

Abstract
Clostridium difficile infection (CDI) symptoms range from diarrhea to severe toxic megacolon and even death. Due to its rapid acquisition of resistance, C. difficile is listed as an urgent antibiotic-resistant threat, and has surpassed methicillin-resistant Staphylococcus aureus (MRSA) as the most common hospital-acquired infection in the USA. To combat this pathogen, a new structural class of pseudo-peptides that exhibit antimicrobial activities could play an important role. Herein we report a set of bis-cyclic guanidine compounds that show potent antibacterial activity against C. difficile with decent selectivity. Eight compounds showed high in vitro potency against C. difficile UK6 with MIC values of 1.0 μg mL−1, and cytotoxic selectivity index (SI) values up to 37. Moreover, the most selective compound is also effective in the treatment of C. difficile-induced disease in a mouse model of CDI, and appears to be a very promising new candidate for the treatment of CDI.
Keywords: antibiotics, bis-cyclic guanidines, Clostridium difficile, drug resistance
Introduction
Clostridium difficile is a Gram-positive, spore-forming, anaerobic and toxigenic microbe. Symptoms of C. difficile infection (CDI) range from uncomplicated diarrhea to pseudomembranous colitis and even toxic megacolon.[1] C. difficile is recognized as the most common cause of hospital-associated diarrhea,[2] and may lead to more related complications,[3] resulting in increasingly infectious morbidity and mortality. More alarmingly, the emergence of hypervirulent strains NAP1/BI/027 has been associated with higher mortality rates in North America and several countries in Europe. [4] Antimicrobial therapeutic options with oral vancomycin and metronidazole are effective for severe and mild-to-moderate CDI, respectively. [4b,5] Treatment options for severe CDI include the use of newly developed antimicrobial agents such as fidaxomicin and fecal microbiota transplantation (FMT), which was identified as an effective treatment for CDI recurrence. [6] However, initial therapy with metronidazole and vancomycin has been associated with increased rates of failure and recurrence. [7] Fidaxomicin is more reliable but more expensive than metronidazole/vancomycin. The US Centers for Disease Control and Prevention has listed C. difficile as an urgent antibiotic-resistant threat. [8] Although C. difficile has not yet developed significant resistance to the antibiotics most used for CDI treatment, it is highly likely that these resistance phenotypes will emerge, as has occurred through the use of clindamycin and the fluoroquinolones. [9]
Novel antibiotics are urgently needed to more effectively treat CDI. Bis-guanidine-related compounds have been reported to bear antiseptic and antibacterial activities; these include hexamidine,[10] norspermidine analogues,[11] teixobactin,[12] brilacidin,[13] and amphipathic xanthone derivatives.[14] These types of compounds display antimicrobial activity against Gram-positive organisms including methicillin-resistant Staphylococcus aureus, methicillin-resistant Staphylococcus epidermidis, and vancomycin-resistant Enterococci faecalis, and relatively weaker activity against Gram-negative organisms such as Klebsiella pneumoniae and Pseudomonas aeruginosa.[15]
We recently discovered a new series of symmetric bis-cyclic guanidine compounds[15] bearing amphipathic structures that could mimic the mechanism of action of host-defense peptides (HDPs).[16] These membrane-active amphipathic compounds showed potent and broad-spectrum activity against both Gram-positive and Gram-negative bacteria. However, to the best of our knowledge, compounds bearing guanidine groups[17] have been rarely explored for bactericidal activities against C. difficile.[18] Herein we report the antibacterial activity of these dimeric cyclic guanidines against C. difficile in vitro and in vivo.
Results and Discussion
The bis-cyclic guanidine library was synthesized by following the same procedure reported previously.[15] Synthesis of compound 13 is shown as an example of the typical synthesis process (Scheme 1). Intermediate R4 was obtained from the readily accessible reagent R1 in a straightforward manner with decent yield. The linear intermediate R5 was obtained by coupling between R4 and diamine (p-phenylenediamine) in the presence of hydroxybenzotriazole (HOBt) and N,N’-dicyclohexylcarbodiimide (DCC) followed by removal of the Boc protecting groups. R5 could be easily cyclized in the presence of cyanogen bromide to furnish the final bis-cyclic guanidine compound 13.
Scheme 1.

Typical synthesis protocol for bis-cyclic guanidine derivative 13: a) hexanal, NaBH3CN, MeOH, AcOH, RT, 3 h; b) Boc2O, NaHCO3, THF, H2O, RT, 5 h; c) LiAlH4, THF, @208C, 30 min; d) glycine benzyl ester, NaBH3CN, MeOH, AcOH, RT, 3 h; e) Boc2O, NaHCO3, THF, H2O, RT, 5 h; f) H2, Pd/C, MeOH, 2 h; g) benzene-1,4-diamine, HOBt, DIPEA, DCC, DMF, RT, 24 h; h) TFA/CH2Cl2 (50:50, v/v), RT, 2 h; i) CNBr, MeCN, RT, 12 h.
The antibacterial potency of these cyclic guanidine dimers on the hypervirulent C. difficile UK6 was assessed and is reported in terms of minimal inhibitory concentration (MIC) values. As shown in Table 1, the majority of these quinoline compounds displayed potent in vitro activity, with MICs in the range of 1.0–4.0 μg mL−1. Compounds 1 and 2, which do not bear hydrophobic groups on the guanidine residues, displayed weak activity; in particular, compound 1 showed a MIC value of 128 μg mL−1. If an ethyl group was installed on the guani-dine to furnish compounds 3 and 4, they exhibited activity against C. difficile; a MIC value of 8.0 μg mL−1 was observed for compound 4. Compound 5 and 6 have a 3-phenylpropyl group attached at the nitrogen atoms of guanidine groups and showed potent antimicrobial activity (MIC =1.0 μg mL−1), due to an enhanced interaction with the bacterial membrane. If a hydrophobic cyclohexane propyl group was attached to the guanidine position to give compounds 7–9, the activities increased with a change in the linker between the two cyclic guanidine rings from p-phenylenediamine to m-phenylenedia-mine, and then 1,6-hexamethylene, with MICs of 4.0, 1.0, and 2.0 μg mL−1, respectively. Compound 10, with an aliphatic C6H13 chain on the guanidine rings, only showed a MIC value of 8 μg mL−1, where the linker was kept as a 1,4-butylene group. However, the activity returned if the linker was replaced with 1,8-octamethylene (11, MIC=2.0 μg mL−1), m-phenylenediamine (12, MIC=1.0 μg mL−1), or p-phenylenediamine (13, MIC=1.0 μg mL−1).
Table 1.
Structures of compounds 1–16 and their antibacterial activity against C. difficile.
| Compd | Structure | logP[a] | MIC [μg mL−1] | Compd | Structure | logP[a] | MIC [μg mL−1] |
|---|---|---|---|---|---|---|---|
| 1 | 1.21 | 128 | 9 | 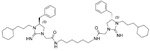 |
6.12 | 2.0 | |
| 2 | 2.28 | 32 | 10 | 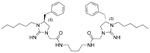 |
4.31 | 8.0 | |
| 3 | 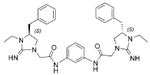 |
3.17 | 32 | 11 | 5.58 | 2.0 | |
| 4 | 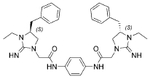 |
3.23 | 8.0 | 12 | 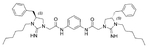 |
5.31 | 1.0 |
| 5 | 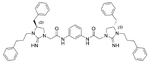 |
5.51 | 1.0 | 13 | 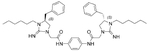 |
5.38 | 1.0 |
| 6 | 6.56 | 1.0 | 14 | 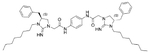 |
5.78 | 1.0 | |
| 7 | 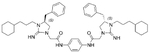 |
5.57 | 4.0 | 15 | 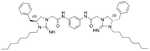 |
5.72 | 1.0 |
| 8 | 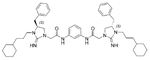 |
5.55 | 1.0 | 16 | 5.01 | 1.0 | |
| vancomycin | - | 0.5 |
Calculated by Alogps version 2.1 software.
Interestingly, replacement of the aliphatic chain on the guanidine rings with chains of increased length (C8H17) did not compromise the activity, showing the same MIC values of 1.0 μg mL−1 for compounds 14 and 15. Replacing the benzyl group (initially starting from a-phenylalanine) with an isobutyl group (initially starting from a-leucine) produced compound 16, which was also potent, with a MIC value of 1.0 μg mL−1, very close to that of the positive control vancomycin under the same assay conditions.
To determinate the cytotoxicity of ten active compounds (MIC <4 μg mL−1) out of sixteen compounds, MTT assays were performed on the human liver cancer cell line HepG2 and the human embryonic kidney cell line HEK293T. As shown in Table 2, 3-phenylpropyl-modified compounds 5 and 6, and cyclohexanepropyl-modified compounds 8 and 9 showed about 20–30-fold selectivity index (SI) values (SI= [CC50 on human cells] / [MIC on C. difficile cells]). Aliphatic side chain bearing compound 11 only displayed tenfold SI; however, compounds 12–15 all had low cytotoxicity against both cell lines, and especially compound 13 had a 37-fold SI against HEK293T cells. Compounds 12 and 13 were also not hemolytic, even at a concentration of 250 μg mL−1.[15] The selectivity decreased slightly if the benzyl group was replaced with an isobutyl group. Overall, compound 13 has the best SI among all the compounds tested.
Table 2.
Cytotoxicity assessment of active compounds (MIC <4 μg mL−1).
| Compd | MIC [μg mL−1] | CC50 [μg mL−1][a] | SI[b] | ||
|---|---|---|---|---|---|
| HEK293T | HepG2 | HEK293T | HepG2 | ||
| 5 | 1 | 20.6 | 36.8 | 20.6 | 36.8 |
| 6 | 1 | 26.5 | 31.7 | 26.5 | 31.7 |
| 8 | 1 | 33.9 | 30.1 | 33.9 | 30.1 |
| 9 | 2 | 42.7 | 40.3 | 21.35 | 20.15 |
| 11 | 2 | 25.1 | 24.0 | 12.55 | 12 |
| 12 | 1 | 32.3 | 33.1 | 32.3 | 33.1 |
| 13 | 1 | 31.8 | 37.3 | 31.8 | 37.3 |
| 14 | 1 | 29.3 | 26.8 | 29.3 | 26.8 |
| 15 | 1 | 25.7 | 25.9 | 25.7 | 25.9 |
| 16 | 1 | 24.3 | 18.1 | 24.3 | 18.1 |
50% cytotoxic centration.
Selectivity index: CC50/MIC.
To further investigate the impact of hydrophobicity on the activity profile, we measured HPLC retention times (Table S1) and determined logP values (Table 1) of all compounds. Generally, the antibacterial activity of compounds increases with longer retention times (tR) if the tR is shorter than 27.25 min. If the tR is longer than 27.25 min, the activity of compounds did not decrease; however, the cytotoxicity of compounds increased (Table 2). A similar trend can be observed by correlating the activity with logP value. Based on these results, we can conclude that the antibacterial activity and selectivity of this compound class could be improved by carefully tuning the balance between hydrophobicity and hydrophilicity, which will aid the future design of HDP mimics.
The efficacy of compound 13 was further evaluated in a mouse model of CDI. As shown in Figure 1a, the C. difficile UK6-challenged control group led to 90% diarrhea, while the administration of compound 13 displayed a significant improvement for overcoming CDI over the entire experimental period (five days). After three days, 90% survival was observed with the administration of compound 13 compared with the control group, in which only 40% of mice survived (Figure 1b). Five days later, 60% of the mice were still alive with the treatment of compound 13, whereas only 40% survival was observed in the C. difficile UK6-challenged control group. These data indicate that compound 13 can improve the diarrhea and survival of mice challenged with C. difficile UK6, a hypervirulent strain.
Figure 1.

In vivo efficacy of compound 13 in a mouse model of C. difficile infection. Two groups of mice (UK6 and UK6 +13, n=10 per group) were challenged with C. difficile spores at 106 per mouse in the absence or presence of compound 13, after pretreatment of antibiotics. The third group mice (n=5) were administered compound 13 only as controls. a) Percent of diarrhea with or without treatment of compound 13. b) Survival rates of mice with or without treatment of compound 13. Results were analyzed by two-way ANOVA. The differences between the UK6 group and the treatment group (UK6+13) are statistically significant, p<0.05.
The amount of C. difficile in fecal samples after treatment was also determined. As shown in Figure 2, one day after infection, the amount of C. difficile in feces from mice treated with compound 13 was 50% less than that of the C. difficile UK6-challenged group. After five days, the amount of C. difficile in fecal samples from the control group continued to increase, while mice treated with compound 13 were observed with significant decrease of C. difficile in fecal samples, down by 80% relative to control levels. This result demonstrates that compound 13 has good efficacy on the inhibition of C. difficile in mice.
Figure 2.

C. difficile in fecal samples from compound 13-treated (UK6 +13) or control (UK6) mice. Two groups of mice (UK6 and UK6 +13, n= 10 per group) were challenged with C. difficile spores at 106 per mouse in the absence or presence of compound 13, after pretreatment of antibiotics. Fecal samples were collected, and C. difficile spores were determined as described in the Experimental Section. C. difficile isolated in fecal matter from mice treated with compound 13 showed a significant decrease relative to that of the UK6-challenged group (two-way ANOVA, p<0.001).
Conclusions
We have reported a series of membrane-active bis-cyclic guani-dines (molecular mass 600–900 Da) that display potency against C. difficile UK6, an emerging hypervirulent bacteria. In vitro studies demonstrated that eight out of sixteen cyclic guanidine dimeric compounds showed MIC values of 1.0 μg mL−1 against C. difficile, very close to that of vancomycin (MIC =0.5 μg Ml−1). Moreover, the cyclic guanidine dimers also revealed significant efficacy in a mouse model of CDI. Further modifications of these compounds may lead to novel potent antibiotics against C. difficile.
Experimental Section
General:
The starting material to synthesize R1 was purchased from Chem-Impex International, Inc. Solvents and other reagents were purchased from either Sigma–Aldrich or Fisher Scientific and were used without further purification. The final products were purified on a Waters Breeze 2 HPLC system, and lyophilized on a Labconco lyophilizer. The purity of the compounds was determined to be >95% by analytical HPLC (1 mLmin−1 flow rate, 5 → 100% linear gradient of solvent B (0.1% TFA in acetonitrile) in A (0.1% TFA in water) over 50 min), and the data are shown in the Supporting Information. The NMR spectra were obtained on a Varian Inova 500 instrument.
Synthesis of the intermediate building block R4:
Compound R1 (TFA salt, 13.6 g, 42.3 mmol) was dissolved in MeOH and treated with TEA (5.8 mL, 42.3 mmol) before adding to a solution of hexanal (5.2 mL, 42.3 mmol) in MeOH and acetic acid (5.1 mL,82.6 mmol). After stirring for 10 min under ice/H2O bath, NaBH3CN (5.6 g, 82.6 mmol) was added portion-wise. The reaction was stirred for 3 h at room temperature before the solvent was removed. The crude mixture was treated with NaHCO3 (aq) and extracted with EtOAc, and the organic layer was separated and evaporated to give a crude oil, which was purified by silica gel column chromatography to give 8.9 g of the desired secondary amine. Boc2O (8 g, 36.6 mmol) was added in the THF/H2O (1:1, v/v) solution of this intermediate containing NaHCO3 (5.1 g, 61 mmol) and allowed to react for 5 h, after which EtOAc was added and the organic layer was collected. The solvent was removed under reduced pressure to give the colorless crude, which was purified by flash column chromatography to give 9.1 g of compound R2. Next, compound R2 was taken in THF and reduced by LiAlH4 (926 mg, 23.2 mol) for 30 min at @208C, then water was added to quench the reaction.The mixture was extracted with EtOAc, and the organic layer was separated and the solvent was removed in vacuo to give the crude R3 (7.2 g), which was used in the next reaction without any further purification. The Boc protecting group was attached using the same procedure for attaching Boc onto compound R2, followed by hydrogenation to remove the benzyl protecting group in MeOH to give the building block R4 (7.5 g) as a white solid after filtration and concentration.
Building block R4 (400 mg, 0.81 mmol), HOBt (249 mg, 1.6 mmol), DIPEA (284 μL, 1.6 mmol), and p-phenylenediamine (53 mg, 0.49 mmol) were dissolved in DMF (3 mL) and then DCC (335 mg, 1.6 mmol) was added. The reaction mixture was stirred at room temperature for 24 h. The afforded byproduct DCU was filtered off and the filtrate was added into water and extracted with EtOAc (☓3). The organic phase was combined and washed with 1m HCl (☓2), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude oil compound was treated with TFA in CH2Cl2 (1:1, v/v) for 2 h to completely remove Boc protecting groups to yield crude compound R5. Subsequently, R5 was dissolved in acetonitrile (3 mL), to which CNBr (4 equiv) was added carefully (caution: very toxic). The reaction was stirred for 12 h at room temperature. A solution of NaOH (1m) was added carefully, followed by a proper amount of sodium hypochlorite to deactivate excess CNBr. The mixture was filtered through a Millipore filter and purified by HPLC purification on a Waters HPLC system, and the desired fraction was lyophilized to give the pure product 13.
The other compounds were synthesized according to the same procedure as compound 13. Various aldehydes were used at the first step to give different compounds with various side chains. The NMR data of compounds 1–5, 12, 13, and 16 are consistent with previously published data.[15] The NMR and HRMS data for other compounds are listed below:
Compound 6: 1H NMR (500 MHz, CD3OD): δ=7.64 (d, J=7.5 Hz, 4H), 7.57 (d, J=7.5 Hz, 4H), 7.18–7.33 (m, 20H), 4.22–4.27 (m, 2H), 4.14 (s, 4H), 3.63 (t, J=9.5 Hz, 2H), 3.51–3.57 (m, 2H), 3.42 (dd, J=9.5, 5.0 Hz, 2H), 3.34 (dd, J=9.0, 5.5 Hz, 2H), 3.09 (dd, J=13.5,4.5 Hz, 2H), 2.85 (dd, J=14.0, 8.5 Hz, 2H), 2.62–2.74 (m, 4H), 1.92–2.06 ppm (m, 4H); 13C NMR (125 MHz, CD3OD):δ =165.1, 158.0, 140.9, 137.2, 136.2, 135.7, 134.3, 129.0 (2C), 128.5 (2C), 128.2 (2C), 128.0 (2C), 126.8, 126.6 (2C), 125.8, 119.8, 58.0, 51.7, 42.3, 37.4, 32.1 (2C), 28.3 ppm; HRMS (ESI) C54H59N8O2 [M+H]+ calcd=851.4755; found=851.4742.
Compound 7: 1H NMR (500 MHz, CD3OD): δ=7.53 (s, 4H), 7.31–7.34 (m, 4H), 7.24–7.28 (m, 6H), 4.27–4.32 (m, 2H), 4.13, 4.10 (ABq, J=18.0 Hz, 4H), 3.68 (t, J=9.0 Hz, 2H), 3.41–3.48 (m, 4H), 3.25 (ddd, J=15.0, 9.5, 5.5 Hz, 2H), 3.15 (dd, J=13.5, 5.0 Hz, 2H), 2.89 (dd, J=13.5, 8.0 Hz, 2H), 1.58–1.76 (m, 14H), 1.15–1.32 (m, 12H), 0.90–0.97 ppm (m, 4H); 13C NMR (125 MHz, CD3OD): δ=165.1, 158.0, 135.8, 134.3, 128.9 (2C), 128.5 (2C), 126.8, 120.1 (2C), 57.9, 51.8, 47.0, 43.1, 37.6, 37.5, 37.3, 33.6, 33.0, 32.9, 26.3, 26.0, 24.0 ppm; HRMS (ESI) C48H67N8O2 [M+H]+ calcd=787.5381; found=787.5374.
Compound 8: 1H NMR (500 MHz, CD3OD): δ=8.04 (s, 1H), 7.31–7.34 (m, 4H), 7.24–7.28 (m, 9H), 4.26–4.32 (m, 2H), 4.14, 4.10 (ABq, J=18.0 Hz, 4H), 3.68 (t, J=9.5 Hz, 2H), 3.41–3.47 (m, 4H), 3.25 (ddd, J=14.5, 9.0, 5.5 Hz, 2H), 3.15 (dd, J=13.5, 5.0 Hz, 2H), 2.89 (dd, J=13.5, 8.0 Hz, 2H), 1.60–1.75 (m, 14H), 1.14–1.32 (m, 12H), 0.90–0.96 ppm (m, 4H); 13C NMR (125 MHz, CD3OD): δ=165.2, 158.0, 138.6, 135.8, 128.9 (2C), 128.5 (2C), 126.8, 120.1 115.3, 58.0, 51.7, 47.0, 43.1, 37.6, 37.3, 33.7, 33.0, 32.9, 26.3, 26.0 (2C), 24.0 ppm; HRMS (ESI) C48H67N8O2 [M+H]+ calcd=787.5381; found=787.5357.
Compound 9: 1H NMR (500 MHz, CD3OD): δ=7.31–7.34 (m, 4H), 7.25–7.27 (m, 6H), 4.24–4.30 (m, 2H), 3.93, 3.89 (ABq, J=17.5 Hz, 4H), 3.60 (t, J=9.5 Hz, 2H), 3.42 (ddd, J=14.5, 9.0, 6.0 Hz, 2H), 3.33 (dd, J=9.5, 6.0 Hz, 2H), 3.21–3.26 (m, 2H), 3.19 (t, J=7.0 Hz, 4H), 3.13 (dd, J=13.5, 5.0 Hz, 2H), 2.85 (dd, J=13.5, 8.0 Hz, 2H), 1.65–1.74 (m, 12H), 1.58–1.62 (m, 2H), 1.50 (p, J=6.5 Hz, 4H), 1.32–1.35 (m, 4H), 1.14–1.31 (m, 12H), 0.85–0.95 ppm (m, 4H); 13C NMR (125 MHz, CD3OD): δ=166.9, 158.0, 135.8, 128.9 (2C), 128.5 (2C), 126.8, 57.9, 51.6, 46.6, 43.1, 39.1, 37.5, 37.3, 33.7, 33.0, 32.9, 28.8, 26.3, 26.1, 26.0 (2C), 24.0 ppm; HRMS (ESI) C48H74N8O2 [M+H]+ calcd=795.6006; found=795.5998.
Compound 10: 1H NMR (500 MHz, CD3OD): δ=7.30–7.34 (m, 4H), 7.24–7.26 (m, 6H), 4.24–4.30 (m, 2H), 3.94, 3.90 (ABq, J=18.0 Hz, 4H), 3.59 (t, J=9.5 Hz, 2H), 3.45 (ddd, J=15.0, 9.0, 6.5 Hz, 2H), 3.34 (dd, J=9.5, 6.0 Hz, 2H), 3.23–3.29 (m, 2H), 3.19–3.23 (m, 4H), 3.13 (dd, J=13.5, 5.0 Hz, 2H), 2.87 (dd, J=14.0, 8.0 Hz, 2H), 1.51–1.54 (m, 4H), 1.28–1.36 (m, 12H), 0.92 ppm (t, J=6.5 Hz, 6H); 13C NMR (125 MHz, CD3OD): δ=167.0, 157.9, 135.8, 129.0 (2C), 128.5 (2C), 126.8, 57.9, 51.6, 46.7, 42.9, 38.7, 37.5, 31.1, 26.6, 26.2, 25.8, 22.1, 12.9 ppm; HRMS (ESI) C40H63N8O2 [M+H]+ calcd=687.5068; found=687.5056.
Compound 11: 1H NMR (500 MHz, CD3OD): δ=7.31–7.34 (m, 4H), 7.24–7.27 (m, 6H), 4.24–4.30 (m, 2H), 3.94, 3.90 (ABq, J=18.0 Hz, 4H), 3.59 (t, J=9.0 Hz, 2H), 3.45 (ddd, J=15.0, 9.0, 6.5 Hz, 2H), 3.34 (dd, J=9.5, 5.5 Hz, 2H), 3.25 (ddd, J=15.0, 9.0, 5.5 Hz, 2H), 3.18 (t, J=7.0 Hz, 4H), 3.14 (dd, J=13.5, 5.0 Hz, 2H), 2.86 (dd, J=13.5, 8.0 Hz, 2H), 1.54–1.69 (m, 4H),1.50 (t, J=6.0 Hz, 4H), 1.30–1.36 (m, 20H), 0.92 ppm (t, J=7.0 Hz, 6H); 13C NMR (125 MHz, CD3OD): δ=166.9, 157.9, 135.8, 128.9 (2C), 128.5 (2C), 126.8, 57.9, 51.5, 46.7, 42.8, 39.2, 37.5, 31.2, 28.9, 26.6, 26.5, 25.8, 22.2, 12.9 ppm; HRMS (ESI) C44H71N8O2 [M+H]+ calcd=743.5694; found=743.5675.
Compound 14: 1H NMR (500 MHz, CD3OD): δ=8.02 (d, J=1.0 Hz, 1H), 7.31–7.34 (m, 4H), 7.24–7.28 (m, 9H), 4.27–4.32 (m, 2H), 4.14, 4.11 (ABq, J=18.0 Hz, 4H), 3.67 (t, J=9.5 Hz, 2H), 3.47 (ddd, J=15.5, 9.5, 7.0 Hz, 2H), 3.42 (dd, J=9.5, 5.5 Hz, 2H), 3.27 (ddd, J=15.0, 9.5, 5.5 Hz, 2H), 3.16 (dd, J=13.5, 5.0 Hz, 2H), 2.89 (dd, J=14.9, 8.5 Hz, 2H), 1.59–1.70 (m, 4H), 1.30–1.34 (m, 20H), 0.91 ppm (t, J=7.0 Hz, 6H); 13C NMR (125 MHz, CD3OD): δ=165.2, 158.0, 138.6, 135.8, 129.0 (2C), 128.5 (2C), 126.8, 115.3, 111.2, 58.0, 51.7, 42.8, 37.5, 31.5, 28.9 (2C), 26.7, 26.1, 22.3, 13.0 ppm; HRMS (ESI) C46H67N8O2 [M+H]+ calcd=763.5381; found=763.5359.
Compound 15: 1H NMR (500 MHz, CD3OD): δ=7.52 (s, 4H), 7.31–7.34 (m, 4H), 7.24–7.28 (m, 6H), 4.27–4.33 (m, 2H), 4.12, 4.09 (ABq, J=18.5 Hz, 4H), 3.67 (t, J=9.5 Hz, 2H), 3.48 (ddd, J=15.5, 9.0, 7.0 Hz, 2H), 3.42 (dd, J=9.5, 5.5 Hz, 2H), 3.25–3.29 (m, 2H), 3.16 (dd, J=14.0, 5.0 Hz, 2H), 2.89 (dd, J=13.5, 8.5 Hz, 2H), 1.58–1.71 (m, 4H), 1.27–1.35 (m, 20H), 0.91 ppm (t, J=6.5 Hz, 6H); 13C NMR (125 MHz, CD3OD): δ=165.0, 158.0, 135.8, 134.3, 129.0 (2C), 128.5 (2C), 126.7, 120.0 (2C), 57.9, 51.7, 47.0, 42.8, 37.5, 31.5, 28.9 (2C), 26.6, 26.0, 22.3, 13.0 ppm; HRMS (ESI) C46H67N8O2 [M+H]+ calcd= 763.5381; found=763.5358.
Minimum inhibitory concentration (MIC) determination:
The antimicrobial activities of the cyclic guanidine dimers against C. difficile UK6 were tested using media and methods recommended by the Clinical and Laboratory Standards Institute for susceptibility testing of anaerobes.[19] Compounds at 5 mgmL−1 were added to wells of 96-well microplates containing UK6 culture at a density of 0.5 McFarland (100 μL per well) in BHIS medium to make final concentrations of extracts ranging from 128 μg mL−1 to 0.5 μg mL−1 at a twofold dilution. The plates were incubated at 378C for 24 h. The MICs were determined as the lowest concentration that completely inhibits the bacterial growth in the wells. Vancomycin was included as a positive control.
MTT assay:
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-dipheynyltetrazolium bromide; Sigma–Aldrich, St. Louis, MO, USA) cell viability assay was performed to evaluate the cytotoxicity of the compounds on human HepG2 and HEK293T cell lines. HepG2 is an immortalized cell line consisting of human liver carcinoma cells. HEK293T is a specific cell line originally derived from human embryonic kidney cells grown in tissue culture. Both cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 4.5 gL−1 glucose, l-glutamine, and sodium pyruvate (Corning; Manassas, VA, USA) containing 10% FBS (Thermo Scientific) and 1% penicillin/streptomycin at 378C in 5% CO2. Cells (104 per well) were plated in 96-well plates. After incubation overnight, cells were treated with the compounds at concentrations from 128 mgmL−1 to 0.125 μg mL−1 or 1% DMSO (as a control reagent) for 24 h at 378°C. Then 10 μL of MTT stock solution (5 mgmL−1) were added to cells in each well, and further incubated for 4 h at 37°C. After careful removal of media from each well without disturbing cells, 100 μL of DMSO was added to each well, and incubated for 15 min at 37°C. Absorbance at 540 nm was read in a Synergy HTX multi-mode reader (BioTek Instruments, Inc., Winooski VT, USA). Data were analyzed using GraphPad Prism 6.0 (GraphPad Software, Inc., La Jolla, CA, USA), and the 50% cytotoxic concentration (CC50) was reported as the extract concentration that decreased cell viability by 50% relative to the untreated control. CC50 values were determined to establish a selectivity index (SI) (SI=CC50/MIC).
Evaluation of compounds in mouse model of C. difficile infection (CDI):
C57BL/6 female mice (six weeks old) were purchased from Charles River Laboratories, MA (USA). During the experiment, mice were housed in groups of five animals per cage under the same conditions. All animal experiments were approved by the institutional committee for animal care and use at the University of South Florida. The experimental design is illustrated in Figure S1. Twenty-five mice were divided into three groups (groups 1–3). Group 1 (n=10) were challenged with spores of C. difficile UK6. Group 2 (n=10) were challenged with spores of C. difficile UK6, and treated with compound 13. Group 3 (n=5) were only treated with compound 13 without infection. The mice were given drinking water containing a mixture of six antibiotics including ampicillin (200 mgkg−1), kanamycin (40 mgkg−1), gentamycin (3.5 mgkg−1), colistin (4.2 mgkg−1), metronidazole (21.5 mgkg−1), and vancomycin (4.5 mgkg−1) for five days, and then received autoclaved water for two days, followed by a single dose of clindamycin (10 mgkg−1) intraperitoneally one day before (day 1) challenge day. On the challenge day (day 0), mice in groups 1 and 2 were challenged with C. difficile UK6 spores at 106 colony-forming units (CFU) by gavage. At 4 h post-challenge, the mice in group 2 were given one dose of compound 13 (50 mgkg−1) orally. From the first day post-challenge (day 1), mice in group 2 received one dose of compound 13 twice a day (50 mgkg−1day−1) for five days. Meanwhile, the mice in group 3 were also given compound 13 at the same time with the same dose to determine the toxicity of the compound to the mice. After C. difficile challenge and/or compound treatment, mice were monitored twice a day during the experiment for weight changes, diarrhea (defined as soft or watery feces) and other symptoms of the disease.
Fecal samples were collected at the first, third, and fifth day post-challenge for C. difficile spore enumeration. Fecal samples were weighed and shocked in 95% ethanol (0.1 g mL−1) for 1 h followed by serial dilution in PBS, spreading on BHI plates supplemented with 10% taurocholic acid, and incubation in an anaerobic chamber. After incubation for 48 h, the colonies on plates in three duplicates for the selected dilutions were counted.
Statistical analysis:
Data were analyzed using GraphPad Prism 6.0 (GraphPad Software, Inc.). Statistical analyses were performed using the Kaplan–Meier survival analysis (survival rate) and twoway ANOVA (results of diarrhea rate and the amount of C. difficile in fecal samples after treatment were expressed as means standard errors); p values less than or equal to 0.05 were considered significant.
Supplementary Material
Acknowledgements
This work was supported in part by the US National Science Foundation (NSF; grant CAREER 1351265) and the US National Institutes of Health (NIH; grants 1R01GM112652–01A1, K01-DK092352, R21-AI113470, R03-DK112004, and R01-AI132711).
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/cmdc.201800240.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].McFarland LV, Nat. Clin. Pract. Gastroenterol. hepatol 2008, 5, 40–48. [DOI] [PubMed] [Google Scholar]
- [2].Redelings MD, Sorvillo F, Mascola L, Emerging Infect. Dis 2007, 13, 1417–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Loo VG, Poirier L, Miller MA, Oughton M, Libman MD, Michaud S, Bourgault A-M, Nguyen T, Frenette C, Kelly M, Vibien A, Brassard P, Fenn S, Dewar K, Hudson TJ, Horn R, René P Monczak Y, Dascal A, N. Engl. J. Med 2005, 353, 2442–2449; [DOI] [PubMed] [Google Scholar]; b) Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, Wilson LE, Winston LG, Cohen JA, Limbago BM, Fridkin SK, Gerding DN, McDonald LC, N. Engl. J. Med 2015, 372, 825–834; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bartlett JG, N. Engl. J. Med 2002, 346, 334–339. [DOI] [PubMed] [Google Scholar]
- [4].a) Brazier JS, Br. J. Biomed. Sci 2008, 65, 39–44; [DOI] [PubMed] [Google Scholar]; b) Warny M, Pepin J,Fang A, Killgore G, Thompson A, Brazier J, Frost E, McDonald LC, Lancet 2005, 366, 1079–1084. [DOI] [PubMed] [Google Scholar]
- [5].Cohen SH, Gerding DN, Johnson S, Kelly CP, Loo VG, McDonald LC, Pepin J, Wilcox MH, Infect. Control. Hosp. Epidemiol 2010, 31, 431–455. [DOI] [PubMed] [Google Scholar]
- [6].a) Koo HL, Garey KW, DuPont HL, Expert Opin. Invest. Drugs 2010, 19, 825–836; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zuo T, Wong SH, Lam K, Lui R, Cheung K, Tang W, Ching JYL, Chan PKS, Chan MCW, Wu JCY, Chan FKL, Yu J, Sung JJY, Ng SC, Gut 2018, 67, 634–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pepin J, Alary ME, Valiquette L, Raiche E, Ruel J, Fulop K, Godin D, Bourassa C, Clin. Infect. Dis 2005, 40, 1591–1597. [DOI] [PubMed] [Google Scholar]
- [8].Antibiotic Resistance Threats in the United States, 2013, Centers for Disease Control and Prevention (CDC), Atlanta, GA (USA), 2013, pp. 50–52; https://www.cdc.gov/drugresistance/threat-report-2013/index.html. [Google Scholar]
- [9].Johanesen PA, Mackin KE, Hutton ML, Awad MM, Larcombe S, Amy JM, Lyras D, Genes 2015, 6, 1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Raulji CM, Clay K, Velasco C, Yu LC, J. Pediatr. Hematol. Oncol 2015, 32, 315–321; [DOI] [PubMed] [Google Scholar]; b) Grare M, Dibama HM, Lafosse S, Ribon A, Mourer M, Regnouf-de-Vains JB, Finance C, Duval RE, Clin. Microbiol. Infect 2010, 16, 432–438. [DOI] [PubMed] [Google Scholar]
- [11].a) Bçttcher T, Kolodkin-Gal I, Kolter R, Losick R, Clardy J, J. Am. Chem. Soc 2013, 135, 2927–2930; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hobley L, Kim SH, Maezato Y, Wyllie S, Fairlamb AH, Stanley-Wall NR, Michael AJ, Cell 2014, 156, 844–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schaberle TF, Hughes DE, Epstein S, Jones M, Lazarides L, Steadman VA, Cohen DR, Felix CR, Fetterman KA, Millett WP, Nitti AG, Zullo AM, Chen C, Lewis K, Nature 2015, 517, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kowalski RP, Romanowski EG, Yates KA, Mah FS, J. Ocul. Pharmacol. Ther 2016, 32, 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Lin S, Koh JJ, Aung TT, Lim F, Li J, Zou H, Wang L, Lakshminarayanan R, Verma C, Wang Y, Tan DT, Cao D, Beuerman RW, Ren L, Liu S, J. Med. Chem 2017, 60, 1362–1378; [DOI] [PubMed] [Google Scholar]; b) Koh JJ, Lin S, Aung TT, Lim F, Zou H, Bai Y, Li J, Lin H, Pang LM, Koh WL, Salleh SM, Lakshminarayanan R, Zhou L, Qiu S, Pervushin K, Verma C, Tan DT, Cao D, Liu S, Beuerman RW, J. Med. Chem 2015, 58, 739–752. [DOI] [PubMed] [Google Scholar]
- [15].Teng P, Nimmagadda A, Su M, Hong Y, Shen N, Li C, Tsai L-Y, Cao J, Li Q, Cai J, Chem. Commun 2017, 53, 11948–11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Konai MM, Samaddar S, Bocchinfuso G, Santucci V, Stella L, Haldar J, Chem. Commun 2018, 54, 4943–4946; [DOI] [PubMed] [Google Scholar]; b) Jennings MC, Ator LE, Paniak TJ, Minbiole KPC, Wuest WM, ChemBioChem 2014, 15, 2211–2215; [DOI] [PubMed] [Google Scholar]; c) Rossiter SE, Fletcher MH, Wuest WM, Chem. Rev 2017, 117, 12415–12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tanaka K, Mikamo H, Nakao K, Ichiishi T, Goto T, Yamagishi Y, Watanabe K, Antimicrob. Agents Chemother 2009, 53, 319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tsutsumi LS, Owusu YB, Hurdle JG, Sun D, Curr. Top. Med. Chem 2014, 14, 152–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hecht DH, Onderdonk OA, Citron DM, Roe-Carpenter D, Cox M, Rosenblatt JE, Jacobus N, Wexler HM, Jenkins SG, Methods for Anti microbial Susceptibility Testing of Anaerobic Bacteria, 7th ed, Wayne, PA (USA), 2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.