 4.7 KCl, 1.2 MgCl2, 2.5 CaCl2, 1.2 NaH2PO4, 24.9 NaHCO3, and 11.5 glucose. Slices were recorded at 36 °C while perfused (1.5 ml/min) with oxygenated (95% O2–5% CO2) aCSF in an immersion chamber for patch clamp studies as previously described13,14,45. During recordings, aCSF solution contained the following synaptic receptor antagonists: the selective NMDA receptor antagonist 2-amino-5-phosphonovaleric acid (APV, 40 μM), the competitive AMPA/kainate glutamate receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μM), the glycine receptor antagonist strychnine (STR, 10 μM), the specific GABAA receptor antagonist gabazine (GBZ, 10 μM), and the nicotinic receptor antagonist mecamylamine (MEC, 10 μM) collectively referred to here as synaptic blockers (SB). We also used the sodium channel blocker tetrodotoxin (TTX, 3 μM). All experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Arkansas for Medical Sciences and were in agreement with the “Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research” (National Research Council, 2003), used by the National Institutes of Health for the care and use of laboratory animals.
Whole-cell patch-clamp recordings
Differential interference contrast optics was used to visualize neurons using an upright microscope (Nikon FN-1, Nikon, USA). Whole-cell recordings were performed using borosilicate glass capillaries pulled on a P-97 puller (Sutter Instrument Company, Novato, CA) and filled with a high-K+ intracellular solution, designed to mimic the intracellular electrolyte concentration, of (in mM): 110 K+-Gluconate; 30 KCl; 10 Hepes; 10 Na2 phosphocreatine; 0.2 EGTA; 2 Mg-ATP; 0.5 Li-GTP; 1 MgCl2. Osmolarity was adjusted to ~270–290 mOsm and pH to 7.3. The pipette resistance was 2–5 MΩ. All recordings were made using a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA) in both current and voltage clamp mode. Digital signals were low-pass filtered at 2 kHz, and digitized at 5 kHz using a Digidata-1440A interface and pClamp10 software (Molecular Devices). We used 1.5 sec-long depolarizing current ramps to study membrane potential oscillations. Current ramps allowed us to gradually change membrane potential from resting values up to 0 mV in current clamp mode13,14,45. Input resistance was calculated using square hyperpolarizing current pulses (−300 pA, 500 msec long). When recorded cells expressed HCN channels, membrane resistance was calculated after the membrane was charged and immediately prior to voltage rectification.
The recorded neurons were localized in the pars compacta in the posterior PPN, which is easily identified in sagittal sections of the brainstem13,14,41,45. The recording region was located mainly in the pars compacta in the posterior PPN, immediately dorsal to the superior cerebellar peduncle. This area of PPN has been shown to have the highest density of cells22,53. We first identified PPN neurons by cell type as previously described41,52,54. Gigaseal formation and further access to the intracellular neuronal compartment were achieved in voltage-clamp configuration mode. Within a short time after rupturing the membrane, the intracellular solution reached equilibrium with the pipette solution without significant changes in either series resistance (ranging 7–15 MΩ) or membrane capacitance values.
Voltage-dependent Ca2+ currents (ICa) were studied using a high-Cs+/QX314 pipette solution (in mM: 110 CsMeSO3, 40 HEPES, 10 TEA-Cl, 12 Na2-phosphocreatine, 0.5 EGTA, 2 Mg-ATP, 0.5 Li-GTP, and 1 MgCl2. pH was adjusted to 7.3 with CsOH). Cesium and TEA-Cl are widely used potassium channel blockers. ICa was recorded in the presence of extracellular synaptic receptor antagonists13,14,45, and the sodium and potassium channel blockers tetrodotoxin TTX (3 μM) and TEA-Cl (25 μM), respectively. Square voltage steps (100 msec long) were used to generate PPN neuronal ICa from a holding potential of −50 mV, and then depolarized up to 40 mV. Setting the holding potential at −50 mV allowed us to inactivate T-type Ca2+ channels, while allowing normal activation of high-threshold P/Q- and N-type Ca2+ channels that have been described by our group to mediate membrane potential oscillations13,14,31,45. Both series resistance and liquid junction potential were compensated (>14 kHz correction bandwidth; equivalent to <10 msec lag). Time course of activation (τON) and deactivation (τOFF) of calcium currents were obtained after fitting individual currents to the function y = y0 + a × exp (−Time (ms)/τ(ON or OFF)). τON was obtained from baseline to the peak ICa achieved during initial pulse depolarization, while τOFF was obtained fitting the deactivation from peak tail currents to baseline14. As shown previously, we used the specific N-type channel blocker ω-Conotoxin-GVIA (ω-CgTx, 2.5 μM) to identify PPN cell types according to the Ca2+ channels type they expressed31–33. If ICa was blocked completely, the cell was classified as N-only, if it was blocked partially, it was classified as N + P/Q, and if it was not affected, it was classified as P/Q-only.
Drug application
Bath-applied drugs were administered to the slice via a peristaltic pump (Cole-Parmer, Vernon Hills, IL), and a three-way valve system such that solutions reached the slice 1.5 min after the start of application. Drugs reached a steady state maximal concentration after 10 min. In this study, we observed maximum effects of histone deacetylase inhibitors after 20 min, and lasted for up to 120 min. The sodium channel blocker TTX, as well as tetraethylammonium (TEA-Cl) were purchased from Sigma Aldrich (St. Louis, MO). KN‐93, a selective inhibitor of Ca2+-calmodulin‐dependent kinase type II (CaMKII), was purchased from Cayman (caymanchem.com). Trichostatin-A [(2E,4E,6 R)-7-(4-(Dimethylamino)phenyl)-N-hydroxy-4,6-dimethyl-7-oxo-2,4-heptadienamide], MS275 [Pyridin-3-yl)methyl 4-(2-aminophenylcarbamoyl)benzylcarbamate], MC1568 [3-[5-(3-(3-Fluorophenyl)−3-oxopropen-1-yl)-1-methyl-1H-pyrrol-2-yl]-N-hydroxy-2-propenamide], and tubastatin A hydrochloride [N-Hydroxy-4-[(1,2,3,4-tetrahydro-2-methyl-5H-pyrido[4,3-b]indol-5-yl)methyl]benzamide hydrochloride] were purchased from TOCRIS (Minneapolis, MN). Stock solution of histone deacetylase (HDAC) inhibitors used in this study were dissolved in DMSO and immediately stored at −30 °C. The final concentration of DMSO in the recording aCSF solution was <0.01%. Control aCSF contained the same DMSO concentration during control, SB + TTX, and pre-drug recording conditions. We based our working concentrations of HDAC inhibitors on previously published reports using neurons in vitro. Trichostatin A (TSA) at 250 nM and 1 μM concentrations were 1.6 and 6.5 times higher, respectively, than IC50 values reported previously in cell cultures50, and 10 times higher than the lowest effective concentration used on dopaminergic neurons in vitro55. The same TSA concentrations were used in hippocampal dissociated neurons in culture4 as well as on organotypic slices in culture5. MS275 500 nM and MC1568 1 μM concentrations were shown to yield maximal effects after pre-incubation in vitro for dopaminergic cell cultures56,57. MC1568 at 1 μM corresponded to a 10 times higher concentration than IC50 values reported using tumor cell lines in culture16,17. Tubastatin A 100 nM and 500 nM concentrations were 10 and 30 times higher, respectively, than IC50 values reported previously15. That is, based on the differential affinities, the final concentrations used were 1 μM for both TSA (6.5x IC50), and MC1568 (10x IC50), and 500 nM for MS275 (10x IC50) and tubastatin A (30x IC50).
Data Analysis
Off-line analyses were performed using Clampfit software (Molecular Devices, Sunnyvale, CA). Peak oscillatory amplitude was analyzed by first filtering each ramp recording and measuring the three highest amplitude oscillations to derive a mean amplitude induced during each ramp. The mean peak frequency of the same three oscillations was filtered and measured to derive a mean frequency of oscillations during the three highest amplitude oscillations in each ramp. The power of each frequency was also analyzed by composing a power spectrum for the frequencies in the entire ramp (bandpass filtered high pass 10 Hz, low pass 100 Hz), giving a measure of peak power for frequency31. Comparisons between groups were carried out using OriginPro 9.1.0 (Originlab.com, MA, USA) (see Tables 1 and 2). We used either Student’s t-test or One-way ANOVA, with Bonferroni post hoc testing for multiple comparisons. F values and degrees of freedom are reported for all linear regression ANOVAs. Differences were considered significant at values of p ≤ 0.05. All results are presented as mean ± SEM.
Acknowledgements
Supported by NIH award P30 GM110702 from the IDeA program at NIGMS to the CTN. In addition, this work was supported by grants from FONCYT-Agencia Nacional de Promoción Científica y Tecnológica; Préstamo BID 1728 OC.AR. PICT-2016-1728 and UBACYT 2014–2017 #20120130101305BA (to Dr. Urbano); FONCYT-Agencia Nacional de Promoción Científica y Tecnológica; Préstamo BID 1728 OC.AR. PICT 2015–2594 (to Dr. Bisagno), the College of Medicine, University of Arkansas for Medical Sciences (startup funding to Dr. Lee).
Author Contributions
Dr. Urbano designed the experiments, performed recordings, performed data analysis and data interpretation, and helped write the article. Dr. Bisagno helped design the experiments, performed data analysis, and data interpretation. Ms Mahaffey helped cut and incubate slices, make solutions, and draft figures. Dr. Lee helped with data analysis and interpretation, as well as writing the article. Dr. Garcia-Rill helped with experimental design, data interpretation, data analysis, and wrote the article.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Francisco J. Urbano and Edgar Garcia-Rill contributed equally.
References
- 1.Cadet JL. Epigenetics of Stress, Addiction, and Resilience: Therapeutic Implications. Mol. Neurobiol. 2016;53:545–560. doi: 10.1007/s12035-014-9040-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broide RS, et al. Distribution of histone deacetylases 1-11 in the rat brain. J. Mol. Neurosci. 2007;31:47–58. doi: 10.1007/BF02686117. [DOI] [PubMed] [Google Scholar]
- 4.Akhtar MW, et al. Histone deacetylases 1 and 2 form a developmental switch that controls excitatory synapse maturation and function. J. Neurosci. 2009;29:8288–8297. doi: 10.1523/JNEUROSCI.0097-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calfa G, et al. HDAC activity is required for BDNF to increase quantal neurotransmitter release and dendritic spine density in CA1 pyramidal neurons. Hippocampus. 2012;22:1493–1500. doi: 10.1002/hipo.20990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Invest. 2014;124:30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Didonna A, Opal P. (2015) The promise and perils of HDAC inhibitors in neurodegeneration. Ann. Clin. Transl. Neurol. 2015;2:79–101. doi: 10.1002/acn3.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL. Pharmaceuticals (Basel) 2010. Clinical Toxicities of Histone Deacetylase Inhibitors; pp. 2751–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Rill E, et al. Gamma band activity in the RAS-intracellular mechanisms. Exp. Brain Res. 2014;232:1509–1522. doi: 10.1007/s00221-013-3794-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Rill E, et al. Implications of gamma band activity in the pedunculopontine nucleus. J. Neural Transm. 2015;123:655–665. doi: 10.1007/s00702-015-1485-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Rill E, Luster B, Mahaffey S, Bisagno V, Urbano FJ. Pedunculopontine arousal system physiology- implications for insomnia. Sleep Sci. 2015;8:92–99. doi: 10.1016/j.slsci.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urbano FJ, et al. Pedunculopontine nucleus gamma band activity- preconscious awareness, waking, and REM sleep. Front. Sleep Chronobiol. 2014;5:210. doi: 10.3389/fneur.2014.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kezunovic N, et al. Mechanism behind gamma band activity in the pedunculopontine nucleus (PPN) Eur. J. Neurosci. 2011;34:404–415. doi: 10.1111/j.1460-9568.2011.07766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kezunovic N, et al. Muscarinic modulation of high frequency activity in pedunculopontine neurons. Front. Neurol. 2013;4:176. doi: 10.3389/fneur.2013.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butler KV, et al. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010;132:10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mai A, et al. Class II (IIa)-selective histone deacetylase inhibitors. 1. Synthesis and biological evaluation of novel (aryloxopropenyl)pyrrolyl hydroxyamides. J. Med. Chem. 2009;48:3344–3353. doi: 10.1021/jm049002a. [DOI] [PubMed] [Google Scholar]
- 17.Nebbioso A, et al. Selective class II HDAC inhibitors impair myogenesis by modulating the stability and activity of HDAC-MEF2 complexes. EMBO Rep. 2009;10:776–782. doi: 10.1038/embor.2009.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venza I, et al. Class II-specific histone deacetylase inhibitors MC1568 and MC1575 suppress IL-8 expression in human melanoma cells. Pigment. Cell Melanoma Res. 2013;26:193–204. doi: 10.1111/pcmr.12049. [DOI] [PubMed] [Google Scholar]
- 19.Di Giorgio E, Gagliostro E, Brancolini C. Selective class IIa HDAC inhibitors: myth or reality. Cell. Mol. Life Sci. 2015;72:73–86. doi: 10.1007/s00018-014-1727-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buzsáki G, Draguhn A. Neuronal oscillations in cortical networks. Science. 2004;304:1926–1929. doi: 10.1126/science.1099745. [DOI] [PubMed] [Google Scholar]
- 21.Garcia-Rill, E. Waking and the Reticular Activating System. pp. 330 (Academic Press, 2015).
- 22.Wang HL, Morales M. Pedunculopontine and laterodorsal tegmental nuclei contain distinct populations of cholinergic, glutamatergic and GABAergic neurons in the rat. Eur. J. Neurosci. 2009;29:340–358. doi: 10.1111/j.1460-9568.2008.06576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakai K, El Mansari M, Jouvet M. Inhibition by carbachol microinjections of presumptive cholinergic PGO-on neurons in freely moving cats. Brain Res. 1990;527:213–223. doi: 10.1016/0006-8993(90)91140-C. [DOI] [PubMed] [Google Scholar]
- 24.Steriade M, et al. Different cellular types in mesopontine cholinergic nuclei related to ponto-geniculo-occipital waves. J. Neurosci. 1990;10:2560–2579. doi: 10.1523/JNEUROSCI.10-08-02560.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kayama Y, Ohta M, Jodo E. Firing of ‘possibly’ cholinergic neurons in the rat laterodorsal tegmental nucleus during sleep and wakefulness. Brain Res. 1992;569:210–220. doi: 10.1016/0006-8993(92)90632-J. [DOI] [PubMed] [Google Scholar]
- 26.Datta S, Siwek DF. Single cell activity patterns of pedunculopontine tegmentum neurons across the sleep-wake cycle in the freely moving rats. J. Neurosci. Res. 2002;70:79–82. doi: 10.1002/jnr.10405. [DOI] [PubMed] [Google Scholar]
- 27.Datta S, Siwek DF, Stack EC. Identification of cholinergic and non-cholinergic neurons in the pons expressing phosphorylated cyclic adenosine monophosphate response element-binding protein as a function of rapid eye movement sleep. Neurosci. 2009;163:397–414. doi: 10.1016/j.neuroscience.2009.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boucetta S, et al. Discharge profiles across the sleep-waking cycle of identified cholinergic, gabaergic, and glutamatergic neurons in the pontomesencephalic tegmentum of the rat. J. Neurosci. 2014;34:4708–4727. doi: 10.1523/JNEUROSCI.2617-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fraix V, et al. Pedunculopontine nucleus area oscillations during stance, stepping and freezing in Parkinson’s disease. PLoS One. 2013;8:e83919. doi: 10.1371/journal.pone.0083919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goetz L, et al. The primate pedunculopontine nucleus region: towards a dual role in locomotion and waking state. J. Neural Transm. 2016;123:667–678. doi: 10.1007/s00702-016-1577-7. [DOI] [PubMed] [Google Scholar]
- 31.D’Onofrio S, et al. Modulation of gamma oscillations in the pedunculopontine nucleus by neuronal calcium sensor protein-1: relevance to schizophrenia and bipolar disorder. J. Neurophysiol. 2015;113:709–719. doi: 10.1152/jn.00828.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luster B, et al. High-Threshold Ca2+ channels behind gamma band activity in the pedunculopontine nucleus (PPN). Physiol. Rep. 2015;3:e12431. doi: 10.14814/phy2.12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luster B, Urbano FJ, Garcia-Rill E. Intracellular mechanisms modulating gamma band activity in the pedunculopontine nucleus (PPN). Physiol. Rep. 2016;4(12):e12787. doi: 10.14814/phy2.12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshida M, et al. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 35.Huang Y, et al. Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J. Neurosci. 2002;22:8422–8428. doi: 10.1523/JNEUROSCI.22-19-08422.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKenzie GJ, et al. Nuclear Ca2+ and CaM kinase IV specify hormonal- and Notch-responsiveness. J. Neurochem. 2005;93:171–185. doi: 10.1111/j.1471-4159.2005.03010.x. [DOI] [PubMed] [Google Scholar]
- 37.Oliveira JM, et al. Mitochondrial-dependent Ca2+ handling in Huntington’s disease striatal cells: effect of histone deacetylase inhibitors. J. Neurosci. 2006;26:11174–11186. doi: 10.1523/JNEUROSCI.3004-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takano, K. et al. Histone deacetylase inhibition prevents cell death induced by loss of tricellular tight junction proteins in temperature-sensitive mouse cochlear cells. PLoS ONE12, e0182291. [DOI] [PMC free article] [PubMed]
- 39.Clayton AL, et al. Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J. 2000;19:3714–3726. doi: 10.1093/emboj/19.14.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khan DH, Davie JR. HDAC inhibitors prevent the induction of the immediate-early gene FOSL1, but do not alter the nucleosome response. FEBS Lett. 2013;587:1510–1517. doi: 10.1016/j.febslet.2013.03.029. [DOI] [PubMed] [Google Scholar]
- 41.Simon C, et al. Gamma band unit activity and population responses in the pedunculopontine nucleus. J. Neurophysiol. 2010;104:463–474. doi: 10.1152/jn.00242.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia-Rill E, et al. Coherence and frequency in the reticular activating system (RAS) Sleep Med. Rev. 2013;17:227–238. doi: 10.1016/j.smrv.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia-Rill E, et al. The 10 Hz Frequency: a fulcrum for transitional brain states. Transl. Brain Rhyth. 2016;1:7–13. [PMC free article] [PubMed] [Google Scholar]
- 44.Garcia-Rill E, et al. (2016) Arousal and the control of perception and movement. Curr. Trends Neurol. 2016;10:53–64. [PMC free article] [PubMed] [Google Scholar]
- 45.Kezunovic N, et al. Gamma band activity in the developing parafascicular nucleus. J. Neurophysiol. 2012;107:772–784. doi: 10.1152/jn.00677.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hyde J, et al. Visualization of fast calcium oscillations in the parafascicular nucleus. Pflugers Arch. 2013;465:1327–1340. doi: 10.1007/s00424-013-1264-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cavelli M, et al. Coherent neocortical gamma oscillations decrease during REM sleep in the rat. Behav. Brain Res. 2015;281:318–325. doi: 10.1016/j.bbr.2014.12.050. [DOI] [PubMed] [Google Scholar]
- 48.Torterolo P, et al. Neocortical 40 Hz oscillations during carbachol-induced rapid eye movement sleep and cataplexy. Eur. J. Neurosci. 2016;43:580–589. doi: 10.1111/ejn.13151. [DOI] [PubMed] [Google Scholar]
- 49.Barbado M, et al. Gene regulation by voltage-dependent calcium channels. Biochimica Biophys. Acta. 2009;1793:1096–1104. doi: 10.1016/j.bbamcr.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 50.Schorge S, Rajakulendran S. The P/Q channel in human disease: untangling the genetics and physiology. WIREs Membr. Transp. Signal. 2012;1:311–320. doi: 10.1002/wmts.22. [DOI] [Google Scholar]
- 51.Jou vet-Mounier D, Astic L, Lacote D. Ontogenesis of the states of sleep in rat, cat, and guinea pig during the first postnatal month. Dev. Psychobiol. 1970;2:216–239. doi: 10.1002/dev.420020407. [DOI] [PubMed] [Google Scholar]
- 52.Garcia-Rill E, et al. The developmental decrease in REM sleep: the role of transmitters and electrical coupling. Sleep. 2008;31:673–690. doi: 10.1093/sleep/31.5.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ye M, et al. Cholinergic modulation of fast inhibitory and excitatory transmission to pedunculopontine thalamic projecting neurons. J. Neurophysiol. 2010;103:2417–2432. doi: 10.1152/jn.01143.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garcia-Rill E, Heister DS, Ye M, Charlesworth A, Hayar A. Electrical coupling: novel mechanism for sleep-wake control. Sleep. 2007;30:1405–1414. doi: 10.1093/sleep/30.11.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hsu CW, et al. Identification of HDAC Inhibitors Using a Cell-Based HDAC I/II Assay. J. Biomol. Screen. 2016;21:643–652. doi: 10.1177/1087057116629381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, et al. HDAC inhibitor trichostatin A-inhibited survival of dopaminergic neuronal cells. Neurosci. Lett. 2009;467:212–216. doi: 10.1016/j.neulet.2009.10.037. [DOI] [PubMed] [Google Scholar]
- 57.Formisano L, et al. MS-275 inhibits aroclor 1254-induced SH-SY5Y neuronal cell toxicity by preventing the formation of the HDAC3/REST complex on the synapsin-1 promoter. J. Pharmacol. Exp. Ther. 2015;352:236–243. doi: 10.1124/jpet.114.219345. [DOI] [PubMed] [Google Scholar]