

Abstract
Purpose:
The current study was designed to address our hypothesis that oxidative stress secondary to the ionizing event upregulates PTEN mRNA and protein in the lungs of C57BL/6J mice through oxidative DNA damage resulting in CpG hypomethylation in the PTEN promoter.
Methods:
Fibrosis-prone C57BL/6J mice were exposed to 0 or 15 Gy of 320 kVp x-rays to the whole thorax. Lung tissue was serially harvested at time points between 1 day and 6 months postirradiation. Tissue levels of PTEN mRNA, total protein, and phosphorylated PTEN, as well as CpG methylation of the PTEN promoter, expression of DNA methyltransferases 1 (Dnmt1) and 3a (Dnmt3a), NADPH oxidase 4 (Nox4) protein expression, and DNA damage levels were measured. The induction of DNA damage and global methylation changes were also examined in hydrogen peroxide (H2O2)–treated human umbilical vein endothelial cells and human bronchial epithelial cells in vitro.
Results:
These experiments demonstrate that PTEN mRNA and protein, Nox4 protein, and DNA damage levels increase continuously from 1 day to 6 months following radiation exposure. Elevated PTEN transcription and translation are likely the result of the observed decrease in CpG methylation of the PTEN promoter region. This finding is not consistent with the observed increase in Dnmt1 and Dnmt3a protein expression, implicating an alternative mechanism as the driving force behind hypomethylation. In vitro results provide evidence that H2O2 can induce DNA damage and affect DNA methylation status. The Mn porphyrin-based superoxide dismutase mimic MnTnHEx-2-PyP5+ exhibited partial rescue from radiation-induced hypomethylation.
Conclusions:
Taken together, these data suggest that reactive oxygen species–induced DNA damage results in hypomethylation of the PTEN promoter, upregulation of PTEN mRNA and protein, and a subsequent increase in apoptosis in irradiated lung tissue.
Keywords: whole thorax irradiation, PTEN, methylation, radiation lung injury
Introduction
The success of clinical radiotherapy in treating tumors in or around the thoracic region is limited by adverse normal tissue complications resulting from the damaging effects of ionizing radiation on healthy tissue [1]. Lung tissue is known to be particularly radiosensitive, with 5%–50% of lung cancer patients experiencing radiation pneumonitis and/or fibrosis after radiotherapy [2]. The risk for normal tissue complications limits the maximum effective dose that can be delivered and has the potential to limit quality of life among cancer survivors [3–5]. Damage to the lungs is also of concern following accidental radiation exposure. Studies examining victims of nuclear accidents have shown that, while whole-body exposure to radiation can cause acute lethal hematopoietic and gastrointestinal syndromes, these syndromes are potentially survivable with intensive supportive care [6]. In such cases, damage to later responding tissues such as the lung becomes a major impediment to survival [7] Although progress has been made in treating the acute symptoms of radiation exposure, no clinical treatments are currently available to prevent or successfully mitigate damage in the lung [8]. The absence of a U.S. Food and Drug Administration–approved therapeutic intervention is the result, in part, of poor understanding of the molecular basis of the progression of injury in late-responding tissues.
Recent studies from our lab have shown that the increase in reactive oxygen species (ROS) observed following radiation exposure induces apoptosis in parenchymal cells in lung tissue [9]. This finding implicates that both the primary radiation-induced ROS production and the inflammation-driven continuous ROS overproduction play a key role in the development of pulmonary injury. We determined that the ROS-mediated apoptosis was the result, at least in part, of upregulation of phosphatase and tensin homolog (PTEN) mRNA and protein expression and subsequent inhibition of PI3K-ILK/Akt survival signaling [9]. The means through which ROS is able to regulate PTEN gene expression remains unknown. In pursuit of an explanation we looked to recent advances in epigenetics, particularly in DNA methylation, which have provided new insights into the regulation of gene expression [10]. Although established patterns of DNA methylation can maintain transcriptional levels of genes through the cell life, the pattern of DNA methylation is observed to be dynamic and responsive to both endogenous and exogenous stimulation [11]. The response of methylation machinery to such signals can cause hypermethylation or hypomethylation of gene promoter sequences, respectively reducing or increasing gene expression [12]. We therefore hypothesized that the upregulation of PTEN expression in irradiated lung may result from hypomethylation of PTEN promoter and that this may be the result of an increase in cellular oxidative stress.
DNA methylation is accomplished by the DNA methyltransferase (Dnmt) catalyzed transfer of a methyl group from S-adenosyl-l-methionine to the 5’ position of a cytosine residue in mammalian DNA [13]. The extent of DNA methylation is regulated by the activity of DNA methyltransferases. At present, three active forms of DNA methyltransferases (Dnmt1, Dnmt3a, and Dnmt3b) have been identified in mammals. Dnmt1 is the predominant mediator maintaining existing methylation patterns in the DNA of somatic cells [14]. Dnmt1 acts primarily on hemimethylated CpG dinucleotides [15]. Dnmt3s, the enzymes responsible for de novo methylation, can methylate hemimethylated and unmethylated CpG at the same rate [14]. In CpG dinucleotides, these enzymes methylate the cytosine base, whereas the guanine base is more likely to be affected by oxidative damage. In fact, 8-hydroxy-2-deoxyguanosine (8-OHdG) is one of the best characterized byproducts of ROS-induced DNA damage [16]. For this reason it is commonly used as a marker of DNA damage. It has been suggested that, although the cytosine residue is not damaged by oxidative stress, the damaged guanine base interferes with methylation at the cytosine residue [17].
In the genomic DNA of mammalian cells, a GC-rich region is located at the 5’ promoter region of all housekeeping genes and many other ubiquitously expressed genes [18]. These regions are crucial for methylation-mediated gene silencing [11]. In this study, we examined CpG-methylation of PTEN promoter from the –809 to +59 area to determine the effect of radiation exposure on PTEN promoter methylation. We used this information to further scrutinize the relationship between oxidative damage and PTEN expression and methylation in irradiated lung tissues. In addition, we studied the role of hydrogen peroxide (H2O2) in inducing DNA damage and PTEN methylation in cultured lung epithelial cells and human umbilical vein endothelial cells (HUVECs). Our findings indicate that, in the presence of oxidative stress, PTEN expression is upregulated as a result of hypo-CpG methylation of the PTEN promoter. We next examined whether targeting oxidative stress using a powerful superoxide dismutase (SOD) mimic with high lipophilicity and bioavailability, Mn(III) meso-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin, MnTnHex-2-PyP5+, can affect the magnitude of methylation. MnTnHex-2-PyP5+, which has previously been shown to mitigate radiation damage in lung tissue [19], accumulates in cellular organelles such as membranes, mitochondria, lysosomes and nuclei. It also accumulates at high levels in all tissues and crosses the blood–brain barrier [20,21].
Materials and Methods
Animals and irradiation
All experiments were performed with prior approval by the Institutional Animal Care and Use Committee (IACUC) at XX.
Experiment 1:
Female C57BL/6J mice, weighing between 22 and 24 g, were housed five animals/cage and maintained under identical conditions with food and water provided ad libitum in accordance with the Guide for Care and Use of Laboratory Animals. Mice were randomized to two groups, one of nonirradiated controls and the other of animals exposed to IR. Radiation was delivered as a single fraction of 15 Gy to the whole thorax using 320 kV x-rays (69 cGy min–1, HVL = 1.0 mm Cu; Therapax 320, Pantak Inc., East Haven, CT). Eight-millimeter-thick lead shielding was used to block radiation to the head, abdomen, and legs. All mice were anesthetized before irradiation with an intraperitoneal injection of a ketamine (100 mg/kg)/xylazine (10 mg/kg) mixture. Mice were humanely euthanized per IACUC regulations at predetermined time points of 1 and 3 days; 1, 3, and 6 weeks; and 6 months postradiation (10 mice for each time point for each group). At the time of sacrifice, lung tissues were harvested, snap frozen in liquid nitrogen, and stored at –80°C.
Experiment 2:
Forty female C57BL/6J mice, weighing between 22 and 24 g, were grouped for saline control (control: sham-radiation, saline injections), MnTnHex-2-PyP5+ subcutaneous injections (Hxyl: 1.6 mg/kg, twice per day for 4 weeks), radiation (Rad: single fraction of 15 Gy to the whole thorax), and radiation plus Hxyl injections (Rad+Hxyl). Each group contained 10 mice. Animals were sacrificed 6 weeks after treatments, and lung tissues were harvested. Radiation was delivered as described in Experiment 1.
RNA and genomic DNA isolation
Snap-frozen mouse (experiment 1 and 2) right lung tissue was homogenized in 2 mL of Trizol reagent (Invitrogen; Carlsbad, CA) using the PowerGen 125 homogenizer (Fisher Scientific; Pittsburgh, PA). The total RNA and genomic DNA were isolated following instructions for use of Trizol.
cDNA synthesis and RT-PCR
Reverse transcription (RT) was performed, per user manual, using SuperScript III Reverse Transcriptase (Invitrogen). Polymerase chain reaction (PCR) amplification was carried out using Platinum Taq DNA polymerase (Invitrogen) and two pairs of primers in the same reaction: one amplifies a 536bp of PTEN gene fragment using the forward primer 5’-GACTTGAAGGTGTATACAGGAAC-3’ and reverse primer 5’-GCTGGCAGACCACAAACTGAG-3’; the other amplifies a 1-kb GAPDH fragment, serving as an internal control, using the forward primer 5’-GGTGAAGGTCGGTGTGAACG-3’ and reverse primer 5’-TGGAGGCCATGTAGGCCATG-3’. The two gene fragments were visualized by electrophoretic separation in 1.5% agarose gel containing 0.5 µg/mL ethidium bromide.
Sodium bisulfite treatment and PCR amplification
The genomic DNA of five mice from each time point was isolated as described above. Bisulfite treatment was used to convert unmethylated cytosine residues to uracil residues. This reaction has been shown to be more than 99% complete. This treatment was accomplished using the EZ methylation kit (ZYMO Research; Orange, CA). Briefly, a total of 600 ng of genomic DNA (total volume 20 µL) was added to 130 µL of ZYMO conversion reagent followed by incubation in a thermal cycler at 98°C for 10 minutes and then 53°C for 5 hours, according to the manufacturer’s manual. After purification, the methylated genomic DNA was subjected to PCR amplification. The 5’-terminal sequence of PTEN gene from –809 to +59 (NM_008960) that contains 79 CpG dinucleotides was analyzed by two pairs of anchored primers. The forward primer 1, 5’-GTTGTAGTTATGATGGAAGTTTGAGAGTTGAG-3’, and reverse primer 1, 5’-TATTAAAAACAAAAAAAAAAAAAAAAACAACTCTCC-3’, amplified the bisulfate-treated PTEN sequence from –809 to –385. The forward primer 2, 5’-GGAGAGTTGTTTTTTTTTTTTTTTTTGTTTTTAATA-3’, and reverse primer 2, 5’-CCATCCTCTTAATATCTCCTTTTATTTCTAC-3’, amplified the bisulfate-treated PTEN sequence from –415 to +59. The amplified two fragments were subsequently separately cloned.
Cloning, sequencing, and calculating of methylation
The amplified PCR fragments were cloned using a PCR Cloning kit (QIAGEN; Valencia, CA) or a pCR8/GW/TOPO TA cloning kit (Invitrogen) according to the user manuals. One microliter of ligation–reaction mixture was incubated with 50 μL of DH5α competent cells on ice for 30 minutes followed by heat shock in a 42°C water bath for 30 seconds. After incubating on ice for 2 minutes, competent cells were incubated with 250 μL of Super Optimal broth with catabolite repression medium at 37°C for 30 minutes and then were plated on two lysogeny broth (LB) agar plates containing 100μg/mL of ampicillin or spectinomycin, depending on which cloning kit was used. The colonies were picked up and grown in 3 mL of LB medium for plasmid extraction. The miniprep of plasmid DNA was performed using the QIAGEN Miniprep kit (QIAGEN). Insertion was verified by digestion with Eco RI. Four positive clones per amplified fragment were sequenced. The methylation ratio of total methylated CpG in each sample was calculated by the followed formula: ratio = total methylation score/clone number. We analyzed the statistical difference in methylation between irradiated and control mice at each time point using the calculated methylation ratio.
Immunohistochemistry and TUNEL staining
Immunohistochemical staining was carried out as previously described [22]. The anti-8-OHdG antibody was purchased from Millipore (Billerica, MA). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was performed according to the user manual with an in situ cell death detection kit purchased from Roche Molecular Biology (Mannheim, Germany). The staining was scored by researchers blinded to the study design.
Tissue homogenization and Western blot
The snap-frozen right lung was homogenized and Western blot was performed as previously described [23]. Antibodies for site-specific phosphorylation of PTEN and total PTEN, Dnmt1, Dnmt3A, Dnmt3B, and the peroxidase-conjugated secondary antibody were purchased from Cell Signaling Technology (Beverley, MA). The blots were visualized and developed using Pico West Chemiluminescent substrate (Pierce Chemical Company; Rockford, IL). To control for loading efficiency, membranes were stripped and reprobed with an antibody against α-tubulin antibody (Sigma–Aldrich; St. Louis, MO). Images were scanned with Adobe photoshop (Adobe; San Jose, CA) and quantified with the National Institutes of Health (NIH) ImageJ tool. Expression and phosphorylation levels were normalized to α-tubulin or unphophorylated protein expression where described and are expressed as relative values (ie, relative density = protein/α-tubulin levels or phophorylated protein/unphophorylated protein). Background correction values were subtracted from each lane to minimize variability across membranes.
Cell culture
HUVECs were obtained from Cambrex Bio Science (Walkersville, MD) and cultured in endothelial cell growth medium-2 supplied with growth factors. BEAS-2B cells, a human bronchial epithelial cell line, were obtained from American Type Culture Collection (ATCC, Manassas, VA) and grown in serum-free bronchial epithelial cell growth medium (Cambrex Bio Science) with 100 units/mL penicillin, and 100 µg/mL streptomycin at 37°C, 5% CO2.
Detection of DNA damage and methylation in H2O2-treated cells
HUVECs and Beas-2B cells were seeded in 12-well plates or 10-cm dishes 24 hours before H2O2 treatment. Cells were then treated with 1 mM of H2O2 for 0, 30, 60, and 120 minutes. Twelve-well plates were fixed, and 8-OHdG levels were evaluated using immunofluorescent labeling as previously described [23]. Cells in 10-cm dishes were collected for DNA isolation using the phenol/chloroform method. The Methylamp Global DNA Methylation Quantification Kit (Epigentex; Brooklyn, NY) was used to detect global methylation in genomic DNA of cells treated with H2O2.
Results
Radiation progressively induced apoptosis in lung tissue
Previous study showed that exposure to a radiation dose of 15 Gy induces apoptosis in type I and II pneumocytes and endothelial cells 6 weeks after irradiation [9]. To investigate whether a dynamic induction of apoptosis in the lung tissue occurs during progression to injury, we looked at apoptosis in lung tissues of C57BL/6J mice isolated at various time points, ranging from 1 day to 6 months after radiation. TUNEL-positive cells increased progressively from 1 day to 6 month following radiation (Fig. 1).
Figure 1.
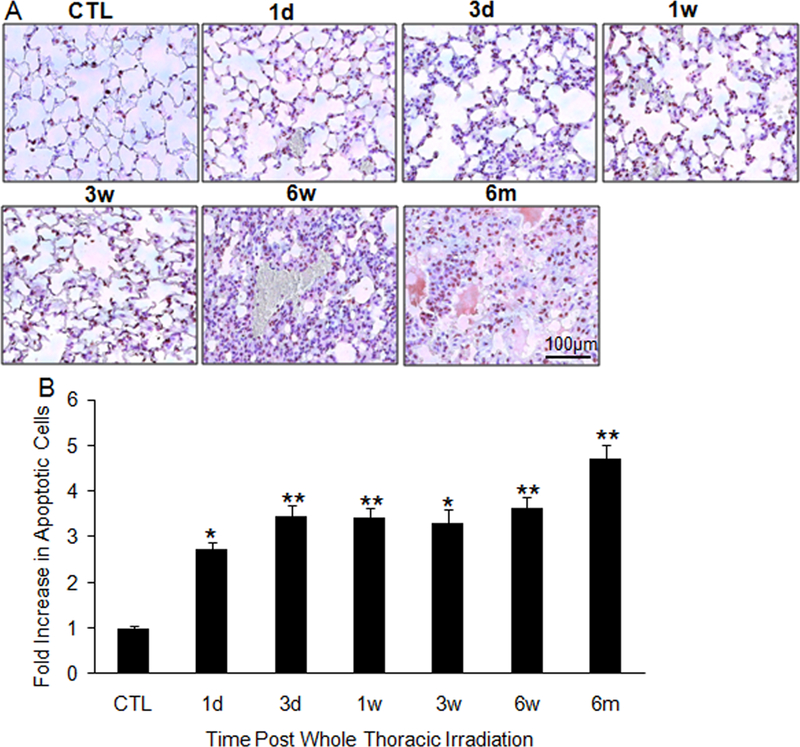
Apoptosis in irradiated lung tissue. (A) TUNEL staining of lung tissue sections collected at 1 and 3 days; 1, 3, and 6 weeks; and 6 months after 15-Gy whole-thorax irradiation. (B) Increases in positive staining in irradiated lung tissue were quantified as a percentage of apoptosis observed in controls. Each image is 400 × magnifications. CTL: control; 1d: 1 day; 3d: 3-day; 1w: 1-week; 3w: 3-week; 6w: 6-week; 6m: 6-month. *P < 0.05; **P < 0.01 vs. age-matched control mice; n = 5.
Radiation upregulates PTEN mRNA and protein expression but not phosphorylation
Although transcriptional regulation, as indicated by promoter activity, is a major modulator of gene expression and protein activity [24,25], protein level and function can also be regulated at posttranslational levels. In the case of PTEN, the phosphorylated protein is more stable than the unphosphorylated protein but is less active [24,26]. We therefore investigated the levels of PTEN mRNA and protein, as well as the phosphorylation status of the protein, in lung tissues of C57BL/6J mice. Significant increases in mRNA expression (Fig. 2A), total protein (Fig. 2B), and protein phosphorylation (Fig. 2C) were observed beginning 1 day after radiation and continuing to the 6-month time point. We further analyzed the ratio of phosphorylated PTEN to total PTEN. A slight decrease in this ratio was observed from 1 day to 6 months after radiation (Fig. 2D), suggesting that the observed increase in phosphorylation may reflect only the increase in total PTEN protein levels. This also suggests that the increase in PTEN protein levels is the result of upregulation of promoter activity, and not the result of phosphorylation-mediated stabilization of the protein.
Figure 2.
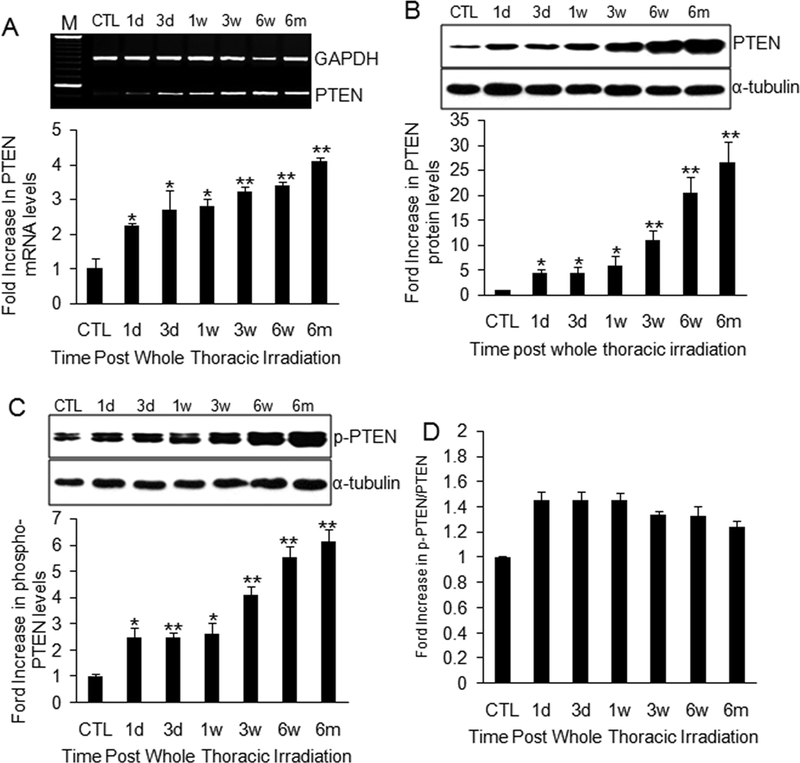
PTEN mRNA and protein expression, and phosphorylated PTEN levels in irradiated lung. (A) PTEN mRNA increased progressively at each time point after radiation. Top panel: Representative electrophoresis of PTEN RT-PCR in agarose gel. M: 100 bp DNA marker. Bottom panel: Semi-quantitative analysis of RT-PCR amplification. (B) Total PTEN protein increased at each postradiation time point. (C) Total levels of phospho-PTEN (phosphorylated PTEN) were also significantly increased in lung tissue after radiation. Top panel: Representative Western blot of total PTEN (B) or phospho-PTEN (C) and α-tubulin protein. Bottom panel: Semi-quantitative analysis of Western blot. (D) The ratio of P-PTEN (phosphorylated PTEN) to total PTEN revealed a slight decrease from 1 day to 6 months postradiation, but no significant differences were observed. CTL: control; 1d: 1 day; 3d: 3-day; 1w: 1-week; 3w: 3-week; 6w: 6-week; 6m: 6-month. *P < 0.05; **P < 0.01 vs. age-matched control mice; n = 6–10.
Radiation induces hypomethylation of the PTEN promoter at CpG dinucleotides
Hypomethylation of a gene promoter contributes to elevated gene expression [12]. Our study revealed a persistent increase in PTEN mRNA after radiation exposure. We therefore investigated whether there is a corresponding reduction in methylation in the PTEN promoter region. The bisulfate-based sequencing method was applied to examine CpG methylation within the PTEN promoter. As shown in Fig. 3, the methylation ratio of PTEN was significantly decreased in irradiated C57BL/6J mice lung tissue when compared with that in sham-irradiated mice. Moreover, the methylation ratio decreased progressively from 1 day to 6 months postradiation (Fig. 3A). We were able to examine the change in methylation at a single CpG dinucleotide and found that methylation was reduced for most CpG dinucleotides in irradiated lung tissue (Fig. 3C) compared to control tissue (Fig. 3B).
Figure 3.
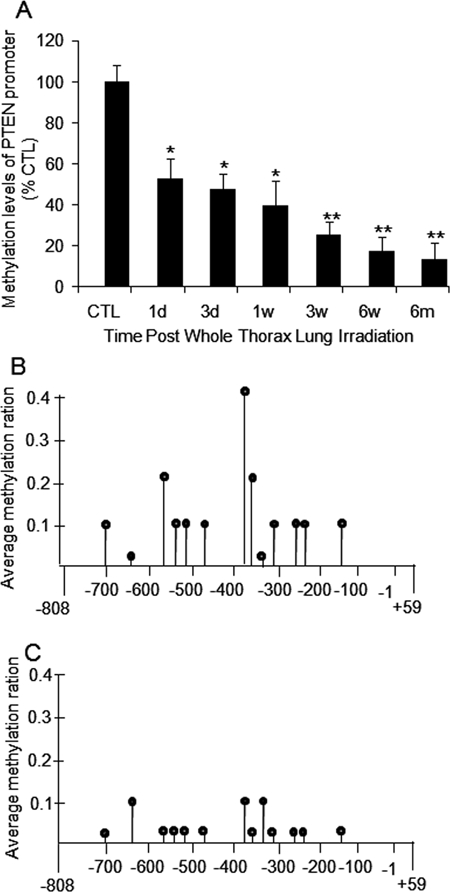
Hypomethylation of PTEN promoter after radiation. (A) Relative CpG methylation in PTEN promoter normalized with age-matched control mice was reduced at all time points following radiation. CTL: control; 1d: 1 day; 3d: 3-day; 1w: 1-week; 3w: 3-week; 6w: 6-week; 6m: 6-month. (B) Average methylation ratio of single CpG methylation in sham-irradiated control mice. (C) Average methylation ratio of single CpG methylation in irradiated mice. *P < 0.05; **P < 0.01 vs. age-matched control mice; n = 5.
Hypomethylation of PTEN promoter is not associated with DNA methyltransferase expression
DNA methyltransferases (Dnmt) are the enzymes that catalyze the transfer of a methyl group to DNA. Three active forms of DNA methyltransferases (Dnmt1, Dnmt3a, and Dnmt3b) have been identified in mammals.15 We investigated the expression of Dnmt1, Dnmt3a, and Dnmt3b from 1 day to 6 months following radiation. We found that protein levels of Dnmt1 (Fig. 4A) and Dnmt3a (Fig. 4B) were significantly reduced 1 day after radiation but had recovered by day 3. A decrease in expression was observed between 1 and 3 weeks; however, expression of both of these proteins was significantly increased from 6 weeks to 6 months postradiation. We were unable to detect a Dnmt3b signal, suggesting that expression of this Dnmt3b may be much lower in lung tissue. Unexpectedly, the changes in Dnmt1 and Dnmt3a did not correlate with hypomethylation of the PTEN promoter or with PTEN mRNA expression, suggesting that PTEN promoter hypomethylation is affected by factors other than methyltransferase activity.
Figure 4.
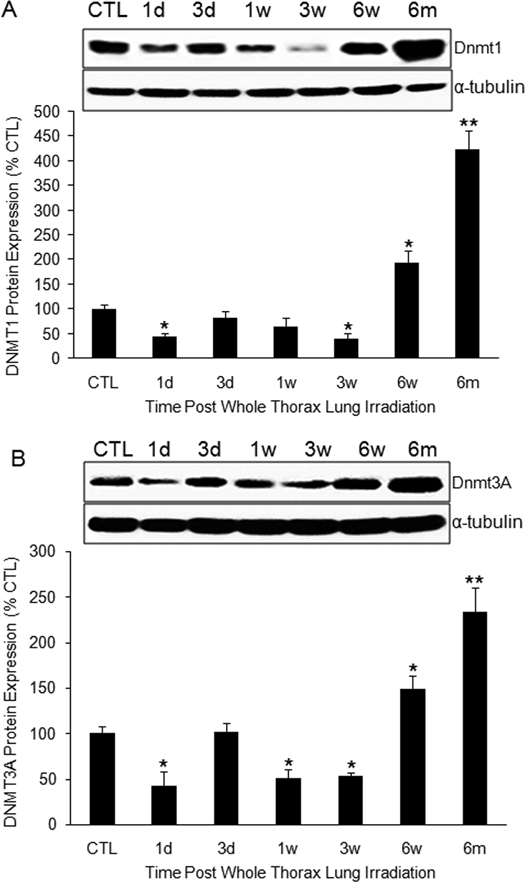
DNA methyltransferases expression after radiation. (A) Dnmt1 protein expression was reduced at early time points in irradiated lung, but increased 6 weeks and 6 months following radiation. (B) Protein levels of Dnmt3A in irradiated lung tissue showed a similar pattern. Top panel in (A) and (B): Representative Western blot of Dnmt1 (A) or Dnmt3 (B) and α-tubulin protein. CTL: control; 1d: 1 day; 3d: 3-day; 1w: 1-week; 3w: 3-week; 6w: 6-week; 6m: 6-month. *P < 0.05; **P < 0.01 vs. age-matched control mice; n = 6–10.
Consistent increase in DNA damage and Nox4 expression after radiation
It is well established that an environment of chronic oxidative stress persists in the irradiated lung [27,28]. High levels of ROS can cause a wide range of DNA lesions [17]. The best characterized byproduct of oxidative DNA damage is 8-OHdG [29]. We therefore assessed oxidative stress by measuring 8-OHdG levels in irradiated and nonirradiated lung tissues using immunohistochemistry. Irradiated lung tissues showed a consistent increase in 8-OHdG levels from postirradiation day 1 to 6 months following exposure (Fig. 5A, 5B). In phagocytic cells, ROS is produced through activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. Nox4 is the only nonphagocytic NADPH oxidase subunit [30,31] and the only subunit upregulated by chronic hypoxia.32 Because of this known relationship, we chose to evaluate Nox4 protein expression in irradiated lung tissues from day 1 to 6 months postradiation. Western blot revealed that Nox4 protein levels increased continuously from 1 day to 6 months after radiation (Fig. 5C).
Figure 5.
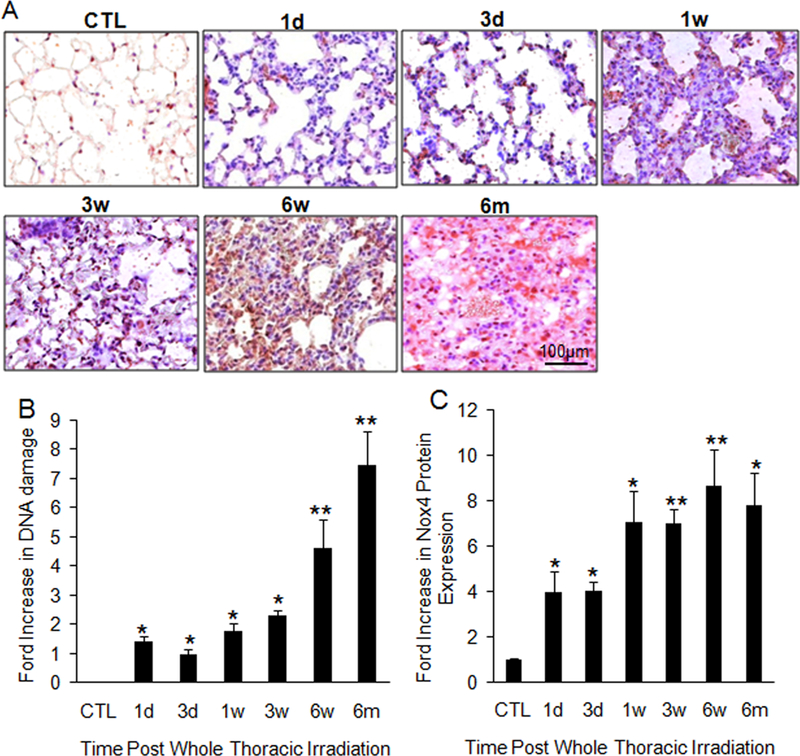
Consistent increase in DNA damage and Nox4 expression after radiation. (A) Immunochemistry of 8-OHdG levels shows an increase in oxidative damage. Each image is 400× magnification. (B) Semiquantitative analysis of 8-OHdG levels shows continuous increase in damage over time (a). n = 5. (C) Nox4 protein levels detected by Western blot show increased expression at all points following radiation. CTL: control; 1d: 1 day; 3d: 3-day; 1w: 1-week; 3w: 3-week; 6w: 6-week; 6m: 6-month. *P < 0.05; **P < 0.01 vs. age-matched control mice; n = 6–10.
H2O2-induced DNA damage alters global methylation in lung epithelial cells and HUVECs
To further explore the relationship between ROS-induced DNA damage and methylation, we measured 8-OHdG levels and global methylation in H2O2-treated HUVECs and BEAS-2B cells. Immunofluorescent staining showed that 8-OHdG levels were increased in both HUVECs and Beas-2B cells 30 minutes after treatment with 1 mM H2O2. The most intense fluorescent signal was observed 120 minutes after treatment in both cell lines (Fig. 6A). We observed significant increases in global methylation in HUVECs (Fig. 6B) 60 minutes after H2O2 treatment and in Beas-2B cells (Fig. 6C) 30 minutes after H2O2 treatment. Interestingly, methylation at 120 minutes was significantly decreased compared to methylation measured at 60 minutes. This suggests that the global level of methylation is elevated in response to the initial oxidative insult but decreases with greater oxidative damage to DNA.
Figure 6.
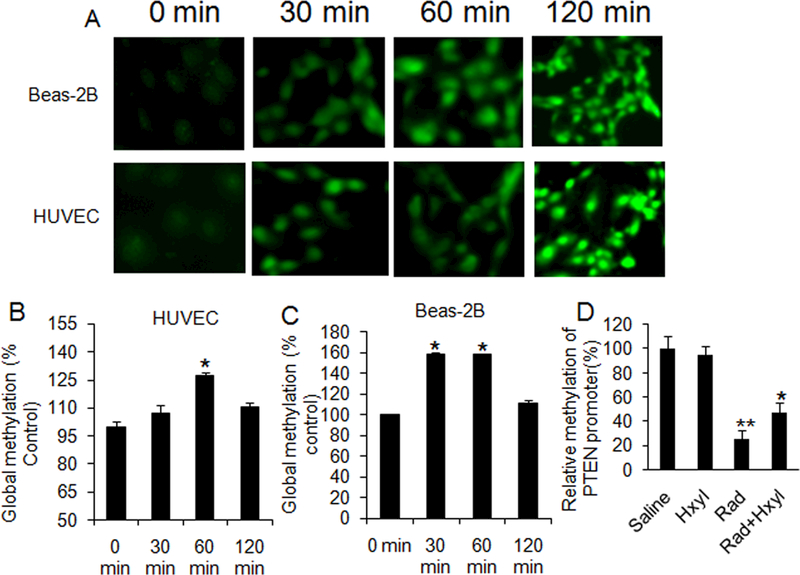
H2O2-induced DNA damage and global methylation in cells and effects of MnTnHex-2-PyP5+ on PTEN promoter methylation. (A) Immunofluorescence of 8-OHdG levels in Beas-2B cells and HUVECs. Cells were incubated with 1 mM of H2O2 for 0, 30, 60, and 120 minutes. Each image is 400× magnification. (B) Global methylation level in HUVECs after treatment with 1 mM of H2O2 for 0, 30, 60, and 120 min. (C) Global methylation level in Beas-2B cells after treatment with 1 mM of H2O2. *P < 0.01 vs. 0 and 120 min; n = 4. (D) Relative PTEN promoter methylation in female C57BL/6J mice treated for 4 weeks with 1.6 mg/kg MnTnHex-2-PyP5+ daily. Radiation-induced oxidative stress led to significant hypomethylation of PTEN promoter, which was partially reversed with MnTnHex-2-PyP5+ (Hxyl). **P < 0.001 vs. control (Sal); *P = 0.051 vs. radiation alone group (Rad); n = 10.
MnTnHex-2-PyP5+significantly reverses PTEN promoter hypomethylation
To determine whether targeting oxidative stress in vivo can reverse the observed hypomethylation of the PTEN promoter in vivo, a SOD mimic with reported ability to suppress radiation damage to normal lung tissue was utilized [19,20]. In this study, radiation induced significant hypomethylation of PTEN promoter in the irradiated lung tissue of female C57BL/6J mice (P < 0.001), while MnTnHex-2-PyP5+ administration partially reversed PTEN promoter methylation (P = 0.051) (Fig. 6D).
Discussion
It is widely accepted that exposure to ionizing radiation causes damage to DNA, resulting in cell death via apoptosis or mitotic catastrophe [33]. Our recent studies have shown that Mn(III) meso-tetrakis(N,N’-duethylimidazolium-2-yl)porphyrin (AEOL 10150), another potent SOD mimic, can mitigate cell apoptosis in rodent lung when given after radiation [9]. These findings are supported by previous work performed by Vorotnikova et al., which demonstrated that two neutral porphyrins are capable of mitigating radiation-induced apoptosis of endothelial cells in vitro [34]. Together, these studies suggest that apoptosis following radiation might be induced through redox-related signaling, the mechanisms of which have not been well described.
Our previous work identified the PI3K/Akt pathway as playing a role in radiation-induced apoptosis [9]. In those studies, we found that PTEN, an antagonist of PI3K/Akt prosurvival signaling, was upregulated at the mRNA and protein level in irradiated murine lungs 6 weeks following radiation exposure. In those studies only a single time point was evaluated.
Here, we sought to determine the mechanism driving this upregulation. We first determined that the increase in apoptosis in irradiated lungs, previously observed at 6 weeks, begins 24 hours postirradiation and increases continuously until the 6-month endpoint of this study (Fig. 1). The increasing number of cells undergoing apoptosis corresponds to elevated PTEN mRNA and protein levels across all of the examined time points (Fig. 2). The minimal decrease in the ratio of phosphorylated PTEN/total PTEN suggests that elevated PTEN protein levels following radiation were not the result of increased stability of the protein but that other mechanisms play a role in the observed upregulation.
Upon examination of 5’-terminal methylation patterns within the PTEN gene, we found that the PTEN promoter was hypomethylated in irradiated tissues (Fig. 3). However, hypomethylation of cytosine bases in the promoter region did not correspond to significant changes in expression of any of the DNA methyltransferases. We found that methylation of CpG dinucleotides in the lungs of irradiated mice decreases progressively from 1 day to 6 months following radiation (Figure 3). Surprisingly, the protein levels of Dnmt1 and Dnmt3a, the two active DNA methyltransferases with expression observed in the lung, were significantly increased from 6 weeks to 6 months (Fig. 4). This discrepancy between methylation and DNA methyltransferase expression suggests that radiation-induced hypomethylation of the PTEN promoter is mediated independently of methyltransferase activity. Taken together, these findings indicate that, following radiation, PTEN expression increases as a result of hypomethylation of the promoter regions of the gene, but that this hypomethylation is not the result of changes in Dnmt expression or, by extension, activity. This suggests that hypomethylation results instead from interference with the ability of Dmnts to interact with the DNA. This supposition is supported by work from Turk et al., who demonstrated that 8-OHdG residues resulting from oxidative damage to DNA interfere with the ability of Dmnts to methylate cytosine residues that are two or three base pairs away in the 5’ direction on the same strand [35]. Our data, together with this finding, suggest that ROS-induced DNA damage is responsible for radiation-induced hypomethylation of PTEN promoter.
Increased NADPH oxidase expression and activity are possible sources of increased ROS production postirradiation [36]. We and others have shown an elevation in NADPH oxidase expression in irradiated tissue [9] and cells [37,38]. Here, we observed that Nox4 expression continues to increase at all time points from 1 day to 6 months postradiation. This rise in protein expression is mirrored by an increase in DNA damage, as measured by the presence of 8-OHdG (Fig. 5). The Nox4 isoform is unique among NADPH oxidases, in that the wild-type protein produces H2O2 rather than superoxide anion (O2–) [39]. The production of H2O2 is thought to be the result of an extra 28 amino acid sequence present only in Nox4 that contains a highly conserved histidine residue [40]. Takac and colleagues hypothesized that the histidine residue might serve as a source of protons to spontaneously dismutate O2– to H2O2 [40].
Because of this specific element of Nox4 and the apparent radiation sensitivity of Nox4 expression, we chose H2O2 treatment as an in vitro model for (at least a part of) radiation-induced oxidative stress (Fig. 6). We found evidence of ROS-induced DNA damage (8-OHdG) in cultured HUVECs and human lung epithelial cells (Beas-2B) treated with 1 mM H2O2. This suggests that Nox4-derived H2O2 may be responsible for some of the oxidative DNA damage that we observed in vivo. Furthermore, the simultaneous loss of Nox4 mRNA and protein expression and mitigation of oxidative DNA damage in animals treated with AEOL10150, a catalytic antioxidant, is highly suggestive of a self-perpetuating relationship between Nox4 expression and ROS overproduction [9].
Radiation-induced upregulation of PTEN expression recently has been demonstrated in vitro [41,42]. In alveolar macrophages, elevated production of ROS following radiation is known to contribute to an increase in PTEN mRNA and protein levels [43]. This observation indicates that, following an ionizing radiation event, the presence of high levels of ROS affects regulation of PTEN mRNA in exposed tissues. Our previous observation of parallel expression of Nox4 and PTEN mRNA levels in irradiated lung tissues [9] further supports a relationship between ROS and transcription or stability of PTEN mRNA.
Previous work has demonstrated that ROS can cause a wide range of DNA lesions, including base modification, deletions, strand breakage, and chromosomal rearrangement [17], that can interfere with the binding of DNA methyltransferases to the DNA, resulting in global hypomethylation [44]. Our in vitro findings indicate that, during the initial period of oxidative stress, cellular response is quite the opposite and DNA is globally hypermethylated. This hypermethylation subsides at later incubation times, when DNA damage is more extensive, suggesting that ROS-induced hypomethylation may be dependent on the level of oxidative DNA damage. The implications of the early hypermethylation phase are as yet unknown, but a link between the degree of hypomethylation and the extent of DNA damage fits in with the late development of pathologic processes in lung tissue. These in vitro findings are supported by our in vivo work which shows that radiation-induced DNA damage increases progressively over time while the level of CpG methylation in PTEN promoter continues to decrease. This continual reduction in PTEN promoter methylation results in a time-dependent increase in PTEN mRNA and protein expression, which likely mediates the latent increase in apoptosis in irradiated lung tissue observed in a previous study [9]. MnTnHex-2-PyP5+, a lipophilic analog with SOD-like activity identical to that of AEOL 10150, was able to partly reverse radiation-induced hypomethylation in PTEN promoter in female C57BL/6J mice (Fig. 6D).
Conclusions
Radiation-induced upregulation of ROS in the lung creates an environment of chronic oxidative stress, exacerbating initial tissue injury through continuous induction of apoptosis. Apoptosis can be mediated through several signaling pathways, one of which is increased expression of PTEN, a negative regulator of the PI3K/Akt survival pathway. Here, we have shown that ROS-induced DNA damage results in hypomethylation of the PTEN promoter region, causing upregulation of PTEN expression. Our results also indicate that radiation-induced oxidative damage to DNA increases over time and that the extent of PTEN promoter hypomethylation is correlated with the level of DNA damage. This suggests not only that the negative regulation of the PI3K/Akt pathway is a key element of radiation-induced normal tissue injury but also that DNA damage-dependent hypomethylation of the PTEN promoter may account for the progressive nature of pulmonary toxicity. These findings suggest that reversing PTEN promoter hypomethylation may be an excellent therapeutic target for mitigation of pulmonary toxicity resulting from accidental or therapeutic radiation exposure.
We acknowledged the limitations in this study. First, in general, the molecular changes based on the measurements of lung homogenate may not accurately reflect the conclusions based on individual cell types. Second, inflammatory phagocytic NADPH oxidases would be expected to play a major role in early ROS-related lung tissue injury. In contrast, cells related to fibrotic resolution stages would play a larger role in late stages. Third, it should be remained that conclusion from the high radiation dose may not reflect the real clinical situation. Fourth, previous studies have demonstrated that PTEN transcription can be regulated by pathways involving p53, NF-𝜅B, Egr-1, PPAR𝛾, and SMADs, while p38 MAPK can regulate these transcriptional factors [45,46]. Importantly, several studies have demonstrated that irradiation upregulated p38 MAPK expression in lung tissues [47,48]. Although p38 MAPK does not directly regulate PTEN transcription, p38 MAPK may regulate PTEN expression through pathways involving p53, NF-𝜅B, Egr-1, PPAR𝛾, and SMADs. Therefore, measuring p38 MAPK and its downstream signal molecules may help us to understand whether PTEN upregulation only came from promoter hypomethylation in this study.
Acknowledgment
This project has been funded by an NIH grant (5U19-AI067798; project PI, Z. Vujaskovic).
Footnotes
Conflict of interest: None
References
- 1.Anscher MS, Chen L, Rabbani Z, Kang S, Larrier N, Huang H, Samulski TV, Dewhirst MW, Brizel DM, Folz RJ, Vujaskovic Z. Recent progress in defining mechanisms and potential targets for prevention of normal tissue injury after radiation therapy. Int J Radiat Oncol Biol Phys 2005; 62: 255–259. [DOI] [PubMed] [Google Scholar]
- 2.Marks L, Bentzen SM, Deasy JO, Kong FM,Bradley JD, Vogelius IS,El Naqa I, Hubbs JL, Lebesque JV, Timmerman RD, Martel MK, Jackson A. Radiation dose-volume effets in the lung. Int J Radiat Oncol Biol Phys 2010; 76(3 suppl): S70–S76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Ruysscher D, Faivre-Finn C, Le Pechoux C, Peeters S, Belderbos J. High-dose re-irradiation following radical radiotherapy for non-small-cell lung cancer. Lancet Oncol 2014; 15: e620–624. [DOI] [PubMed] [Google Scholar]
- 4.Ding NH, Li JJ, Sun LQ. Molecular mechanisms and treatment of radiation-induced lung fibrosis. Curr Drug Targets 2013; 14: 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avanzo M, Trovo M, Furlan C, Barresi L, Linda A, Stancanello J, Andreon L, Minatel E, Bazzocchi M, Trovo MG, Capra E. Normal tissue complication probability models for severe acute radiological lung injury after radiotherapy for lung cancer. Phys Med 2015; 31: 1–8. [DOI] [PubMed] [Google Scholar]
- 6.Mahmood J, Jelveh S, Calveley V, Zaidi A, Doctrow SR, Hill RP. Mitigation of lung injury after accidental exposure to radiation. Radiat Res 2011; 176: 770–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waselenko JK, MacVittie TJ, Blakely WF, Pesik N, Wiley AL, Dickerson WE, Tsu H, Confer DL, Coleman CN, Seed T, Lowry P, Armitage JO, Dainiak N; Strategic National Stockpile RadiationWorking Group. Medical Management of the Acute Radiation Syndrome: Recommendations of the Strategic National Stockpile Radiation Working Group. Ann Intern Med 2004; 140: 1037–1051. [DOI] [PubMed] [Google Scholar]
- 8.DiCarlo AL, Hatchett RJ, Kaminski JM, Ledney GD, Pellmar TC, Okunieff P, Ramakrishnan N. Medical Countermeasures for Radiation Combined Injury: Radiation with Burn, Blast, Trauma and/or Sepsis. Report of an NIAID Workshop, March 26– 27, 2007 Radiation Research 2008; 169: 712–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Zhang X, Rabbani ZN, Jackson IL, Vujaskovic Z. Oxidative Stress Mediates Radiation Lung Injury by Inducing Apoptosis. Int J Radiat Oncol Biol Phys 2012; 83: 740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bird A Perceptions of epigenetics. Nature 2007; 447: 396–398. [DOI] [PubMed] [Google Scholar]
- 11.Bird A DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16: 6–21. [DOI] [PubMed] [Google Scholar]
- 12.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediator. Trends Biochem Sci 2006; 31: 89–97. [DOI] [PubMed] [Google Scholar]
- 13.Li E, Zhang Y A methylation in mammals. Cold Spring Harb Perspect Biol 2014; 6: a019133.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luczak MW, Jagodzinski PP. The role of DNA methylation in cancer development. Folia Histochem Cytobiol 2006; 44: 143–154. [PubMed] [Google Scholar]
- 15.Trasler JM Epigenetics in spermatogenesis. Mol Cell Endocrinol 2009; 306: 33–36. [DOI] [PubMed] [Google Scholar]
- 16.Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res 2004; 567: 1–61. [DOI] [PubMed] [Google Scholar]
- 17.Donkena KV, Young CY, Tindall DJ. Oxidative stress and DNA methylation in prostate cancer. Obstet Gynecol Int 2010; 2010: 302051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takai D, Jones PA. The CpG island searcher: a new WWW resource. In Silico Biol 2003; 3: 235–240. [PubMed] [Google Scholar]
- 19.Gauter-Fleckenstein B, Reboucas JS, Fleckenstein K, Tovmasyan A, Owzar K, Jiang C, Batinic-Haberle I, Vujaskovic Z. Robust rat pulmonary radioprotection by a lipophilic Mn N-alkylpyridylporphyrin, MnTnHex-2-PyP(5+). Redox biology 2014; 2: 400–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Batinic-Haberle I, Tovmasyan A, Roberts ER, Vujaskovic Z, Leong KW, Spasojevic I. SOD therapeutics: latest insights into their structure-activity relationships and impact on the cellular redox-based signaling pathways. Antioxid Redox Signal 2014; 20: 2372–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weitner T, Kos I, Sheng H, Tovmasyan A, Reboucas JS, Fan P, Warner DS, Vujaskovic Z, Batinic-Haberle I, Spasojevic I. Comprehensive pharmacokinetic studies and oral bioavailability of two Mn porphyrin-based SOD mimics, MnTE-2-PyP(5+) and MnTnHex-2-PyP(5+). Free Radic Biol Med 2013; 58: 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rabbani ZN, Salahuddin FK, Yarmolenko P, Batinic-Haberle I, Thrasher BA, Gauter-Fleckenstein B, Dewhirst MW, Anscher MS, Vujaskovic Z. Low molecular weight catalytic metalloporphyrin antioxidant AEOL 10150 protects lungs from fractionated radiation. Free Radic Res 2007; 41: 1273–1282. [DOI] [PubMed] [Google Scholar]
- 23.Mi J, Zhang X, Rabbani ZN, Liu Y, Reddy SK, Su Z, Salahuddin FK, Viles K, Giangrande PH, Dewhirst MW, Sullenger BA, Kontos CD, Clary BM. RNA aptamer-targeted inhibition of NF-kappa B suppresses non-small cell lung cancer resistance to doxorubicin. Mol Ther 2008; 16: 66–73. [DOI] [PubMed] [Google Scholar]
- 24.Gericke A, Munson M, Ross AH. Regulation of the PTEN phosphatase. Gene 2006; 374: 1–9. [DOI] [PubMed] [Google Scholar]
- 25.Maehama T, Okahara F, Kanaho Y. The tumour suppressor PTEN: involvement of a tumour suppressor candidate protein in PTEN turnover. Biochem Soc Trans 2004; 32(Pt2): 343–347. [DOI] [PubMed] [Google Scholar]
- 26.Dahia PL. PTEN, a unique tumor suppressor gene. Endocr Relat Cancer 2000; 7: 115–129. [DOI] [PubMed] [Google Scholar]
- 27.Anscher MS, Thrasher B, Rabbani Z, Teicher B, Vujaskovic Z. Antitransforming growth factor-beta antibody 1D11 ameliorates normal tissue damage caused by high-dose radiation. Int J Radiat Oncol Biol Phys 2006; 65: 876–881. [DOI] [PubMed] [Google Scholar]
- 28.Fleckenstein K, Gauter-Fleckenstein B, Jackson IL, Rabbani Z, Anscher M, Vujaskovic Z. Using biological markers to predict risk of radiation injury. Semin Radiat Oncol 2007; 17: 89–98. [DOI] [PubMed] [Google Scholar]
- 29.Franco R, Schoneveld O, Georgakilas AG, Panayiotidis MI. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett 2008; 266: 6–11. [DOI] [PubMed] [Google Scholar]
- 30.Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 2001; 269: 131–140. [DOI] [PubMed] [Google Scholar]
- 31.Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic Biol Med 2007; 43: 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mittal M, Roth M, König P, Hofmann S, Dony E, Goyal P, Selbitz AC, Schermuly RT, Ghofrani HA, Kwapiszewska G, Kummer W, Klepetko W, Hoda MA, Fink L,Hänze J, Seeger W, Grimminger F, Schmidt HH, Weissmann N. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res 2007; 101: 258–267. [DOI] [PubMed] [Google Scholar]
- 33.Stewart FA, Dorr W. Milestones in normal tissue radiation biology over the past 50 years: from clonogenic cell survival to cytokine networks and back to stem cell recovery. Int J Radiat Biol 2009; 85: 574–586. [DOI] [PubMed] [Google Scholar]
- 34.Vorotnikova E, Rosenthal RA, Tries M, Doctrow SR, Braunhut SJ. Novel synthetic SOD/catalase mimetics can mitigate capillary endothelial cell apoptosis caused by ionizing radiation. Radiat Res 2010; 173: 748–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turk PW, Laayoun A, Smith SS, Weitzman SA. DNA adduct 8-hydroxyl-2‚Ä≤-deoxyguanosine (8-hydroxyguanine) affects function of human DNA methyltransferase. Carcinogenesis 1995; 16: 1253–1255. [DOI] [PubMed] [Google Scholar]
- 36.Spitz DR, Azzam EI, Li JJ. Gius D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: A unifying concept in stress response biology. Cancer and Metastasis Reviews 2004; 23: 311–322. [DOI] [PubMed] [Google Scholar]
- 37.Collins-Underwood JR, Zhao W, Sharpe JG, Robbins ME. NADPH oxidase mediates radiation-induced oxidative stress in rat brain microvascular endothelial cells. Free Radic Biol Med 2008; 45: 929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park S, Ahn JY, Lim MJ, Kim MH, Yun YS, Jeong G, Song JY. Sustained expression of NADPH oxidase 4 by p38 MAPK-Akt signaling potentiates radiation-induced differentiation of lung fibroblasts. J Mol Med 2010; 88: 807–816. [DOI] [PubMed] [Google Scholar]
- 39.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Fórró L, Schlegel W, Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J 2007; 406: 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takac I, Schröder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, Shah AM, Morel F, Brandes RP. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem 2011; 286:13304–13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhuang HQ, Wang J, Yuan ZY, Zhao LJ, Wang P, Wang CL. The drug-resistance to gefitinib in PTEN low expression cancer cells is reversed by irradiation in vitro. J Exp Clin Cancer Res 2009; 28: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Escriva M, Peiró S, Herranz N, Villagrasa P, Dave N, Montserrat-Sentís B, Murray SA, Francí C, Gridley T, Virtanen I, García de Herreros A. Repression of PTEN phosphatase by Snail1 transcriptional factor during gamma radiation-induced apoptosis. Mol Cell Biol 2008; 28: 1528–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flaherty DM, Monick MM, Hinde SL. Human alveolar macrophages are deficient in PTEN. The role of endogenous oxidants. J Biol Chem 2006; 281: 5058–5064. [DOI] [PubMed] [Google Scholar]
- 44.Wu Q, Ni X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr Drug Targets 2015; 16: 13–19. [DOI] [PubMed] [Google Scholar]
- 45.Jerde TJ. Phosphatase and Tensin Homologue: Novel Regulation by Developmental Signaling. J Signal Transduct 2015; 2015: 282567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lei YY, Wang WJ, Mei JH, Wang CL. Mitogen-activated protein kinase signal transduction in solid tumors. Asian Pac J Cancer Prev 2014; 15: 8539–8548. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, Zhang YY, Cheng J, Zhang JL, Li BS. Preventive and therapeutic effects of quercetin on experimental radiation induced lung injury in mice. Asian Pac J Cancer Prev 2015; 16: 2909–2914. [DOI] [PubMed] [Google Scholar]
- 48.Ryu SY, Do SH, Chung JY, Kim TH, Kim SH, Choi CY, Jeong KS, Park SJ. Activation of MAP kinases during progression of radiation-induced pneumonitis in rats. Hum Exp Toxicol 2011;30:876–83. [DOI] [PubMed] [Google Scholar]