

Abstract
Background & Aims
Antibiotic (ABx) therapy is associated with increased risk for Crohn’s disease but underlying mechanisms are unknown. We observed high fecal serine protease activity (PA) to be a frequent side effect of ABx therapy. The aim of the present study was to unravel whether this rise in large intestinal PA may promote colitis development via detrimental effects on the large intestinal barrier.
Methods
Transwell experiments were used to assess the impact of high PA in ABx-treated patients or vancomycin/metronidazole-treated mice on the epithelial barrier. Serine protease profiling was performed using liquid chromatography–mass spectrometry/mass spectrometry analysis. The impact of high large intestinal PA on the intestinal barrier in wild-type and interleukin (IL)10-/- mice and on colitis development in IL10-/- mice was investigated using vancomycin/metronidazole with or without oral serine protease inhibitor (AEBSF) treatment.
Results
The ABx-induced, high large intestinal PA was caused by significantly increased levels of pancreatic proteases and impaired epithelial barrier integrity. In wild-type mice, the rise in PA caused a transient increase in intestinal permeability but did not affect susceptibility to chemically induced acute colitis. In IL10-/- mice, increased PA caused a consistent impairment of the intestinal barrier associated with inflammatory activation in the large intestinal tissue. In the long term, the vancomycin/metronidazole-induced lasting increase in PA aggravated colitis development in IL10-/- mice.
Conclusions
High large intestinal PA is a frequent adverse effect of ABx therapy, which is detrimental to the large intestinal barrier and may contribute to the development of chronic intestinal inflammation in susceptible individuals.
Keywords: Epithelial Barrier, Serine Proteases, Gut Microbiota, Inflammatory Bowel Diseases
Abbreviations used in this paper: ABx, antibiotics; AEBSF, 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride; cecal-sup, cecal-supernatants; ctr, control; DSS, dextran sulfate sodium; GF, germ-free; IBD, inflammatory bowel diseases; IL, interleukin; LC-MS/MS, liquid chromatography–mass spectrometry/mass spectrometry; PA, protease activity; PBS, phosphate-buffered saline; PMSF, phenylmethane-sulfonylfluoride; stool-sup, stool-supernatants; SPF, specific pathogen-free; TEER, transepithelial electrical resistance; V/M, vancomycin/metronidazole; WT, wild-type
Graphical abstract

See editorial on page 347.
Summary.
Antibiotic therapies eliminate antiproteolytic bacteria in the large intestine, resulting in an increase in proteolytic activity, which is detrimental to the epithelial barrier function. In inflammatory bowel disease–susceptible individuals, high proteolytic activity promotes colitis development in the long term.
Inflammatory bowel diseases (IBD), including Crohn's disease and ulcerative colitis, are idiopathic diseases that are thought to be the consequence of a dysregulated intestinal immune response toward the intestinal microbiota.1, 2, 3 IBD are characterized by chronic inflammation, impaired intestinal barrier function, and microbial dysbiosis.4, 5, 6, 7 The highly complex etiology of IBD, comprising approximately 200 susceptibility loci8 and abundant environmental factors,9 results in an individual-specific pathogenesis from unknown initial triggers to the manifestation of the chronic inflammation. Interestingly, recent studies suggest a critical role of the microbial ecosystem in the initiation and progression of IBD.10, 11, 12, 13
The intestinal microbiota of healthy individuals consists of highly diverse communities of microbes performing important functions, such as the production of short-chain fatty acids, metabolism of bile acids,14 pathogen exclusion,15 modulation of the intestinal immune system,16 and degradation of pancreatic proteases in the large intestine.17 In consequence, antibiotic (Abx)- or infection-induced microbial dysbiosis may exert a significant impact on the onset and progression of chronic metabolic or chronic inflammatory diseases in susceptible organisms.18, 19, 20, 21 Perturbations of the intestinal microbial ecosystem by ABx therapies were found to be associated with functional intestinal disorders, such as irritable bowel syndrome,22, 23 colorectal cancer,24, 25 overweight, and asthma.26 In the context of IBD, several clinical studies have already revealed that early and frequent ABx therapies, especially metronidazole or fluoroquinolone treatments, are associated with increased risk for Crohn's disease.27, 28 However, the causal role of ABx therapies in the disease development and the mechanisms underlying this potential serious long-term adverse effect of ABx on the intestinal immune homeostasis remain unknown.
Interestingly, it has long been known that ABx treatments can result in increased luminal serine protease activity (PA) in the large intestine of rodents29, 30 or patients.31, 32 This increase in PA is assumed to be caused by the ABx-mediated eradication of yet unknown intestinal bacteria that normally inactivate the high load of pancreatic proteases on entry of the chyme into the large intestine.33, 34 The finding that ex vivo exposure of murine colonic mucosa to high PA fecal supernatants from patients with diarrhea-predominant irritable bowel syndrome can induce a serine protease-dependent increase of the mucosal permeability35 indicates that the ABx-mediated rise in PA may be rather detrimental to the large intestinal barrier. In the context of IBD, reduced intestinal barrier functions and increased translocation of luminal antigens into the mucosal tissue are known to promote chronic inflammation.36, 37, 38, 39 In view of these data, we hypothesized that the ABx-increased PA in the large intestine is a relevant risk factor for the development of colitis in susceptible organisms.
Methods
Ethics Statement
The breeding and experimental use of mice in the animal facilities of the Technische Universität München (School of Life Sciences Weihenstephan) was approved by the local institution in charge (Regierung von Oberbayern; approval number 55.2-1-54-2531-99-13 and 55.2-1-54-2532-17-2015).
Stool Sample Collection From Patients
Samples were collected before and after ABx therapy at the Capio Hospital General de Catalunya in Barcelona40 and the Medical University of Graz (ethical approval number 17-199 ex 05/06) and stored at -20°C until analysis. Patient characteristics are summarized in Table 1.
Table 1.
Patient Characteristics
| Classification | ABx name | ABx therapy duration, d | Indication | Fold increase in PA | Diarrhea (after ABx) |
|---|---|---|---|---|---|
| Fluoroquinolone (+Imidazole) | Levofloxacin | 7 | Bronchitis | 7.57 | No |
| Levofloxacin | 7 | Bronchitis | 0.30 | No | |
| Levofloxacin | 7 | Bronchitis | 3.04 | No | |
| Levofloxacin | 7 | Bronchitis | 2.35 | No | |
| Levofloxacin | 7 | Bronchitis | 5.50 | No | |
| Levofloxacin | 7 | Bronchitis | 1.83 | No | |
| Ciprofloxacin | 7 | UC | 13.59 | No | |
| Levofloxacin | 7 | Bronchitis | 0.32 | No | |
| Levofloxacin+metronidazole | 7 | Clostridium difficile infection | 8.27 | No | |
| β-Lactam | Amoxicillin+clavulanate | 7 | Bacteremia | 12.74 | No |
| Amoxicillin+clavulanate | 7 | Bronchitis | 0.96 | No | |
| Amoxicillin+clavulanate | 7 | Bacteremia | 1.43 | No | |
| Amoxicillin+clavulanate | 7 | Bronchitis | 1.02 | No | |
| Amoxicillin+clavulanate | 7 | Urinary infection | 2.23 | No | |
| Amoxicillin+clavulanate | 7 | Bronchitis | 0.26 | No | |
| Amoxicillin+clavulanate | 7 | Bronchitis | 0.37 | No | |
| Cephalosporin (+macrolide) | Azithromycin | 2 | CD | 6.10 | No |
| Ceftriaxone+azithromycin | 7 | Pneumonia | 0.41 | No | |
| Ceftriaxone+azithromycin | 7 | Bronchitis | 1.29 | No | |
| Ceftriaxone | 7 | Urinary infection | 0.51 | No | |
| Rifamycin | Rifaximin | 3 | IBS | 10.49 | No |
| Rifaximin | 3 | IBS | 0.34 | No | |
| Rifaximin | 3 | IBS | 0.45 | No | |
| Rifaximin | 3 | IBS | 2.49 | No | |
| Rifaximin | 3 | IBS | 2.17 | No | |
| Rifaximin | 3 | IBS | 0.82 | Yes | |
| Rifaximin | 3 | IBS | 0.77 | No | |
| Rifaximin | 3 | IBS | 1.85 | No | |
| Rifaximin | 3 | IBS | 0.92 | Yes | |
| Rifaximin | 3 | IBS | 0.73 | Yes | |
| Rifaximin | 3 | IBS | 0.64 | Yes | |
| Rifaximin | 3 | IBS | 5.36 | No |
ABx, antibiotic; CD, Crohn’s disease; IBS, inflammatory bowel syndrome; PA, protease activity; UC, ulcerative colitis.
Experimental Design (Wild-Type and Interleukin 10-/- Mice)
In all experiments, mice were fed with vancomycin/metronidazole (V/M)-containing chow mash or control (ctr) mash ad libitum for the indicated period of time. V/M mash (Fluka, Seelze, Germany) was prepared by mixing a vancomycin (0.25 g/L; Fluka)/metronidazole (1.0 g/L; Sigma-Aldrich, Steinheim, Germany) solution with chow powder (ssniff-Spezialdiäten GmbH, Soest, Germany) in a 1:1 volume (mL)/weight (g) ratio.
In the short-term experiments, 8-week-old wild-type (WT) (C57BL/6, male and female) or interleukin (IL) 10-/- mice (129/SvEv, male and female) (n ≥5/group) were treated with V/M for either 2 days, 7 days, or left untreated. The protease inhibitor 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (300 mg/kg body weight) (AEBSF; Sigma-Aldrich) or water was applied by oral gavage starting from 1 day before V/M treatment where indicated. Fresh fecal pellets were sampled daily from all mice. Fluorescein isothiocyanate dextran (4 kDa; Sigma-Aldrich) was applied by oral gavage 4 hours before mice were sacrificed by CO2.
In the long-term experiments, WT (male) and IL10-/- mice (male and female) (n ≥5/group) were treated with V/M for 7 days at the age of 4 and 8 weeks or left untreated. Fresh fecal pellets were sampled on a weekly basis. At 12 weeks of age, WT mice were exposed to 1.5% dextran sulfate sodium (DSS; Sigma-Aldrich) in drinking water for 7 days and sacrificed at Day 9. The severity of DSS-induced colitis was monitored by daily determination of the disease activity index. From IL10-/- mice, facial blood was collected every second week from the age of 10–16 weeks. IL10-/- mice were sacrificed at 16 weeks of age.
Colonization of Germfree Mice
Cecal microbiota from untreated or V/M-treated specific pathogen-free (SPF) mice was prepared as described12 and orally administered to germ-free (GF) mice (10 weeks of age). Mice were sacrificed 2 weeks after the transfer.
Preparation of Cecal Content/Stool-Supernatants
Cecal content/stool-supernatants (cecal-sup/stool-sup, 10% wt/vol) were generated from the cecal contents of WT, IL10-/- mice, or patient stool. A total of 100 mg of patient stool or murine gut contents from the ileum, cecum, colon, or feces were homogenized and dissolved in sterile phosphate-buffered saline (PBS, 10% wt/vol) using sterile glass beads and vigorous vortexing. Supernatant was generated by a 2-step centrifugation (5 min, 1000 rpm, 4°C) followed by centrifugation of the resulting supernatant (10 min, 6000 rpm, 4°C). Whenever indicated, the supernatants were incubated with the serine protease inhibitor phenylmethane-sulfonylfluoride (PMSF; 5 mM; Sigma-Aldrich). The supernatants were stored at -20°C.
Protease Activity Assay
The tryptic activity in gut content supernatants was analyzed using the Protease Assay Kit (Calbiochem, EMD Millipore Corp, Billerica, MA) according to the manufacturerś instructions. In short, 10 μL of the respective supernatant were incubated with FTC-casein solution (1 hour, 37°C). Fluorescence was subsequently measured using an Infinite 200 PRO plate reader (Tecan Group Ltd, Männedorf, Switzerland).
Visualization of Active Serine Proteases
Gut content supernatants were incubated with ActivX TAMRA-FP Probes (1 μM; Thermo Fisher Scientific, Rockford, IL) (30 min, RT, in the dark). The mixture was diluted with 2X sodium dodecyl sulfate buffer, boiled (5 min, 95°C), and subjected to gel electrophoresis on 15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Labelled proteins were detected using a Typhoon Trio+ scanner (GE Health Care Science, Buckinghamshire, UK).
Ex Vivo Incubation Assay
Fresh cecal bacteria (1 × 109) isolated from SPF- or V/M-treated mice were incubated with cecal-sup (50 μL) from GF mice for 24 hours at 37°C. To heat-inactivate bacteria, bacteria were boiled for 20 minutes at 95°C before incubation with cecal-sup. After the incubation for 24 hours, 10 μL of the supernatants were used for the PA assay.
Liquid Chromatography–Mass Spectrometry/Mass Spectrometry Analysis
Proteome analysis of cecal-sup from ctr, V/M-treated, and GF mice was performed using liquid chromatography–mass spectrometry/mass spectrometry (LC-MS/MS) analysis. Cecal-sup was diluted 1:1 with 2×NuPAGE LDS Sample Buffer (Invitrogen, Darmstadt, Germany) and proteins were reduced by 10 mM dithiothreitol and alkylated by 55 mM iodoacetamide. Samples were then run into a 4%–12% NuPAGE gel (Invitrogen) for about 1 cm to concentrate the sample before in-gel tryptic digestion. In-gel trypsin digestion was performed according to standard procedures.41
Peptides generated by in-gel trypsin digestion were dried in a vacuum concentrator and then dissolved in 20 μL 0.1% formic acid before LC–MS/MS analysis. LC–nanoESI-MS/MS was performed by coupling a nanoLC-Ultra (Eksigent Technologies, Dublin, CA) to an LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, Schwerte, Germany). For each analysis, 10 μL of dissolved peptides was delivered to a trap column (ReproSil-pur C18-AQ, 5 μm, Dr. Maisch, Ammerbuch, Germany, 20 mm × 75 μm, self-packed) at a flow rate of 5 μL/min in 100% solvent A (0.1% formic acid in high-performance liquid chromatography grade water). After 10 minutes of loading and washing, peptides were transferred to an analytical column (ReproSil-gold C18-AQ, 3 μm, Dr. Maisch, Ammerbuch, Germany, 400 mm × 75 μm, self-packed) and separated using a 210-minute gradient from 2% to 32% of solvent B (0.1% formic acid in acetonitrile) at 300 nL/min flow rate. Both LC solvents contained 5% (vol/vol) dimethyl sulfoxide to boost the nanoESI response of peptides.42 The LTQ Orbitrap Velos was operated in data-dependent mode, automatically switching between MS and MS/MS. Full-scan MS spectra (360–1300 m/z) were acquired in the Orbitrap at 30,000 resolution (at m/z 400) after accumulation of precursor ions to a target value of 1,000,000 for a maximum time of 100 ms. Internal lock mass calibration was performed using an ion signal (m/z 401.922720) present in dimethyl sulfoxide containing solvents.42 Tandem mass spectra were generated for up to 10 peptide precursors by higher energy collision-induced dissociation (target value of 40,000, max 100 ms accumulation time, isolation width 2.0 Th) at a normalized collision energy of 30% and fragment ions were recorded at a resolution of 7500 in the Orbitrap. To maximize the number of precursors targeted for analysis, dynamic exclusion was enabled with 1 repeat count in 10-second and 20-second exclusion time (mass tolerance ± 10 ppm).
Label-free quantification was performed using MaxQuant (version 1.5.3.30)43 by searching MS data against a mus musculus UniProt reference database (version 03.05.2016, 24864 entries) using the search engine Andromeda.44 Carbamidomethylated cysteine was used as fixed modification; variable modifications included oxidation of methionine and N-terminal protein acetylation. Trypsin/P was specified as proteolytic enzyme with up to 2 allowed miscleavage sites. Precursor tolerance was set to 4.5 ppm and fragment ion tolerance was set to 20 ppm. Label-free quantification,45 match-between-runs, and intensity-based absolute quantification options were enabled and results were filtered for a minimal length of 7 amino acids, 1% peptide, and protein FDR and reverse identifications.
MaxQuant results were imported into the MaxQuant associated software suite Perseus (v.1.5.6.0).46 iBAQ intensities were filtered for at least 2 valid values per experimental group and at least 2 peptides for identification per protein. Missing values were imputed from normal distribution (width 0.3, downshift 1.8). A 2-sided Student t test was performed to assess statistical significance. Protein P values were corrected for multiple testing using a permutation based 5 % FDR cutoff (250 randomizations, s0 of 3.37).
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD007914.
Colonic Epithelial Cell (PTK6 cell) Culture, Transepithelial Electrical Resistance Measurement, and Permeability Assay
PTK6 cells (colonic epithelial cells, 1.5 × 105 cells/well) were seeded on Transwells (polyester membrane with pore size of 0.4 μm, Corning Inc, Kennebunk, ME) and cultured with RPMI-1640 medium including insulin transferrin selenium A and fetal calf serum (5% CO2, 37°C) until the transepithelial electrical resistance (TEER) reached a value >1.0 kΩ. PTK6 cells were then stimulated with cecal-sup/stool-sup (10% wt/vol), which was prepared in RPMI-1640 medium without fetal calf serum on the apical side (fresh medium without fetal calf serum on basolateral side). TEER was measured on a heating block (Thermo Fisher Scientific) to maintain a constant temperature at 37°C. After the TEER measurements, the medium on the basolateral side was replaced by fresh Krebs buffer and the one on the apical side was replaced by fresh Krebs buffer supplemented with fluorescein (500 μg/mL). The PTK6 cells were subsequently incubated for 30 minutes at 37°C. The level of translocation of fluorescein through the epithelial layer was determined via the measurement of fluorescence in the Krebs buffer on the basolateral side (emission at 538 nm).
Ussing Chamber Analysis
The permeability and TEER of cecal/colonic tissue from V/M-treated/untreated WT or IL10-/- mice were measured by Ussing chamber analysis. Segments of the cecum and the distal colon (1 cm each) were cut open, gut contents were carefully removed, and the tissue was mounted in the Ussing chamber. The mounted tissues were connected to a voltage clamp apparatus and incubated in Krebs buffer (95% O2, 5% CO2, 37°C). The tissues were equilibrated for 20 minutes and TEER was measured (for 2 hours, every 30 minutes) by Acquire & Analyze 2.3 software. To assess the intestinal permeability, fluorescein (50 mg/mL) was loaded to the apical side after the TEER measurement. The Krebs buffer on the basolateral side was collected and the level of fluorescence was determined (emission at 538 nm).
To investigate the impact of high PA on large intestinal tissue from ctr SPF mice ex vivo, the mucosal side of the tissue sections was exposed to cecal-sup (10% wt/vol) from SPF or GF mice ± PMSF (5 mM) for 4 hours and TEER was measured every hour. To assess the impact of the respective cecal-sup on the permeability, fluorescein was added to the apical chamber for 30 minutes after the last TEER measurement and the level of basolateral fluorescein was measured as described previously.
Tissue Processing, Hematoxylin-Eosin, and Immunofluorescence Staining
Cecal and colonic tissue was fixed in formaldehyde (4%) and embedded in paraffin. For hematoxylin-eosin staining, the severed sections (4 μm) were stained with hematoxylin-eosin (0.2%) before mounting. The stained sections were visualized by Zeiss Axioskop 40 microscope (Zeiss).
For the immunofluorescence staining of CD3+ T cells, antigen unmasking was performed by boiling tissue sections (4 μm) in 10 mM citrate buffer (pH 6, 900 W, 23 minutes) in a pressure cooker. Then, the sections were incubated in blocking buffer including 5% rabbit serum for 1 hour at RT after a rinse with PBS. The sections were incubated overnight with the primary antibody (CD3, 1:200, Sigma-Aldrich) at 4°C and then rinsed with PBS. The sections were incubated with fluorescence-conjugated secondary antibodies (antirabbit AF594, 1:200, Life Technologies, Carlsbad, CA) for 1 hour at RT. All sections were counterstained with DAPI (1:2000, Sigma-Aldrich) and fluorescence measurements were performed using Flouview FV10i microscope (Olympus, Shinjuku, Japan).
Histopathology
Histopathologic inflammation was blindly assessed by an independent pathologist using an established scoring system (score from 0 to 12).47, 48 Images were acquired by Touch microscope V. precipoint (PreciPoint GmbH, Freising, Germany).
Disease Activity Index
The disease activity index in DSS-treated mice was determined by daily monitoring of weight loss, stool consistency, and occult blood (scores range from 0 to 4 as described in Table 2). The disease activity index is the average of these individual scores/mouse/day.
Table 2.
Disease Activity Score
| Score | Weight loss (%) | Stool consistency | Blood in stool |
|---|---|---|---|
| 0 | 0 | Normal | No blood |
| 1 | Lower than 5 | — | — |
| 2 | Lower than 10 | Slightly changed | Positive |
| 3 | Lower than 15 | — | Positive for 2 d |
| 4 | Lower than 20 | Diarrhea | Positive for more than 2 d |
Quantitative Real-Time Polymerase Chain Reaction
RNA from cecal and colonic tissue was extracted with RA1 buffer (Macherey-Nagel, Düren, Germany) using NucleoSpin RNAII kit (Macherey-Nagel GmbH). cDNA was synthesized using 500 ng total RNA, random hexamers, and MMLV reverse transcriptase Point Mutant Synthesis System (Promega, Fitchburg, WI). For the real-time polymerase chain reaction, the amplification was performed with Universal Probe Library system in a Light Cycler 480 system (Roche Diagnostics, Mannheim, Germany). The primers and probes are described in Table 3.
Table 3.
Primer Sequences and UPL Probe IDs for Quantitative Polymerase Chain Reaction Analysis
| Gene | Forward primer | Reverse primer | Probe |
|---|---|---|---|
| IFNγ | 5’-cctttggaccctctgacttg | 5’-agcgttcattgtctcagagcta | 63 |
| IL-1β | 5’-tgtaatgaaagacggcacacc | 5’-tcttctttgggtattgcttgg | 78 |
| C3 | 5’-accttacctcggcaagtttct | 5’-ttgtagagctgctggtcagg | 76 |
| CD3 | 5’-cttgtacctgaaagctcgagtg | 5’-gatgattatggctactgctgtca | 10 |
| GAPDH | 5’-tccactcatggcaaattcaa | 5’-tttgatgttagtggggtctcg | 9 |
Serum Amyloid A and Complement C3 Enzyme-Linked Immunosorbent Assay
Plasma serum amyloid A and fecal complement C3 levels were measured by enzyme-linked immunosorbent assay (Immunology Consultants Laboratory Inc, Portland, OR) according to the manufacturer instructions.
16S Ribosomal RNA Gene Amplicon Analysis
Metagenomic DNA from the cecal contents of repeatedly V/M-treated WT and IL10-/- mice was isolated using a modified protocol according to Godon et al.49 Briefly, snap-frozen samples were thawed on ice, mixed with 600 μL stool DNA stabilizer (Stratec, Beringen, Switzerland), and transferred into autoclaved 2-mL screw-cap tubes containing 500-mg, 0.1-mm diameter silica/zirconia beads. After addition of 250 μL 4 M guanidine thiocyanate in 0.1 M Tris (pH 7.5) and 500 μL 5 % N-lauroyl sarcosine in 0.1 M PBS (pH 8.0), samples were incubated at 70°C and 700 rpm for 60 minutes. A FastPrep instrument (MP Biomedicals, Santa Ana, CA) fitted with a 24 × 2 mL cooling adaptor filled with dry ice was used for cell disruption. The program was run 3 times for 40 seconds at 6.5 M/s. After each run, the cooling adapter was refilled with dry ice. An amount of 15 mg polyvinylpyrrolidone was added and samples were vortexed, followed by 30-minute centrifugation at 15.000 × g and 4°C. Approximately 650 μL of the supernatant were transferred into a new 2-mL tube, which was centrifuged again for 3 minutes at 15.000 × g and 4°C. Subsequently, 500 μL of the supernatant was transferred into a new 2-mL tube and 50 μg of RNase was added. After 20 minutes at 37°C and 700 rpm, gDNA was isolated using the NucleoSpin gDNA Clean-up Kit from Macherey-Nagel according to the manufacturer protocol. DNA was eluted from columns twice using 40 μL Elution buffer and concentration was measured with NanoDrop (Thermo Fisher Scientific). Samples were stored at -20°C.
Library preparation and sequencing were performed as described in detail previously.50 Samples were processed in a semiautomated manner using a Biomek-4000 robot (Beckman-Coulter, Brea, CA). Briefly, the V3-V4 region of 16S rRNA genes was amplified (25 cycles) following a 2-step protocol51 using primers 341F-785R.52 After purification using the AMPure XP system (Beckmann), sequencing was carried out with pooled samples in paired-end modus (PE275) using a MiSeq system (Illumina, San Diego, CA) according to the manufacturer instructions and 25% (vol/vol) PhiX standard library.
Data were analyzed as described in detail previously.50 Raw reads were processed using an in-house developed pipeline (www.imngs.org)53 based on UPARSE.54 In brief, sequences were demultiplexed, trimmed to the first base with a quality score <3, and then paired. Sequences with less than 380 and more than 480 nucleotides and paired reads with an expected error >3 were excluded from the analysis. Remaining reads were trimmed by 10 nucleotides on each end to avoid GC bias and nonrandom base composition. The presence of chimeras was tested using UCHIME.55 Operational taxonomic units were clustered at 97% sequence similarity, and only those with a relative abundance >0.5% in at least 1 sample were kept. Taxonomies were assigned at 80% confidence level using the RDP classifier.56
Downstream analysis was performed in the R programming environment using Rhea (https://lagkouvardos.github.io/Rhea/).57 Operational taxonomic unit tables were normalized to account for differences in sequence depth. ß-diversity was computed based on generalized UniFrac distances.58 α-diversity was assessed on the basis of species richness and Shannon effective diversity.59 P values were calculated using analysis of variance on ranks and corrected for multiple comparisons according to the Benjamini-Hochberg method. Only taxa with a prevalence of at least 30% samples in one given group were considered for statistical analysis.
Statistics
Data are presented as mean ± SD. Statistical significance was determined by unpaired Student t test or analysis of variance with 1-way analysis of variance followed by Tukey posttest using Graph-Pad Prism (Graph Pad, version 6.01). P < .05 was considered as statistically significant.
Results
High Fecal Protease Activity in Antibiotic-Treated Patients Impairs the Epithelial Barrier
To assess the impact of frequently prescribed ABx therapies on the large intestinal PA, patient stool sampled before and after ABx treatments was analyzed, demonstrating a major (>5-fold) increase in PA in 25% of the investigated patients (Figure 1A, Table 1). Pattern analysis of active serine proteases revealed a major increase in low-molecular-weight proteases (<25 kDa) in the stool of these patients (Figure 1B), indicating high levels of pancreatic proteases, such as trypsin (∼23.5 kDa) and chymotrypsin (∼24.8 kDa) on ABx treatment. To assess the impact of the high stool PA on the epithelial barrier, polarized PTK6 cells were apically exposed to stool-sup from patients that showed a rise in fecal PA on ABx therapy. Whereas the epithelial barrier was not affected by exposure to the low PA present in stool-sup from patients before ABx therapy, the high PA in stool-sup from the same patient after ABx treatment significantly increased epithelial permeability and reduced TEER. The detrimental effect of high PA stool-sup from ABx-treated patients on the epithelial barrier was abrogated by prior serine protease inhibition via PMSF (Figure 1C). Of note, apical exposure to PMSF alone did not alter epithelial barrier functions (data not shown).
Figure 1.

High stool PA is a frequent adverse effect of ABx treatment in patients and impairs the epithelial barrier ex vivo. (A) Serine protease activity in stool-supernatant of patients before and after ABx treatment. Red line (n = 9), fluoroquinolone (+imidazole) antibiotic; blue line (n = 7), β-lactam antibiotic; green line (n = 4), cephalosporin (+macrolide) antibiotic; black line (n = 12), rifamycine antibiotic. (B) Staining of active serine proteases in stool-supernatants of patients before and after ABx treatment (TAMRA-FP sodium dodecyl sulfate–polyacrylamide gel electrophoresis). (C) Fluorescein translocation and TEER in polarized PTK6 cells on apical exposure to stool-supernatants from the same patient before versus after the respective ABx treatment ± PMSF-inhibition.
Eradication of Antiproteolytic Bacteria by Vancomycin/Metronidazole Treatment Results in Excessive Large Intestinal Protease Activity and Impairs the Epithelial Barrier Ex Vivo
To investigate the physiological relevance of ABx-increased PA, oral application of V/M was used in different experimental setups. V/M treatment of colonized mice resulted in large intestinal PA levels comparable with GF mice (Figure 2A), indicating highly efficient eradication of antiproteolytic bacteria by this ABx treatment. The V/M-mediated loss of anti-PA in the large intestinal microbiota was confirmed via an ex vivo incubation assay (Figure 2B) and gnotobiotic studies (Figure 2C). In the latter case, GF mice exposed to cecal microbiota from SPF mice showed normalized low PA levels 2 weeks after colonization, whereas cecal microbiota from V/M-treated mice was not able to mediate this effect.
Figure 2.

Eradication of antiproteolytic bacteria by V/M treatment results in excessive large intestinal PA and impairs the epithelial barrier ex vivo. (A) Comparison of the level of luminal serine protease activity in the ileum (il), cecum (ce), colon (co), and feces (fe) of untreated SPF (ctr), V/M-treated (V/M), and GF mice. (B) Bacteria were isolated from fresh cecum content of SPF or V/M-treated mice, and cecal-sup from GF mice was incubated for 24 hours with buffer (ctr), the respective freshly isolated bacteria, or heat-killed (h.) bacteria. The remaining serine protease activity in GF cecal-sup was measured after the incubation. (C) Protease activity in cecal-sup of ctr mice, GF mice, GF mice associated with cecal microbiota from either ctr SPF mice (ctr->GF), or V/M-treated SPF mice for 2 weeks (V/M->GF). (D) Staining of active serine proteases in cecal-sup of ctr, V/M, or GF mice (TAMRA-FP sodium dodecyl sulfate–polyacrylamide gel electrophoresis). (E) Volcano plot showing all murine proteases (red) and protease inhibitors (PI, blue) that were detected via LC-MS/MS analysis in cecal-supernatants of ctr, V/M, or GF mice (n = 3/group). The log2 intensity (depicted by the dot size) indicates the abundance of the respective protein. The proteins on the right side of the volcano blots are significantly higher abundant in V/M-treated or GF mice, respectively, compared with control mice. (F) Fluorescein translocation and TEER in polarized PTK6 cells on apical exposure to cecal-sup from ctr mice versus V/M-treated mice ± PMSF inhibition. (G) Colonic tissue sections from SPF mice were apically exposed to cecal-sup from control SPF (ctr) mice or cecal-sup from GF mice ± PMSF in an Ussing chamber setup. Translocation of apically applied fluorescein to the basolateral side and TEER of the respective tissue sections were measured.
Analogous to the observation in stool from ABx-treated patients, cecal content from V/M-treated mice showed a strong increase in active, low-molecular-weight serine proteases (<25 kDa) (Figure 2D). LC-MS/MS analysis of cecal-sup revealed significantly higher levels of mostly pancreatic serine proteases, such as trypsin and chymotrypsin-like elastase in V/M-treated (trypsin, 22-fold; chymotrypsin-like elastase, 9-fold increased) and GF mice (trypsin, 85-fold; chymotrypsin-like elastase, 28-fold increased) compared with untreated SPF mice (Figure 2E, Supplementary Table 1). Of note, the abundance of bacterial proteases was found to be very low in comparison with host-derived proteases in ctr and V/M-treated mice, and none of the detected bacterial proteases was significantly increased on V/M-treatment (data not shown). Analogous to the results using patient stool-sup (Figure 1C), apical exposure of PTK6 cells to cecal-sup from V/M-treated (Figure 2F) or GF (data not shown) mice resulted in significantly impaired epithelial barrier functions, whereas pretreatment with PMSF abrogated this detrimental effect. Furthermore, apical exposure of large intestinal tissue sections from SPF mice to high PA cecal-sup from GF mice resulted in significantly increased permeability and reduced TEER (Figure 2G), which was again abrogated by PMSF pretreatment of the cecal-sup. These findings clearly indicate that excess levels of pancreatic proteases are indeed detrimental to the large intestinal barrier function.
Excessive Large Intestinal Protease Activity Results in Transient Impairment of the Intestinal Barrier But No Significant Disturbance of the Intestinal Immune Homeostasis in Wild-Type Mice
V/M treatment of adult WT mice resulted in a rapid (Day 2) and lasting (Day 14: 7 days after discontinuation of ABx treatment) increase in large intestinal PA (Figure 3A–C). The rapid increase in PA was associated with significantly increased plasma levels of orally administered fluorescein isothiocyanate dextran (Figure 3D) and reduced ex vivo barrier functions of large intestinal tissue sections (Figure 3E). The integrity of the intestinal barrier was almost reestablished at Day 14 (Figure 3D and E) despite the lasting increase in PA, suggesting yet unknown adaptation mechanisms to restore the integrity of the intestinal barrier toward constantly increased PA. Interestingly, the V/M-induced transient leakage of the intestinal barrier affected neither the expression level of proinflammatory cytokines, such as IL1β (Figure 3F) or tumor necrosis factor (data not shown), nor markers for immune cell infiltration, such as CD3 (Figure 3F). This suggests an only minor impact of the high PA and the transiently increased permeability on the large intestinal immune homeostasis in WT mice.
Figure 3.

Excessive large intestinal PA results in transient impairment of the intestinal barrier in WT mice. (A) Experimental schedule: untreated WT mice (ctr), V/M-treated WT mice at Day 2 of the treatment (V/M), and V/M-treated WT mice 7 days after discontinuation of the treatment (post-V/M) were investigated. (B, C) Fecal and cecal serine protease activity. (D) Plasma concentration of fluorescein isothiocyanate (FITC) dextran 4 hours after oral gavage of FITC dextran (4 kDa). (E) Ussing chamber analyses of fluorescein translocation and TEER in cecal tissue sections. (F) mRNA expression levels of IL1β and CD3 in colonic tissue. (G) Experimental schedule: V/M-treated WT mice were cotreated (oral gavage) with the serine protease inhibitor AEBSF (V/M AEBSF) or H2O (V/M wat), starting from 1 day before V/M treatment. (H, I) Fecal and cecal serine protease activity. (J) Plasma concentration of FITC dextran 4 hours after oral gavage of FITC dextran (4 kDa). (K) Ussing chamber analyses of fluorescein translocation and TEER in cecal tissue sections.
Importantly, the rise in PA was found to be causal for the V/M-induced impairment of the intestinal barrier, because oral coadministration of the serine protease inhibitor AEBSF resulted in significantly reduced PA (Figure 3 G–I) and significant protection from the V/M-induced increase in mucosal permeability (Figure 3J and K).
To investigate whether the observed increase in PA and the concomitant transient impairment of the intestinal barrier results in increased vulnerability of the large intestinal immune homeostasis in the long term, WT mice underwent early and repeated V/M treatment followed by exposure to DSS 3 weeks after cessation of the second ABx treatment (Figure 4A). During each V/M course, microbial diversity was strongly reduced with Lactobacillaceae and Enterobacteriaceae being the dominant taxa (>99% relative abundance) (Figure 4B). Three weeks after cessation of the respective V/M course, the microbial composition was close to the pretreatment state at the level of dominant family composition. However, microbial diversity was still significantly reduced, which was mostly caused by the lasting eradication of families belonging to the order Bacteroidales, such as Bacteroidaceae, Porphyromonadaceae, and Prevotellaceae (Figure 4C–E), and the PA was still strongly increased in at least 50% of the mice (Figure 4F). However, these V/M-induced alterations did not promote the development of subsequent DSS-induced colitis (Figure 4G–K).
Figure 4.

Excessive large intestinal PA does not disturb the intestinal immune homeostasis in WT mice. (A) Long-term experimental setup: WT mice were left untreated (ctr) or repeatedly treated with V/M (2 times for 7 days at the age of 4 and 8 weeks) (V/M) before acute DSS colitis was induced at the age of 12 weeks (n = 6/group). (B) Overview on the V/M-mediated and DSS-mediated compositional dynamics in the intestinal microbiota of V/M-treated mice compared with ctr mice (family level). (C) Overview on the long-term impact of V/M treatment on the phylogenetic makeup (beta-diversity) of cecal microbiota and its response to DSS treatment. (D) Richness of the intestinal microbiota. (E) Relative abundance of Bacteroidaceae, Prevotellaceae, and Porphyromonadaceae in untreated versus V/M-treated WT mice before and after exposure to DSS. The number of mice in which the respective bacterial family was detected (prevalence) is depicted above the respective figure. Color code: purple = during V/M treatment, light red = post V/M treatment (after first and second V/M treatments), red = DSS treatment. (F) Kinetic of the fecal serine protease activity in untreated (ctr) versus V/M-treated (V/M) WT mice. (G) Body weight development in ctr and V/M-treated WT mice. (H) The left panel shows the development of the disease activity index after induction of acute colitis by DSS. The right panel shows representative hematoxylin-eosin stainings including the mean histopathological score of colonic tissue from DSS-treated ctr versus DSS-treated V/M-pretreated mice at Day 8 after the induction of DSS colitis. (I–K) Organ weight in DSS-treated ctr and V/M-pretreated WT mice. DAI, disease activity index.
Excessive Large Intestinal Protease Activity Results in Lasting Impairment of the Intestinal Barrier in Interleukin 10-/- Mice
Analogous to WT mice, V/M induced a rapid and lasting increase in PA in IL10-/- mice (Figure 5A–C), which was associated with significantly increased permeability of the intestinal barrier in vivo (Figure 5D) and significantly increased permeability of the large intestinal barrier ex vivo (Figure 5E). In contrast to WT mice, IL10-/- mice still showed a significant impairment of the intestinal barrier after the cessation of the ABx treatment (Figure 5D and E), indicating that the intestinal mucosa in IL10-/- mice is not able to cope with the persistently increased PA. In addition, IL10-/- mice showed significantly increased expression levels of IL1β (Figure 5F), tumor necrosis factor (data not shown), and CD3 (Figure 5F) already 7 days after cessation of the ABx treatment, suggesting that the increased permeability of the intestinal barrier triggers an inflammatory reaction in the large intestinal tissue. Importantly, oral coapplication of AEBSF during V/M-treatment was again found to reduce the rise in PA (Figure 5G–I) and to protect the intestinal barrier function (Figure 5J and K), demonstrating that the high PA is causal for the impaired intestinal barrier function in V/M-treated IL10-/- mice.
Figure 5.

Excessive large intestinal PA results in lasting impairment of the intestinal barrier in IL10-/-mice. (A) Experimental setup: Untreated IL10-/- mice (ctr), V/M-treated IL10-/- mice at Day 2 of the treatment (V/M), and V/M-treated IL10-/- mice 7 days after discontinuation of the V/M treatment (post-V/M) were investigated. (B, C) Fecal and cecal serine protease activity. (D) Plasma concentration of fluorescein isothiocyanate dextran (4 kDa) 4 hours after oral gavage. (E) Ussing chamber analyses of cecal and colonic tissue. (F) mRNA expression level of IL1β and CD3 in colonic tissue. (G) Experimental schedule: V/M-treated IL10-/- mice were orally gavaged with the serine protease inhibitor AEBSF (V/M-AEBSF) or H2O (V/M-wat), starting from 1 day before V/M treatment. (H, I) Fecal and cecal serine protease activity. (J) Plasma concentration of fluorescein isothiocyanate dextran (4 kDa) 4 hours after oral gavage. (K) Ussing chamber analyses of fluorescein translocation and TEER in colonic tissue sections. FITC, fluorescein isothiocyanate.
Excessive Large Intestinal Protease Activity Accelerates Colitis Development in Interleukin 10-/- Mice
To determine the relevance of V/M-increased PA for the development of colitis in genetically susceptible organisms, IL10-/- mice underwent early and repeated V/M treatment with or without oral AEBSF cotreatment during the respective V/M course (Figure 6A). The V/M-induced alterations in the intestinal microbial ecosystem of IL10-/- mice were similar to the ones observed in WT mice (Figure 6B–E). The V/M-mediated rise in PA was significantly reduced by oral coadministration of AEBSF (Figure 7A). From the age of 14 weeks on, V/M-treated but not V/M-AEBSF cotreated IL10-/- mice showed significantly higher blood serum amyloid A and fecal complement component 3 (C3) levels compared with untreated control animals, indicating that the high PA accelerates colitis development (Figure 7B and C). Indeed, expression levels of inflammation markers, such as C3, IL1β, and interferon-γ, were significantly increased in the large intestinal tissue of V/M-treated IL10-/- mice compared with untreated or V/M-AEBSF cotreated IL10-/- mice (Figure 7D). Immunofluorescence analysis revealed significantly increased numbers of CD3+ T cells in the large intestinal tissue of V/M-treated IL10-/- mice compared with untreated and V/M-AEBSF cotreated IL10-/- mice (Figure 7E). Histopathologic analysis revealed significantly increased colitis severity in V/M-treated IL10-/- mice compared with untreated and V/M-AEBSF cotreated IL10-/- mice (Figure 7F). These findings demonstrate that the ABx-mediated rise in PA aggravates colitis in IL10-/- mice. Interestingly, in contrast to untreated IL10-/- mice, most V/M-treated IL10-/- mice (∼80%) and some V/M-AEBSF cotreated IL10-/- mice (∼30%) unexpectedly developed macroscopically visible large intestinal tumors already at 16 weeks of age (Figure 7G), indicating the ABx-mediated rise in PA to accelerate colitis-associated tumorigenesis.60
Figure 6.

Impact of repeated V/M treatment on the microbial ecosystem in IL10-/-mice. (A) Long-term experimental setup: IL10-/- mice were left untreated or underwent repeated V/M treatment (2 times for 7 days at the age of 4 and 8 weeks) ± oral gavage of the serine protease inhibitor AEBSF (V/M-AEBSF) from 1 day before the beginning of the respective V/M treatment to Day 7. (B) Kinetic of the fecal microbiota composition in untreated versus V/M-treated IL10-/- mice (family level). (C) MetaNMDS plot showing fecal microbiota profiles (beta diversity) of untreated versus V/M-treated IL10-/- mice. (D) Microbial richness in ctr and V/M IL10-/- mice. Color code: purple = during V/M treatment, light red = post V/M treatment (after first and second V/M treatments). (E) Relative abundance of Bacteroidaceae, Prevotellaceae, and Porphyromonadaceae in untreated versus V/M-treated IL10-/- mice. The number of mice, in which the respective bacterial family was detected (prevalence) is depicted above the respective figure.
Figure 7.

Excessive large intestinal PA accelerates colitis development in IL10-/-mice. (A) Kinetic of the fecal serine protease activity in IL10-/- mice. (B, C) Development of plasma serum amyloid A levels (systemic inflammation marker) and fecal complement component 3 (C3) levels (marker for intestinal inflammation) in IL10-/- mice. (D) mRNA expression levels of C3, IL1β, and interferon-γ in large intestinal tissue of IL10-/- mice. (E) T-cell infiltration in the proximal colon was determined by immunofluorescence staining of CD3+ cells. Quantification of CD3+ T cells per mm2 at 6 representative regions in tissue sections per mouse. (F) The left panel shows representative pictures of hematoxylin-eosin-stained colonic tissue and the right panel shows the histopathologic score of individual IL10-/- mice. (G) The left panel shows representative pictures of hematoxylin-eosin-stained tumors that were exclusively observed in the cecum and colon of V/M-treated IL10-/- mice. The right panel shows the prevalence of tumor formation in the respective treatment group. IFN, interferon; SAA, serum amyloid A.
Discussion
The present study is the first to provide experimental evidence for the assumption that ABx therapies can trigger the development of chronic intestinal inflammation in genetically susceptible individuals. Excessive large intestinal PA, which is a frequently observed consequence of ABx therapy in patients, was identified to cause this severe adverse effect.
It has long been known that ABx treatment can abrogate the ability of the large intestinal microbiota to inactivate the high load of pancreatic proteases in the small intestinal content,61, 62 resulting in a major increase in large intestinal PA in rodents and human beings.29, 30, 31, 32 However, the physiological consequences of this adverse effect of ABx have been unknown. In our patient cohort, approximately a quarter of all patients showed a major (>5-fold) rise in stool PA on ABx treatment. All tested ABx classes mediated a major PA increase in some patients but not in others, demonstrating individual-specific vulnerability of the intestinal microbiota toward ABx therapies,40, 63, 64 which is likely caused by the high interindividual variability of human microbial ecosystems. The fluoroquinolone ABx levofloxacin tended to confer a higher risk for a major PA rise than amoxicillin or rifaximin; however, this tendency needs to be confirmed in larger patient cohorts. In mice, oral treatment with V/M was consistently found to mediate an almost total loss of antiproteolytic activity in the large intestinal microbiota, indicating that the ABx mixture eradicates all antiproteolytic taxa. The inability of the intestinal microbiota from V/M-treated mice to inactivate serine proteases ex vivo or on transfer into GF mice supports the latter assumption. However, although our data are in line with published literature suggesting specific strains of the order Bacteroidales to play a role in the physiological inactivation of pancreatic proteases,34, 61, 65 the identity of relevant antiproteolytic species/strains remains to be elucidated. The identification and isolation of antiproteolytic bacteria is challenging because the capacity to inactivate/degrade pancreatic proteases seems to be a rather rare bacterial function and none of the already investigated simplified microbial consortia (SIHUMIx,62 altered Schaedler flora,66 Oligo-MM community67; data not shown) are able to normalize the PA.
Although relevant microbes and molecular mechanisms remain to be discovered, the results of the present study demonstrate that the antiproteolytic activity of the large intestinal microbial ecosystem is of major importance for the integrity of the intestinal barrier. The rise in PA on V/M treatment resulted in significantly impaired intestinal barrier functions in WT and IL10-/- mice. Interestingly, the integrity of the intestinal barrier was rapidly restored in WT mice, despite constantly increased PA. In addition, the transient impairment of the intestinal barrier did not trigger a significant inflammatory activation of the large intestinal tissue in WT mice. It can be assumed that the observed adaptation of the large intestinal barrier to high PA levels is a physiological feedback mechanism to increased PA levels (as observed in diarrhea),68, 69 protecting the organism from lasting excess penetration of luminal microbes or microbe-associated molecular patterns. However, in contrast to healthy WT mice, colitis-susceptible IL10-/- mice were not able to restore the integrity of the intestinal barrier. Shortly after cessation of the ABx treatment, the lasting impairment of the intestinal barrier was paralleled by increased expression of inflammation markers in the large intestinal tissue, suggesting that the penetration of luminal antigens triggers proinflammatory mechanisms in genetically susceptible IL10-/- mice. The lasting impairment of the intestinal barrier may therefore be caused by the observed inflammatory activation of the large intestinal tissue, possibly affecting epithelial proliferation70, 71 and/or formation of tight and adherens junction complexes,72, 73 or vice versa. However, it can be assumed that these acute detrimental effects of the high PA on the large intestinal homeostasis cannot be controlled in colitis-susceptible IL10-/- mice, resulting in the observed high PA-mediated acceleration and aggravation of colitis development in V/M-treated IL10-/- mice. The finding that V/M-treated WT mice, despite lasting alterations in the intestinal microbial composition, high PA levels, and transiently reduced intestinal barrier functions, neither developed spontaneous colitis nor showed higher susceptibility toward DSS-induced colitis supports the assumption that the intestinal barrier and immune functions in healthy organisms are well able to adapt to ABx-induced acute and long-term changes in the intestinal microbial ecosystem. This finding is in line with the fact that most ABx courses do not result in the development of chronic intestinal inflammation in patients.
In summary, the present study unravels significant detrimental effects of ABx-induced excess PA levels on the intestinal barrier and immune function in colitis-susceptible mice. Because a substantial fraction of ABx-treated patients shows an analogous rise in PA, it is highly probable that ABx treatment is not only associated28 but indeed an independent risk factor for the development of chronic intestinal inflammation in IBD-susceptible individuals. It may be speculated that the high PA-mediated impairment of the large intestinal barrier might also play a role in the development of other chronic diseases that have been found to be associated with ABx therapies, such as irritable bowel syndrome22, 23 or colorectal cancer.25, 26 However, the pathophysiological impact of the rise in PA on the intestinal barrier in ABx-treated patients remains to be elucidated. With regard to IBD patients, the observed detrimental effects of ABx-induced high PA on the intestinal homeostasis, the lack of consistent clinical data supporting a protective effect of ABx therapy in IBD,74 and the finding that ABx are associated with a more severe disease course and aggravated IBD-related microbial dysbiosis13 suggest that ABx therapies rather contribute to the progression of the chronic inflammation. Finally, the present results support the development of innovative antiproteolytic strategies targeting the large intestinal PA (eg, oral administration of encapsulated serine protease inhibitors, commensal antiproteolytic bacteria, or genetically modified bacteria expressing protease inhibitors) to protect the large intestinal mucosa from adverse effects of ABx-induced or diarrhea-induced35, 38, 69 high PA.
Acknowledgements
The authors thank Sigrid Kisling and Silvia Pitariu for histopathologic scoring; Alexandra Buse, Felicitas Firlus, and Caroline Ziegler for outstanding technical assistance; and Valentina Schüppel for support with Ussing chamber assays.
Hongsup Yoon and Gabriele Hörmannsperger designed the experiments. Hongsup Yoon, Monika Schaubeck, and Gabriele Hörmannsperger performed experiments. Suchita Panda, Chaysavanh Manichanh, Patrizia Kump, and Andreas Blesl provided patient stool samples. Ilias Lagkouvardos and Thomas Clavel performed 16S rRNA gene amplicon analysis. Andreas Blesl, Christina Ludwig, Hannes Hahne, and Bernhard Küster performed LC-MS/MS analysis. Hongsup Yoon and Gabriele Hörmannsperger wrote the manuscript. Monika Schaubeck, Ilias Lagkouvardos, Thomas Clavel, and Dirk Haller provided intellectual input and reviewed the manuscript intensively. All authors approved the final version of the manuscript.
Footnotes
Conflicts of interest The authors disclose no conflicts.
Funding This work was financed by the Deutscher Akademischer Austauschdienst, and supported by the Technische Universität München Graduate School.
Contributor Information
Dirk Haller, Email: dirk.haller@tum.de.
Gabriele Hörmannsperger, Email: gabriele.hoermannsperger@tum.de.
Supplementary Material
Supplementary Table 1.
Abundance of Proteases and Protease Inhibitors in Cecal-Sup of Untreated, V/M-Treated, and GF WT Mice (Determined Via LC-MS/MS Analysis)
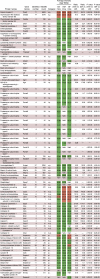 |
NOTE. The number in the columns with color code is mean intensity of proteins (log2 intensity based absolute quantitation) as described by the dot size in Figure 2D. The ratio is the fold change of the respective protein in V/M or GF mice compared with untreated (ctrl) mice. The P value is the significance level of the abundance difference. Color code: red = high intensity; green = low intensity.
a.p., aspartyl protease; c.p., cysteine protease; iBAQ, intensity-based absolute quantitation; m.p., metalloprotease; NaN, not detected; s.p., serine protease; t.p., threonine protease.
References
- 1.Sartor R.B. Genetics and environmental interactions shape the intestinal microbiome to promote inflammatory bowel disease versus mucosal homeostasis. Gastroenterology. 2010;139:1816–1819. doi: 10.1053/j.gastro.2010.10.036. [DOI] [PubMed] [Google Scholar]
- 2.Sartor R.B. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 3.Jostins L., Ripke S., Weersma R.K., Duerr R.H., McGovern D.P., Hui K.Y., Lee J.C., Schumm L.P., Sharma Y., Anderson C.A., Essers J., Mitrovic M., Ning K., Cleynen I., Theatre E., Spain S.L., Raychaudhuri S., Goyette P., Wei Z., Abraham C., Achkar J.P., Ahmad T., Amininejad L., Ananthakrishnan A.N., Andersen V., Andrews J.M., Baidoo L., Balschun T., Bampton P.A., Bitton A., Boucher G., Brand S., Büning C., Cohain A., Cichon S., D'Amato M., De Jong D., Devaney K.L., Dubinsky M., Edwards C., Ellinghaus D., Ferguson L.R., Franchimont D., Fransen K., Gearry R., Georges M., Gieger C., Glas J., Haritunians T., Hart A., Hawkey C., Hedl M., Hu X., Karlsen T.H., Kupcinskas L., Kugathasan S., Latiano A., Laukens D., Lawrance I.C., Lees C.W., Louis E., Mahy G., Mansfield J., Morgan A.R., Mowat C., Newman W., Palmieri O., Ponsioen C.Y., Potocnik U., Prescott N.J., Regueiro M., Rotter J.I., Russell R.K., Sanderson J.D., Sans M., Satsangi J., Schreiber S., Simms L.A., Sventoraityte J., Targan S.R., Taylor K.D., Tremelling M., Verspaget H.W., De Vos M., Wijmenga C., Wilson D.C., Winkelmann J., Xavier R.J., Zeissig S., Zhang B., Zhang C.K., Zhao H., International IBD Genetics Consortium (IIBDGC), Silverberg M.S., Annese V., Hakonarson H., Brant S.R., Radford-Smith G., Mathew C.G., Rioux J.D., Schadt E.E., Daly M.J., Franke A., Parkes M., Vermeire S., Barrett J.C., Cho J.H. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khor B., Gardet A., Xavier R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strober W., Fuss I., Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maloy K.J., Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 7.Heller F., Florian P., Bojarski C., Richter J., Christ M., Hillenbrand B., Mankertz J., Gitter A.H., Bürgel N., Fromm M., Zeitz M., Fuss I., Strober W., Schulzke J.D. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Liu J.Z., van Sommeren S., Huang H., Ng S.C., Alberts R., Takahashi A., Ripke S., Lee J.C., Jostins L., Shah T., Abedian S., Cheon J.H., Cho J., Dayani N.E., Franke L., Fuyuno Y., Hart A., Juyal R.C., Juyal G., Kim W.H., Morris A.P., Poustchi H., Newman W.G., Midha V., Orchard T.R., Vahedi H., Sood A., Sung J.Y., Malekzadeh R., Westra H.J., Yamazaki K., Yang S.K., International Multiple Sclerosis Genetics Consortium, International IBD Genetics Consortium, Barrett J.C., Alizadeh B.Z., Parkes M., Bk T., Daly M.J., Kubo M., Anderson C.A., Weersma R.K. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979–986. doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ananthakrishnan A.N. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol. 2015;12:205–217. doi: 10.1038/nrgastro.2015.34. [DOI] [PubMed] [Google Scholar]
- 10.Kaser A., Zeissig S., Blumberg R.S. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huttenhower C., Kostic A.D., Xavier R.J. Inflammatory bowel disease as a model for translating the microbiome. Immunity. 2014;40:843–854. doi: 10.1016/j.immuni.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schaubeck M., Clavel T., Calasan J., Lagkouvardos I., Haange S.B., Jehmlich N., Basic M., Dupont A., Hornef M., von Bergen M., Bleich A., Haller D. Dysbiotic gut microbiota causes transmissible Crohn's disease-like ileitis independent of failure in antimicrobial defense. Gut. 2016;65:225–237. doi: 10.1136/gutjnl-2015-309333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gevers D., Kugathasan S., Denson L.A., Vázquez-Baeza Y., Van Treuren W., Ren B., Schwager E., Knights D., Song S.J., Yassour M., Morgan X.C., Kostic A.D., Luo C., González A., McDonald D., Haberman Y., Walters T., Baker S., Rosh J., Stephens M., Heyman M., Markowitz J., Baldassano R., Griffiths A., Sylvester F., Mack D., Kim S., Crandall W., Hyams J., Huttenhower C., Knight R., Xavier R.J. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tremaroli V., Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- 15.Buffie C.G., Pamer E.G. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol. 2013;13:790–801. doi: 10.1038/nri3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hooper L.V., Gordon J.I. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–1118. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- 17.Macfarlane G.T., Macfarlane S. Human colonic microbiota: ecology, physiology and metabolic potential of intestinal bacteria. Scand J Gastroenterol Suppl. 1997;222:3–9. doi: 10.1080/00365521.1997.11720708. [DOI] [PubMed] [Google Scholar]
- 18.Schulfer A.F., Battaglia T., Alvarez Y., Bijnens L., Ruiz V.E., Ho M., Robinson S., Ward T., Cox L.M., Rogers A.B., Knights D., Sartor R.B., Blaser M.J. Intergenerational transfer of antibiotic-perturbed microbiota enhances colitis in susceptible mice. Nat Microbiol. 2018;3:234–242. doi: 10.1038/s41564-017-0075-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willing B.P., Russell S.L., Finlay B.B. Shifting the balance: antibiotic effects on host-microbiota mutualism. Nat Rev Microbiol. 2011;9:233–243. doi: 10.1038/nrmicro2536. [DOI] [PubMed] [Google Scholar]
- 20.Dalal S.R., Chang E.B. The microbial basis of inflammatory bowel diseases. J Clin Invest. 2014;124:4190–4196. doi: 10.1172/JCI72330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nobel Y.R., Cox L.M., Kirigin F.F., Bokulich N.A., Yamanishi S., Teitler I., Chung J., Sohn J., Barber C.M., Goldfarb D.S., Raju K., Abubucker S., Zhou Y., Ruiz V.E., Li H., Mitreva M., Alekseyenko A.V., Weinstock G.M., Sodergren E., Blaser M.J. Metabolic and metagenomic outcomes from early-life pulsed antibiotic treatment. Nat Commun. 2015;6:7486. doi: 10.1038/ncomms8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maxwell P.R., Rink E., Kumar D., Mendall M.A. Antibiotics increase functional abdominal symptoms. Am J Gastroenterol. 2002;97:104–108. doi: 10.1111/j.1572-0241.2002.05428.x. [DOI] [PubMed] [Google Scholar]
- 23.Mendall M.A., Kumar D. Antibiotic use, childhood affluence and irritable bowel syndrome (IBS) Eur J Gastroenterol Hepatol. 1998;10:59–62. doi: 10.1097/00042737-199801000-00011. [DOI] [PubMed] [Google Scholar]
- 24.Wang J.L., Chang C.H., Lin J.W., Wu L.C., Chuang L.M., Lai M.S. Infection, antibiotic therapy and risk of colorectal cancer: a nationwide nested case-control study in patients with type 2 diabetes mellitus. Int J Cancer. 2014;135:956–967. doi: 10.1002/ijc.28738. [DOI] [PubMed] [Google Scholar]
- 25.Dik V.K., van Oijen M.G., Smeets H.M., Siersema P.D. Frequent use of antibiotics is associated with colorectal cancer risk: results of a nested case-control study. Dig Dis Sci. 2016;61:255–264. doi: 10.1007/s10620-015-3828-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korpela K., Salonen A., Virta L., Kekkonen R.A., Forslund K., Bork P., de Vos W.M. Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nat Commun. 2016;7:10410. doi: 10.1038/ncomms10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hviid A., Svanström H., Frisch M. Antibiotic use and inflammatory bowel diseases in childhood. Gut. 2011;60:49–54. doi: 10.1136/gut.2010.219683. [DOI] [PubMed] [Google Scholar]
- 28.Ungaro R., Bernstein C.N., Gearry R., Hviid A., Kolho K.L., Kronman M.P., Shaw S., Van Kruiningen H., Colombel J.F., Atreja A. Antibiotics associated with increased risk of new-onset Crohn's disease but not ulcerative colitis: a meta-analysis. Am J Gastroenterol. 2014;109:1728–1738. doi: 10.1038/ajg.2014.246. [DOI] [PubMed] [Google Scholar]
- 29.Genell S., Gustafsson B.E. Impaired enteric degradation of pancreatic endopeptidases in antibiotic-treated rats. Scand J Gastroenterol. 1977;12:801–809. doi: 10.3109/00365527709181723. [DOI] [PubMed] [Google Scholar]
- 30.Carlstedt-Duke B., Gustafsson B.E., Midtvedt T. Clindamycin-induced alterations in intestinal microflora-associated characteristics in rats. Scand J Gastroenterol. 1985;20:92–98. doi: 10.3109/00365528509089639. [DOI] [PubMed] [Google Scholar]
- 31.Bohe M., Borgstrom A., Genell S., Ohlsson K. Determination of immunoreactive trypsin, pancreatic elastase and chymotrypsin in extracts of human feces and ileostomy drainage. Digestion. 1983;27:8–15. doi: 10.1159/000198913. [DOI] [PubMed] [Google Scholar]
- 32.Borgstrom A., Genell S., Ohlsson K. Elevated fecal levels of endogenous pancreatic endopeptidases after antibiotic treatment. Scand J Gastroenterol. 1977;12:525–529. doi: 10.3109/00365527709181329. [DOI] [PubMed] [Google Scholar]
- 33.Macfarlane G.T., Allison C., Gibson S.A., Cummings J.H. Contribution of the microflora to proteolysis in the human large intestine. J Appl Bacteriol. 1988;64:37–46. doi: 10.1111/j.1365-2672.1988.tb02427.x. [DOI] [PubMed] [Google Scholar]
- 34.Carroll I.M., Ringel-Kulka T., Ferrier L., Wu M.C., Siddle J.P., Bueno L., Ringel Y. Fecal protease activity is associated with compositional alterations in the intestinal microbiota. PLoS One. 2013;8:e78017. doi: 10.1371/journal.pone.0078017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gecse K., Róka R., Ferrier L., Leveque M., Eutamene H., Cartier C., Ait-Belgnaoui A., Rosztóczy A., Izbéki F., Fioramonti J., Wittmann T., Bueno L. Increased faecal serine protease activity in diarrhoeic IBS patients: a colonic lumenal factor impairing colonic permeability and sensitivity. Gut. 2008;57:591–599. doi: 10.1136/gut.2007.140210. [DOI] [PubMed] [Google Scholar]
- 36.Antalis T.M., Shea-Donohue T., Vogel S.N., Sears C., Fasano A. Mechanisms of disease: protease functions in intestinal mucosal pathobiology. Nat Clin Pract Gastroenterol Hepatol. 2007;4:393–402. doi: 10.1038/ncpgasthep0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeissig S., Bürgel N., Günzel D., Richter J., Mankertz J., Wahnschaffe U., Kroesen A.J., Zeitz M., Fromm M., Schulzke J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn's disease. Gut. 2007;56:61–72. doi: 10.1136/gut.2006.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kiesslich R., Duckworth C.A., Moussata D., Gloeckner A., Lim L.G., Goetz M., Pritchard D.M., Galle P.R., Neurath M.F., Watson A.J. Local barrier dysfunction identified by confocal laser endomicroscopy predicts relapse in inflammatory bowel disease. Gut. 2012;61:1146–1153. doi: 10.1136/gutjnl-2011-300695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hansen K.K., Sherman P.M., Cellars L., Andrade-Gordon P., Pan Z., Baruch A., Wallace J.L., Hollenberg M.D., Vergnolle N. A major role for proteolytic activity and proteinase-activated receptor-2 in the pathogenesis of infectious colitis. Proc Natl Acad Sci U S A. 2005;102:8363–8368. doi: 10.1073/pnas.0409535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panda S., El khader I., Casellas F., López Vivancos J., García Cors M., Santiago A., Cuenca S., Guarner F., Manichanh C. Short-term effect of antibiotics on human gut microbiota. PLoS One. 2014;18:9. doi: 10.1371/journal.pone.0095476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shevchenko A., Tomas H., Havlis J., Olsen J.V., Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 42.Hahne H., Pachl F., Ruprecht B., Maier S.K., Klaeger S., Helm D., Médard G., Wilm M., Lemeer S., Kuster B. DMSO enhances electrospray response, boosting sensitivity of proteomic experiments. Nat Methods. 2013;10:989–991. doi: 10.1038/nmeth.2610. [DOI] [PubMed] [Google Scholar]
- 43.Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 44.Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 45.Cox J., Hein M.Y., Luber C.A., Paron I., Nagaraj N., Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 47.Rutgeerts P., Van Assche G., Vermeire S., D'Haens G., Baert F., Noman M., Aerden I., De Hertogh G., Geboes K., Hiele M., D'Hoore A., Penninckx F. Ornidazole for prophylaxis of postoperative Crohn's disease recurrence: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2005;128:856–861. doi: 10.1053/j.gastro.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 48.Katakura K., Lee J., Rachmilewitz D., Li G., Eckmann L., Raz E. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest. 2005;115:695–702. doi: 10.1172/JCI22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Godon J.J., Zumstein E., Dabert P., Habouzit F., Moletta R. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit Rdna sequence analysis. Appl Environ Microbiol. 1997;63:2802–2813. doi: 10.1128/aem.63.7.2802-2813.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lagkouvardos I., Klaring K., Heinzmann S., Platz S., Scholz B., Engel K.H., Schmitt-Kopplin P., Haller D., Rohn S., Skurk T., Clavel T. Gut metabolites and bacterial community networks during a pilot intervention study with flaxseeds in healthy adult men. Mol Nutr Food Res. 2015;59:1614–1628. doi: 10.1002/mnfr.201500125. [DOI] [PubMed] [Google Scholar]
- 51.Berry D., Ben Mahfoudh K., Wagner M., Loy A. Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl Environ Microbiol. 2011;77:7846–7849. doi: 10.1128/AEM.05220-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., Horn M., Glöckner F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lagkouvardos I., Joseph D., Kapfhammer M., Giritli S., Horn M., Haller D., Clavel T. IMNGS: A massive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci Rep. 2016;6:33721. doi: 10.1038/srep33721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Edgar R.C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 55.Edgar R.C., Haas B.J., Clemente J.C., Quince C., Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Q., Garrity G.M., Tiedje J.M., Cole J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lagkouvardos I., Fischer S., Kumar N., Clavel T. Rhea: a transparent and modular R pipeline for microbial profiling based on 16S rRNA gene amplicons. PeerJ. 2017;5:e2836. doi: 10.7717/peerj.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen J., Bittinger K., Charlson E.S., Hoffmann C., Lewis J., Wu G.D., Collman R.G., Bushman F.D., Li H. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics. 2012;28:2106–2113. doi: 10.1093/bioinformatics/bts342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jost L. Partitioning diversity into independent alpha and beta components. Ecology. 2007;88:2427–2439. doi: 10.1890/06-1736.1. [DOI] [PubMed] [Google Scholar]
- 60.Berg D.J., Davidson N., Kühn R., Müller W., Menon S., Holland G., Thompson-Snipes L., Leach M.W., Rennick D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98:1010–1020. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramare F., Hautefort I., Verhe F., Raibaud P., Iovanna J. Inactivation of tryptic activity by a human-derived strain of Bacteroides distasonis in the large intestines of gnotobiotic rats and mice. Appl Environ Microbiol. 1996;62:1434–1436. doi: 10.1128/aem.62.4.1434-1436.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Becker N., Kunath J., Loh G., Blaut M. Human intestinal microbiota: characterization of a simplified and stable gnotobiotic rat model. Gut Microbes. 2011;2:25–33. doi: 10.4161/gmic.2.1.14651. [DOI] [PubMed] [Google Scholar]
- 63.DuPont H.L. Review article: the antimicrobial effects of rifaximin on the gut microbiota. Aliment Pharmacol Ther. 2016;43(Suppl 1):3–10. doi: 10.1111/apt.13434. [DOI] [PubMed] [Google Scholar]
- 64.Dethlefsen L., Relman D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Midtvedt T., Zabarovsky E., Norin E., Bark J., Gizatullin R., Kashuba V., Ljungqvist O., Zabarovska V., Möllby R., Ernberg I. Increase of faecal tryptic activity relates to changes in the intestinal microbiome: analysis of Crohn's disease with a multidisciplinary platform. PLoS One. 2013;8:e66074. doi: 10.1371/journal.pone.0066074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Norin E., Midtvedt T. Intestinal microflora functions in laboratory mice claimed to harbor a “normal” intestinal microflora. Is the SPF concept running out of date? Anaerobe. 2010;16:311–313. doi: 10.1016/j.anaerobe.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 67.Brugiroux S., Beutler M., Pfann C., Garzetti D., Ruscheweyh H.J., Ring D., Diehl M., Herp S., Lötscher Y., Hussain S., Bunk B., Pukall R., Huson D.H., Münch P.C., McHardy A.C., McCoy K.D., Macpherson A.J., Loy A., Clavel T., Berry D., Stecher B. Genome-guided design of a defined mouse microbiota that confers colonization resistance against Salmonella enterica serovar Typhimurium. Nat Microbiol. 2016;2:16215. doi: 10.1038/nmicrobiol.2016.215. [DOI] [PubMed] [Google Scholar]
- 68.Tooth D., Garsed K., Singh G., Marciani L., Lam C., Fordham I., Fields A., Banwait R., Lingaya M., Layfield R., Hastings M., Whorwell P., Spiller R. Characterisation of faecal protease activity in irritable bowel syndrome with diarrhoea: origin and effect of gut transit. Gut. 2014;63:753–760. doi: 10.1136/gutjnl-2012-304042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Róka R., Rosztóczy A., Leveque M., Izbéki F., Nagy F., Molnár T., Lonovics J., Garcia-Villar R., Fioramonti J., Wittmann T., Bueno L. A pilot study of fecal serine-protease activity: a pathophysiologic factor in diarrhea-predominant irritable bowel syndrome. Clin Gastroenterol Hepatol. 2007;5:550–555. doi: 10.1016/j.cgh.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 70.Kaiser G.C., Polk D.B. Tumor necrosis factor alpha regulates proliferation in a mouse intestinal cell line. Gastroenterology. 1997;112:1231–1240. doi: 10.1016/s0016-5085(97)70135-5. [DOI] [PubMed] [Google Scholar]
- 71.Ruemmele F.M., Gurbindo C., Mansour A.M., Marchand R., Levy E., Seidman E.G. Effects of interferon gamma on growth, apoptosis, and MHC class II expression of immature rat intestinal crypt (IEC-6) cells. J Cell Physiol. 1998;176:120–126. doi: 10.1002/(SICI)1097-4652(199807)176:1<120::AID-JCP14>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 72.Bruewer M., Samarin S., Nusrat A. Inflammatory bowel disease and the apical junctional complex. Ann N Y Acad Sci. 2006;1072:242–252. doi: 10.1196/annals.1326.017. [DOI] [PubMed] [Google Scholar]
- 73.Capaldo C.T., Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta. 2009;1788:864–871. doi: 10.1016/j.bbamem.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sartor R.B. Therapeutic manipulation of the enteric microflora in inflammatory bowel diseases: antibiotics, probiotics, and prebiotics. Gastroenterology. 2004;126:1620–1633. doi: 10.1053/j.gastro.2004.03.024. [DOI] [PubMed] [Google Scholar]