

Abstract
Most of the associated pathologies in Gulf War Illness (GWI) have been ascribed to chemical and pharmaceutical exposures during the war. Since an increased number of veterans complain of gastrointestinal (GI), neuroinflammatory and metabolic complications as they age and there are limited options for a cure, the present study was focused to assess the role of butyrate, a short chain fatty acid for attenuating GWI-associated GI and metabolic complications. Results in a GWI-mouse model of permethrin and pyridostigmine bromide (PB) exposure showed that oral butyrate restored gut homeostasis and increased GPR109A receptor copies in the small intestine (SI). Claudin-2, a protein shown to be upregulated in conditions of leaky gut was significantly decreased following butyrate administration. Butyrate decreased TLR4 and TLR5 expressions in the liver concomitant to a decrease in TLR4 activation. GW-chemical exposure showed no clinical signs of liver disease but a significant alteration of metabolic markers such as SREBP1c, PPAR-α, and PFK was evident. Liver markers for lipogenesis and carbohydrate metabolism that were significantly upregulated following GW chemical exposure were attenuated by butyrate priming in vivo and in human primary hepatocytes. Further, Glucose transporter Glut-4 that was shown to be elevated following liver complications were significantly decreased in these mice after butyrate administration. Finally, use of TLR4 KO mice completely attenuated the liver metabolic changes suggesting the central role of these receptors in the GWI pathology. In conclusion, we report a butyrate specific mechanistic approach to identify and treat increased metabolic abnormalities in GWI veterans with systemic inflammation, chronic fatigue, GI disturbances, metabolic complications and weight gain.
Keywords: Permethri, Pyridostigmine bromide, Gut dysbiosis, TLR4, Claudin-2, Cytokines
1. Introduction
Gulf War Illness (GWI) has been characterized as a chronic multisymptom illness with pathology that includes neuronal inflammation leading to cognitive deficiencies, chronic fatigue, joint and muscle pain and gastrointestinal complications (White et al., 2016; Kerr, 2015; Gulf War and Health, 2016). GWI research has identified toxicant chemical exposures in the war theater including sarin nerve gas, pyridostigmine bromide (PB) anti-nerve gas pills, insecticides and insect repellents, to be prime reasons for most symptoms reported by the veterans (White et al., 2016). A study of military pesticide applicators from the GW recently reported increased cognitive decrements in attention, memory and information processing speed in veterans with combined exposures to PB, pesticides and insect repellents (Sullivan et al., 2018). Animal models of GWI have also shown chronic impairment in learning and memory, fatigue and gastrointestinal dysfunction when exposed to GW-relevant chemicals including PB, pesticides and the insect repellent permethrin (Abdullah et al., 2016; Parihar et al., 2013). GWI has emerged as a primarily neuroimmune disorder with greater inflammatory effects noted when GW-relevant toxicants were combined with corticosterone in animal models to mimic the physical and mental stressors of war deployment (Koo et al., 2018). However the basis of gastrointestinal complications and parallel networks for neuronal complications have remained largely elusive (Georgopoulos et al., 2017; O’Callaghan et al., 2015; O’Callaghan et al., 2016). Though the larger epidemiological studies list GI complications as a major health problem in only a subset of GW veterans, there are hardly any reports that show hepatic metabolic disturbances as a widely reported symptom (White et al., 2016; Kerr, 2015; Haley et al., 2013). This may be due to the silent nature of the presentation of these symptoms and unknown links to a wider systemic complication in GW veterans following sedentary lifestyles/physical disabilities/diet/aging over a longer period of time (Coughlin et al., 2011). However, with increased incidences of obesity and persistence of symptoms in GW-veterans and the US population in general, the causes, outcomes, and extent of metabolic disturbances can no longer be ignored. The long-ranging implications of such silent changes in the GI tract and the liver following chemical exposure form the basis of the present study.
We have shown recently the unambiguous role of the gut microbiome in causing neuronal inflammation largely due to gut leaching and systemic endotoxemia (Alhasson et al., 2017). The altered gut bacterial signature obtained following Gulf War chemical exposure caused a TLR4-linked inflammatory surge in the GI tract and could be traced in the frontal cortex (Alhasson et al., 2017). The observations reported paved the way for newer investigations into wider systemic inflammatory complications in extraneuronal organs that might not have a clear phenotype yet may be a basis for multisymptom illness as described in GWI.
GWI is also characterized by the presence of chronic fatigue. Most classifications in the past have listed chronic fatigue as one of the most widely reported symptom burdens in GW veterans (Kerr, 2015; Gulf War and Health, 2016; Coker, 1996). Interestingly, changes in the microbiome of affected patients with chronic fatigue syndrome/fibromyalgia have been strongly associated with the causes of this illness (Nagy-Szakal et al., 2017; Morris et al., 2016; Giloteaux et al., 2016). Chronic fatigue is also strongly linked to widescale changes both in the gut bacteria and the systemic metabolism with the latter believed to have roots in the liver though skeletal muscles also play a major role (Naviaux et al., 2016). Persistent alteration of liver metabolism following changes in gut microbiome and its subsequent effects on systemic metabolism may affect the development of chronic fatigue via the altered availability of NADPH, ATP, and cofactors for various biochemical pathways (Naviaux et al., 2016).
Most chronic liver diseases like fatty liver disease and biliary fibrosis are silent in a presentation at the clinic and remain asymptomatic until it reaches an irreversible stage (Milic & Stimac, 2012). Nonalcoholic fatty liver disease or cholestatic liver disease have chronic fatigue as one of the symptoms (Schreuder et al., 2008; Zakharia et al., 2018). The fatigue associated with these silent liver complications have been assigned to lipotoxicity, insulin and leptin resistance, endocrinopathies and metabolic syndrome (Kaltsas et al., 2010). Interestingly, we have shown that environmental chemicals alter liver metabolism by increasing glucose transporters in the liver fibroblasts, elevating expressions of PPAR-α, PFK and decreasing PPAR-γ levels with a concomitant rise in leptin (Seth et al., 2013a). These changes in the liver have been associated with an altered microbiome following consumption of a diet rich in high fat and low fiber for a long period of time at least in the murine models (Chiu et al., 2017; Marra & Svegliati-Baroni, 2018).
With strong emphasis on the altered microbiome being associated with pathologies of inflammatory bowel disease, chronic liver disease, chronic fatigue syndrome and neuronal complications (all have an inflammatory component) via the gut-brain axis, it is important that studies be focused on bacterial metabolites within the gut that might be linked to some or all the pathways that link these systemic complications. Interestingly, GWI patients present significant symptoms that resemble some or all these in the clinics. Thus bacterial metabolites like butyric acid, propionic acid and acetate need to be considered as molecules of interest in treating multiple symptoms of GWI since these molecules have shown promising results in the clinic to cure IBD and dysbiosis related complications (Sun et al., 2017; Zhang et al., 2016; Imhann et al., 2018). Further, Butyrate-producing bacteria such as Roseburia species supplementation rescued patients from IBD (Imhann et al., 2018). Butyrate, in particular, is a short chain fatty acid (SCFA) that primarily interacts with the GPR109A and is an immunosuppressant widely known to increase T-regulatory cells in the intestine and a prominent HDAC1 inhibitor (Sekhavat et al., 2007; Singh et al., 2014).
The present study tests the hypothesis that GW chemical exposure causes a decrease in butyrate-producing bacteria and concomitant butyrate priming in the gut through oral supplementation attenuates GI inflammation, gut leaching and metabolic abnormalities in the liver and higher systemic leptin levels. The study uses state of the art genomic approaches and an oral priming by butyrate for elucidating genus-specific changes in gut bacteria, and human hepatocytes treated with an insect repellent permethrin, (used in Gulf War theater) for mechanistic investigations.
2. Materials and methods
2.1. Materials
Pyridostigmine bromide (PB), Permethrin (Per), Lipopolysaccharides (LPS), Corticosterone and Sodium butyrate (NaBT) were purchased from Sigma-Aldrich (St. Louis, MO). Anti-claudin-2, anti-occludin, anti-TLR4, anti-flotillin, anti-HMGBl, anti-Leptin and anti-IL1β primary antibodies were purchased from Abeam (Cambridge, MA). Anti-TLR5 primary antibody was purchased from Santacruz Biotechnology (Dallas, TX). Species-specific biotinylated conjugated secondary antibodies and Streptavidin-HRP (Vectastain Elite ABC kit) were purchased from Vector Laboratories (Burlingame, CA). Fluorescence-conjugated (Alexa Fluor) secondary antibodies, ProLong Gold antifade mounting media with DAPI were purchased from Thermofisher Scientific (Grand Island, NY) and all other chemicals which were used in this project purchased from sigma only if otherwise specified. Paraffin-embedding of tissue sections on slides were done by AML laboratories (Baltimore, MD). Microbiome analysis was done by Second Genome, the microbiome company (San Francisco, CA).
2.2. Animals
Adult wild-type male (C57BL/6J mice) and adult mice that contained the disrupted TLR4 gene (TLR4 KO) (B6·B10ScN-Tlr4lps-del / JthJ) were purchased from the Jackson Laboratories (Bar Harbor, ME). Mice were implemented in accordance with NIH guideline for human care and use of laboratory animals and local IACUC standards. All procedures were approved by The University of South Carolina at Columbia, SC. Mice were housed individually and fed with chow diet at 22–24 °C with a 12-h light/ 12-h dark cycle. All mice were sacrificed after animal experiments had been completed. Right after anesthesia, blood from the mice was drawn using cardiac puncture, in order to preserve serum for the experiments. The mice liver was collected for further experiments immediately after terminal euthanasia. Fecal pellets and luminal contents were collected from the animals, followed by dissection of the small intestine. The tissues were fixed using 10% neutral buffered formalin. Distal segments of small intestines were used for the staining and visualizations.
2.3. Rodent model of Gulf War Illness (GWI)
Mice were exposed to Gulf War chemicals based on established rodent models of Gulf War Illness with some modifications (O’Callaghan et al., 2015; Zakirova et al., 2015). The treated wild-type mice group (GWI) and treated TLR4 KO mice group (TLR4 KO) were dosed triweekly for one week with PB (2 mg/kg) and Permethrin (200 mg/kg) via the oral route. After completion of PB/Permethrin dosages, mice were administered corticosterone intraperitoneally (i.p.) with a dose of 100 μg/mice/day for 5 days of the week for one week. The dose of corticosterone was selected from the study which exposed mice to 200mg/L of corticosterone through drinking water. The i.p. dose of corticosterone had similar immunosuppression as examined by low splenic T cell proliferation (data not shown). The vehicle control group (Veh) of mice received saline injections and vehicle for oral gavage in the same paradigm. Another group of wild-type mice was exposed with PB, Permethrin and corticosterone similar to GWI group of mice and co-exposed with sodium butyrate (GWI + NaBT) 10 mg/kg via the oral route.
2.4. Microbiome analysis
Fecal pellets and luminal contents were collected from the animals of each group after sacrifice and then sent to Second Genome and School of Medicine, the University of South Carolina for microbiome analysis. The second Genome performed nucleic acid isolation with the MoBio PowerMag Microbiome kit (Carlsbad, CA) according to manufacturer’s guidelines and optimized for high-throughput processing V3-V4 sequencing and bioinformatic analysis.
2.5. Cell culture
Freshly isolated primary human hepatocytes were obtained from Liver Tissue Cell Distribution System, University of Minnesota, Minneapolis, MN. Plated hepatocytes were maintained in DMEM media supplemented with 10% FBS until treated. Cells were then serum starved in DMEM supplemented with 1.5% FBS for 8h and exposed to vehicle control and chemicals. Cells were then treated with vehicle (Veh Cont), LPS (1 μM), LPS + NaBT (LPS 1 μM and Sodium Butyrate 0.2mg/mL) for 24 h. After experiment cells were harvested for mRNA extraction and gene expression analysis.
2.6. Laboratory methods
2.6.1. Immunohistochemistry
The distal part of small intestine was collected from mice and fixed in 10% neutral buffered formalin. The fixed tissues swiss rolled, paraffin embedded and cut in 5 μM thick section. These sections were subjected to deparaffinization using a standard protocol. Epitope retrieval solution and steamer (IHC-Word, Woodstock, MD) were used for epitope retrieval for deparaffinized sections. 3% H2O2 was used for the recommended time to block the endogenous peroxidase. After serum blocking, the tissue was incubated overnight at 4.0 °C with primary antibody IL1β. Species-specific biotinylated conjugated secondary antibodies and streptavidin conjugated with HRP were used to implement antigen-specific immunohistochemistry. 3,3′-Diaminobenzidine (DAB) (Sigma Aldrich, St Louis, MD) was used as a chromogenic substrate. Mayer’s Hematoxylin solution (Sigma Aldrich) was used as a counterstain. Sections were washed between the steps using phosphate buffered saline 1×. Finally, stained sections were mounted with Simpo-mount (GBI laboratories, Mukilteo, WA). Tissue sections were observed using Olympus BX51 microscope (Olympus, America). Cellsens software from Olympus America (Center Valley, PA) was used for morphometric analysis of images.
2.6.2. Immunofluorescence staining
Paraffin-embedded distal part of the small intestine or liver sections were deparaffinized using a standard protocol. Epitope retrieval solution and steamer were used for epitope retrieval of sections. Primary antibodies such as anti-Claudin-2, anti-Occludin, anti-GPR109A, anti-TLR4, anti-Flotillin, and anti-TLR5 were used at the recommended dilution. Species-specific secondary antibodies conjugated with Alexa Fluor (633-red and 488-green) were used at advised dilution. In the end, the stained sections were mounted using Prolong gold antifade reagent with DAPI. Sections were observed under-Olympus fluorescence microscope using 20×, 40× or 60× objective lenses.
2.6.3. Real-time quantitative PCR
mRNA expression in small intestine, liver, and human primary hepatocytes was examined by quantitative real-time PCR analysis. Total RNA was isolated from each 25 mg liver tissue or 15 mg small intestine tissue or 1 × 106 primary human hepatocytes cell by homogenization in TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions and purified with the use of RNeasy mini kit columns (Qiagen, Valencia, CA). cDNA was synthesized from purified RNA (1 μg) using iScript cDNA synthesis kit (Bio-rad, Hercules, CA) following the manufacturer’s standard protocol. Real-time qPCR (qRTPCR) was performed with the gene-specific primers using SsoAdvanced SYBR Green Supermix and CFX96 thermal cycler (Bio-rad, Hercules, CA). Threshold Cycle (Ct) values for the selected genes were normalized against respective samples internal control (18S). Each reaction was carried out in triplicates for each gene and for each sample. The relative fold change was calculated by the 2-ΔΔCt method. The sequences for the primers used for Real-time PCR are provided in Table 1.
Table 1.
Real-time PCR primer sequences.
| Genea | Primer sequence (5′ to 3′ orientation) |
|---|---|
| MM_IL-1β | Sense: CCTCGGCCAAGACAGGTCGC Antisense: TGCCCATCAGAGGCAAGGAGGA |
| MM_MCP-1 | Sense: CACAGTTGCCGGCTGGAGCAT Antisense: GTAGCAGCAGGTGAGTGGGGC |
| MM_TNF-α | Sense: CAACGCCCTCCTGGCCAACG Antisense: TCGGGGCAGCCTTGTCCCTT |
| MM_SREBP1c | Sense: GGAACAGACACTGGCCGA Antisense: AAGTCACTGTCTTGGTTGTTGAT |
| MM_PPAR-α | Sense: AGACCTTCGGCAGCTGGTCAC Antisense: GTGGCAACGGCCTGCCATCT |
| MM_PPAR-γ | Sense: TTCGCTGATGCACTGCCTAT Antisense: GGAATGCGAGTGGTCTTCCA |
| MM_GLUT-1 | Sense: CCTGTCTCTTCCTACCCAACC Antisense: GCAGGAGTGTCCGTGTCTTC |
| MM_GLUT-4 | Sense: CACCGGCAGCCTCTTATCAT Antisense: CACCGAGACCAACGTGAAGA |
| MM_PFK | Sense: GCCGTGAAACTCCGAGGAA Antisense: GTTGCTCTTGACAATCTTCTCATCAG |
| Hs_SREBP1c | Sense: CATGGATTGCACTTTCGAA Antisense: GGCCAGGGAAGTCACTGTCTT |
| Hs_PPAR-γ | Sense: GGCTTCATGACAAGGGAGTTTC Antisense: AACTCAAACTTGGGCTCCATAAAG |
MM: Mouse specific primers, Hs: Human specific primers.
2.6.4. Elisa
Serum Leptin and serum HMGB1 was estimated using ELISA kits from Abclonal Biotechnologies (Woburn, MA) following manufacturer protocol. Serum IL1β was estimated using an ELISA kit from ProteinTech (Rosemont, IL) following manufacturer protocol.
2.6.5. Serum biochemistry tests
Biochemical analysis of mouse serum was done for ALT, urea Nitrogen, creatinine, cholesterol, triglycerides and glucose from the University of Georgia college of veterinary medicine.
2.7. Statistical analysis
Prior to initiation of the study, we conducted calculations for each experimental condition with appropriate preliminary data to confirm that the sample number is sufficient to achieve a minimum statistical power of 0.80 at an alpha of 0.05. All in vivo and in vitro experiments were repeated three times with 3 mice per group (N = 3; data from each group of three mice were pooled). Student’s t-test was used to compare means between two groups at the termination of treatment. A one-way ANOVA was applied as needed, to evaluate differences among treatment groups followed by the Bonferroni post-hoc correction for intergroup comparisons.
3. Results
3.1. Butyrate production is key to gut health in GW-chemical exposure and microbial dysbiosis
We have shown previously that GW chemical exposure caused a significant alteration in microbial population when compared to untreated controls with significant increases in Firmicutes-Bacteriodetes ratio, a trend that is uniformly observed in IBD, neuroinflammation and metabolic syndrome. The changes were consistent with the neuroinflammatory phenotype in the mouse model of GWI. On in-depth analysis of the microbial data, we found that GW-chemical exposed group showed a marked decrease in Lactobacillus, and Bifidobacterium sp., the genus being responsible for producing the short chain fatty acid butyrate. Interestingly butyrate has been shown to attenuate IBD and resists proinflammatory changes in the small intestine (Fig. 1A) (p < 0.05). The two genus showed a > 5 fold (log scale) decrease in abundance (Fig. 1A). Butyrate priming through oral gavage and its presence during exposure significantly elevated the levels of Bifidobacterium, butyrogenic bacteria Roseburia sp. and Lactobacillus (i, ii and iii) (Fig. 1B) (p < 0.05) when compared to GWI alone with the first two genera showing an increase up to > 60% when compared to GWI. The percentages noted in the figure are compared to the overall abundance of all genus detected in the metagenomic analysis. The comparisons between GWI and GWI + NaBT groups were done using GWI as the base line. Such a comparison showed a > 60% increase of these genus in GWI + NaBT group when compared to GWI alone (Fig. 1B). The observations in Fig. 1A led to the rationale for using Butyrate as a viable molecule for attenuating microbiome-associated inflammatory phenotype and the subsequent changes observed in the GWI model. Butyrate exerts its actions via binding to the niacin receptor. GPR109A has been recently discovered to bind butyrate and stimulate the activation of Treg cells thus suppressing TH17 mediated proinflammatory events (Singh et al., 2014). Our results showed that there is a significant decrease in the protein levels of GPR109A in GW-chemical exposed group when compared to untreated controls (Fig. 2A and D) (p < 0.05). Butyrate presence in the intestine via feeding GW-exposed mice through an oral gavage significantly increased GPR109A protein levels in the villi regions when compared to GWI-group (Fig. 2A and D) (p < 0.05) suggesting that butyrate presence resisted the decrease in GPR109A protein levels thus helping butyrate to exert its actions in the dysbiosis-affected small intestine and restore gut-epithelial cell integrity and metabolic homeostasis.
Fig. 1.
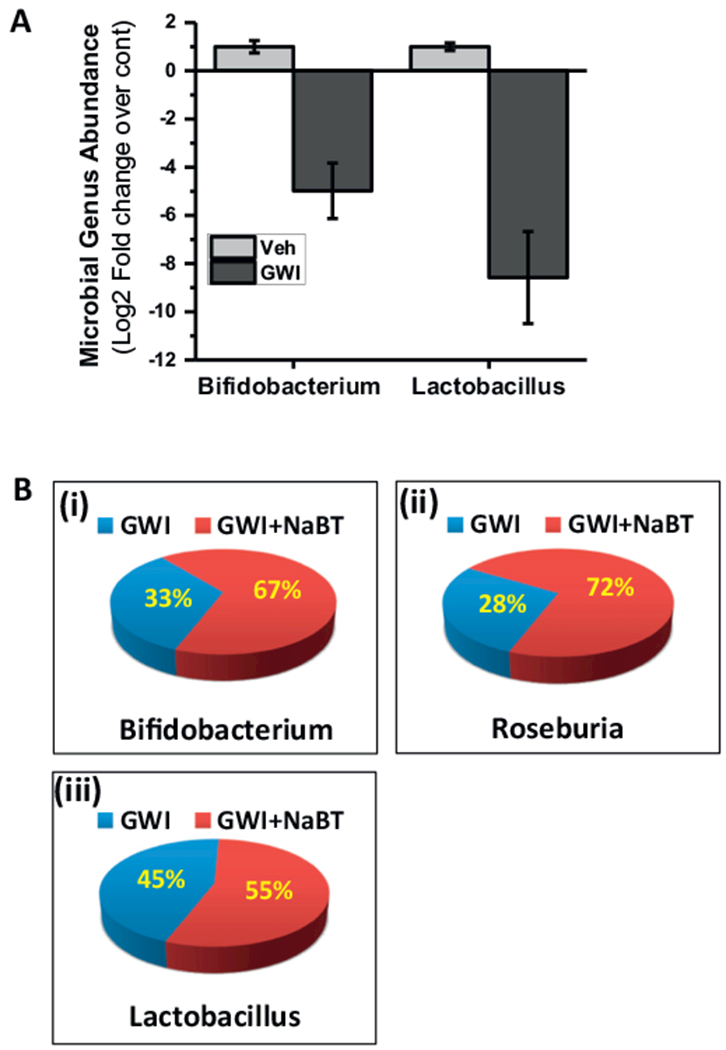
Gut microbiome alteration in mice model of Gulf War Illness (GWI). A. The proportional abundance of microbial genera: Graphical representation of the most abundant taxa of bacteria at the genus level. Groups compared are gulf war illness group (wild-type mice exposed to gulf war chemicals) (GWI, n = 3) and control group fed with vehicle (Veh, n = 3) (p-value: < 0.05). B. Percentage abundance of gut bacteria Bifidobacterium (i), Roseburia (ii), and Lactobacillus (iii) in a group of mice co-exposed with Gulf war chemicals and Sodium butyrate (GWI + NaBT, n = 3) as compared with GWI mice (n = 3) (p-value: < 0.05).
Fig. 2.
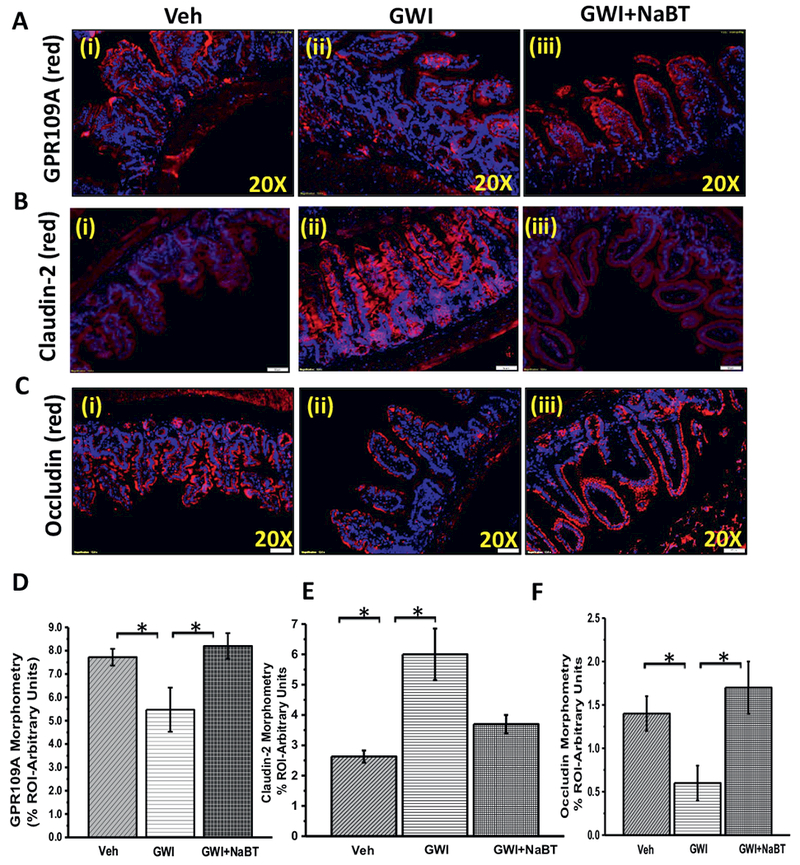
Change in gut microbiome in GWI alter niacin receptor (GPR109A) and tight junction proteins in the intestine. A. The expression pattern of butyrate and niacin receptor GPR109A was assessed by immunofluorescence microscopy. The representative images showed immunoreactivity of GPR109A in the distal part of the small intestine of veh control group of mice (veh, n = 3), gulf war illness group of mice (GWI, n = 3) and a group of mice co-exposed with GWI and sodium butyrate (GWI + NaBT, n = 3). B and C. The expression pattern of Claudin-2 and Occludin (tight junction proteins) was assessed by immunofluorescence microscopy. Tissue levels of Claudin-2 (B) and Occludin (C) in Vehicle control group of mice (Veh, n = 3), gulf war chemical treated group of mice (GWI, n = 3) and a group of mice co-exposed with GWI and sodium butyrate (GWI + NaBT, n = 3) was assessed by immunofluorescent microscopy after labeling the protein with the red fluorescent secondary antibody and counterstained with DAPI (blue). D–F. The bar diagram shows the quantitative morphometric analysis of fluorescence intensities of GPR109A (D), Claudin-2 (E), and Occludin (F) immunoreactivity in the region of interest (ROI) in the small intestine. *(p < 0.05). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. Butyrate priming through oral route restores tight junction protein levels
The epithelial tight junction determines the paracellular water and ion movement in the intestine and also prevents uptake of larger molecules, including antigens, in an uncontrolled manner where Claudin-2 and Occludin play a major role and are perceived as a marker for leaky gut (Luettig et al., 2015). Our results from immunofluorescence microscopy for the immunoreactivity of Claudin-2 showed a significant increase in GW-chemical exposed group when compared to untreated controls (Fig. 2B and E) (p < 0.05), thus confirming our previously reported data. Butyrate presence in GW-chemical exposed group showed a significant decrease in that group when compared to GW-chemical exposed group alone suggesting a parallel role of butyrate in Claudin-2 protein levels in the small intestine (Fig. 2B and E) (p < 0.05). The results also showed that butyrate priming nearly restored the Claudin-2 levels to untreated controls (Fig. 2B). Similarly, the protein level of another tight junction protein Occludin was significantly decreased in GW-chemical exposed groups (Fig. 2C and F) and sodium butyrate treatment significantly restored the levels of Occludin in the intestine. The results suggested that Butyrate may have a previously unconfirmed role in modulating Claudin-2 and Occludin proteins in the small intestine though ILlOA-dependent repression of Claudin-2 has been shown (Zheng et al., 2017).
3.3. Butyrate priming in the intestine attenuates proinflammatory phenotype in the intestine via a decrease in TLR4 activation
Since gut leaching was predominant in GWI mouse model and resulted in endotoxemia, we studied whether butyrate priming helped in attenuating the proinflammatory microenvironment in the small intestine (Alhasson et al., 2017). Results showed that butyrate administration through an oral route decreased TLR4 colocalization (as shown by white circles), a hallmark of its activation in GW-chemical exposed group when compared to GW-group alone (Fig. 3A and B) (p < 0.05). Notably, the results also confirmed our earlier observations of an increased TLR4 trafficking to lipid rafts in GW chemical exposed group when compared to untreated controls (Fig. 3A and B) (p < 0.05). TLR4 activation was followed by increased IL-1β protein levels in the villi regions but not in crypts of GW chemical exposed group when compared to untreated controls (Fig. 4A and B) (p < 0.05). Also, butyrate priming significantly decreased the IL-1β levels in the same regions when compared to GW-Chemical exposed group (Fig. 4A and B). Gene expressions of IL-1β, monocyte chemoattractant MCP-1 and TNF-α were significantly decreased in Butyrate administered group when compared to GW-chemical exposed group (Fig. 4C) (p < 0.05). Interestingly, serum IL-1β significantly increased in GW-chemical exposed groups (66.71 ± 1.98pg/mL) as compared to vehicle control group (39.95 ± 1.8pg/mL) (Fig. 4D). However, Butyrate exposure to the GW-Chemical exposed mice showed a significant decrease in the serum IL-1β (34.56 ± 1.26pg/mL) (Fig.4D). The results suggested that butyrate presence helped attenuate intestinal inflammation primarily from a TLR4 pathway however it could not rule out other parallel inflammatory pathways in the gut such as histone deacetylases.
Fig. 3.
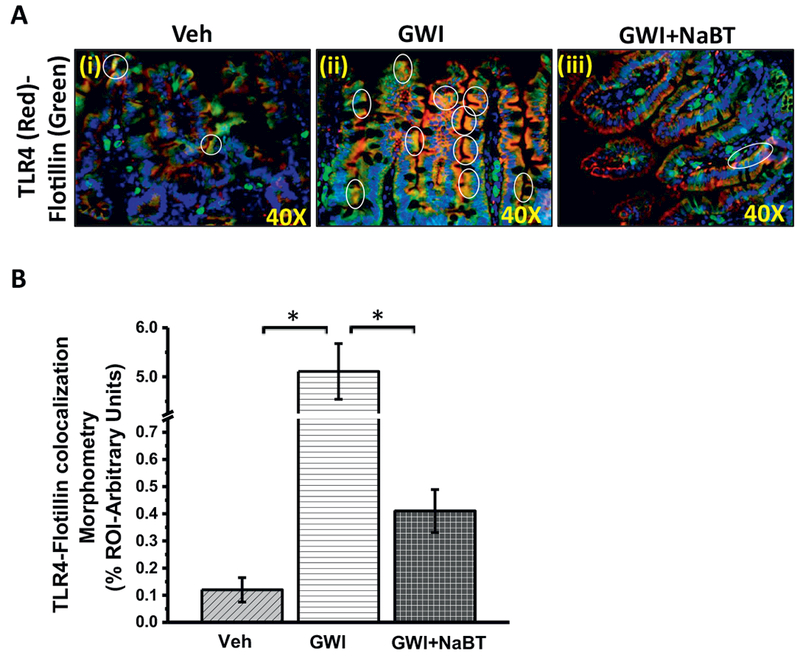
Sodium butyrate priming in a rodent model of GWI attenuates TLR4 activation in the small intestine. A. Immunofluorescence microscopy of small intestine showing TLR4 (red) trafficking to the lipid rafts (green) of the small intestine tissue, an essential process for TLR4 activation causing a co-localization of TLR4 in flotillin-rich rafts (yellow). Representative images of TLR4-flotillin co-localization in the small intestine of vehicle control group of mice (Veh, n = 3), gulf war chemical treated group of mice (GWI, n = 3) and a group of mice co-exposed with GWI and sodium butyrate (GWI + NaBT, n = 3) shown by white circles covering the yellow spots created by an overlay of red (TLR4) and green (Flotillin). Images were taken at higher magnification (40× oil). B. Graphical representation of the quantitative morphometric analysis of colocalization events in the region of interest (ROI) in the small intestine. Images for analysis were randomly chosen in different microscopic fields. Data is represented as Mean ± SEM and *(p < 0.05). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 4.
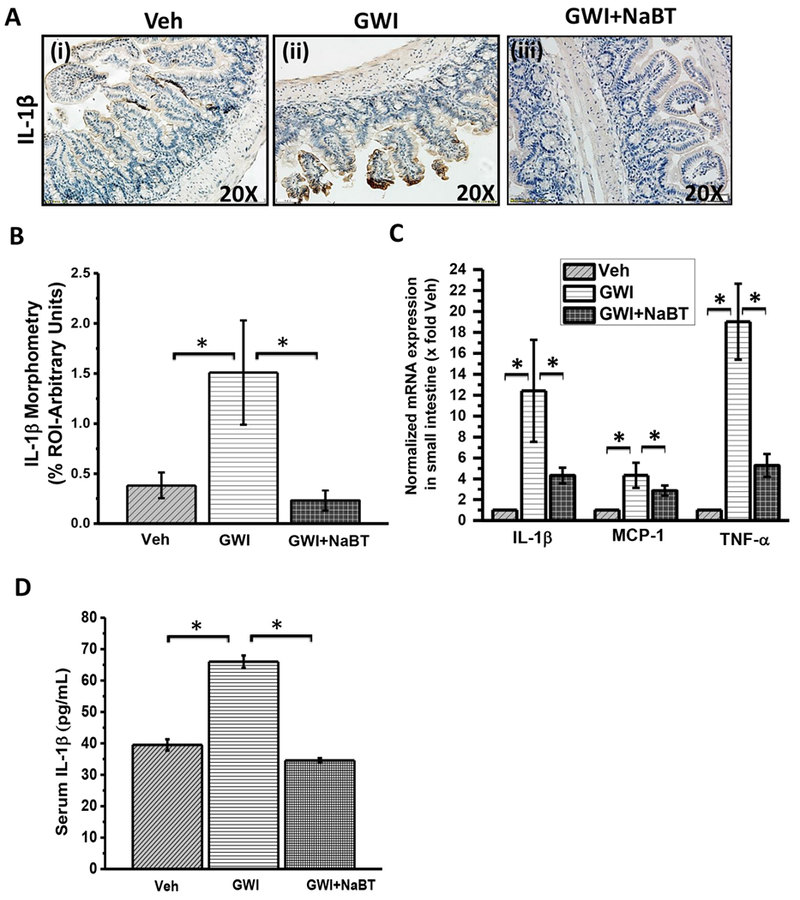
Sodium butyrate priming in a rodent model of GWI improves proinflammatory phenotype in small intestine mediated by the TLR4 pathway. A. Small intestine tissue slices were probed for IL-1β immunoreactivity in vehicle control group of mice (Veh, n = 3), gulf war chemical treated group of mice (GWI, n = 3) and a group of mice co-exposed with GWI and sodium butyrate (GWI + NaBT, n = 3) using immunohistochemistry. Specific immunoreactivity to IL-1β is evident by dark brown spots. B. Graphical representation of morphometric analysis of the IL-1β immunoreactivity in tissue slices. Data normalized against vehicle control (veh) *(p < 0.05). C. Quantitative real-time PCR (qRTPCR) analysis of inflammatory markers in the small intestine. mRNA expression of IL-1β, MCP-1, and TNF-α was analyzed in the samples of vehicle control group of mice (Veh, n = 3), gulf war chemical treated group of mice (GWI, n = 3) and a group of mice co-exposed with GWI and sodium butyrate (GWI + NaBT, n = 3). Normalized mRNA expression is represented as a fold change of vehicle control (veh) on Y-axis. Data points represented with Mean ± SEM *(p < 0.05). D. Graphical representation of serum IL-1β in pg/mL of the samples of vehicle control group of mice (Veh, n = 3), gulf war chemical treated group of mice (GWI, n = 3) and a group of mice co-exposed with GWI and sodium butyrate (GWI + NaBT, n = 3). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Similar to pathogen-associated molecular patterns (PAMPs), that can trigger a proinflammatory response, sterile inflammation can be triggered by endogenous molecules from a necrotic or damaged cell that can activate several proinflammatory pathways including TLR4 (Garcia-Martinez et al., 2015). Such endogenous molecules are collectively called Damage-associated molecular patterns or DAMPs. We have shown previously that HMGB1 and leptin can be released from several organ systems and can trigger a proinflammatory cascade (Chatterjee et al., 2013; Chandrashekaran et al., 2016; Chandrashekaran et al., 2017). We quantified the released HMGB1 and leptin in mouse serum using competitive ELISA techniques. Result showed that GW-chemical exposed groups had significantly higher levels of HMGB1 and leptin in the serum when compared to untreated controls (Fig. 5A and B) (p < 0.05). Butyrate priming significantly decreased the level of HMGB1 while a decrease in leptin levels was not significant when compared to GW-chemical exposed group (Fig. 5A and B) (p < 0.05). The results suggested that circulatory DAMPs can be soluble mediators of ectopic inflammatory events distant to the small intestine while butyrate priming may attenuate such effects and help identify therapeutic targets in the systemic inflammatory phenotype seen in GWI.
Fig. 5.

Sodium butyrate treatment in GWI improves circulatory DAMPs. Similar to the pathogen-associated molecular pattern (PAMPs), the endogenous molecules called damage-associated molecular patterns or DAMPs (such as HMGB1) are linked with proinflammatory responses in distal organs. A. Western blot analysis of serum high mobility group box 1 protein (HMGB1) and serum adipokine leptin from samples of vehicle control group of mice (Veh, n = 3), gulf war chemical treated group of mice (GWI, n = 3) and a group of mice co-exposed with GWI and sodium butyrate (GWI + NaBT, n = 3). Ponceau S staining was done to see the equal loading of serum proteins and used for normalization of protein expression. B–C. Graphical representation of morphometric analysis of HMGB1 and leptin western blot bands. The data was normalized to a total serum protein (Ponceau S). Y-axis depicts the HMGBl/Ponceau S ratio (B) and leptin/Ponceau S ratio (C) from Veh, GWI and GWI + NaBT groups. *p < 0.05 is considered statistically significant. D-E. Graphical representation of serum HMGB1 (D) and serum leptin (E) in ng/mL of the samples of vehicle control group of mice (Veh, n = 3), gulf war chemical treated group of mice (GWI, n = 3) and a group of mice co-exposed with GWI and sodium butyrate (GWI + NaBT, n = 3).
3.4. Increased TLR activation in the liver is attenuated by butyrate priming in the intestine
TLR activation is observed in organ systems following gut dysbiosis (Reichardt et al., 2017; Henao-Mejia et al., 2012). TLRs especially TLR4, TLR2, and TLR5 have been shown to increase tissue inflammation (Henao-Mejia et al., 2012). Interestingly, TLR-induced metabolic deregulation is increasingly seen as an important event in metabolic syndrome (Boutagy et al., 2016). Our results showed that GW chemical exposed group showed a significantly increased TLR4 activation (trafficking to lipid rafts) in the liver especially in the sinusoidal cells (white circles) (Fig. 6A and B). TLR4 trafficking significantly decreased in butyrate administered group when compared to GW-chemical exposed group (Fig. 6A and B). Interestingly, butyrate administration markedly increased TLR4 protein (red) levels in the liver but could not be observed in the rafts of the membrane (yellow), a sign that TLR4 protein was increased but the activation was attenuated by butyrate administration (Fig. 6A). TLR5 a protein that is activated the following binding with flagellin also increased in the liver of GW exposed mice but was significantly decreased in the butyrate administered group (Fig. 6C and D). The results suggested that increased circulatory levels of DAMPs or a leaky gut-associated flagellin might have resulted in activation of TLR4 and TLR5 in the liver of GW chemical exposed group but was blocked by the presence of butyrate.
Fig. 6.
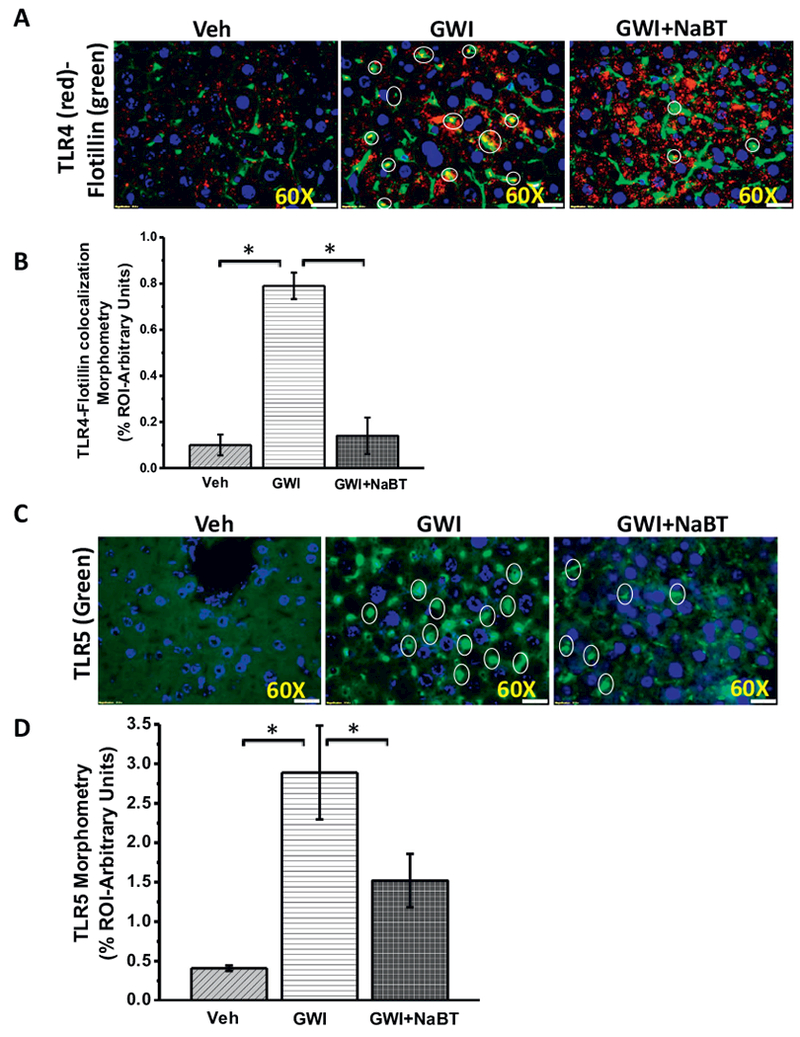
Sodium butyrate treatment in a rodent model of GWI attenuates TLR4 activation in Liver. A. Immunofluorescence microscopy of liver slices showing TLR4 (red) trafficking to the lipid rafts (green), an essential process for TLR4 activation causing a co-localization of TLR4 in flotillin-rich rafts (yellow). Representative images of TLR4-flotillin co-localization in the liver of vehicle control group of mice (Veh, n = 3), gulf war chemical treated group of mice (GWI, n = 3) and a group of mice co-exposed with GWI and sodium butyrate (GWI + NaBT, n = 3) shown by white circles covering the yellow spots created by an overlay of red (TLR4) and green (Flotillin). Images were taken at higher magnification (60× oil). B. Graphical representation of the quantitative morphometric analysis of colocalization events in the liver. Images for morphometric analysis were randomly chosen in different microscopic fields. Data is represented as Mean ± SEM *(p < 0.05). C. Tissue levels of TLR5 immunoreactivity in vehicle control group of mice (Veh, n = 3), gulf war chemical treated group of mice (GWI, n = 3) and a group of mice co-exposed with GWI and sodium butyrate (GWI + NaBT, n = 3) mouse liver samples as observed by immunofluorescent microscopy after labeling the TLR5 protein with the green fluorescent secondary antibody and counterstained by DAPI (blue). D. The bar diagram shows the quantitative morphometric analysis of fluorescence intensities of TLR5 immunoreactivity in the liver tissue. Images for morphometric analysis of TLR5 were randomly chosen in different microscopic fields. Data is represented as Mean ± SEM *(p < 0.05). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.5. TLR4 activation is associated with metabolic changes and inflammatory response in the liver but the phenotypic liver injury is predominantly absent
The liver is the principal organ for gluconeogenesis, lipogenesis and cholesterol metabolism (Bechmann et al., 2012). Recent studies have put a great deal of emphasis on liver metabolic reprogramming in conditions of metabolic syndrome that include hepatic expression of lipid and glucose metabolism markers, hepatic insulin and leptin resistance (Seth et al., 2013a; Chen et al., 2012). Interestingly, the fatty liver disease is associated with a long-term metabolic alteration (often years to manifest) and inflammatory response in the liver till the disease phenotype surfaces and is rightly called a silent disease (Milic & Stimac, 2012; Doycheva et al., 2017). We studied both the changes in hepatic metabolic markers and the inflammatory response arising from a TLR4 activation following GW-chemical exposure and microbial dysbiosis to ensure whether we can detect early responses in the liver that can manifest into liver disease years later. Results showed that hepatic SREBP1c, a molecule predominantly responsible for lipogenesis was upregulated following GW-chemical exposure when compared to untreated controls (Fig. 7A) (p < 0.05) (Geidl-Flueck & Gerber, 2017). Butyrate administration significantly decreased SREBP1c gene expression in the hepatic lobule when compared to GW-chemical exposed group (Fig. 7A) (p < 0.05). PPAR-α is a transcription factor and a major regulator of lipid metabolism in the liver (Badman et al., 2007). PPAR-α is activated under conditions of energy deprivation and is necessary for the process of ketogenesis (Badman et al., 2007). PPAR-α was significantly upregulated in the GW-chemical exposed group when compared to untreated controls while butyrate administration significantly decreased and restored the PPAR-α levels when compared to GW-chemical exposed group (Fig. 7A) (p < 0.05). PPAR-γ is an important player in liver fat metabolism and is known to be increased in benign steatosis but is significantly down-regulated in models of liver injury (Seth et al., 2013b). Our results showed that PPAR-γ was significantly decreased in GW chemical exposed group when compared to untreated controls and butyrate priming reversed this downregulation when compared to GW-chemical exposed group (Fig. 7A) (p < 0.05). The results were in agreement with our previous studies in a liver metabolic disease that had similar decreases in PPAR-γ (Seth et al., 2013b).
Fig. 7.

TLR4 activation is associated with metabolic changes and inflammatory response in the liver but the phenotypic liver injury is predominantly absent. A. Quantitative real-time PCR (qRTPCR) analysis principle carbohydrate metabolic markers (PFK, GLUT-1, and GLUT-4) and fat metabolic markers (SREBP1c, PPAR-α, and PPAR-γ) in the liver tissue. mRNA expression of SREBP1c, PPAR-α, PPAR-γ and PFK, GLUT-1, GLUT-4 and B. mRNA expression analysis of inflammatory marker IL-1β, MCP-1, TNF-α, and Kupffer cell activation marker CD68 were analyzed in the liver sample of vehicle control group of mice (Veh, n = 3), gulf war chemical treated group of mice (GWI, n = 3) and a group of mice co-exposed with GWI and sodium butyrate (GWI + NaBT, n = 3). Normalized mRNA expression is represented as a fold change of Vehicle control (veh) on Y-axis. Data points represented with Mean ± SEM *(p < 0.05). C. mRNA expression of SREBP1c, PPAR-γ were analyzed in the primary human hepatocytes cells treated with lipopolysaccharide (LPS) and Co-treated with LPS and sodium butyrate (LPS + NaBT). Normalized mRNA expression is represented as a fold change of Vehicle control (Veh Cont) on Y-axis. Data points represented with Mean ± SEM *(p < 0.05). D. Representative Hematoxylin and Eosin stained (H&E) images of liver sections showed liver pathophysiology of vehicle control group of mice (Veh, n = 3), gulf war chemical treated a group of mice (GWI, n = 3). Images were taken at 10× magnification.
We have shown previously that liver metabolic disorders triggered by environmental contaminants can increase the expression of Phosphofructokinase (PFK) (Seth et al., 2013b). PFK, a rate-limiting enzyme in the glycolytic pathway was significantly upregulated in GW chemical exposed group when compared to untreated controls while butyrate administration significantly restored the PFK levels (Fig. 7A) (p < 0.05). Hepatic class I glucose transporters (GLUT) have limited role in the liver but recent studies show their importance in hepatic disease states (Karim et al., 2012). We and others have shown that GLUT-1 and GLUT-4 are regulated by leptin and purinergic signaling and they are upregulated in fatty liver disease primarily in hepatic stellate cells (Chandrashekaran et al., 2016; Tang & Chen, 2010). Our results show that both GLUT-1 and GLUT-4 were upregulated in the GW chemical exposed groups when compared to untreated controls however butyrate administration significantly decreased the GLUT-1 and GLUT-4 levels when compared to GW-chemical exposed groups (Fig. 7A) (p < 0.05). Further, to investigate the role of GW-Chemical exposure in exacerbating the inflammatory response in liver, hepatic mRNA expression profiles of interleukin (IL)-lp, monocyte chemotactic protein 1 (MCP-1), tumor necrosis factor (TNF)-α and Kupffer cell activation marker CD68 were analyzed. Results indicated that there was a significant increase in the mRNA expression profiles of IL-1β, MCP-1, TNF-α and CD68 in GW Chemical exposed mice livers compared with vehicle treated mice livers (Fig. 7B) (p < 0.05). Interestingly, mice groups co-exposed with GW chemicals and sodium butyrate showed significantly decreased level of IL-1β, MCP-1, and CD68 but not TNF-α. The results suggested a similar role of higher leptin and/or heightened inflammation in causing the increase but remained to be seen whether it was cell or organ specific.
The liver has multiple cell types and includes cells of epithelial, endothelial, fibroblast and macrophage lineages. They perform multiple functions including metabolic, cellular defense and wound healing. The liver lobule comprises of 90% hepatocytes which are epithelial in origin and is a center for most of the metabolic functions. We used human primary hepatocytes, primed with lipopolysaccharide (LPS) (concentrations found in our previous study (Alhasson et al., 2017)) to study the effects of metabolic dysregulation if any due to GW-chemical exposure. Results showed that LPS primed hepatocytes showed a significant increase in lipogenesis mediator SREBP1c while butyrate co-exposure decreased these levels (Fig. 7C) (p < 0.05). Similar to our in vivo data, LPS primed hepatocytes showed a significant increase in PPAR-γ gene expression when co-exposed to butyrate while LPS only or untreated controls showed no change in the PPAR-γ levels (Fig. 7C) (p < 0.05).
Hematoxylin and Eosin stains of liver tissue sections obtained from GW-chemical exposed group showed no signs of lipid accumulation or macrophage infiltration or Mallory body formation signifying the absence of advanced stage inflammatory foci or liver disease (Fig. 7D). Though histopathology of the liver section from each mice group clearly showed that there was no sign of liver damage or development of Nonalcoholic steatohepatitis, we resorted to clinical chemistry analysis for more detailed outcomes. To confirm such observations we performed clinical chemistry analysis of mouse serum samples for ALT, BUN, creatinine, total cholesterol, triglyceride and serum glucose. The clinical chemistry data showed (Table 2) that there was no significant difference in serum ALT (showed a marked increase in GWI group but was not significant between groups), BUN, creatinine and total cholesterol upon GW Chemical exposure as compared to vehicle control group. However, co-exposure with sodium butyrate decreased the levels of ALT, BUN but not the total cholesterol (Table 2). The triglyceride levels were significantly increased in GWI chemical exposed mice groups as compared to untreated control groups, while co-exposure with sodium butyrate caused the triglyceride level to be decreased significantly when compared to GWI group (p < 0.05) suggesting a slow but incremental risk of fatty liver in the future (Table 2). The glucose levels showed no significant difference between groups (data not shown).
Table 2.
Serum chemistry analysis.
| Serum chemistry | Vehicle (n = 3) | GWI (n = 3) | GWI + NaBT (n = 3) |
|---|---|---|---|
| ALT (U/L) | 6 | 10 | 6 |
| BUN (mg/dl) | 32 | 28 | 24 |
| Creatinine (mg/dl) | ND | ND | ND |
| Cholesterol (mg/dl) | 78 | 82 | 98 |
| Triglycerides (mg/dl) | 84 | 102 | 84 |
ND = Creatinine result is less than the linearity of the assay 0.2 mg/dL.
3.6. TLR4 drives the metabolic alterations in GW-chemical exposed liver
TLR4 induced downstream proinflammatory signaling has been found to aid in insulin resistance (Razolli et al., 2015). Prolonged insulin resistance has been shown to cause metabolic disturbances in the liver, skeletal muscle and adipose tissue (Boutagy et al., 2016). Since microbiome associated gut leaching and systemic endotoxemia were reported in the mouse model of GWI and the present study found metabolic changes in the liver, we studied the direct role of TLR4 in causing the metabolic changes. The results showed that TLR4 knockout (TLR4 KO) mice had decreased TLR5 expression in the liver when compared to GW-chemical exposed group (Fig. 8A and B) (p < 0.05). TLR4 KO mice had significantly decreased expression of Class I glucose transporter GLUT-4, PFK, PPAR-α, and lipogenesis mediator SREBP1c when compared to GW-chemical exposed group while GLUT-1 showed no change in the expression suggesting GLUT-1 might not be regulated by TLR4 (Fig. 8C) (p < 0.05). The results suggested that TLR4 activation following systemic endotoxemia might be responsible for the ectopic metabolic alterations in the liver but is unable to present any significant changes in liver disease phenotype. The results also are in agreement with epidemiological studies where veterans deployed in GW don’t report liver abnormalities in the clinics based on the typical symptoms.
Fig. 8.
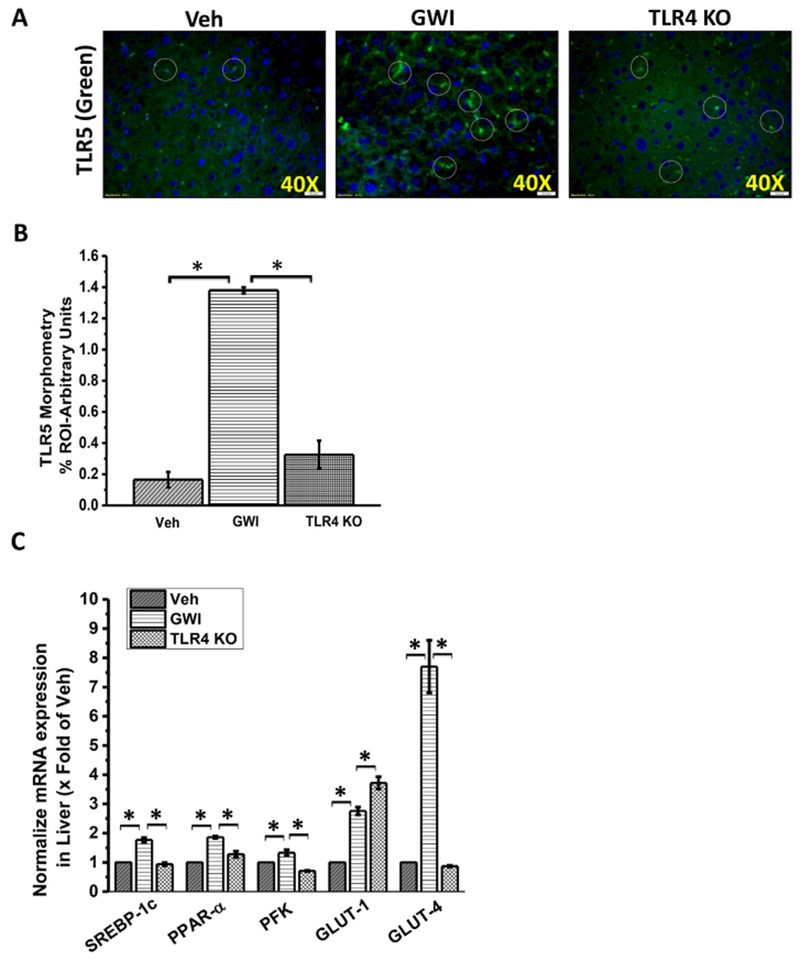
TLR4 drives the metabolic alterations in GW-chemical exposed liver. A. Tissue levels of TLR5 in Gulf War chemical treated a group of wild-type mice (GWI, n = 3) and a group of TLR4 knockout mice (TLR4 KO, n = 3). Mouse liver samples as observed by immunofluorescence microscopy after labeling the TLR5 protein with green fluorescent secondary antibody and nuclear counterstaining by DAPI (blue). Images were taken at 60× (oil) magnification. B. The bar diagram showed the quantitative morphometric analysis of fluorescence intensities of TLR5 immunoreactivity in the liver tissue. *(p < 0.05). C. mRNA expression analysis of principle carbohydrate metabolic markers (PFK, GLUT-1, and GLUT-4) and fat metabolic markers (SREBP1c, PPAR-α) in the liver tissue of vehicle, GWI and TLR4 KO mice. Normalized mRNA expression is represented as a fold change of vehicle control (veh) on Y-axis. Data points represented with Mean ± SEM *(p < 0.05). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
Epidemiological studies have shown a strong correlation between GW toxicant exposures and cognitive/neurological complications but there are also reports of chronic fatigue, gastrointestinal disturbances and occasional cases of metabolic syndrome (White et al., 2016). Our study shows that microbial dysbiosis owing to GW-chemical exposure causes a significant decrease in healthy gut bacteria like Bifidobacterium and Lactobacillus (LeBlanc et al., 2017). Interestingly, they are a class of bacteria that generate butyrate in the gut (LeBlanc et al., 2017). Recent studies have shown a beneficial effect of butyrate in preclinical studies involving colitis and IBD (Sun et al., 2017). The above results prompted us to use sodium butyrate administration through an oral route as a priming agent throughout the chemical exposure time so that a restored butyrate in the gut could prevent and prime the gut against the dysbiosis, inflammatory leaching, and generation of systemic mediators in the small intestine. Results also showed the role of butyrate in increasing the levels of the butyrogenic bacteria, increasing the expression of butyrate receptor GPR109A, decreasing Claudin- 2 and decreasing TLR4 activation. We have shown recently that GW chemical exposure causes gut dysbiosis, the disintegration of gut membrane causing leaching and systemic endotoxemia (Alhasson et al., 2017) that eventually led to TLR4 activation. We also showed a causal role of dysbiosis to the neuroinflammation in frontal cortex thus raising a possibility of the existence of a “Gut-Brain-Axis” in GWI similar to other pathological conditions (Alhasson et al., 2017). This axis may act in parallel to some of the direct toxic effects of GW chemical exposures on the brain tissue (O’Callaghan et al., 2015; Zakirova et al., 2015). Sodium Butyrate priming, as shown in our data might reverse the pathology associated with GW chemical exposure since it restored gut health, reversed gut barrier integrity and decreased SI inflammation (decreased IL-1β) while increasing the possibility of increased butyrate binding to GPR109A due to higher availability of this receptor in SI. Notably, butyrate priming also decreased the release of HMGB1 and leptin though slightly in circulation albeit from the intestinal epithelial cells but other sources like liver cannot be ruled out. The source might be the damaged epithelial cells in the small intestine since the potential generation of free radical species has been shown before and oxidative stress in the intestinal epithelial cells and macrophages could release HMGB1 (Tang et al., 2012; Sappington et al., 2002). On the other hand, HMGB1 release due to gut integrity changes also causes oxidative stress and cell necrosis as have been reported in other studies (Sappington et al., 2002). Though leptin is primarily released from adipocytes and liver, chemical/food-induced leptin release have been shown in the gut and has been traced in duodenal juice (Sobhani et al., 2000; Guilmeau et al., 2003). Thus our finding of increased leptin in circulation following GW chemical exposure might be a result of the leaky gut or liver though the exact source remains to be determined at this time. The release of both leptin and HMGB1 and its modulation by butyrate priming in the gut points to the intestine as a source of these inflammatory mediators along with endotoxin and has tremendous implication determining ectopic/endocrine pathology of GWI.
In spite of well-coordinated symptom reporting in GWI about chronic fatigue in most of the studies, the causes of such chronic fatigue have been limited to abnormalities in neurological pathways or mitochondrial dysfunction without an organ-specific definition (Koslik et al., 2014). Presence of symptoms related to metabolic syndrome or liver diseases is rare (Kelsall et al., 2009). Interestingly, fatigue is also associated with metabolic syndrome and various liver diseases (Carey et al., 2015; Sweet et al., 2017; Jensen et al., 2017). Though we hardly see literature reporting liver abnormalities in GWI, asymptomatic metabolic abnormalities in the liver (as evident in silent liver diseases like NAFLD), can contribute to chronic fatigue. These facts mentioned above led us to examine the liver pathology likely affected by higher circulatory mediators like endotoxin, leptin, and HMGB1. Owing to the tremendous role of the liver in carbohydrate metabolism, we focused on the role of circulatory HMGB1 and leptin on (a) hepatic TLR4 activation and (b) alterations in both lipid and carbohydrate metabolism. Our results showed a significant increase in TLR4 trafficking to the lipid rafts, a hallmark of activation of the TLR4 pathway in the liver following exposure to GW-chemicals. Also, there was a subsequent increase in TLR5 levels in the liver with both TLR4 activation and TLR5 levels showing decreases after butyrate priming. These results assume huge significance since TLR4 activation has been found to cause insulin resistance, uptake of free fatty acids for triglyceride production in macrophages and sterol biosynthesis (Boutagy et al., 2016; Feingold et al., 2012). On the other hand, stearic acid has been shown to promote TLR4 mediated inflammation (Song et al., 2006; Anderson et al., 2012). Our results of increased expression of genes such as SREBP1c and PPAR-α which play a major role in liver lipogenesis (cholesterol biosynthesis and import) might be the result of the increased TLR4 activation and the subsequent cascade of events that alter liver metabolism following GW-chemical exposure. Interestingly, both SREBP1c and LXRs control lipid metabolism and it remains to be seen whether an increase in SREBP1c in the GW-chemical exposed liver was an adaptive way to suppress a chronic TLR4 activation thus mounting an anti-inflammatory response as is seen in some studies (Iizuka & Horikawa, 2008; Oishi et al., 2017). There are numerous reports which find increased glucose metabolism following NF-kB activity which is downstream of TLR4 activation (Mauro et al., 2011). Our results of increased expression of phosphofructokinase, a rate-limiting enzyme for glycolysis show that a TLR4 mediated mechanism might play a role in driving a glycolytic pathway in the liver. Notably, isolated hepatocytes when stimulated with TLR4 ligand LPS or GW-chemical Permethrin did not show an increase in PFK but exhibited a 3-fold increase in SREBP1c over vehicle control suggesting that hepatocytes along with macrophages may be targets of TLR4 activation thus playing a vital role in the reprogramming of lipid metabolism. Further, Class I glucose transporters GLUT-1 and GLUT-4 was elevated in the GW-chemical exposed liver. The result assumes significance since inflammation in the liver has been shown to increase glucose uptake in hepatic stellate cells in a mouse model of fatty liver disease (Chandrashekaran et al., 2016). Our results of decreased inflammation and subsequent metabolic disturbances in the liver following butyrate priming may shed some light on the inhibitory role of butyrate on histone deacetylase activity (Khan & Jena, 2014). Butyrate is a known HDAC inhibitor (Khan & Jena, 2015). Studies show that butyrate can act as an HDAC inhibitor and decrease NFκb activity (Segain, 2000). We found a decrease in NFxb activity following butyrate administration but was found to be insignificant (data not shown). Also, butyrate can act independently of TL4 activation by inhibiting HDACs (Chang et al., 2014). Future studies should target the extensive role of butyrate in HDAC inhibiton in Gulf War Illness that may be independent of TLR4 activation. Importantly, the results of altered expressions of the metabolic genes failed to induce any histological changes that support inflammatory or metabolic liver disease following GW-chemical exposure. This is of high significance since liver diseases take years to manifest and most remain asymptomatic (silent) thus evading most clinical observations. It remains to be seen whether the unavailability of reports related to liver complications in GWI is due to the silent nature of the manifestations that are only limited to changes in the expressions of metabolic genes and would perhaps take years to show any phenotypic disease. Finally, our studies with TLR4 gene-deficient mice exposed to GW chemicals reversed the levels of TLR5 and expressions for SREBP1c, PPAR-α, PFK and GLUT-4 emphasizing the fact that TLR4 activation was indeed responsible for the metabolic reprogramming in the liver. Further, the reversal of TLR4-inducible systemic release of DAMPS and metabolic changes in the liver bodes well for a potential use of this compound for a gut-targeted therapy in GWI veterans.
In summary, we show that GW-chemical exposure in mice and subsequent systemic inflammation following a dysbiosis in the gut could cause significant changes in the way the liver metabolizes lipid and carbohydrate with no detectable pathology while butyrate resists those changes. The study will help us advance our efforts to scrutinize clinical symptom reporting in the liver and re-evaluate the way we approach the therapeutic aspect of GWI by targeting multiple physiological pathways. Uses of short-chain fatty acids or probiotics can help in such pursuits.
Acknowledgement
The authors gratefully acknowledge the technical services of Benny Davidson at the IRF, University of South Carolina School of Medicine and AML Labs (Baltimore MD), and University of Georgia college of veterinary medicine for support in clinical blood chemistry test. We also thank the Instrumentation resource facility (IRF) at the University of South Carolina for equipment usage and consulting services.
Grant support
This work was supported by a pilot funding received from the Gulf War Illness Research Consortium to Saurabh Chatterjee (Parent DOD grant # W81XWH-13-2-0072, PI: Dr. Kimberly Sullivan) and P01AT003961 (Project 4) to Saurabh Chatterjee, P01AT003961, P20GM103641, R01AT006888, R01ES019313, R01MH094755 and VA Merit Award BX001357 to Mitzi Nagarkatti and Prakash S. Nagarkatti.
Footnotes
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Abdullah L, Evans JE, Joshi U, Crynen G, Reed J, Mouzon B, et al. , 2016. Translational potential of long-term decreases in mitochondrial lipids in a mouse model of Gulf War Illness. Toxicology 372, 22–33. [DOI] [PubMed] [Google Scholar]
- Alhasson F, Das S, Seth R, Dattaroy D, Chandrashekaran V, Ryan CN, et al. , 2017. Altered gut microbiome in a mouse model of Gulf War illness causes neuroinflammation and intestinal injury via leaky gut and TLR4 activation. PLoS One 12, e0172914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EK, Hill AA, Hasty AH, 2012. Stearic acid accumulation in macrophages induces toll-like receptor 4/2-independent inflammation leading to endoplasmic reticulum stress-mediated apoptosis. Arterioscler. Thromb. Vase. Biol 32, 1687–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E, 2007. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab 5, 426–437. [DOI] [PubMed] [Google Scholar]
- Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A, 2012. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol 56, 952–964. [DOI] [PubMed] [Google Scholar]
- Boutagy NE, McMillan RP, Frisard MI, Hulver MW, 2016. Metabolic endotoxemia with obesity: is it real and is it relevant? Biochimie 124, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey EJ, Ali AH, Lindor KD, 2015. Primary biliary cirrhosis. Lancet 386, 1565–1575. [DOI] [PubMed] [Google Scholar]
- Chandrashekaran V, Das S, Seth RK, Dattaroy D, Alhasson F, Michelotti G, et al. , 2016. Purinergic receptor X7 mediates leptin induced GLUT4 function in stellate cells in nonalcoholic steatohepatitis. Biochim. Biophys. Acta 1862, 32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrashekaran V, Seth RK, Dattaroy D, Alhasson F, Ziolenka J, Carson J, et al. , 2017. HMGB1-RAGE pathway drives peroxynitrite signaling-induced IBD-like inflammation in murine nonalcoholic fatty liver disease. Redox Biol 13, 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PV, Hao L, Offermanns S, Medzhitov R, 2014. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. U. S. A. Ill, 2247–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Ganini D, Tokar EJ, Kumar A, Das S, Corbett J, et al. , 2013. Leptin is key to peroxynitrite-mediated oxidative stress and Kupffer cell activation in experimental non-alcoholic steatohepatitis. J. Hepatol 58, 778–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Choi SS, Michelotti GA, Chan IS, Swiderska-Syn M, Karaca GF, et al. , 2012. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 143 (1319–29 e1–11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CC, Ching YH, Li YP, Liu JY, Huang YT, Huang YW, et al. , 2017. Nonalcoholic fatty liver disease is exacerbated in high-fat diet-fed Gnotobiotic mice by colonization with the gut microbiota from patients with nonalcoholic Steatohepatitis. Nutrients 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coker WJ, 1996. A review of Gulf War illness. J. R. Nav. Med. Serv 82, 141–146. [PubMed] [Google Scholar]
- Gulf war and health In: Cory-Slechta D, Wedge R (Eds.), Update of Health Effects of Serving in the Gulf War, 2016. vol. 10 National Academy of Sciences, Washington DC. [Google Scholar]
- Coughlin SS, Kang HK, Mahan CM, 2011. Selected health conditions among overweight, obese, and non-obese veterans of the 1991 Gulf War: results from a survey conducted in 2003–2005. Open Epidemiol. J 4, 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doycheva I, Watt KD, Rifai G, Abou Mrad R, Lopez R, Zein NN, et al. , 2017. Increasing burden of chronic liver disease among adolescents and young adults in the USA: a silent epidemic. Dig. Dis. Sci 62, 1373–1380. [DOI] [PubMed] [Google Scholar]
- Feingold KR, Shigenaga JK, Kazemi MR, McDonald CM, Patzek SM, Cross AS, et al. , 2012. Mechanisms of triglyceride accumulation in activated macrophages. J. Leukoc. Biol 92, 829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Martinez I, Shaker ME, Mehal WZ, 2015. The rapeutic opportunities in damage-associated molecular pattern-driven metabolic diseases. Antioxid. Redox Signal 23, 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geidl-Flueck B, Gerber PA, 2017. Insights into the hexose liver metabolism-glucose versus fructose. Nutrients 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgopoulos AP, James LM, Carpenter AF, Engdahl BE, Leuthold AC, Lewis SM, 2017. Gulf War illness (GWI) as a neuroimmune disease. Exp. Brain Res 235, 3217–3225. [DOI] [PubMed] [Google Scholar]
- Giloteaux L, Goodrich JK, Walters WA, Levine SM, Ley RE, Hanson MR, 2016. Reduced diversity and altered composition of the gut microbiome in individuals with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome 4, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilmeau S, Buyse M, Tsocas A, Laigneau JP, Bado A, 2003. Duodenal leptin stimulates cholecystokinin secretion: evidence of a positive leptin-cholecystokinin feedback loop. Diabetes 52, 1664–1672. [DOI] [PubMed] [Google Scholar]
- Haley RW, Charuvastra E, Shell WE, Buhner DM, Marshall WW, Biggs MM, et al. , 2013. Cholinergic autonomic dysfunction in veterans with Gulf War illness: confirmation in a population-based sample. JAMA Neurol 70, 191–200. [DOI] [PubMed] [Google Scholar]
- Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. , 2012. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka K, Horikawa Y, 2008. ChREBP: a glucose-activated transcription factor involved in the development of metabolic syndrome. Endocr. J 55, 617–624. [DOI] [PubMed] [Google Scholar]
- Imhann F, Vich Vila A, Bonder MJ, Fu J, Gevers D, Visschedijk MC, et al. , 2018. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 67, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen O, Bernklev T, Jelsness-Jorgensen LP, 2017. Fatigue in type 1 diabetes: a systematic review of observational studies. Diabetes Res. Clin. Pract 123, 63–74. [DOI] [PubMed] [Google Scholar]
- Kaltsas G, Vgontzas A, Chrousos G, 2010. Fatigue, endocrinopathies, and metabolic disorders. PM & R 2, 393–398. [DOI] [PubMed] [Google Scholar]
- Karim S, Adams DH, Lalor PF, 2012. Hepatic expression and cellular distribution of the glucose transporter family. World J. Gastroenterol 18, 6771–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsall HL, McKenzie DP, Sim MR, Leder K, Forbes AB, Dwyer T, 2009. Physical, psychological, and functional comorbidities of multisymptom illness in Australian male veterans of the 1991 Gulf War. Am. J. Epidemiol 170, 1048–1056. [DOI] [PubMed] [Google Scholar]
- Kerr KJ, 2015. Gulf War illness: an overview of events, most prevalent health outcomes, exposures, and clues as to pathogenesis. Rev. Environ. Health 30, 273–286. [DOI] [PubMed] [Google Scholar]
- Khan S, Jena G, 2014. Sodium butyrate, a HDAC inhibitor ameliorates eNOS, iNOS and TGF-betal-induced fibrogenesis, apoptosis and DNA damage in the kidney of juvenile diabetic rats. Food Chem. Toxicol 73, 127–139. [DOI] [PubMed] [Google Scholar]
- Khan S, Jena G, 2015. The role of butyrate, a histone deacetylase inhibitor in diabetes mellitus: experimental evidence for therapeutic intervention. Epigenomics 7, 669–680. [DOI] [PubMed] [Google Scholar]
- Koo BB, Michalovicz LT, Calderazzo S, Kelly KA, Sullivan K, Killiany RJ, et al. , 2018. Corticosterone potentiates DFP-induced neuroinflammation and affects high-order diffusion imaging in a rat model of Gulf War Illness. Brain Behav. Immun 67, 42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koslik HJ, Hamilton G, Golomb BA, 2014. Mitochondrial dysfunction in Gulf War illness revealed by 31 Phosphorus magnetic resonance spectroscopy: a case-control study. PLoS One 9, e92887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc JG, Chain F, Martin R, Bermudez-Humaran LG, Courau S, Langella P, 2017. Beneficial effects on host energy metabolism of short-chain fatty acids and vitamins produced by commensal and probiotic bacteria. Microb. Cell Factories 16, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luettig J, Rosenthal R, Barmeyer C, Schulzke JD, 2015. Claudin-2 as a mediator of leaky gut barrier during intestinal inflammation. Tissue Barriers 3, e977176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra F, Svegliati-Baroni G, 2018. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol 68 (2), 280 10.1016/j.jhep.2017.11.014 (Epub 2017 Nov 14. Review, PMID: 29154964). [DOI] [PubMed] [Google Scholar]
- Mauro C, Leow SC, Anso E, Rocha S, Thotakura AK, Tornatore L, et al. , 2011. NF-kappaB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol 13, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milic S, Stimac D, 2012. Nonalcoholic fatty liver disease/steatohepatitis: epidemiology, pathogenesis, clinical presentation and treatment. Dig. Dis 30, 158–162. [DOI] [PubMed] [Google Scholar]
- Morris G, Berk M, Carvalho AF, Caso JR, Sanz Y, Maes M, 2016. The role of microbiota and intestinal permeability in the pathophysiology of autoimmune and Neuroimmune processes with an emphasis on inflammatory bowel disease type 1 diabetes and chronic fatigue syndrome. Curr. Pharm. Des 22, 6058–6075. [DOI] [PubMed] [Google Scholar]
- Nagy-Szakal D, Williams BL, Mishra N, Che X, Lee B, Bateman L, et al. , 2017. Fecal metagenomic profiles in subgroups of patients with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome 5, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naviaux RK, Naviaux JC, Li K, Bright AT, Alaynick WA, Wang L, et al. , 2016. Metabolic features of chronic fatigue syndrome. Proc. Natl. Acad. Sci. U. S. A 113, E5472–E5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan JP, Kelly KA, Locker AR, Miller DB, Lasley SM, 2015. Corticosterone primes the neuroinflammatory response to DFP in mice: potential animal model of Gulf War Illness. J. Neurochem 133, 708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan JP, Michalovicz LT, Kelly KA, 2016. Supporting a Neuroimmune basis of Gulf War Illness. EBioMedicine 13, 5–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi Y, Spann NJ, Link VM, Muse ED, Strid T, Edillor C, et al. , 2017. SREBP1 contributes to resolution of pro-inflammatory TLR4 signaling by reprogramming fatty acid metabolism. Cell Metab 25, 412–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parihar VK, Hattiangady B, Shuai B, Shetty AK, 2013. Mood and memory deficits in a model of Gulf War Illness are linked with reduced neurogenesis, partial neuron loss, and mild inflammation in the hippocampus. Neuropsychopharmacology 38, 2348–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razolli DS, Moraes JC, Morari J, Moura RF, Vinolo MA, Velloso LA, 2015. TLR4 expression in bone marrow-derived cells is both necessary and sufficient to produce the insulin resistance phenotype in diet-induced obesity. Endocrinology 156, 103–113. [DOI] [PubMed] [Google Scholar]
- Reichardt F, Chassaing B, Nezami BG, Li G, Tabatabavakili S, Mwangi S, et al. , 2017. Western diet induces colonic nitrergic myenteric neuropathy and dysmotility in mice via saturated fatty acid- and lipopolysaccharide-induced TLR4 signalling. J. Physiol 595, 1831–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sappington PL, Yang R, Yang H, Tracey KJ, Delude RL, Fink MP, 2002. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology 123, 790–802. [DOI] [PubMed] [Google Scholar]
- Schreuder TC, Verwer BJ, van Nieuwkerk CM, Mulder CJ, 2008. Nonalcoholic fatty liver disease: an overview of current insights in pathogenesis, diagnosis and treatment. World J. Gastroenterol 14, 2474–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segain JP, Raingeard de la Bletiere D, Bourreille A, Leray V, Gervois N, Rosales C, et al. , 2000. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease. Gut 47, 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekhavat A, Sun JM, Davie JR, 2007. Competitive inhibition of histone deacetylase activity by trichostatin a and butyrate. Biochem. Cell Biol 85, 751–758. [DOI] [PubMed] [Google Scholar]
- Seth RK, Kumar A, Das S, Kadiiska MB, Michelotti G, Diehl AM, et al. , 2013a. Environmental toxin-linked nonalcoholic steatohepatitis and hepatic metabolic reprogramming in obese mice. Toxicol. Sci 134, 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RK, Kumar A, Das S, Kadiiska MB, Michelotti G, Diehl AM, et al. , 2013b. Environmental toxin-linked nonalcoholic steatohepatitis and hepatic metabolic reprogramming in obese mice. Toxicol. Sci 134 (2), 291–303. 10.1093/toxsciAftl04 Epub 2013 May 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. , 2014. Activation of Gprl09a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40, 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhani I, Bado A, Vissuzaine C, Buyse M, Kermorgant S, Laigneau JP, et al. , 2000. Leptin secretion and leptin receptor in the human stomach. Gut 47, 178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MJ, Kim KH, Yoon JM, Kim JB, 2006. Activation of toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem. Biophys. Res. Commun 346, 739–745. [DOI] [PubMed] [Google Scholar]
- Sullivan K, Krengel M, Bradford W, Stone C, Thompson TA, Heeren T, et al. , 2018. Neuropsychological functioning in military pesticide applicators from the Gulf War: effects on information processing speed, attention and visual memory. Neurotoxicol. Teratol 65, 1–13. [DOI] [PubMed] [Google Scholar]
- Sun M, Wu W, Liu Z, Cong Y, 2017. Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J. Gastroenterol 52, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet PH, Khoo T, Nguyen S, 2017. Nonalcoholic fatty liver disease. Prim. Care 44, 599–607. [DOI] [PubMed] [Google Scholar]
- Tang Y, Chen A, 2010. Curcumin prevents leptin raising glucose levels in hepatic stellate cells by blocking translocation of glucose transporter-4 and increasing glucokinase. Br. J. Pharmacol 161, 1137–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT, 2012. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol. Rev 249, 158–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RF, Steele L, O’Callaghan JP, Sullivan K, Binns JH, Golomb BA, et al. , 2016. Recent research on gulf war illness and other health problems in veterans of the 1991 gulf war: effects of toxicant exposures during deployment. Cortex 74, 449–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharia K, Tabibian A, Lindor KD, Tabibian JH, 2018. Complications, symptoms, quality of life and pregnancy in cholestatic liver disease. Liver Int 38 (3), 399–411. http://dx.doi.org/10.llll/liv.13591. Epub 2017 Oct 3. [DOI] [PubMed] [Google Scholar]
- Zakirova Z, Crynen G, Hassan S, Abdullah L, Horne L, Mathura V, et al. , 2015. A chronic longitudinal characterization of neurobehavioral and Neuropathological cognitive impairment in a mouse model of gulf war agent exposure. Front. Integr. Neurosci 9, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhou Q, Dorfman RG, Huang X, Fan T, Zhang H, et al. , 2016. Butyrate inhibits interleukin-17 and generates Tregs to ameliorate colorectal colitis in rats. BMC Gastroenterol 16, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Kelly CJ, Battista KD, Schaefer R, Lanis JM, Alexeev EE, et al. , 2017. Microbial-derived butyrate promotes epithelial barrier function through IL-10 receptor-dependent repression of Claudin-2. J. Immunol 199, 2976–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]