

Abstract
Stereoselective thioallylation of alkynes under possible gold redox catalysis was accomplished with high efficiency (as low as 0.1% catalyst loading, up to 99% yield) and broad substrate scope (various alkynes, inter- and intramolecular fashion). The gold(I) catalyst acts as both a π-acid for alkyne activation and a redox catalyst for AuI/III coupling, whereas the sulfonium cation generated in situ functions as a mild oxidant. This novel methodology provides an exciting system for gold redox catalysis without the need for a strong oxidant.
Keywords: alkynes, gold redox catalysis, sulfonium cations, thioallylation, vinyl gold intermediates
The past two decades have witnessed rapid growth of homogenous gold catalysis.[1] Owing to relativistic effects,[2] gold complexes exhibit superior capabilities in activating π-bonds of alkenes, allenes, and especially alkynes. Although the vinyl gold complex generated from gold-catalyzed nucleophilic addition to an alkyne is well-known,[3] it was not of interest to the synthetic community for quite a while, since rapid protodeauration is the dominant decomposition pathway in most cases. However, vinyl gold complexes have received more and more attention as versatile intermediates for subsequent transformations, including halogenation,[4] radical addition,[5] and transmetalation.[6]
For a long time, the oxidation of AuI to AuIII species was considered challenging owing to the high oxidation potential (1.40 eV). The reluctance of AuI species to undergo oxidative addition, which is usually the entry point for metal-catalyzed cross-coupling reactions, significantly limits their synthetic applications.[7] However, an alternative solution for AuI oxidation to AuIII was discovered in the use of strong oxidants, such as Selectfluor or PhI(OAc)2.[8] This new reaction mode thus unleashes numerous new and unique opportunities for gold catalysis. For example, the stronger AuIII catalyst can activate alkenes towards nucleophilic attack, and the resulting alkyl–gold(III) intermediate further undergoes transmetalation and reductive elimination to yield the cross-coupling product.[9] Similar AuI/III reactivity can also be achieved by using a diazonium salt as an oxidant under photochemical or basic conditions (Scheme 1A).[10]
Scheme 1.

Gold redox catalysis. EWG=electron-withdrawing group, IPr=N,N’-bis(2,6-diisopropylphenyl)imidazol-2-ylidene, PIDA=phenyliodine(III) diacetate, R.E.=reductive elimination, Tf=trifluoromethanesulfonyl.
Overall, this new gold redox catalysis offers an effective route for alkene difunctionalization. However, two major limitations of this methodology are the requirement of strong oxidants and the competing reactivity between AuI and AuIII cations as π-acids. As a result, only very few successful examples of alkyne difunctionalization have been reported on the basis of this methodology.[11] Thus, the search for milder oxidants to promote this gold redox catalysis is highly desirable.[7f] Herein, we report a successful thioallylation of alkynes under possible gold redox catalysis. A cationic AuI catalyst effectively promoted the nucleophilic addition of an allyl sulfide to the alkyne; subsequently, the resulting vinyl gold intermediate could be oxidized by the allylsulfonium cation generated in situ to provide a vinyl thioether in a stereoselective fashion. This novel thioallylation reaction is highly efficient (as low as 0.1% catalyst loading, up to 99% yield, gram-scale conversion) with broad substrate scope (Scheme 1B). To the best of our knowledge, this reaction is the first example of gold redox catalysis involving AuI π-acid activation followed by vinyl-gold oxidation with a mild oxidant, which represents an innovative strategy for alkyne difunctionalization through gold redox catalysis.
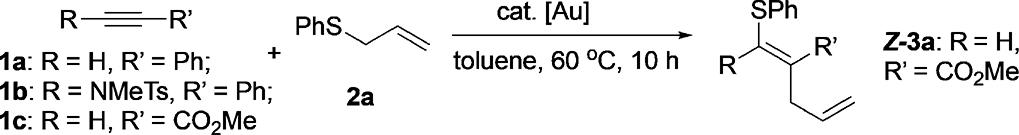
Our interest in utilizing a sulfonium cation as a potential oxidant for gold redox catalysis was initiated by our recent investigation on gold-catalyzed thioalkyne activation.[12] In that study, we discovered that thioalkynes could react with allyl sulfides to form ketenedithioacetals, though in moderate yields with low E/Z selectivity (Figure 1). To investigate the key allyl-transfer process from the proposed intermediate A,[13] we conducted a cross-over experiment between a thioalkyne and two different allyl sulfides.
Figure 1.

Intermolecular thioallylation suggested by cross-over experiment. DCE=1,2-dichlorethane, Tol=p-tolyl.
Interestingly, a significant amount of cross-over products were observed. It is intriguing to us that this reaction proceeds through an intermolecular allyl-transfer process. Two possible mechanisms are proposed herein. First, the allyl group attached to the sulfonium cation can form a C–C bond directly with the σ- or π-bond of the vinyl–gold(I) species to generate the thioallylation product. Alternatively, the allyl sulfonium cation can serve as a mild oxidant for the vinyl– gold(I) species to generate a transient AuIII intermediate, which delivers the same product through reductive elimination. The latter mechanism is very exciting to us because it represents a novel method to achieve gold redox catalysis. To further explore the detailed mechanism and scope of this thioallylation reaction, we conducted reactions between different alkynes and allyl phenyl sulfide (2a) under various conditions of gold catalysis (Table 1).
Table 1:
Screening of the reaction conditions.
 | ||||
|---|---|---|---|---|
| Entry | 1 | Conditions | Conv. [%] | Yield [%] (Z/E) |
| 1 | 1a | 5% JohnPhosAuNTf2 | <5 | – |
| 2 | 1b | 5% JohnPhosAuNTf2 | 100 | messy |
| 3 | 1c | 5% JohnPhosAuNTf2 | 100 | 79 (3:2) |
| 4 | 1c | 5% PPh3AuNTf2 | 100 | 80 (3:2) |
| 5 | 1c | 5% RuPhosAuNTf2 | 100 | 80 (3:1) |
| 6 | 1c | 5% IPrAuNTf2 | 100 | 96 (Z only) |
| 7 | 1c | 5% IPrAuCl | <10 | <5 |
| 8 | 1c | other [Au] catalysts | <90% yield[a] | |
| 9 | 1c | 5% IPrAuNTf2 (other solvents) | 40–90% yield[a] | |
| 10 | 1c | other metal catalysts (Ag, Cu, Fe, Pd, Rh, Ir, Zn, La, etc.) | <5% conversion[a] | |
| 11 | 1c | 1% IPrAuNTf2 (c=0.5m, 608C) | 100 | 98 (Z only) |
| 12 | 1c | 0.1% IPrAuNTf2 (c=2.0m, 608C), 48 h, gram scale | 100 | 95 (Z only) |
| 13 | 1c | 1% IPrAuNTf2 (c=0.5 m, 408C, 48 h) | 93 | 88 (Z only) |
Reaction conditions: The catalyst (5 mol%) was added to a solution of alkyne 1 (0.15 mmol) and allyl sulfide 2a (0.1 mmol) in toluene (1 mL), and the reaction mixture was kept at 60°C for 10 h. Conversion and yield were determined by 1H NMR spectroscopy with dimethyl sulfone as the internal standard.
See the Supporting Information.
Under gold catalysis with JohnPhosAuNTf2 (5 mol%) in toluene at 608C, the reaction of 2a and phenylacetylene 1a gave almost no conversion; most of the starting material was recovered, suggesting phenylacetylene was not reactive enough under these conditions. The more electron-rich ynamide 1b gave messy reaction mixtures with no clear product identified. We then turned our attention to the carbonyl-activated alkyne 1c owing to more facile nucleophilic addition. To our delight, the thioallylation product 3a was observed in 79% yield as a 3:2 Z/E mixture. Further screening revealed IPrAuNTf2 as the optimal gold catalyst, which gave product 3a as a single isomer. The alkene was later confirmed to have the Z configuration by NMR spectroscopy.[14] This result suggested a possible mechanism involving trans addition of the sulfide to the gold(I)-activated alkyne, followed by subsequent allyl transfer. Toluene was optimal for this reaction; other solvents provided inferior results. Notably, other metal catalysts, including Ag, Cu, Fe, Pd, Rh, Ir, Zn, and La complexes, were inactive for this reaction (see details in the Supporting Information), which highlighted the unique reactivity of gold catalysts for this transformation. Finally, an increase in the reaction concentration to 0.5m gave complete conversion (10 h) and excellent yield (98%) even with a catalyst loading of only 1 mol%. A further increase in the concentration to 2m allowed a gramscale synthesis of 3a in excellent yield with only 0.1 mol% of the catalyst, although an extended reaction time was required (48 h). Lowering of the reaction temperature to 408C resulted in incomplete conversion.
We tested the scope of this reaction under the optimized conditions (Table 2). Various aryl allyl sulfides were suitable for this reaction, giving excellent yields in all cases regardless of substitution on the phenyl group (products 3a–j). Methylbranched allyl sulfides successfully participated in this reaction, and the desired products 3l and 3m were each formed as a single isomer in excellent yield. Notably, the structure of 3m clearly demonstrated the exclusive SN2’ addition of the vinyl gold intermediate to the allylsulfonium cation. The transformation of alkyl-substituted allyl sulfides also worked well, providing the desired products 3n–p in good to excellent yields. Alkynes bearing other electronwithdrawing groups were also tested. Whereas reactions of a benzylic ester and a carboxylic acid provided products 3q and 3t in excellent yields, ketone 3s and amides 3u and 3v were obtained in only moderate yields, presumably owing to catalyst deactivation by substrate coordination. Phenyl- and alkyl-substituted (R’ =Me or n-Bu) internal propiolates led to almost no reaction even over an extended reaction time (48 h), probably as a result of the low reactivity of these internal alkynes. However, a diester-substituted internal alkyne (dimethyl acetylenedicarboxylate) was converted into 3r in 74% yield. No sulfide conversion was observed when benzyl methyl sulfide was used, which highlighted the unique reactivity of allyl sulfides for this transformation. Notably, in all cases only the Z isomer was observed. The alkene geometry was ambiguously confirmed by the X-ray crystal structure of 3c.
Table 2:
Scope of the reaction of carbonyl-activated alkynes.
 |
Reaction conditions: The catalyst (1 mol%) was added to a solution of alkyne 1 (0.45 mmol) and allyl sulfide 2 (0.3 mmol) in toluene (0.6 mL), and the reaction mixture was kept at 608C for 10 h. [a] Starting with transcrotyl phenyl sulfide. [b] The reaction was carried out with 2 mol% of the catalyst. [c] The reaction was carried out with 0.3 mmol of the alkyne and 0.45 mmol of the sulfide. Bn=benzyl.
To further identify whether a AuI/III process was involved during the allyl transfer, the reaction between 1c and 2a was monitored by mass spectrometry (Figure 2). An ion at m/ z 819.3 was detected and identified as intermediate B based on its collision-induced dissociation (CID) data. Importantly, an ion at m/z 859.4 corresponding to AuIII intermediate C ([M@H]+ ion) was clearly observed, and its structure was further confirmed by CID. Intermediate D was also present in the mass spectrum (see the Supporting Information). Furthermore, careful interpretation of the mass data revealed another AuIII ion at m/z 735.1, which corresponds to an allyl– AuIII–SPh ion (see the Supporting Information). In conclusion, the MS data largely supports the AuI/III pathway; herein, a tentative mechanism involving the formation of a vinyl gold species (intermediate B) and subsequent allyl transfer to form AuIII intermediate C is proposed. This mechanism was further backed up by additional MS evidence with a different gold catalyst and sulfide (see the Supporting Information). More efforts will be made to further confirm this intriguing AuI/III mechanism, including NMR analysis and a computational study.
Figure 2.

Mechanistic study by mass spectrometry.
This novel thioallylation method provides rapid access to highly functionalized alkenes in a stereoselective fashion. To further expand the reaction scope, we tested several terminal alkynes with electron-deficient aromatic substituents, including C6F5- and 4-pyridinal-substituted alkynes. Unfortunately, these substrates gave no reaction under the gold redox conditions. We then turned our attention to internal alkynes with electron-withdrawing substituents. Notably, as compared with NO2-, CN-, and CF3-modified alkynes, haloalkynes are easy to prepare from readily available starting materials. However, the application of these compounds in synthesis is rather limited to ynamide preparation[15] and Cardiot–Chodkiewicz-type coupling reactions.[16] The potential synthetic utility of haloalkynes has been neglected to a large extent.[17] To our delight, haloalkynes 4 with Br or Cl substitution underwent the thioallylation well with a slightly increased catalyst loading (2%). The desired tetrasubstituted alkenes 5 were prepared successfully in good to excellent yields, exclusively as the Z isomers (Table 3).
Table 3:
Scope of the reaction of haloalkynes.
 |
Reaction conditions: The catalyst (2 mol%) was added to a solution of alkyne 4 (0.3 mmol) and allyl sulfide 2 (0.2 mmol) in toluene (0.4 mL), and the reaction mixture was kept at 60°C for 12 or 24 h.
As compared with the carbonyl-activated alkynes, reactions of haloalkynes, especially bromoalkynes, required longer reaction times, presumably owing to the decreased reactivity of the alkynes. Different aryl allyl sulfides underwent this transformation in good to excellent yields, with electron-withdrawing or electron-donating groups (EWG/ EDG) on the arene (products 5a–h). In general, excellent yields were observed with EWG-substituted aryl sulfides, whereas EDG-substituted sulfides gave lower yields owing to incomplete conversion (around 90% conversion). Similar good to excellent yields were observed with different aryl substituents on the bromoalkyne (products 5i–l). Alkylsubstituted allyl sulfides and bromoalkynes were also suitable substrates for this transformation, thus indicating its broad scope (products 5m–p). Notably, no cyclopropane ring opening was observed in the synthesis of 5o, which ruled out a radical reaction pathway. Chloroalkynes were also prepared and subjected to the reaction conditions. As expected, chloroalkynes showed superior reactivity over bromoalkynes (a faster reaction) to give the desired tetrasubstituted vinyl chlorides 5q–x in excellent yields in most cases. Moreover, a single alkene isomer was obtained in all cases. Comprehensive NMR analysis for 5a, 5c, and 5p confirmed that the alkene had exclusively the Z configuration, as observed for the carbonyl-activated alkyne substrates, which is consistent with the proposed mechanism (see the Supporting Information). The efficient synthesis of the tetrasubstituted alkenes in a stereoselective fashion highlights the unique advantage of this novel gold redox approach.
As mentioned in Table 1, simple internal alkynes failed to participate in this transformation, presumably owing to low reactivity toward nucleophilic addition. To further extend the reaction scope to regular alkynes, we proposed to facilitate this reaction in an intramolecular fashion. Alkynes 6 bearing a pendant allyl sulfide were subjected to the reactionconditions. To our delight, dihydrothiophenes 7 were obtained through a 5-endo-dig cyclization in excellent yields, even with a catalyst loading as low as 0.1% (Table 4). This intramolecular thioallylation of the corresponding terminal alkyne gave 7a in excellent yield. Good to excellent yields were also observed for aryl-substituted substrates (products 7b–i, 7k). Interestingly, aryl alkynes with a strongly electron withdrawing substituent reacted much slower (products 7c, 7d, and 7i), suggesting more difficult AuI oxidation. Notably, heterocyclic substituents, such as thiophene and unprotected indole, were also welltolerated in this transformation (products 7k, 7l). The ultralow catalyst loading (TON around 1000) greatly highlights the efficiency and practicality of this transformation.
Table 4:
Intramolecular cyclization with regular alkynes.
 |
Reaction conditions: The catalyst (0.1 mol%) was added to a solution of alkyne 6 (0.3 mmol) in toluene (0.3 mL), and the reaction mixture was kept at 608C for 6 h. [a] Reaction time: 24 h.
To further showcase the synthetic utility of this transformation, we aimed to convert the thioallylation products into value-added synthetic intermediates. By treatment with TFA/TFAA, thioflavone 8 was successfully formed from acid 3t through a Friedel–Crafts-type cyclization (Figure 3A). The sulfide 5a could be efficiently converted into sulfone 9 by oxidation with mCPBA at 0°C without affecting the pendant allyl group (Figure 3B). The vinyl bromide motif in 5a proved to be a useful synthetic handle for cross-coupling reactions as well, as the desired Suzuki coupling product 10 was obtained in excellent yield (Figure 3C). These results clearly demonstrate the versatile synthetic utility of the thioallylation substrates.
Figure 3.

Synthetic utility of the products. DCM=dichloromethane, mCPBA=m-chloroperbenzoic acid, TFA=trifluoroacetic acid, TFAA= trifluoroacetic anhydride.
In conclusion, we have reported herein an efficient and stereoselective thioallylation of alkynes, possibly enabled by gold redox catalysis. The AuI/III catalytic cycle is proposed with a sulfonium cation as a mild oxidant, which was confirmed by a mass spectrometry study. This reaction displayed broad substrate scope. Different alkynes, including carbonyl-activated alkynes and haloalkynes, are well-suited for this transformation; intramolecular cyclization reactions proved feasible too. Subsequent transformations of the thioallylation products further indicated the synthetic utility of this reaction. Mechanistic details as well as the expansion of this mild gold redox strategy to other sulfides and alkynes are currently under investigation in our group.
Supplementary Material
Acknowledgements
We are grateful to the NSF (CHE-1665122 and CHE1709075), the NIH (1R01GM120240–01), and the NSFC (21629201) for financial support.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201802540
References
- [1].For reviews, see: Hashmi ASK, Gold Bull. 2004, 37, 51–65; Fürstner A, Davies P, Angew. Chem. Int. Ed 2007, 46, 3410–3449;Angew. Chem 2007, 119, 3478–3519; Hashmi ASK, Chem. Rev 2007, 107, 3180–3211; Jiménez-NúñlÇez E, Echavarren AM, Chem. Rev 2008, 108, 3326–3350; Gorin DJ, Sherry BD, Toste FD, Chem. Rev 2008, 108, 3351–3378; Dorel R, Echavarren AM, Chem. Rev 2015, 115, 9028–9072; Asiri AM, Hashmi ASK, Chem. Soc. Rev 2016, 45, 4471–4503.
- [2].a) Gorin DJ, Toste FD, Nature 2007, 446, 395–403; [DOI] [PubMed] [Google Scholar]; b) Pernpointner M, Hashmi ASK, J. Chem. Theory Comput 2009, 5, 2717–2725; [DOI] [PubMed] [Google Scholar]; c) Leyva-Perez A, Corma A, Angew. Chem. Int. Ed 2012, 51, 614–635; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 636–658. [Google Scholar]
- [3].Several vinyl gold species have been isolated; see: Akana JA, Bhattacharyya KX, Muller P, Sadighi JP, J. Am. Chem. Soc 2007, 129, 7736–7737; Liu L-P, Xu B, Mashuta MS, Hammond GB, J. Am. Chem. Soc 2008, 130, 17642–17643; Weber D, Tarselli MA, Gagne MR, Angew. Chem. Int. Ed 2009, 48, 5733–5736;Angew. Chem. 2009, 121, 5843–5846; Hashmi ASK, Shuster AM, Rominger F, Angew. Chem. Int. Ed 2009, 48, 8247–8249;Angew. Chem. 2009, 121, 8396–8398.
- [4].a) Buzas A, Gagosz F, Org. Lett. 2006, 8, 515–518; [DOI] [PubMed] [Google Scholar]; b) Yu M, Zhang G, Zhang L, Org. Lett 2007, 9, 2147–2150; [DOI] [PubMed] [Google Scholar]; c) Wang D, Ye X, Shi X, Org. Lett 2010, 12, 2088–2091; [DOI] [PubMed] [Google Scholar]; d) Wang T, Shi S, Rudolph M, Hashmi ASK, Adv. Synth. Catal 2014, 356, 2337–2342. [Google Scholar]
- [5].Peng H, Akhmedov NG, Liang Y, Jiao N, Shi X, J. Am. Chem. Soc 2015, 137, 8912–8915. [DOI] [PubMed] [Google Scholar]
- [6].For gold/metal dual catalysis, see: Hirner J, Shi Y, Blum S, Acc. Chem. Res 2011, 44, 603–613; Hashmi ASK, Lothschgtz C, Dopp R, Ackermann M, Becker JDB, Rudolph M, Scholz C, Rominger F, Adv. Synth. Catal 2012, 354, 133–147; Garc&a-Dom&nguez P, Nevado C, J. Am. Chem. Soc 2016, 138, 3266–3269.
- [7].For examples of the oxidative addition of AuI to alkyl halides, see: Tamaki A, Kochi JK, J. Organomet. Chem 1972, 40, C81–C84; Tamaki A, Kochi JK, J. Chem. Soc. Dalton Trans 1973, 23, 2620–2626; Tamaki A, Kochi JK, Inorg. Nucl. Chem. Lett 1973, 9, 1175–1177; Tamaki A, Kochi JK, J. Organomet. Chem 1974, 64, 411–425; Johnson A, Puddephatt RJ, J. Organomet. Chem 1975, 85, 115–121; Levin MD, Toste FD, Angew. Chem. Int. Ed 2014, 53, 6211–6215;Angew. Chem. 2014, 126, 6325–6329; Winston MS, Wolf WJ, Toste FD, J. Am. Chem. Soc 2014, 136, 7777–7782.
- [8].For reviews on AuI/III oxidative coupling, see: Hopkinson MN, Gee AD, Gouverneur V, Chem. Eur. J 2011, 17, 8248–8262; Wegner HA, Auzias M, Angew. Chem. Int. Ed 2011, 50, 8236–8247;Angew. Chem. 2011, 123, 8386–8397; for pioneer studies on AuI/III cross-coupling, see: Zhang G, Peng Y, Cui L, Zhang L, Angew. Chem. Int. Ed 2009, 48, 3112–3115;Angew. Chem. 2009, 121, 3158–3161;19322861 Hashmi ASK, Ramamurthi TD, Rominger F, J. Organomet. Chem 2009, 694, 592–597; Pazˇicky´ M, Loos A, Ferreira MJ, Serra D, Vinokunov N, Rominger F, J-kel C, Hashmi ASK, Limbach M, Organometallics 2010, 29, 4448–4458.
- [9].For early examples of gold(III)-mediated nucleophilic addition– homocoupling sequences, see: Hashmi ASK, Blanco MC, Fisher D, Bats JW, Eur. J. Org. Chem 2006, 1387–1389; Wegner HA, Ahles S, Neuburger M, Chem. Eur. J 2008, 14, 11310–11313; Fustero S, Bello P, Fernandez B, del Pozo C, Hammond GB, J. Org. Chem 2009, 74, 7690–7696; for gold(I)catalyzed nucleophilic addition–cross-coupling sequences, see: Zhang G, Cui L, Wang Y, Zhang L, J. Am. Chem. Soc 2010, 132, 1474–1475; Melhado AD, Brenzovich WE Jr., Lacktner AD, Toste FD, J. Am. Chem. Soc 2010, 132, 8885–8887; Brenzovich WE Jr., Benitez D, Lacktner AD, Shunatona HP, Tkatchouk E, Goddard III WA, Toste FD, Angew. Chem. Int. Ed 2010, 49, 5519–5522;Angew. Chem 2010, 122, 5651–5654.
- [10].For photoredox conditions with gold catalysts and photoactive dyes, see: Sahoo B, Hopkinson MN, Glorius F, J. Am. Chem. Soc 2013, 135, 5505–5508; Shu X, Zhang M, He Y, Frei H, Toste FD, J. Am. Chem. Soc 2014, 136, 5844–5847; Hopkinson MN, Tlahuext-Aca A, Glorius F, Acc. Chem. Res 2016, 49, 2261–2272; for photoredox conditions with photoactive gold catalysts only and no additional dyes, see: Xie J, Shi S, Zhang T, Mehrkens N, Rudolph M, Hashmi ASK, Angew. Chem. Int. Ed 2015, 54, 6046–6050;Angew. Chem. 2015, 127, 6144–6148; Xie J, Zhang T, Chen F, Mehrkens N, Rominger F, Rudolph M, Hashmi ASK, Angew. Chem. Int. Ed 2016, 55, 2934–2938;Angew. Chem 2016, 128, 2987–2991; Huang L, Rudolph M, Rominger F, Hashmi ASK, Angew. Chem. Int. Ed 2016, 55, 4808–4813;Angew. Chem 2016, 128, 4888–4893; Xie J, Li J, Weingand V, Rudolph M, Hashmi ASK, Chem. Eur. J. 2016, 22, 12646–12650; Huang L, Rominger F, Rudolph M, Hashmi ASK, Chem. Commun 2016, 52, 6435–6438; Witzel S, Xie J, Rudolph M, Hashmi ASK, Adv. Synth. Catal. 2017, 359, 1522–1528; for basic conditions, see: Cai R, Lu M, Aguilera EY, Xi Y, Akmedov NG, Petersen JL, Chen H, Shi X, Angew. Chem. Int. Ed 2015, 54, 8772–8776;Angew. Chem 2015, 127, 8896–8900;26119925 Dong B, Peng H, Motika SE, Shi X, Chem. Eur. J 2017, 23, 11093–11099.
- [11].For Meyer–Schuster-type rearrangement, see: a) Ref. [8c]; Yu Y, Yang W, Pflasterer D, Hashmi ASK, Angew. Chem. Int. Ed 2014, 53, 1144–1147;Angew. Chem 2014, 126, 1162–1165; Um J, Yun H, Shin S, Org. Lett 2016, 18, 484–487; Tlahuext-Aca A, Hopkinson MN, Garza-Sanchez RA, Glorius F, Chem. Eur. J 2016, 22, 5909–5913; for a Sonogashira-type coupling reaction, see: Tlahuext-Aca A, Hopkinson MN, Sahoo B, Glorius F, Chem. Sci 2016, 7, 89–93; for oxyarylation and aminoarylation, see: f) Ref. [10f]; Wang Z, Tan T, Wang C, Yuan D, Zhang T, Zhu P, Zhu C, Zhou J, Ye L, Chem. Commun 2017, 53, 6848–6851.
- [12].Ye X, Wang J, Ding S, Hosseyni S, Wojtas L, Akhmedov NG, Shi X, Chem. Eur. J 2017, 23, 10506–10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].The Hashmi group reported an intramolecular oxyallylation reaction in an analogous allyl 2-ethynylphenyl ether system; a mechanism involving [3,3] sigmatropic rearrangement was proposed, which was supported by a cross-over experiment and a computational study: Hashmi ASK, Graf K, Ackermann M, Rominger F, Chem Cat Chem 2013, 5, 1200–1204; Ackermann M, Bucher J, Rappold M, Graf K, Rominger F, Hashmi ASK, Chem. Asian J 2013, 8, 1786–1794; Nunes dos Santos Comprido L, Klein JEMN, Knizia G, Kastner J, Hashmi ASK, Chem. Eur. J 2017, 23, 10901–10905.
- [14].Hayakawa K, Kamikawaji Y, Kanematsu K, Tetrahedron Lett. 1982, 23, 2171–2174. [Google Scholar]
- [15].a) Dekorver KA, Li H, Lohse AG, Hayashi R, Lu Z, Zhang Y, Hsung RP, Chem. Rev 2010, 110, 5064–5106; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Evano G, Coste A, Jouvin K, Angew. Chem. Int. Ed 2010, 49, 2840–2859; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2902–2921. [Google Scholar]
- [16].Sindhu KS, Thankachan AP, Sajitha PS, Anikumar G, Org. Biomol. Chem 2015, 13, 6891–6905. [DOI] [PubMed] [Google Scholar]
- [17].Wu W, Jiang H, Acc. Chem. Res 2014, 47, 2483–2504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.