

Abstract
Ischemic stroke and cerebral infarction triggered by the blockage of blood supply can cause damage to the brain via a complex series of pathological changes. Recently, diverse therapies have emerged as promising candidates for the treatment of stroke. These treatments exert therapeutic effects by acting on diverse target molecules and cells in different time windows from the acute to chronic phases. Here, using immunohistochemistry, we show pathophysiological changes in the brain microenvironment at the hyperacute (within 6 h), acute (1~3 days), subacute (7 days), and chronic (1 month) phases following ischemic injury. Ischemic injury in rats was induced by occluding the middle cerebral artery and was validated by magnetic resonance imaging. The progression of damage to the brain was evaluated by immunohistochemistry for NeuN+ neurons, GFAP+ astrocytes, and Iba1+ microglia, and by the emergence of the cell death-related molecules such as AIF, FAF1, and activated caspase-3. Our data regarding the spatial and temporal information on pathophysiological changes may warrant the investigation of the timing of administration of therapeutic treatments in preclinical studies with an animal model of stroke.
Keywords: Brain ischemia, Cell death, Stem cell therapy, Antibody staining, Chronic stroke
Graphical Abstract
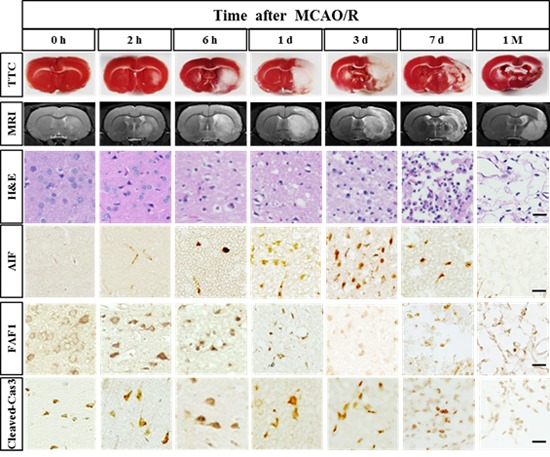
INTRODUCTION
Ischemic stroke and cerebral infarction result from insufficient blood and oxygen supply to the brain. Blockage of the blood supply to the brain causes a cascade of pathological changes associated with energy failure, excessive glutamate release, elevated intracellular Ca2+ levels, generation of free radicals, blood-brain barrier (BBB) disruption, inflammation, and eventually massive cell death via necrosis and apoptosis [1]. The cells in the central portion of the ischemic tissue, also known as the infarct core, have been known to be afflicted by irreversible damage, and the cells around the infarct core, called the penumbra, also undergo delayed cell death within several hours or days [2,3].
Over the past several decades, the development of imaging technology, including magnetic resonance imaging (MRI), has allowed the visualization of temporal and spatial changes in brain lesions [4,5]. Many studies related to stroke lesions have revealed pathophysiological changes in the brain, including cell death-related pathways; however, most studies have been limited to specific times and areas.
In this study, we aimed to analyze the spatiotemporal expression profiles of cell death-related molecules in a commonly utilized rat stroke model of transient occlusion and reperfusion of middle cerebral artery (MCAO/R). We obtained MR and histological images from the same animals to minimize potential variances. In addition to microscopic images of the most representative regions, we provide the entire brain images with high resolution in the Supplementary Figures, in which the region of interest can be magnified to microscopic levels. This study showed that the pathophysiological changes in the whole brain including the penumbra and core at a specific time after an ischemic injury may provide useful information for determining the optimal administration route and therapeutic time window for diverse therapies.
MATERIALS AND METHODS
Animals
Eight-week-old male Sprague-Dawley rats (ORIENT, Seongnam, South Korea) weighing 280~300 g were used for all experiments. Rats were allowed free access to food and water before the experiments. All animal protocols were approved by the Institutional Animal Care and Use Committee at the Ajou University School of Medicine (Registration No. 2015-0012).
Focal ischemic stroke model
The middle cerebral artery occlusion/reperfusion (MCAO/R) stroke model was generated using intraluminal vascular occlusion as previously reported [6]. Briefly, the animals were anesthetized with 5% isoflurane in 70% N2O and 30% O2 and anesthesia was maintained with 3% isoflurane. The right common carotid artery, internal carotid artery (ICA), and external carotid artery (ECA) were exposed. After cutting the ECA, a 4-0 monofilament nylon suture with a round, coated tip (filament size 4-0, diameter 0.19 mm, length 30 mm; diameter of coated tip 0.35 +/− 0.02 mm, length 5~6 mm, LMS Korea, Seongnam, South Korea) was inserted into the ECA stump and gently advanced to the bifurcating point of ICA and MCA. Two hours later, the animals were re-anesthetized and re-perfused by removing the suture to resume blood supply via the ICA. The ECA stump was permanently ligated. The body temperature of animals was maintained using a heating pad (FHC, Bowdoinham, ME) during the entire procedure. At indicated times following the surgery, the animals were subject to MRI analysis and then immediately sacrificed for histological analysis to ensure the direct comparison between MR images and histological data. The animals that showed incomplete infarction as tested by MRI or those that suddenly died immediately after the surgery (<10%) were excluded in the study. The overall survival rate up to 1 month was approximately 72%. During the 1st week after surgery, the animals were fed with wet mashed food on the floor. Animals were monitored daily to ensure survival, condition, and body weight.
Evaluation of damage to the brain
MRI analysis, triphenyl tetrazolium chloride (TTC) staining, and hematoxylin and eosin (H&E) staining were performed with the same animals to evaluate damage to the brain at the following time points: 0 h, 2 h, 6 h, 1 day, 3 days, 7 days, and 1 month (28~33 days) following ischemia/reperfusion. At least four animals were used per group.
MRI experiments were performed with a 9.4-T animal MRI scanner (Biospec 94/30; Bruker BioSpin MRI GmbH, Germany). T2-weighted images were acquired with the following parameters: repetition time=5,000 ms, echo time=20 ms, average=2, acquisition matrix=256×256; 10 slices with 1-mm slice thickness, and flip angle=90°. In total, 10 slices were scanned to cover the whole rat brain. MR images were reconstituted with a Digital Imaging and Communications in Medicine software (DICOM Viewer R3.0 SP3, Philips Healthcare, Amsterdam, Netherlands).
Immediately after the MRI tests, animals were sacrificed to obtain 2-mm-thick coronal brain slices using a mold (CellPoint Scientific, Rockville, MD); these slices were incubated in 2% TTC in phosphate buffered saline (PBS) (pH 7.0) at 37℃ for 30 min. The TTC-stained brain slices were fixed in 10% formalin in PBS for 24 h, photographed, and further processed for immunohistochemical analysis.
Immunohistochemistry of death-related molecules
For immunohistochemistry, the 5th brain coronal slice of previously stained TTC was embedded in paraffin. Serial 5-µm-thick paraffin sections were deparaffinized in xylene and hydrated in a series of graded alcohols and distilled water. Antigen retrieval was performed by boiling the samples with a 10-mM sodium citrate buffer (pH 6.0) for 30 min at 98℃ and then treating them with 0.3% hydrogen peroxide (H2O2). After blocking in 10% normal serum for 1 h, the sections were incubated with primary antibodies at 4℃ overnight. Primary antibodies used were rabbit anti-NeuN (1:200; ABN78, EMD Millipore, Bedford, MA), rabbit anti-glial fibrillary acidic protein (GFAP, 1:500; G9269, Sigma Aldrich, St. Louis, MI), rabbit anti-ionized calcium binding adaptor molecule 1 (Iba1, 1:3000; 019-19741, WAKO, Richmond, VA), mouse anti-CD68 (1:200; MCA341R, Bio-rad, Hercules, CA), mouse anti-apoptosis inducing factor (AIF, 1:500; sc-13116, SantaCruz Biotechnology, Santa Cruz, CA), rabbit anti-Cleaved Caspase-3 (1:200; 9661, Cell signaling technology, Billerica, MA), and goat anti-FAS-associated factor 1 (FAF1, 1:200; sc-1885, SantaCruz). Next, the sections were washed twice with 1% Triton X-100 in PBS and incubated for 1 h with 1:500 dilution of biotinylated goat anti-rabbit IgG (BA-1000, Vector Lab, Burlingame, CA) or biotinylated horse anti-mouse IgG (BA-2000, Vector Lab) in the blocking buffer, and the reaction was amplified for 30 min with avidin-biotin complex, using an ABC kit (PK4000, Vector Lab). The slides were treated with 3,3′-diaminobenzidine (DAB, D-5637, Sigma Aldrich) and then dehydrated through a graded series of alcohol, mounted in Shandon synthetic mountant (6769007, Thermo Scientific, Cheshire, UK) and cover-slipped. Sections without primary antibodies were processed in parallel as a negative control to monitor the nonspecific peroxidase staining of secondary antibodies in the damaged tissue.
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining was performed according to the manufacturer's protocol (In Situ Cell Death Detection Kit, 11684817910; Roche, Indianapolis, IN).
Quantification
All bright-field images were acquired using a slide scanner microscope (Axio Scan.Z1, Zeiss, Jena, Germany). Cells in a boxed area of 1 mm2 were manually counted in each field of view by an investigator blinded to the groups. The number of Iba- and CD68-positive cells were counted in the ischemic core of the cortex and striatum and the penumbra of the mediodorsal cortex. The positively stained cells for cleaved caspase-3 and TUNEL were counted in the ischemic core of the cortex and striatum. The number of immunoreactive cells was counted in one section from each animal. Four sections from each group were averaged for statistical analyses. Results are expressed as mean counts per mm2±standard error of the mean for each area.
Statistical analysis
Data were analyzed and plotted using GraphPad Prism (Graph-Pad Software, San Diego, California, USA). Statistical analysis was performed using one-way ANOVA, followed by Tukey post hoc test for pairwise comparisons. p<0.05 was considered statistically significant.
RESULTS
Focal cerebral ischemia was induced by 2-h occlusion of the MCA and reperfusion. MRI tests immediately followed by TTC staining were carried out to evaluate the infarction. Animals with 0 h reperfusion following a 2 h occlusion showed no obvious infarct in both MRI and TTC tests. The infarct became evident in the striatum at 2 h after the reperfusion (4 h after the onset of ischemia) and gradually increased to the entire area of the striatum and dorsolateral cortex at 6 h. This condition is consistent with previous reports, which demonstrated that the striatum is more vulnerable to death in this stroke model because it is supplied by the lenticulostriate artery, a deep branch of MCA, whereas the cortex is supplied by MCA branches and collateral circulation on the border [7]. The severity of ischemic injury was highest between day 1 and 3 and apparently reduced from day 7 onwards (Fig. 1A). This decrease in infarct size can be attributed to infiltration of CD68-positive cells in the ischemic core, since TTC staining reveals the mitochondrial respiratory enzymes in all cell types (see Fig. 3).
Fig. 1. Time-dependent progression of cerebral infarct after ischemic stroke. (A) Representative TTC-stained brain sections and the corresponding MR images from rats at the indicated post-stroke time points. (B) H&E staining of TTC-stained brain sections. Cell body shrinkage and perineuronal vacuolization were detected in the ischemic core from the early time points. Note the cell body shrinkage with darkly stained pyknotic nuclei in the striatum at 0 h (arrows). (C) Immunohistochemical analysis of NeuN-positive neurons. Neuronal degeneration initiated at 0 h (just after the reperfusion) in the striatum and 2 h in the cortex. The boxed areas in the full-size brain images are magnified in the representative image. Scale bar, 20 µm.

Fig. 3. Immunohistochemistry for Iba1 and CD68 in the ischemic rat brain. (A) Iba1-immunostaining. Within the 1st day, the microglia underwent morphological changes to an activated amoeboid form in the core and penumbra. In the subacute phase (3~7 days), a mixed population of microglia/macrophages increased in the core and penumbra. (B) Quantitative measurement of Iba1-positive cells in the boxed area of 1 mm2. (C) CD68 immunostaining. Note the absence of CD68-positive cells in the sham group. The boxed areas in the full-size brain images are magnified to the representative image. (D) Quantitative measurement of CD68-positive cells in the boxed area of 1 mm2. Data are presented as means±SEM per mm2 obtained from four animals per group (*p<0.05, ***p<0.001, compared to the value of the sham group; #p<0.05, ##p<0.01, ###p<0.001, compared to the values of the 0 h time point). Scale bar, 20 µm.

After the TTC staining, the brain slices were fixed and embedded in the paraffin. Several 5 µm-thick paraffin sections were obtained and subject to histological analyses. In the low magnification of full-sized brain images in the H&E staining, the infarct was first detected at 2 h in the striatum and then in the overlying dorsolateral cortex at 6 h after reperfusion. However, microscopic analysis of the same brain slice revealed that degeneration actually occurred at 0 h (just after reperfusion) in the striatum and 2 h after the reperfusion in the cortex, as evidenced by darkly stained pyknotic nuclei, cell body shrinkage, and perineuronal vacuolization (arrows in Fig. 1B). At day 3, as the darkly stained nuclei increased in number, the neuropil in the background was more vacuolized and decomposed (Fig. 1B). On day 7, the area of ischemic core became hypercellular and packed with infiltrating immune cells, while the surrounding neuropil adopted a sponge-like appearance with a cavity (Fig. 1B). Tissue clearance of the ischemic core further progressed and the growing cavity areas remained at 1 month (Fig. 1B).
Immunostaining with the anti-NeuN antibody showed perinuclear immunoreactivity of the sham-operated animals (Fig. 1C). NeuN staining revealed that neuronal degeneration began at 0 h in the striatum and at 2 h in the cortex. NeuN immunoreactivity was changed in the ischemic core similar to that observed in the H&E staining until day 3 (Fig. 1C). Between day 7 and the end of 1 month, NeuN immunoreactivity completely disappeared in the ischemic tissues. The results are consistent with the previous finding that striatal infarction is mostly necrotic and occurs rapidly, while cortical infarction is more delayed and contains a greater degree of apoptotic cell death than observed in the striatal infarction [7,8].
GFAP immunostaining showed the typical staining of stellate, branched astrocytes in the sham group (Fig. 2). At 2 h after reperfusion, GFAP immunoreactivity began to decrease in the core region and eventually disappeared on day 7. Such rapid astrocytic death has been previously reported [9]. In the penumbra of the cortex, the GFAP immunoreactivity increased from 6 h and exhibited hypertrophied morphology between day 1 and 3. At day 7, astrocytes in the penumbra of the cortex were densely packed and formed a stream with elongated (straight) processes pointing toward the ischemic core, i.e., a feature of astroglial scar formation. At 1 month, the astrocytes exhibited overlapping processes in the compact scar border, suggesting the maturation of astroglial scar tissue (Fig. 2).
Fig. 2. Immunohistochemistry for GFAP in the ischemic rat brain. GFAP immunoreactivity started to decrease in the core region from 2 h reperfusion and disappeared on day 7. Astrocytes in the penumbra formed astroglial scar between day 7 and 1 month. The boxed areas in the full-size brain images are magnified in the representative image. Scale bar, 20 µm.

Activated microglia have been well established to transform into phagocytic cells that release a variety of cytotoxic substances following brain injury, which subsequently induce infiltration of circulating lymphocytes and macrophages [10]. In the sham group, Iba1-immunostaining showed resting microglia with highly branched, thin, and elongated processes (Fig. 3A). Within 1 day after ischemic injury, microglia underwent morphological changes to reach an activated amoeboid state in the core and penumbra rather than just an increase in number (Fig. 3B). In the subacute phase (days 3~7), most microglia in the lesion sites were strongly activated with round, amoeboid shapes, which are indistinguishable from those of infiltrating macrophages. The mixed microglia/macrophage population indicates the breakdown of BBB and massive infiltration of circulating immune cells. At 1 month, microglia were predominantly found in the penumbra as Iba1-positive cells (Fig. 3A). Quantitative analysis indicated that the Iba1 expression in the penumbra prominently increased from day 3, peaked at day 7, and then gradually declined at 1 month (Fig. 3B).
CD68 is a common marker for reactive microglia and macrophages [11]. Immunostaining with anti-CD68 antibody further supported our finding of microglial activation in the core and penumbra (compared in Fig. 3A and C). Unlike Iba1-staining, CD68 immunoreactivity was barely detectable in the sham-operated animals but became evident at 2~6 h after reperfusion in all affected areas, where Iba1-positive microglia displayed amoeboid morphology (Fig. 3C). In the subacute phase (days 3~7), CD68-positive cells appeared as a mixed microglia/macrophage population in the core and penumbra. Quantitative analysis indicated that the number of CD68-positive cells steadily increased up to day 7 in the core and penumbra (Fig. 3D). At 1 month when most ischemic tissues dissolved, the number of CD68-positive cells decreased to the level similar to that at 6 h (Fig. 3D).
Next, we analyzed the spatiotemporal distribution of cell death-related proteins following ischemic stroke. Apoptosis can be triggered by intracellular (caspase-independent) and extracellular (caspase-dependent) stimuli. The intracellular pathway relies on the mitochondrial outer membrane permeabilization, resulting in the release of a number of proapoptotic proteins into the cytosol, including AIF. In the extracellular pathway, binding of ligands to a death receptor leads to the activation of caspase-8, which can directly activate caspase-3. FAS-associated factor 1 (FAF1) enhances FAS-mediated apoptosis via the interaction with caspase-8 and FAS-associated death domain protein [12,13]. Activation of caspase-3 results in mitochondrial membrane permeabilization, chromatin condensation, DNA fragmentation, and eventually cell death [1,2,3].
Activated caspase-3 was minimally detected in the sham group, but egan to increase from 0 h reperfusion in the core regions of the striatum and cortex (Fig. 4A). The TUNEL-positive cells were not detected in the sham group but appeared in the striatum and cortex at 0 h (Fig. 4C). Quantitative analyses indicated that both activated caspase-3- and TUNEL-positive cells were gradually increased, peaked on day 1, and then decreased from day 7 onwards (Fig. 4C and D). The expression of these two death markers eventually decreased to the levels of sham group at 1 month.
Fig. 4. Spatiotemporal profiles of the activated caspase-3 and TUNEL in the ischemic rat brain. (A) Immunostaining for activated caspase-3. Levels of caspase-3 activation in the core were significantly increased at 6 h and remained elevated 3 days after reperfusion. (B) Quantitative measurement of activated caspase-3 in the boxed area of 1 mm2. (C) The TUNEL-positive cells were not detected in the sham group but increased from 0 h reperfusion. The boxed areas in the full-size brain images are magnified to the representative image. (D) Quantitative measurement of TUNEL-positive cells in the boxed area of 1 mm2. Data are presented as means±SEM per mm2 obtained from four animals per group (*p<0.05, ***p<0.001, compared to the value of the sham group; #p<0.05, ###p<0.001, compared to the values of the 0 h time point). Scale bar, 20 µm.

Similarly, AIF immunoreactivity was not detectable in the sham group, but first detected at 0~2 h only in the striatum (Fig. 5A). The AIF immunoreactivity significantly increased between day 1 and 3 in both the striatum and cortex. Such immunoreactivity decreased from day 7 and no significant immunoreactivity was found in the core region at 1 month. By contrast, AIF immunoreactivity was mainly observed in the cytoplasm in the penumbra from day 1 after reperfusion and was observed in the astroglia-enriched region at 1 month (Fig. 5A).
Fig. 5. Spatiotemporal profiles of AIF and FAF-1 in the ischemic rat brain. (A) The AIF-immunoreactivity increased from day 1 to day 3 and decreased slightly at 7 days after reperfusion. (B) FAF1 was found in the cytoplasm of the sham group but immediately found in the condensed cells/nuclei in the striatum (arrowheads) at 0 h. As the ischemic damages progressed, FAF1 immunoreactivity was diffused into the nuclei (arrows). The boxed areas in the full-size brain images are magnified in the representative image. Scale bar, 20 µm.

By comparison, FAF-1 immunoreactivity was detected in the cytoplasm in the sham group but immediately found in condensed cells/nuclei (arrowheads in Fig. 5B) in the striatum from 0 h. FAF-1 immunoreactivity became diffused to the nuclei at very early time points at 0~2 h after the reperfusion in the cortex and then remained in the condensed nuclei (arrows in Fig. 5B). In the penumbra, FAF-1 was localized to the cytoplasm in most cells, while nuclear FAF-1 was detected in some cells (arrows in Fig. 5B). The data indicate that damage to the brain can be evaluated based on the appearance of immunoreactivity against cell death-related molecules in animal models of stroke.
DISCUSSION
Numerous studies have shown histological lesions with MR images, however, most are limited to specific time and regions. Additionally, there have been no systematic studies on newly discovered cell death-related factors with respect to the images of whole brain lesions. Establishing the spatial and temporal distribution of neuronal necrosis should provide useful guidelines for time limits during which therapeutic intervention may be efficacious.
Ischemic stroke triggers a cascade of events leading to rapid neuronal damage and death [1,2]. At very early time points, such as immediately after 2-h of occlusion (0 h reperfusion), when the infarct is not evident in TTC-stained slices, neuronal degeneration proven by the presence of pyknotic nuclei starts in the striatum (Fig. 1A and B). The infarct progresses to the dorsolateral cortex overlying the striatum at 6 h. Our results are consistent with previous findings that striatal infarction is mostly necrotic and occurs rapidly owing to the nature of lenticulostriate artery, i.e., the lack of collateral circulation, whereas cortical infarction is slightly delayed due to the collateral circulation [7,8].
Activated caspase-3 and TUNEL-positive cells were significantly increased in both the striatum and cortex from an early time point (0 h reperfusion). By comparison, elevation of AIF- and nuclear FAF1-positive cells was detected only in the striatum at 0 h and then in the cortex in the delayed time period. AIF is generally believed to be present in the mitochondria of virtually every type of mammalian cells tested [14,15], and AIF-induced cell death processes are known to be associated with its translocation from the mitochondria to nuclei [16]. Compared to such in vitro studies, immunohistochemical studies on the AIF expression in the brain have been limited. AIF is barely detectable in the normal brain but its expression is dramatically increased in the nuclei in the injured brain [17]. In contrast to such immunohistochemical data, western analysis has revealed the presence of AIF in the cytosolic fraction in the normal brain and its nuclear localization after brain injury [18]. We also could not detect the AIF in the sham group with the antibody that could recognize AIF in the cytoplasm of HeLa cells (Fig. 5A) (Supplementary Fig. 1). Thus, the apparent absence of AIF immunoreactivity in the sham group suggests that AIF in the cytosolic/mitochondrial compartment is masked in the normal brain or was inaccessible under our experimental conditions. FAF1 is also known to be localized in the mitochondrial compartment in the normal condition but it translocates into the nucleus owing to oxidative stress, where it binds to Poly [ADP-ribose] polymerase 1 (PAPR1) to positively regulate the enzymatic activity of the PARP1 both at the cellular level and in a mouse model of PD [19]. Unlike AIF, FAF1 is detectable in the cytoplasm in the sham group but it translocates to the nuclear compartment at 0 h in the infarct core (arrows) and pyknotic nuclei (arrowheads) (Fig. 5B). The subcellular location of FAF1 can be a useful tool for assessing the severity of early cell death in animal stroke models. Taken together, the emergence of immunoreactivity for cell death-related molecules precedes morphological changes in Iba1-positive microglia observed at 6 h. The results are consistent with the previous report that activated caspase-8 and caspase-3/7 trigger microglia activation [20].
The only causal therapy for ischemic stroke is endovascular recanalization of the occluded vessels and reperfusion of ischemic brain parenchyma using thrombolysis agents and thrombectomy devices [21]. Even if cerebral blood flow resumes quickly enough to prevent subsequent cell death, a large population of initially surviving neurons may die within the first few hours after reperfusion. Based on the understanding of pathophysiological changes with respect to the post-ischemic time, antiapoptotic drugs may be beneficial in prohibiting the propagation of apoptosis if given early enough, and administration of anti-inflammatory drugs may be essential to block the secondary inflammatory damages caused by earlier cell death.
Stem cell-based approaches are promising potential therapies to restore brain function after ischemic stroke. Stem cells could exert their neuroprotective effects through anti-inflammation, anti-apoptosis, promotion of angiogenesis and neurogenesis, and formation of new neuronal circuitry [22]. Many laboratories including ours have shown that stem cells can effectively treat stroke via anti-inflammatory functions if given at postischemic day 3 [23]. Our study indicates the anti-inflammatory function of stem cells is effective on day 3 when circulating CD68 + cells are heavily infiltrated into the ischemic nucleus (Fig. 3). Although clinical and preclinical studies on ischemic stroke have produced promising results, several questions remain regarding the possible mechanisms of action of the stem cells and the optimal treatment protocol including the administration route and timing. These parameters may vary with respect to the transplantable cell types [24,25] and the pathogenic microenvironment in the ischemic brain [26,27]. With the rapid development of stem cell therapy, the Stem Cells as an Emerging Paradigm in Stroke (STEPS) committees have set out guidelines focusing on preclinical studies that are considered important parts of a development program to support clinical testing on cell therapies [28]. In STEP3, updated in 2014, the guideline for clinical studies on stem cell therapies for chronic stroke was added [29]. For the treatment of chronic stroke, STEPS3 suggested that the efficacy of stem cell treatment should be proven in animals for at least one month after ischemic injury. From the perspective that the microenvironment of the brain at the chronic stage is destructive, stem cell therapy should at least aim to improve the microenvironment of the brain at the chronic stage. Our results provide information on the spectral changes in the ischemic brain from disease onset to the chronic phase.
ACKNOWLEDGEMENTS
This research was supported by grants from the National Research Foundation (NRF-2015M3A9B4067067; and NRF-2017R1D1A1B03030656); and the Ministry of Food and Drug Safety (18172MFDS182). We would like to thank the Center for Neuroscience Imaging Research (Sungkyunkwan University, Suwon, Korea) for allowing us to use the MRI device. The authors declare no conflicts of interest. All authors have reviewed the contents of the manuscript being submitted, approved of its contents, and validated the accuracy of the data.
SUPPLEMENTARY MATERIALS
Subcellular localization of AIF in HeLa cells.
H&E staining of MCAO/R brain.
NeuN immunostaining of MCAO/R brain.
GFAP immunostaining of MCAO/R brain.
Iba1 immunostaining of MCAO/R brain.
CD68 immunostaining of MCAO/R brain.
Cleaved caspase-3 immunostaining of MCAO/R brain.
TUNEL staining of MCAO/R brain.
AIF immunostaining of MCAO/R brain.
FAF1 immunostaining of MCAO/R brain.
References
- 1.Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331–e339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- 2.Heiss WD. The ischemic penumbra: correlates in imaging and implications for treatment of ischemic stroke. The Johann Jacob Wepfer Award 2011. Cerebrovasc Dis. 2011;32:307–320. doi: 10.1159/000330462. [DOI] [PubMed] [Google Scholar]
- 3.Li V, Bi X, Szelemej P, Kong J. Delayed neuronal death in ischemic stroke: molecular pathways. In: Balestrino M, editor. Advances in the preclinical study of ischemic stroke. Rijeka: In Tech; 2012. pp. 117–144. [Google Scholar]
- 4.Leigh R, Knutsson L, Zhou J, van Zijl PC. Imaging the physiological evolution of the ischemic penumbra in acute ischemic stroke. J Cereb Blood Flow Metab. 2017 doi: 10.1177/0271678X17700913. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen F, Suzuki Y, Nagai N, Peeters R, Marchal G, Ni Y. Dynamic susceptibility contrast-enhanced perfusion MR imaging at 1.5 T predicts final infarct size in a rat stroke model. J Neurosci Methods. 2005;141:55–60. doi: 10.1016/j.jneumeth.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 6.Kim SS, Yoo SW, Park TS, Ahn SC, Jeong HS, Kim JW, Chang DY, Cho KG, Kim SU, Huh Y, Lee JE, Lee SY, Lee YD, Suh-Kim H. Neural induction with neurogenin1 increases the therapeutic effects of mesenchymal stem cells in the ischemic brain. Stem Cells. 2008;26:2217–2228. doi: 10.1634/stemcells.2008-0108. [DOI] [PubMed] [Google Scholar]
- 7.Garcia JH, Liu KF, Ho KL. Neuronal necrosis after middle cerebral artery occlusion in Wistar rats progresses at different time intervals in the caudoputamen and the cortex. Stroke. 1995;26:636–643. doi: 10.1161/01.str.26.4.636. [DOI] [PubMed] [Google Scholar]
- 8.Carmichael ST. Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx. 2005;2:396–409. doi: 10.1602/neurorx.2.3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu D, Smith CL, Barone FC, Ellison JA, Lysko PG, Li K, Simpson IA. Astrocytic demise precedes delayed neuronal death in focal ischemic rat brain. Brain Res Mol Brain Res. 1999;68:29–41. doi: 10.1016/s0169-328x(99)00063-7. [DOI] [PubMed] [Google Scholar]
- 10.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87:779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perego C, Fumagalli S, De Simoni MG. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J Neuroinflammation. 2011;8:174. doi: 10.1186/1742-2094-8-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryu SW, Lee SJ, Park MY, Jun JI, Jung YK, Kim E. Fas-associated factor 1, FAF1, is a member of Fas death-inducing signaling complex. J Biol Chem. 2003;278:24003–24010. doi: 10.1074/jbc.M302200200. [DOI] [PubMed] [Google Scholar]
- 13.Park MY, Ryu SW, Kim KD, Lim JS, Lee ZW, Kim E. Fas-associated factor-1 mediates chemotherapeutic-induced apoptosis via death effector filament formation. Int J Cancer. 2005;115:412–418. doi: 10.1002/ijc.20857. [DOI] [PubMed] [Google Scholar]
- 14.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 15.Daugas E, Nochy D, Ravagnan L, Loeffler M, Susin SA, Zamzami N, Kroemer G. Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 2000;476:118–123. doi: 10.1016/s0014-5793(00)01731-2. [DOI] [PubMed] [Google Scholar]
- 16.Piacentini M, Farrace MG, Piredda L, Matarrese P, Ciccosanti F, Falasca L, Rodolfo C, Giammarioli AM, Verderio E, Griffin M, Malorni W. Transglutaminase overexpression sensitizes neuronal cell lines to apoptosis by increasing mitochondrial membrane potential and cellular oxidative stress. J Neurochem. 2002;81:1061–1072. doi: 10.1046/j.1471-4159.2002.00898.x. [DOI] [PubMed] [Google Scholar]
- 17.Cao G, Clark RS, Pei W, Yin W, Zhang F, Sun FY, Graham SH, Chen J. Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J Cereb Blood Flow Metab. 2003;23:1137–1150. doi: 10.1097/01.WCB.0000087090.01171.E7. [DOI] [PubMed] [Google Scholar]
- 18.Sabirzhanov B, Stoica BA, Zhao Z, Loane DJ, Wu J, Dorsey SG, Faden AI. miR-711 upregulation induces neuronal cell death after traumatic brain injury. Cell Death Differ. 2016;23:654–668. doi: 10.1038/cdd.2015.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu C, Kim BS, Kim E. FAF1 mediates regulated necrosis through PARP1 activation upon oxidative stress leading to dopaminergic neurodegeneration. Cell Death Differ. 2016;23:1873–1885. doi: 10.1038/cdd.2016.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A, Cano J, Brundin P, Englund E, Venero JL, Joseph B. Caspase signalling controls microglia activation and neurotoxicity. Nature. 2011;472:319–324. doi: 10.1038/nature09788. [DOI] [PubMed] [Google Scholar]
- 21.Linfante I, Cipolla MJ. Improving reperfusion therapies in the era of mechanical thrombectomy. Transl Stroke Res. 2016;7:294–302. doi: 10.1007/s12975-016-0469-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marei HE, Hasan A, Rizzi R, Althani A, Afifi N, Cenciarelli C, Caceci T, Shuaib A. Potential of stem cell-based therapy for ischemic stroke. Front Neurol. 2018;9:34–40. doi: 10.3389/fneur.2018.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoo SW, Chang DY, Lee HS, Kim GH, Park JS, Ryu BY, Joe EH, Lee YD, Kim SS, Suh-Kim H. Immune following suppression mesenchymal stem cell transplantation in the ischemic brain is mediated by TGF-β. Neurobiol Dis. 2013;58:249–257. doi: 10.1016/j.nbd.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 24.Hess DC, Wechsler LR, Clark WM, Savitz SI, Ford GA, Chiu D, Yavagal DR, Uchino K, Liebeskind DS, Auchus AP, Sen S, Sila CA, Vest JD, Mays RW. Safety and efficacy of multipotent adult progenitor cells in acute ischaemic stroke (MASTERS): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2017;16:360–368. doi: 10.1016/S1474-4422(17)30046-7. [DOI] [PubMed] [Google Scholar]
- 25.Steinberg GK, Kondziolka D, Wechsler LR, Lunsford LD, Coburn ML, Billigen JB, Kim AS, Johnson JN, Bates D, King B, Case C, McGrogan M, Yankee EW, Schwartz NE. Clinical outcomes of transplanted modified bone marrow-derived mesenchymal stem cells in stroke: a phase 1/2a study. Stroke. 2016;47:1817–1824. doi: 10.1161/STROKEAHA.116.012995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosado-de-Castro PH, Pimentel-Coelho PM, da Fonseca LM, de Freitas GR, Mendez-Otero R. The rise of cell therapy trials for stroke: review of published and registered studies. Stem Cells Dev. 2013;22:2095–2111. doi: 10.1089/scd.2013.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kenmuir CL, Wechsler LR. Update on cell therapy for stroke. Stroke Vasc Neurol. 2017;2:59–64. doi: 10.1136/svn-2017-000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Savitz SI, Chopp M, Deans R, Carmichael T, Phinney D, Wechsler L STEPS Participants. Stem cell therapy as an emerging paradigm for stroke (STEPS) II. Stroke. 2011;42:825–829. doi: 10.1161/STROKEAHA.110.601914. [DOI] [PubMed] [Google Scholar]
- 29.Savitz SI, Cramer SC, Wechsler L STEPS 3 Consortium. Stem cells as an emerging paradigm in stroke 3: enhancing the development of clinical trials. Stroke. 2014;45:634–639. doi: 10.1161/STROKEAHA.113.003379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Subcellular localization of AIF in HeLa cells.
H&E staining of MCAO/R brain.
NeuN immunostaining of MCAO/R brain.
GFAP immunostaining of MCAO/R brain.
Iba1 immunostaining of MCAO/R brain.
CD68 immunostaining of MCAO/R brain.
Cleaved caspase-3 immunostaining of MCAO/R brain.
TUNEL staining of MCAO/R brain.
AIF immunostaining of MCAO/R brain.
FAF1 immunostaining of MCAO/R brain.