

Abstract
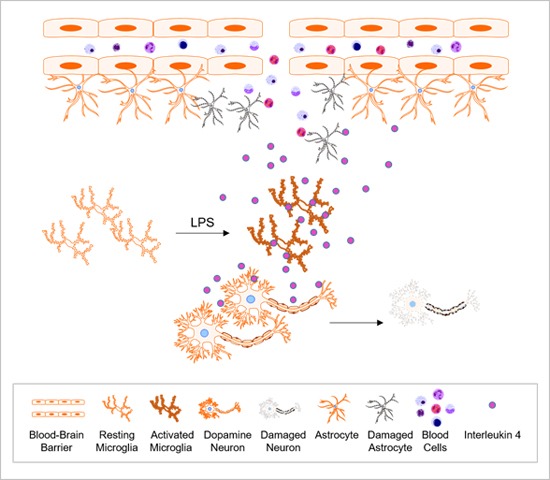
The present study investigated the effects of interleukin (IL)-4 on dopamine (DA) neurons in the substantia nigra (SN) in vivo of lipopolysaccharide (LPS)-treated rat. Tyrosine hydroxylase immunohistochemistry showed a significant loss of nigral DA neurons at 3 and 7 day post-LPS. In parallel, IL-4 immunoreactivity was upregulated as early as 1 day, reached a peak at 3 day and remained elevated at 7 day post-LPS. IL-4 immunoreactivity was detected exclusively in microglia. IL-4 neutralizing antibody (NA) significantly increased survival of DA neurons in LPS-treated SN in vivo by inhibiting microglial activation and production of proinflammatory mediator such as IL-1β as assessed by immunihistochemical, RT-PCR and ELISA analysis, respectively. Accompanying neuroprotection are IL-4NA effects on decreased disruption of blood-brain barrier and astrocytes. The present data suggest that endogenously expressed IL-4 from reactive microglia may be involved in the neuropathological processes of degeneration of DA neurons occurring in Parkinson's disease.
Keywords: Interleukin-4, Parkinson disease, Substantia nigra, Dopaminergic neurons, Lipopolysaccharides
Graphical Abstract
INTRODUCTION
Parkinson's disease (PD) is a common neurodegenerative disorder characterized by progressive degeneration of nigrostriatal dopamine (DA) neurons and a concomitant motor dysfunction [1]. Although the etiology of PD is still largely unknown, accumulating evidence suggests that progression of PD is associated with neuroinflammation [2,3]. Postmortem analyses of brains from patients with PD and experimental animal models of PD indicate that reactive microglia is common features of the PD brain [4,5]. Proinflammatory cytokines released by reactive microglia contribute to degeneration of DA neurons in the substantia nigra pars compacta (SNpc) [6]. However, how these cytokines can lead to the substantial loss of DA neurons in the SN is a major unanswered question.
Cytokines that act as a potent regulator of neuroinflammation may have dual functions in the brain; beneficial vs harmful. Interlukin (IL)-4, a well known anti-inflammatory cytokine, plays beneficial roles in human patients with sepsis [7] and multiple sclerosis [8]. IL-4 was found to play a vital role in the regulation of brain cleanup and repair after stroke as an endogenous defense mechanism [9]. In the IL-4 deficient mice, infarction volume and neurological deficit are increased after transient focal cerebral ischemia [10]. AAV-mediated expression of IL-4 suppressed gliosis and β-amyloidosis in beta-amyloid precursor protein+presenilin-1 Tg mice, resulting in enhanced neurogenesis and improved spatial learning [11]. We previously showed that IL-4 may regulate neuroinflammation by inducing death of CD11b+-reactive microglia and increase neuronal survival in the cerebral cortex of LPS-treated rat [12]. On the contrary, IL-4 was expressed in CD11b+-reactive microglia and contributed to degeneration of neurons in the thrombin and amyloid beta (Aβ)-treated hippocampus in vivo [13,14].
Here we report that endogenously expressed IL-4 within reactive microglia mediates degeneration of DA neurons by increasing expression of CD68+ cells and proinflammatory cytokine such as IL-1β, disruption of blood-brain barrier (BBB) and astrocytes in the SN of LPS-treated rat. Our results suggest that IL-4 could play the detrimental roles in neurodegenerative diseases such as PD in which neuroinflammation and damage of BBB and astrocytes are implicated.
MATERIALS AND METHODS
Stereotaxic surgery and drug injection
All experiments were performed in accordance with approved animal protocols and guidelines established by Kyung Hee University and Korea Institute Toxicology. Animal surgery was processed as previously described [13,15] with some modifications. Sprague Dawley rats (230~280 g) were anesthetized by injection of chloral hydrate (360 mg/kg, i.p.) and positioned in a stereotaxic apparatus. And received a unilateral administration of PBS or LPS into the right SN (anteroposterior −5.2 mm, mediolateral −2.1 mm, dorsoventral −7.8 mm from bregma), according to the atlas of Paxinos and Watson (1998). All injections were made using a Hamilton syringe equipped with a 30s gauge beveled needle and attached to a syringe pump (KD Scientific, MA, USA). Infusions were made at a rate of 0.2 µl/min for LPS (5 µg/3 µl in sterile PBS; Sigma, Saint Louis, USA) and for PBS as a control. For neutralization of IL-4, some of animals received LPS with anti-murine IL-4-neutralizing antibody (IL-4NA; 1 µg/µl; R&D Systems) or nonspecific goat IgG (gIgG; 1 µg/µl; R&D Systems) as a control. After injection, the needle was left in place for an additional 5 min before slowly retracted.
Tissue preparation and immunohistochemistry
Brain tissues were prepared for immunohistochemical staining as previously described with some modifications [15,16]. In brief, animals were anesthetized with chloral hydrate (360 mg/kg, intraperitoneal injection) at the indicated time points after stereotaxic surgery and transcardially perfused. Brains were frozen sectioned using a sliding microtome into 40 µm coronal sections and collected in six separate series for immunohistochemical analysis. Six to eight sections per animal were assessed by immunohistochemistry and immunofluorescence throughout the all Fig. 1, 2, 3, 4, 5. Immunohistochemistry was performed using the avidin-biotin staining technique as previously described with some modifications. Free-floating serial sections were rinsed in PBS twice for 15 min and then quenched for 5 min at room temperature (RT) in PBS containing 3% H2O2. Sections were then rinsed in PBS twice for 15 min in and blocked for 30 min at RT in PBS containing 5% normal serum (Vector Laboratories, CA, USA), 0.2% triton X-100 (Sigma) and 1% bovine serum albumin (BSA) (Sigma). Sections were then rinsed in PBS containing 0.5% BSA twice for 15 min. Next, the sections were incubated overnight with gentle shaking at RT with PBS containing 0.5% BSA and the following primary antibodies: mouse anti-neuron-specific nuclear protein (NeuN; 1:200; Merck millipore, CA, USA) for general neurons, and rabbit anti-tyrosine hydroxylase (TH; 1:2000; Pel-Freez, Brown Deer, WI) for dopaminergic neurons; mouse anti-CD11b (1:500; Serotec, Oxford, UK), which recognizes complement receptor 3; mouse anti-CD68 (1:100; Serotec), which recognizes specific for glycosylated lysosomal antigen for microglia; rabbit anti-ionized calcium binding adaptor molecule 1 (Iba1; Wako, Osaka, Japan) for microglia and macrophage, glial fibrillary acidic protein (GFAP; Sigma) for astrocyte. Sections were then rinsed in PBS containing 0.5% BSA twice for 15 min and incubated for 1 h at RT in biotin-conjugated mouse (1:400; KPL, MD, USA) or rabbit (1:400, Vector Laboratories) secondary antibody. Sections were rinsed again in PBS containing 0.5% BSA and incubated for 1 h at RT in avidin-biotin complex (Vector Laboratories). After rinsing twice in 0.1M phosphate buffer (PB), the signal was visualized by incubating sections in 0.05% 3,3' diaminobenzidine (Sigma) in 0.1M PB containing 0.003% H2O2. Sections were then rinsed in 0.1M PB, mounted on coated slides, and viewed under a bright-field microscope (Olympus Optical, Tokyo, Japan). To analyze the GFAP immunonegative (i.n.) area, CellSense Standard (version 1.16; Olympus) was used. Sections of SN region were obtained and total GFAP-i. n. area was drawn using ‘closed polygon’ under measure tool. For Nissl staining, sections of the SN tissue was mounted on coated slides, dried for 1 h at RT, stained with 0.5% cresyl violet (Sigma), dehydrated, coverslipped, and then analyzed under a bright-field microscope (Olympus Optical).
Fig. 1. LPS induces degeneration of DA neurons and microglial activation in the SN in vivo. PBS (3 µl) or LPS (5 µg/3 µl) was unilaterally injected into the SN. At 7 d post-injection, the coronal sections (40 µm) were selected and processed for TH (A, B, E, F), NeuN (C, G) immunohistochemical staining or Nissl staining (D, H). (B, F) Higher magnifications of (A, E), respectively. (I) Stereological counting results of TH+ and Nissl+ neurons in the SN at 7 d post-injection. The SN tissues were immunostained with CD11b (J, K, P, Q), Iba-1 (L, M R, S), and CD68 (N, O, T, U) antibodies for microglia in the SN. (K, M, O, Q, S, U) Higher magnifications of (J, L, N, P, R, T), respectively. Dotted lines indicate SNpc where DA neurons were degenerated. Scale bars: A, E, 250 µm; B, F, 25 µm; C, D, G, H, 50 µm. J, L, N, P, R, T, 500 µm; K, M, O, Q, S, U, 50 µm. SNpc, substantia nigra pars compacta; SNr, substantia nigra reticulata. Data are presented as the means±SEM of four to seven animals per group. *p<0.001, significantly different from PBS (ANOVA and Bonferroni analyses).

Fig. 2. LPS induces IL-4 expression in the microglia in the SN in vivo. (A~D) Immunofluorescence staining of IL-4 in the SN at 1 d (B), 3 d (C), and 7 d (D) after intranigral injection of LPS or PBS as a control (A, 1d). (E~L) At 3 d post injection, the SN tissues were immunostained simultaneously with IL-4 (Red) and TH (E, I; green), CD11b (F, J; green), Iba-1 (G, K; green) or CD68 (H, L; green). (E~H) PBS injected SN. (I~L) LPS injected SN. Arrows indicate IL-4+ cells merged with CD11b+, Iba-1+, and CD68+ cells. (M) Quantification of IL-4 expression in TH+, CD11b+, Iba-1+ or CD68+cells in the SN. Scale bars: A, E, 250 µm; B, F, 25 µm; C, D, G, H, 50 µm. J, L, N, P, R, T, 500 µm; K, M, O, Q, S, U, 50 µm. SNpc, substantia nigra pars compacta; SNr, substantia nigra reticulata. Data are presented as the means±SEM of four to eight animals per group. *p<0.05, **p<0.01, significantly different from PBS (ANOVA and Bonferroni analyses). Scale bar: A~D, 100 mm; E~L, 25 mm.

Fig. 3. LPS induces degeneration of DA neurons and microglial activation in the SN in vivo. LPS (5 µg/3 µl) was unilaterally injected into the SN in the presence of gIgG (1 µg) or IL-4NA (1 µg). At 7 d post-injection, the coronal sections were selected and processed for TH (A, B, E, F), NeuN (C, G) immunohistochemical staining or Nissl staining (D, H). (B, F) Higher magnifications of (A, E), respectively. (I) Stereological counting results of TH+ and Nissl+ neurons in the SN at 7 d post-injection. The SN tissues were immunostained with CD11b (J, K, P, Q), Iba-1 (L, M R, S), and CD68 (N, O, T, U) antibodies for microglia in the SN. (K, M, O, Q, S, U) higher magnifications of (J, L, N, P, R, T), respectively. Dotted lines indicate SNpc where DA neurons were degenerated. Scale bars: A, E, 250 µm; B, F, 25 µm; C, D, G, H, 50 µm. J, L, N, P, R, T, 500 µm; K, M, O, Q, S, U, 50 µm. Data are presented as the means±SEM of four to five animals per group. *p<0.001, significantly different from LPS with gIgG (ANOVA and Bonferroni analyses).

Fig. 4. Neutralization of IL-4 downregulates IL-1β expression in the SN in vivo. Animals were intranigrally infused with LPS (5 µg/3 µl) or PBS as a control in the presence or absence of gIgG or IL-4NA and sacrificed 1 d later for immunohistochemical analysis (A~C), RT-PCR (D, E), and ELISA (F) of the SN. Photomicrographs (A) and the quantification (B) of CD68+ cells in the SN. (C) Colocalization of IL-1b (red) within CD11b+ cells (green). Arrows indicate merged cells. Representative photograph (D) and histogram for the quantification (E) of RT-PCR shows that IL-4NA decrease mRNA expression of IL-1β in the SN. (F) The amounts of IL-1b in the SN were measured using a ELISA technique. Data are presented as the means±SEM of 3 to 5 animals per group. *p<0.01, **p<0.001, compared with PBS; #p<0.01, compared with LPS or LPS with gIgG (ANOVA and Student-Newman-Keuls analysis). Scale bars: A, C: 50 µm.

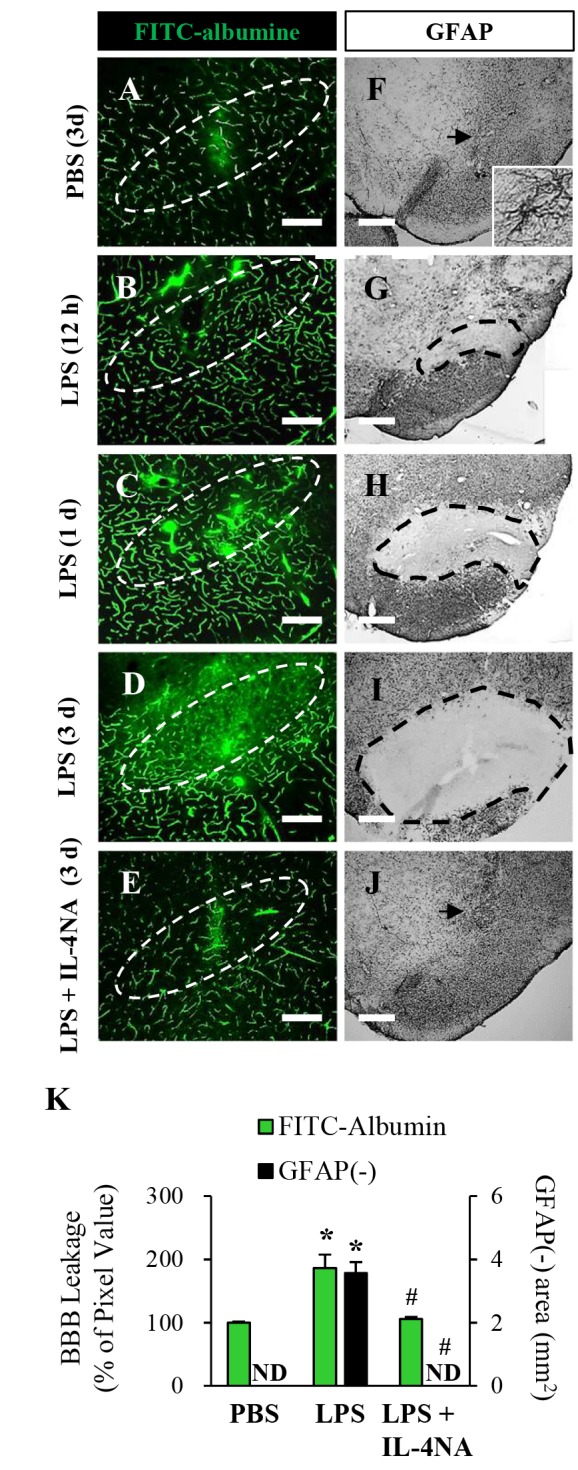
Fig. 5. Neutralization of IL-4 prevents disruption of BBB and astrocytes in the SN in vivo. Animals were administered LPS or PBS in the presence or absence of gIgG or IL-4NA and sacrificed at indicated time point. (A~E) Focal leakage of the BBB was demonstrated by FITC-albumin microangiography. The dashed lines indicate the SNpc. (F~J) The SN tissues were immunostained with anti-GFAP antibody for astrocyte. Dotted lines indicate the lacking area of GFAP staining in the SN. Arrows indicate syringe track. (K) Quantification of BBB leakage density and GFAP immuno negative (i.n.) area at 3 day. Data are presented as the means±SEM of Three to five animals per group. *p<0.01, significantly different from PBS; #p<0.01, significantly different from LPS or LPS with gIgG (ANOVA and Student-Newman-Keuls analysis). Scale bars: A~E, 500 µm; F~J, 200 µm.
Immunofluorescence staining
For immunofluorescence staining, tissue sections were processed as previously described with some modifications [17]. Briefly, free-floating sections were mounted on coated slides and dried for 30 min at RT. After washing in PBS, sections were incubated in PBS containing 5% normal serum, 0.2% triton X-100 and 1% BSA for 30 min and rinsed three times with PBS containing 0.5% BSA. Then the sections were incubated in goat anti-Interleukin-4 (IL-4; 1:1000, R&D Systems, MN, USA) at RT. The next day, tissues were rinsed and incubated with Alexa Fluor 594-conjugated chicken anti-goat IgG (1:2000, Invitrogen, OR, USA). For double-immunofluorescence staining, the sections were incubated in a following primary antibodies combination. A rabbit anti-TH, a mouse anti-CD11b, a rabbit anti-Iba1, a mouse anti-CD68 (1:100) and either a goat anti-IL-4 overnight at 4℃. A goat anti-IL-1β (1:200; R&D systems, MN, USA), and either a mouse anti-CD11b. After washing in PBS containing 0.5% BSA, the sections were incubated simultaneously with a mixture of following secondary antibodies for 1 h at RT. Alexa Fluor 594-conjugated chicken anti-goat IgG (1:5000; Invitrogen) with a Alexa Fluor 488-conjugated donkey anti-mouse IgG (1:5000; Invitrogen) or anti-rabbit IgG (1:5000; Invitrogen); Alexa Fluor 488-conjugated donkey anti-mouse IgG with Alexa Fluor 594-conjugated chicken anti-goat IgG or anti-rabbit IgG. After washing slides were coverslipped with Vectashield medium (Vector Laboratories). Stained samples were analyzed under confocal laser-scanning microscope (Carl Zeiss, Germany). To analyze the localization of different antigens in double-stained samples, images were obtained from the same area and merged using interactive software. Imaging data were analyzed in Image J (National Institutes of Health). ImageJ with co-localization plugin was used to quantify immunofluorescence and with colour deconvolution plugin was used to quantify chromogenic signal intensity on image.
Stereological cell counts of DA neurons
As previously described [17,18], the total number of TH+ neurons were counted in the each groups using the optical fractionator method performed on a brightfield microscope (Olympus Optical, BX51) using Stereo Investigator software (MBF Bioscience). This unbiased stereological method of cell counting is not affected by either the reference volume (SNpc) or the size of the counted elements (neurons).
FITC-labeled albumin assay
Tissues were prepared as previously described [19] with some modifications. FITC-labeled albumin (Sigma) assay was performed for visualization of BBB leakage. After LPS or PBS injections, rats were transcardially perfused with Hank's Balanced Salt Solution containing heparin (10 U/ml) and then immediately by 10 ml FITC-labeled albumin (5 mg/ml, in 0.1 M PBS buffer) injected at a rate of 1.5 ml/min. Brains were dissected from the skull and postfixed overnight in buffered 4% PFA at 4℃. After fixation, the brains were cut into 40 µm slices using a sliding microtome. Sections were mounted on silane-coated slides, and the FITC-labeled albumin contained vessels that were examined by confocal microscopy (Carl Zeiss). To determine the total area for FITC-labeled albumin leakage, 3 images of SN region were obtained, thresholded using Image J, quantified, and normalized by value of PBS injected SN.
Polymerase chain reaction (RT–PCR)
Brain tissues from the ipsilateral SN were dissected at one day after LPS or PBS injection with IL-4NA or gIgG, and total RNA was extracted in a single step using RNAzol B (Tel-Test, Friendswood, TX) following the instructions of the manufacturer. Total RNA was reverse transcribed into cDNA using AMV reverse transcriptase (Promega, WI, USA) and random primers (Promega). The primer sequences used in this study were as follows: 5′-TGATGTTCCCATTAGACAGC-3′ (forward) and 5′-GAGGTGCTGATGTACCAGTT-3′ (reverse) for IL-1β (378 bp); and 5′-TCCCTCAAGATTGTCAGCAA-3′ (forward) and 5′-AGATCCACAACGGATACATT-3′ (reverse) for glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 308 bp). The PCR amplification consisted of 30 cycles of denaturation at 94℃ for 30 s, annealing at 50℃ for 30 s and extension at 72℃ for 30 s. PCR products were separated by electrophoresis on 1.5% agarose gels, after which the gels were stained with ethidium bromide and photographed. For semiquantitative analyses, the photographs were scanned using the Computer Imaging Device and accompanying software (Fujifilm, Tokyo, Japan).
Measurement of IL-1β
The production of IL-1β from rat SN was determined by enzyme-linked immunosorbent assay (ELISA) techniques. Tissues were homogenized in 100 µl ice-cold RIPA buffer (60 mM NaCl, 0.1% sodium dodecyl sulfate, 1% Nonidet P-40, 0.5% sodium deoxycholic acid, 50 mM Tris, pH 8.0) and centrifuged at 14,000 g at 4℃ for 15 min. Equal amounts of protein (25 µg) from each sample were placed in ELISA kit strips coated with the appropriate antibody. ELISA was then performed according to the manufacturer's instructions (Thermo Fisher Scientific, Fremont, CA).
Statistical analysis
All values are expressed as the mean±SEM. Statistical significance (p<0.05 for all analyses) was assessed by ANOVA using Instat 5.01 (GraphPad Software, San Diego, CA, USA).
RESULTS
LPS induces neurodegeneration and microglial activation in the SN in vivo
LPS (5 µg) or PBS as a control were unilaterally injected into the rat SN. 7 d later, brain sections were processed for immunostaining for TH to detect DA neurons and NeuN to detect general neurons, and Nissl staining in the SN. In the LPS-treated SN, the significant loss of TH+ cells (Fig. 1E, F), NeuN+ cells (Fig. 1G), and Nissl+ cells (Fig. 1H) were observed, compared to the PBS-treated SN (Fig. 1A~D), respectively. When quantified by stereological counting, LPS decreased the number of TH+ and Nissl+ cells in the SN by 62% and 73%, respectively, compared to PBS control (Fig. 1I).
To determine microglial activation, sections were immunostained with the antibodies against CD11b, ionized calcium binding adaptor molecule 1 (Iba1) and CD68 as described [20]. In the PBS-injected SN, CD11b+ (Fig. 1J, K) and Iba1+ (Fig. 1L, M) cells exhibited the typical ramified morphology of resting microglia. In the LPS-lesioned SN, the majority of CD11b+ (Fig. 1P, Q) and Iba1+ (Fig. 1R, S) cells displayed an activated morphology, including larger cell bodies with short, thick, or no processes, where DA neurons were degenerated. CD68+ phagocytic cells were detected in the LPS-lesioned SN (Fig. 1T, U), whereas few of CD68+ cells were detected in the PBS-treated SN (Fig. 1N, O).
LPS induces endogenous expression of IL-4 in the SN in vivo
As we previously showed LPS-induced IL-4 expression in the rat cerebral cortex [12], we investigated if LPS could induce IL-4 expression in the LPS-lesioned rat SN. After unilateral injection of LPS or PBS as a control, brain sections were processed for immunostaining for IL-4. In the PBS-injected SN, IL-4+ cells were undetectable (Fig. 2A). In the LPS-lesioned SN, substantial number of IL-4+ cells were observed as early as 1 d post-LPS (Fig. 2B), dramatically increased at 3 d post-LPS (Fig. 2C), which was detectable at 7 d post-LPS (Fig. 2D).
To verify the cell type, which is expressing IL-4 in the SN, brain sections were processed for immunostaining with antibodies against IL-4 and TH (Fig. 2E, I), IL-4 and CD11b (Fig. 2F, J), IL-4 and Iba1 (Fig. 3G, K), or IL-4 and CD68 (Fig. 3H, L) at 3 d after intranigral injection of PBS as a control (Figs. 2E~H) or LPS (Fig. 2I~L) and both images were merged. The levels of IL-4 expression were significantly increased within CD11b+ (Fig. 2J, M), Iba1+ (Fig. 2K, M), and CD68+ (Fig. 2L, M) cells in the LPS-treated SN, compared to the PBS-treated SN (Fig. 2F~H, M). Expression of IL-4 were undetectable in TH+ neurons in the both LPS and PBS-treated SN (Fig. 2E, I, M). These results indicate that IL-4 expression was localized exclusively within activated microglia.
IL-4 contributes to LPS-induced neurodegeneration and microglial activation in the SN in vivo
To investigate the potential function of IL-4 in the LPS-lesioned rat SN, IL-4NA or gIgG as a control were co-injected with LPS into the SN. Pharmacological inhibition of IL-4 function by IL-4NA significantly attenuated LPS-induced neurotoxicity, which was demonstrated by increasing the number of TH+ (Fig. 3E, F, I) and Nissl+ (Fig. 3H, I) cells in the SN, compared to the gIgG-treated control (Fig. 3A, B, D, I). Additional NeuN staining demonstrated the significant increase in NeuN+ cells in IL-4NA-treated LPS-lesioned SN (Fig. 3G), compared with the gIgG-treated LPS-lesioned SN (Fig. 3C).
To determine the IL-4 actions on microglia, sections were immunostained with antibodies against CD11b, Iba1 and CD68. IL-4NA-treated LPS-lesioned SN exhibited the significant inhibition of microglial activation as determined by immunohistochemical staining of CD11b+ (Fig. 3P, Q), Iba-1+ (Fig. 3R, S) and CD68+ (Fig. 3T, U) cells when compared with gIgG-treated LPS-lesioned SN as a control (Fig. 3J~O), respectively.
IL-4 contributes to the expression of proinflammatory cytokine in the SN in vivo
As IL-4 seemed to induce microglial activation, we investigated if it could exert neurotoxicity by regulating the expression of IL-1β within activated microglia. Immunohistochemical staining showed that IL-4NA treatment significantly decreased the number of CD68+ cells in the SN by 43% at 1d post injection (Fig. 4A, B). Additional immunohistochemical analysis demonstrated a colocalization of IL-1β within CD11b+ cells in the SN at 1 d after LPS (Fig. 4C). The mRNA levels of IL-1β were further confirmed by RT-PCR analysis. The results demonstrated that IL-4NA treatment significantly attenuated the mRNA expression of IL-1β by 56% in the LPS-lesioned SN, compared with the gIgG-treated LPS-lesioned SN (Fig. 4D, E). Similar to RT-PCR data, ELISA analysis demonstrated that LPS enhanced the expression of IL-1β protein in the SN at 1 d after LPS compared with the PBS control. In the IL-4NA-treated LPS-lesioned SN, the amount of IL-1β were significantly decreased compared with the LPS-lesioned SN (Fig. 4F), indicating that IL-4 regulates IL-1β expression in the LPS-lesioned SN in vivo.
IL-4 induces disruption of both BBB and astrocytes in the SN in vivo
As IL-1β produced by microglia can alter BBB permeability in vivo [21,22], we wonder if IL-4 could affect BBB in the LPS-lesioned rat SN. To test this, LPS or PBS as a control was unilaterally injected into the rat SN and BBB leakage was assessed by measuring the levels of FITC-labeled albumin. At 3 d post PBS, FITC-labeled albumin was confined to the blood vessels, indicating that the BBB was intact (Fig. 5A, K). In contrast, the diffusion of FITC-labeled albumin into the SN from multiple blood vessels was clearly detected as early as at 1 d and gradually increased up to at 3 d in the LPS-lesioned SN (Fig. 5B~D). Pharmacological inhibition of IL-4 function by IL-4NA almost completely blocked the diffusion of FITC-labeled albumin in the SN at 3 d post-LPS (Fig. 5E, K), compared with the LPS-lesioned SN (Fig 5D, K).
As astrocytes are closely associated with maintenance and formation of BBB [23], the effects of IL-4 on astrocyte were further investigated. SN tissues adjacent to those used to BBB leakage analysis in Fig. 5A~E were immunostained with antibody against GFAP to detect astrocytes. At 3 d post-PBS, the GFAP+ cells displayed ramified morphology as a resting astrocytes in the SN (Fig. 5F). In contrast, area lacking of GFAP+ cells [GFAP(−) area] was observed as early as at 12 h and gradually and significantly increased up to at 3 d in the LPS-lesioned SN (Fig. 5G~J). Pharmacological inhibition of IL-4 function by IL-4NA almost completely reduced the GFAP(−) area in the SN at 3 d post-LPS (Fig. 5E, K), compared with the LPS-lesioned SN (Fig 5D, K) or PBS control SN (Fig. 5F, K).
DISCUSSION
The present study is the first demonstration that the detrimental role of IL-4 on degeneration of DA neurons in the LPS-treated SN are associated with reactive microglia, upregulation of proinflammatory cytokines and disruption of BBB and astrocytes.
Reactive microglia found in brain of patients with PD [24], exacerbates disease progression and loss of DA neurons [25,26]. In our previous reports, CD11b+-reactive microglia was inhibited by capsaicin in the SN of 1-methy-4-phenylpyridinium (MPP+)-treated rat, resulting in survival of DA neurons [27]. Inhibition of Mac-1+-, Iba-1+-, and CD68+-reactive microglia by ethyl pyruvate [28] and inhibition of CD68+-reactive microglia by cannabinoid receptor agonists [29] resulted in DA neuron survival in the SN of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice. In case of LPS-treated rat SN, fluoxetine prevents the degeneration of DA neurons by inhibiting expression of Mac-1+- and CD68+-reactive microglia [30]. The present study demonstrates that LPS significantly upregulated the expression of IL-4 and CD11b+-, Iba-1+- and CD68+-reactive microglia in the SN. In contrast, IL-4NA dramatically decreased expression of CD11b+-, Iba-1+- and CD68+-reactive microglia and the number of CD68+ cells and rescued DA neurons. These data support the hypothesis that IL-4 has the capacity to induce microglial activation, resulting in the degeneration of DA neurons in the SN of LPS-treated rat.
Astrocytes are the most abundant glial cells in the brain, provide structural and metabolic support [31] and produce various neurotrophic molecules, which is essential for the development and survival of DA neurons [32]. Astrocytes are well known to maintain the BBB integrity [23], that has shown to be disrupted in patients with PD [33]. BBB protects the brain from potential neurotoxic molecules in circulation via tightly regulating ion balance, nutrient transport, the entry of plasma components and blood cells [23]. Increasing evidence shows the BBB disruption in patients with PD [33,34] and animal models of PD [35,36,37,38]. Many studies including ours showed that BBB damage contributes to the degeneration of DA neurons in the SN of MPTP-treated mice [19,20,29] and LPS-treated rats [39,40]. The present data demonstrate that LPS increases the diffusion of FITC-labeled albumin into the SN from multiple blood vessels and damage of GFAP+ astrocytes. The pharmacological inhibition of IL-4 by IL-4NA mitigates the LPS-induced disruption of BBB and astrocytes and rescues DA neurons (Fig. 5), suggesting that IL-4 induces disruption of BBB and astrocytes, resulting in degeneration of DA neuros in the SN in vivo. Nevertheless, as our data did not provide the direct and clear evidence of how IL-4 could regulate disruption of BBB and astrocytes, further study should be required to answer these questions in association with specificity and relevance to PD pathogenesis.
IL-1β is significantly upregulated in the cerebrospinal fluid of patients with PD [41]. Accumulating evidence from several studies including ours have demonstrated that IL-1β closely associated with degeneration of DA neurons and BBB disruption in the MPTP [29,42] and LPS model of PD [35,43]. In the 6-hydroxydopamine-treated rat, a subtoxic dose of LPS increased secretion of IL-1β which may contribute degeneration of DA neuron in the SN and motor symptoms in vivo. Inhibition of IL-1β by dexamethasone, a well-known anti-inflammatory steroid, or adenovirus expressing the natural endogenous inhibitor of IL-1 reversed these effects [44]. Ater ischemia-reperfusion, transporting of IL-1β across the BBB was increased [45], and inhibition of IL-1β by neutralizing antibody attenuates ischemia-reperfusion-induced increased in BBB permeability [46]. The current results showed that IL-4NA attenuated mRNA expression of IL-1β (Fig 4D~F) and BBB disruption (Fig. 5A~E, K) in the SN of LPS-treated rat. It is therefore likely that the observed neurotoxicity of IL-4 is associated with its role to upregulate IL-1β which is possibly related to BBB damage in the LPS-treated SN in vivo.
The current results are incompatible to our previous report that IL-4 enhances neuronal survival after traumatic spinal cord injury and in LPS-treated cortex in vivo by regulating neuroinflammation [12,47]. Hühner and colleagues have recently reported that exogenous IL-4 protects MPP+-induced degeneration of midbrain DA neurons in vitro, but endogenous IL-4 has no effect on MPTP-induced degeneration of nigrostriatal dopamine neurons in IL-4 knock-out mice in vivo [48]. On the other hand in the present study, IL-4 contributes to microglial activation, production of IL-1β, and disruption of BBB and astrocytes which subsequently led to the degeneration of DA neurons in the LPS-treated SN in vivo. This is similar to findings that IL-4 is involved in the production of proinflammatory cytokines and oxidative stress, leading to neurodegeneration in the thrombin and Aβ1-42-treated CA1 layer of hippocampus in vivo [13,14]. These apparent inconsistency of IL-4 roles is thought to be due to different insults (LPS vs Aβ1-42 vs thrombin vs traumatic injury vs MPTP) and/or target areas (SN vs cortex vs hippocampus vs spinal cord).
IL-4 and IL-13 partially share their receptors that heterodimerize the IL-4 receptor alpha chain (IL-4Rα) with IL-13Rα1 forming a complex capable of binding IL-4 or IL-13 [49,50]. IL-13Ra1 is expressed in DA neurons and contributes to oxidative stress and lack of IL-13Rα1 delays the loss of DA neurons during restraint stress and LPS-induced acute stress in mice [51,52]. Although the present study did not provide the effect of IL-4 which could be specific and relevant to the pathogenesis of PD, further study involving expression of IL-4R and/or IL-13R on DA neurons could be useful to validate if IL-4 effects can be specific and relevant to the pathogenesis of PD.
Finally, neuroinflammation can be produced by reactive microglia, proinflammatory cytokines, disruption of BBB and astrocytes, which induces onset and/or progression of degeneration of DA neurons occurring in PD. Therefore, it is likely that endogenously expressed IL-4 might serve as the neurotoxic molecule by enhancing PD-related inflammatory processes in the LPS-treated rat model of PD.
ACKNOWLEDGEMENTS
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government [NRF-2016R1A2B4010692], NRFM3C7A1031105 and 2018R1A6A03025124.
References
- 1.Jankovic J. Progression of Parkinson disease: are we making progress in charting the course? Arch Neurol. 2005;62:351–352. doi: 10.1001/archneur.62.3.351. [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson's disease and its potential as therapeutic target. Transl Neurodegener. 2015;4:19. doi: 10.1186/s40035-015-0042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joe EH, Choi DJ, An J, Eun JH, Jou I, Park S. Astrocytes, microglia, and Parkinson's disease. Exp Neurobiol. 2018;27:77–87. doi: 10.5607/en.2018.27.2.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304:1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- 5.Croisier E, Moran LB, Dexter DT, Pearce RK, Graeber MB. Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J Neuroinflammation. 2005;2:14. doi: 10.1186/1742-2094-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sawada M, Imamura K, Nagatsu T. Role of cytokines in inflammatory process in Parkinson's disease. J Neural Transm. 2006;(Suppl):373–381. doi: 10.1007/978-3-211-45295-0_57. [DOI] [PubMed] [Google Scholar]
- 7.Wu HP, Wu CL, Chen CK, Chung K, Tseng JC, Liu YC, Chuang DY. The interleukin-4 expression in patients with severe sepsis. J Crit Care. 2008;23:519–524. doi: 10.1016/j.jcrc.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 8.Urcelay E, Santiago JL, Mas A, Martínez A, de Las Heras V, Arroyo R, de la Concha EG. Role of interleukin 4 in Spanish multiple sclerosis patients. J Neuroimmunol. 2005;168:164–167. doi: 10.1016/j.jneuroim.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 9.Zhao X, Wang H, Sun G, Zhang J, Edwards NJ, Aronowski J. Neuronal interleukin-4 as a modulator of microglial pathways and ischemic brain damage. J Neurosci. 2015;35:11281–11291. doi: 10.1523/JNEUROSCI.1685-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiong X, Barreto GE, Xu L, Ouyang YB, Xie X, Giffard RG. Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke. 2011;42:2026–2032. doi: 10.1161/STROKEAHA.110.593772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kiyota T, Okuyama S, Swan RJ, Jacobsen MT, Gendelman HE, Ikezu T. CNS expression of anti-inflammatory cytokine interleukin-4 attenuates Alzheimer's disease-like pathogenesis in APP+PS1 bigenic mice. FASEB J. 2010;24:3093–3102. doi: 10.1096/fj.10-155317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park KW, Lee DY, Joe EH, Kim SU, Jin BK. Neuroprotective role of microglia expressing interleukin-4. J Neurosci Res. 2005;81:397–402. doi: 10.1002/jnr.20483. [DOI] [PubMed] [Google Scholar]
- 13.Nam JH, Park KW, Park ES, Lee YB, Lee HG, Baik HH, Kim YS, Maeng S, Park J, Jin BK. Interleukin−13/−4-induced oxidative stress contributes to death of hippocampal neurons in aβ1-42-treated hippocampus in vivo. Antioxid Redox Signal. 2012;16:1369–1383. doi: 10.1089/ars.2011.4175. [DOI] [PubMed] [Google Scholar]
- 14.Park KW, Baik HH, Jin BK. Interleukin-4-induced oxidative stress via microglial NADPH oxidase contributes to the death of hippocampal neurons in vivo. Curr Aging Sci. 2008;1:192–201. doi: 10.2174/1874609810801030192. [DOI] [PubMed] [Google Scholar]
- 15.Chung ES, Bok E, Chung YC, Baik HH, Jin BK. Cannabinoids prevent lipopolysaccharide-induced neurodegeneration in the rat substantia nigra in vivo through inhibition of microglial activation and NADPH oxidase. Brain Res. 2012;1451:110–116. doi: 10.1016/j.brainres.2012.02.058. [DOI] [PubMed] [Google Scholar]
- 16.Choi SH, Lee DY, Kim SU, Jin BK. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: role of microglial NADPH oxidase. J Neurosci. 2005;25:4082–4090. doi: 10.1523/JNEUROSCI.4306-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nam JH, Park ES, Won SY, Lee YA, Kim KI, Jeong JY, Baek JY, Cho EJ, Jin M, Chung YC, Lee BD, Kim SH, Kim EG, Byun K, Lee B, Woo DH, Lee CJ, Kim SR, Bok E, Kim YS, Ahn TB, Ko HW, Brahmachari S, Pletinkova O, Troconso JC, Dawson VL, Dawson TM, Jin BK. TRPV1 on astrocytes rescues nigral dopamine neurons in Parkinson's disease via CNTF. Brain. 2015;138:3610–3622. doi: 10.1093/brain/awv297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 19.Chung YC, Kim YS, Bok E, Yune TY, Maeng S, Jin BK. MMP-3 contributes to nigrostriatal dopaminergic neuronal loss, BBB damage, and neuroinflammation in an MPTP mouse model of Parkinson's disease. Mediators Inflamm. 2013;2013:370526. doi: 10.1155/2013/370526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung YC, Kim SR, Jin BK. Paroxetine prevents loss of nigrostriatal dopaminergic neurons by inhibiting brain inflammation and oxidative stress in an experimental model of Parkinson's disease. J Immunol. 2010;185:1230–1237. doi: 10.4049/jimmunol.1000208. [DOI] [PubMed] [Google Scholar]
- 21.Wong D, Dorovini-Zis K, Vincent SR. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp Neurol. 2004;190:446–455. doi: 10.1016/j.expneurol.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Jin S, Sonobe Y, Cheng Y, Horiuchi H, Parajuli B, Kawanokuchi J, Mizuno T, Takeuchi H, Suzumura A. Interleukin-1β induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS One. 2014;9:e110024. doi: 10.1371/journal.pone.0110024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cabezas R, Avila M, Gonzalez J, El-Bachá RS, Báez E, García-Segura LM, Jurado Coronel JC, Capani F, Cardona-Gomez GP, Barreto GE. Astrocytic modulation of blood brain barrier: perspectives on Parkinson's disease. Front Cell Neurosci. 2014;8:211. doi: 10.3389/fncel.2014.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 25.Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T. Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann Neurol. 2005;57:168–175. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
- 26.Perry VH. Innate inflammation in Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a009373. doi: 10.1101/cshperspect.a009373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park ES, Kim SR, Jin BK. Transient receptor potential vanilloid subtype 1 contributes to mesencephalic dopaminergic neuronal survival by inhibiting microglia-originated oxidative stress. Brain Res Bull. 2012;89:92–96. doi: 10.1016/j.brainresbull.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 28.Huh SH, Chung YC, Piao Y, Jin MY, Son HJ, Yoon NS, Hong JY, Pak YK, Kim YS, Hong JK, Hwang O, Jin BK. Ethyl pyruvate rescues nigrostriatal dopaminergic neurons by regulating glial activation in a mouse model of Parkinson's disease. J Immunol. 2011;187:960–969. doi: 10.4049/jimmunol.1100009. [DOI] [PubMed] [Google Scholar]
- 29.Chung YC, Shin WH, Baek JY, Cho EJ, Baik HH, Kim SR, Won SY, Jin BK. CB2 receptor activation prevents glial-derived neurotoxic mediator production, BBB leakage and peripheral immune cell infiltration and rescues dopamine neurons in the MPTP model of Parkinson's disease. Exp Mol Med. 2016;48:e205. doi: 10.1038/emm.2015.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung YC, Kim SR, Park JY, Chung ES, Park KW, Won SY, Bok E, Jin M, Park ES, Yoon SH, Ko HW, Kim YS, Jin BK. Fluoxetine prevents MPTP-induced loss of dopaminergic neurons by inhibiting microglial activation. Neuropharmacology. 2011;60:963–974. doi: 10.1016/j.neuropharm.2011.01.043. [DOI] [PubMed] [Google Scholar]
- 31.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baquet ZC, Bickford PC, Jones KR. Brain-derived neurotrophic factor is required for the establishment of the proper number of dopaminergic neurons in the substantia nigra pars compacta. J Neurosci. 2005;25:6251–6259. doi: 10.1523/JNEUROSCI.4601-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gray MT, Woulfe JM. Striatal blood-brain barrier permeability in Parkinson's disease. J Cereb Blood Flow Metab. 2015;35:747–750. doi: 10.1038/jcbfm.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartels AL, Willemsen AT, Kortekaas R, de Jong BM, de Vries R, de Klerk O, van Oostrom JC, Portman A, Leenders KL. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson's disease, PSP and MSA. J Neural Transm Vienna. 2008;115:1001–1009. doi: 10.1007/s00702-008-0030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Banks WA, Gray AM, Erickson MA, Salameh TS, Damodarasamy M, Sheibani N, Meabon JS, Wing EE, Morofuji Y, Cook DG, Reed MJ. Lipopolysaccharide-induced blood-brain barrier disruption: roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J Neuroinflammation. 2015;12:223. doi: 10.1186/s12974-015-0434-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kortekaas R, Leenders KL, van Oostrom JC, Vaalburg W, Bart J, Willemsen AT, Hendrikse NH. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol. 2005;57:176–179. doi: 10.1002/ana.20369. [DOI] [PubMed] [Google Scholar]
- 37.Olmedo-Díaz S, Estévez-Silva H, Orädd G, Af Bjerkén S, Marcellino D, Virel A. An altered blood-brain barrier contributes to brain iron accumulation and neuroinflammation in the 6-OHDA rat model of Parkinson's disease. Neuroscience. 2017;362:141–151. doi: 10.1016/j.neuroscience.2017.08.023. [DOI] [PubMed] [Google Scholar]
- 38.Park HJ, Shin JY, Kim HN, Oh SH, Song SK, Lee PH. Mesenchymal stem cells stabilize the blood-brain barrier through regulation of astrocytes. Stem Cell Res Ther. 2015;6:187. doi: 10.1186/s13287-015-0180-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villarán RF, Espinosa-Oliva AM, Sarmiento M, De Pablos RM, Argüelles S, Delgado-Cortés MJ, Sobrino V, Van Rooijen N, Venero JL, Herrera AJ, Cano J, Machado A. Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: potential risk factor in Parkinson's disease. J Neurochem. 2010;114:1687–1700. doi: 10.1111/j.1471-4159.2010.06879.x. [DOI] [PubMed] [Google Scholar]
- 40.Hernández-Romero MC, Delgado-Cortés MJ, Sarmiento M, de Pablos RM, Espinosa-Oliva AM, Argüelles S, Bández MJ, Villarán RF, Mauriño R, Santiago M, Venero JL, Herrera AJ, Cano J, Machado A. Peripheral inflammation increases the deleterious effect of CNS inflammation on the nigrostriatal dopaminergic system. Neurotoxicology. 2012;33:347–360. doi: 10.1016/j.neuro.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 41.Blum-Degen D, Müller T, Kuhn W, Gerlach M, Przuntek H, Riederer P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer's and de novo Parkinson's disease patients. Neurosci Lett. 1995;202:17–20. doi: 10.1016/0304-3940(95)12192-7. [DOI] [PubMed] [Google Scholar]
- 42.Chen X, Lan X, Roche I, Liu R, Geiger JD. Caffeine protects against MPTP-induced blood-brain barrier dysfunction in mouse striatum. J Neurochem. 2008;107:1147–1157. doi: 10.1111/j.1471-4159.2008.05697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu XL, Wang P, Liu YH, Xue YX. Effects of poly (ADPribose) polymerase inhibitor 3-aminobenzamide on blood-brain barrier and dopaminergic neurons of rats with lipopolysaccharide-induced Parkinson's disease. J Mol Neurosci. 2014;53:1–9. doi: 10.1007/s12031-013-0175-5. [DOI] [PubMed] [Google Scholar]
- 44.Pott Godoy MC, Tarelli R, Ferrari CC, Sarchi MI, Pitossi FJ. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson's disease. Brain. 2008;131:1880–1894. doi: 10.1093/brain/awn101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patra A, Chen X, Sadowska GB, Zhang J, Lim YP, Padbury JF, Banks WA, Stonestreet BS. Neutralizing anti-interleukin-1β antibodies reduce ischemia-related interleukin-1β transport across the blood-brain barrier in fetal sheep. Neuroscience. 2017;346:113–125. doi: 10.1016/j.neuroscience.2016.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen X, Sadowska GB, Zhang J, Kim JE, Cummings EE, Bodge CA, Lim YP, Makeyev O, Besio WG, Gaitanis J, Threlkeld SW, Banks WA, Stonestreet BS. Neutralizing anti-interleukin-1β antibodies modulate fetal blood-brain barrier function after ischemia. Neurobiol Dis. 2015;73:118–129. doi: 10.1016/j.nbd.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee SI, Jeong SR, Kang YM, Han DH, Jin BK, Namgung U, Kim BG. Endogenous expression of interleukin-4 regulates macrophage activation and confines cavity formation after traumatic spinal cord injury. J Neurosci Res. 2010;88:2409–2419. doi: 10.1002/jnr.22411. [DOI] [PubMed] [Google Scholar]
- 48.Hühner L, Rilka J, Gilsbach R, Zhou X, Machado V, Spittau B. Interleukin-4 protects dopaminergic neurons in vitro but is dispensable for MPTP-induced neurodegeneration in vivo. Front Mol Neurosci. 2017;10:62. doi: 10.3389/fnmol.2017.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hilton DJ, Zhang JG, Metcalf D, Alexander WS, Nicola NA, Willson TA. Cloning and characterization of a binding subunit of the interleukin 13 receptor that is also a component of the interleukin 4 receptor. Proc Natl Acad Sci U S A. 1996;93:497–501. doi: 10.1073/pnas.93.1.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mori S, Maher P, Conti B. Neuroimmunology of the interleukins 13 and 4. Brain Sci. 2016;6:E18. doi: 10.3390/brainsci6020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morrison BE, Marcondes MC, Nomura DK, Sanchez-Alavez M, Sanchez-Gonzalez A, Saar I, Kim KS, Bartfai T, Maher P, Sugama S, Conti B. Cutting edge: IL-13Rα1 expression in dopaminergic neurons contributes to their oxidative stress-mediated loss following chronic peripheral treatment with lipopolysaccharide. J Immunol. 2012;189:5498–5502. doi: 10.4049/jimmunol.1102150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mori S, Sugama S, Nguyen W, Michel T, Sanna MG, Sanchez-Alavez M, Cintron-Colon R, Moroncini G, Kakinuma Y, Maher P, Conti B. Lack of interleukin-13 receptor α1 delays the loss of dopaminergic neurons during chronic stress. J Neuroinflammation. 2017;14:88. doi: 10.1186/s12974-017-0862-1. [DOI] [PMC free article] [PubMed] [Google Scholar]